Dual Specificity Phosphatase 6 Protects Neural Stem Cells from β-Amyloid-Induced Cytotoxicity through ERK1/2 Inactivation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
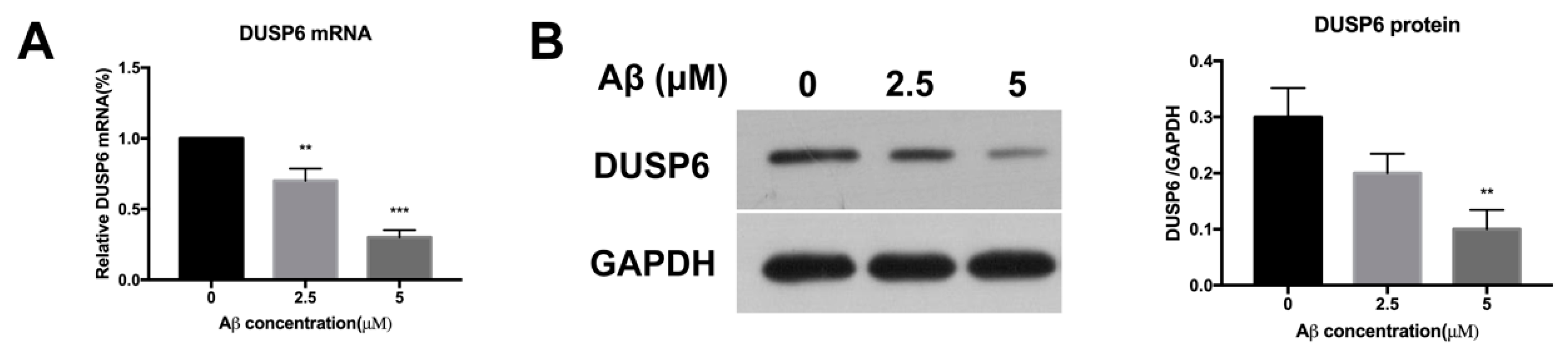
2.1. β-amyloid Treatment Reduced DUSP6 Expression at Different Concentrations
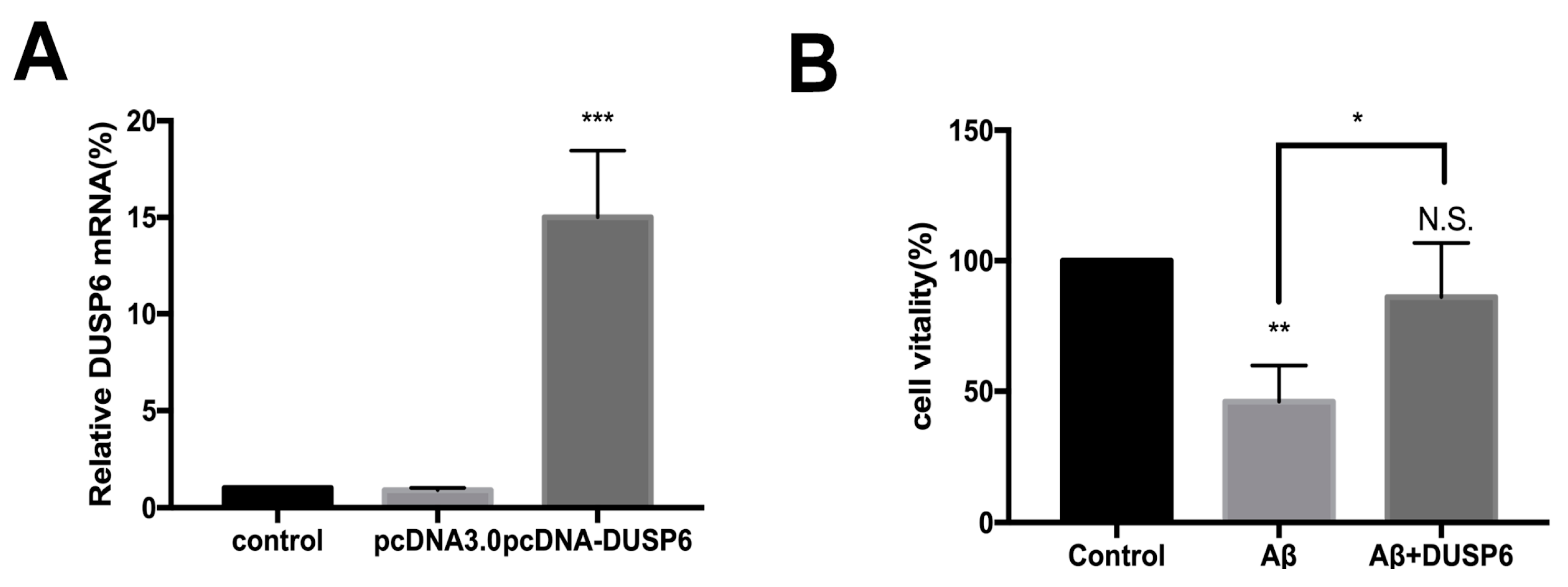
2.2. DUSP6 Prevented the beta-amyloid25–35-Induced Decrease in Neural Stem Cell Viability
2.3. DUSP6 Restored Reduced Intracellular Reactive Oxigen Species and Malondialdehyde Level in Neural Stem Cells After beta amyloid25–35 Treatment
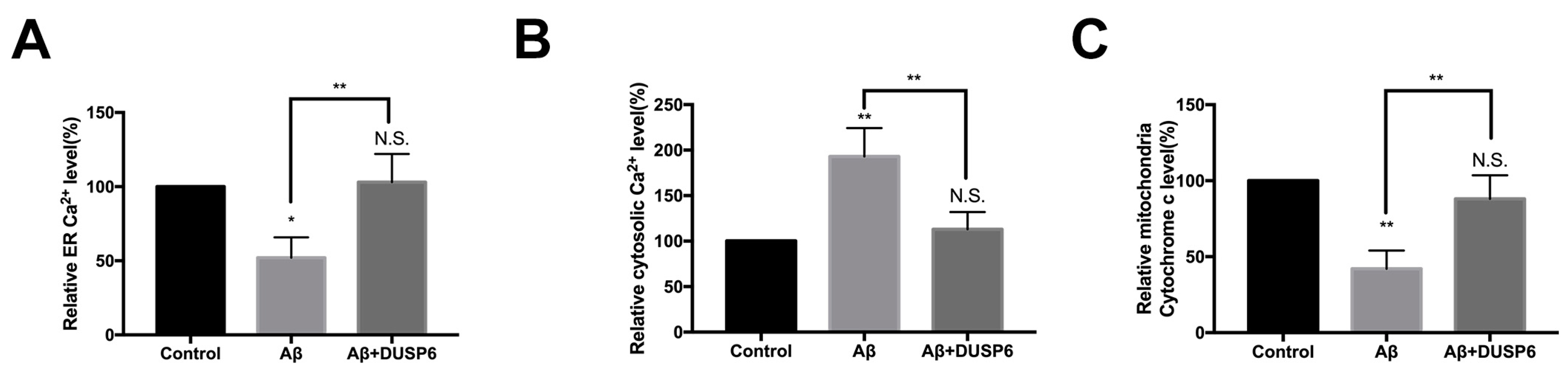
2.4. DUSP6 Reversed Aβ-Induced Effect on ER Calcium and Mitochondrial Cytochrome c Homeostasis in Neural Stem Cells
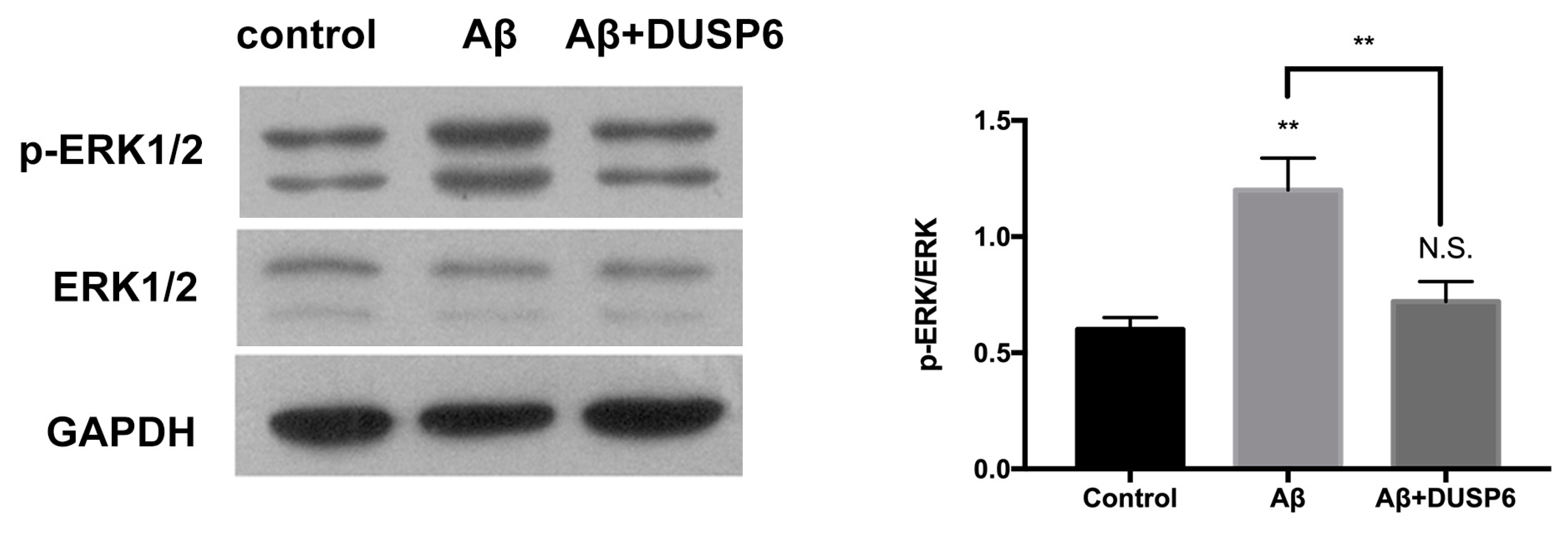
2.5. DUSP6 Regulated ERK1/2 Activation in Aβ25–35-Exposed Neural Stem Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment
4.2. CCK-8 Assay for Cell Viability
4.3. Cell Transfection
4.4. Quantitative Real-Time PCR (RT-PCR)
4.5. Measurement of Oxidative Stress
4.6. Detection of Ca2+ Levels
4.7. Enzyme-Linked Immunosorbent Assay (ELISA)
4.8. Western Blot Analysis
4.9. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cavedo, E.; Chiesa, P.A.; Houot, M.; Ferretti, M.T.; Grothe, M.J.; Teipel, S.J.; Lista, S.; Habert, M.O.; Potier, M.C.; Dubois, B.; et al. Sex differences in functional and molecular neuroimaging biomarkers of Alzheimer’s disease in cognitively normal older adults with subjective memory complaints. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2018, 14, 1204–1215. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Lee, G.; Mortsdorf, T.; Ritter, A.; Zhong, K. Alzheimer’s disease drug development pipeline: 2017. Alzheimer’s Dement. 2017, 3, 367–384. [Google Scholar] [CrossRef] [PubMed]
- Amemori, T.; Jendelova, P.; Ruzicka, J.; Urdzikova, L.M.; Sykova, E. Alzheimer’s disease: Mechanism and approach to cell therapy. Int. J. Mol. Sci. 2015, 16, 26417–26451. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Bylykbashi, E.; Chatila, Z.K.; Lee, S.W.; Pulli, B.; Clemenson, G.D.; Kim, E.; Rompala, A.; Oram, M.K.; Asselin, C.; et al. Combined adult neurogenesis and BDNF mimic exercise effects on cognition in an Alzheimer’s mouse model. Science 2018, 361, eaan8821. [Google Scholar] [CrossRef] [PubMed]
- Tournier, B.B.; Tsartsalis, S.; Rigaud, D.; Fossey, C.; Cailly, T.; Fabis, F.; Pham, T.; Gregoire, M.C.; Kovari, E.; Moulin-Sallanon, M.; et al. TSPO and amyloid deposits in sub-regions of the hippocampus in the 3xTgAD mouse model of Alzheimer’s disease. Neurobiol. Disease 2018, 121, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Voit, R.; Seiler, J.; Grummt, I. Cooperative action of Cdk1/cyclin B and SIRT1 is required for mitotic repression of rRNA synthesis. PLoS Geneti. 2015, 11, e1005246. [Google Scholar] [CrossRef] [PubMed]
- El-Magd, M.A.; Khalifa, S.F.; Fa, A.A.; Badawy, A.A.; El-Shetry, E.S.; Dawood, L.M.; Alruwaili, M.M.; Alrawaili, H.A.; Risha, E.F.; El-Taweel, F.M.; et al. Incensole acetate prevents β-amyloid-induced neurotoxicity in human olfactory bulb neural stem cells. Biomed. Pharmacother. 2018, 105, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Liao, W.; Huang, Y.; Jiang, M.; Chen, J.; Wang, M.; Lin, H.; Guan, S.; Liu, J. Neuroprotective effect of dual specificity phosphatase 6 against glutamate-induced cytotoxicity in mouse hippocampal neurons. Biomed. Pharmacother. 2017, 91, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Banzhaf-Strathmann, J.; Benito, E.; May, S.; Arzberger, T.; Tahirovic, S.; Kretzschmar, H.; Fischer, A.; Edbauer, D. MicroRNA-125b induces tau hyperphosphorylation and cognitive deficits in Alzheimer’s disease. EMBO J. 2014, 33, 1667–1680. [Google Scholar] [CrossRef] [PubMed]
- Bhore, N.; Wang, B.J.; Chen, Y.W.; Liao, Y.F. Critical roles of dual-specificity phosphatases in neuronal proteostasis and neurological diseases. Int. J. Mol. Sci. 2017, 18, 1963. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S. Progression of Alzheimer’s disease, tau propagation, and its modifiable risk factors. Neurosci. Res. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, S.; Guillemin, G.J.; Abiramasundari, R.S.; Essa, M.M.; Akbar, M.; Akbar, M.D. The Role of reactive oxygen species in the pathogenesis of Alzheimer’s Disease, Parkinson’s Disease, and Huntington’s Disease: A Mini Review. Oxid. Med. Cell. Longev. 2016, 2016, 8590578. [Google Scholar] [CrossRef] [PubMed]
- Van Giau, V.; An, S.S.A.; Hulme, J.P. Mitochondrial therapeutic interventions in Alzheimer’s disease. J. Neurol. Sci. 2018, 395, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Saido, T.C. Critical review: Involvement of endoplasmic reticulum stress in the Aetiology of Alzheimer’s disease. Open Biol. 2018, 8, 180024. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Sun, Q.; Chen, S. Oxidative stress: A major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson’s disease and Alzheimer’s disease. Prog. Neurobiol. 2016, 147, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Zeeshan, H.M.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic reticulum stress and associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Y.; Du, L.; Xu, C.; Cao, J.; Wang, Q.; Liu, Q.; Fan, F. Radiation-induced cytochrome c release and the neuroprotective effects of the pan-caspase inhibitor z-VAD-fmk in the hypoglossal nucleus. Exp. Ther. Med. 2014, 7, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Arkell, R.S.; Dickinson, R.J.; Squires, M.; Hayat, S.; Keyse, S.M.; Cook, S.J. DUSP6/MKP-3 inactivates ERK1/2 but fails to bind and inactivate ERK5. Cell. Signal. 2008, 20, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2016 Alzheimer’s disease facts and figures. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2016, 12, 459–509. [Google Scholar] [CrossRef]
- Morris, G.P.; Clark, I.A.; Vissel, B. Questions concerning the role of amyloid-β in the definition, aetiology and diagnosis of Alzheimer’s disease. Acta Neuropathol. 2018, 136, 663–689. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.L.; Li, W.W.; Zhu, C.; Jin, W.S.; Zeng, F.; Liu, Y.H.; Bu, X.L.; Zhu, J.; Yao, X.Q.; Wang, Y.J. Clinical research on Alzheimer’s Disease: Progress and perspectives. Neurosci. Bull. 2018, 34, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Sugaya, K.; Vaidya, M. Stem cell therapies for neurodegenerative diseases. Adv. Exp. Med. Biol. 2018, 1056, 61–84. [Google Scholar] [PubMed]
- Berry, B.J.; Smith, A.S.T.; Young, J.E.; Mack, D.L. Advances and current challenges associated with the use of human induced pluripotent stem cells in modeling neurodegenerative disease. Cells Tissues Organs 2018, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, T.; Oda, Y.; Horiuchi, J.; Yin, J.C.; Morimoto, T.; Saitoe, M. Mg2+ block of Drosophila NMDA receptors is required for long-term memory formation and CREB-dependent gene expression. Neuron 2012, 74, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Jiang, M.; Li, M.; Jin, C.; Xiao, S.; Fan, S.; Fang, W.; Zheng, Y.; Liu, J. Magnesium Elevation Promotes Neuronal Differentiation While Suppressing Glial Differentiation of Primary Cultured Adult Mouse Neural Progenitor Cells through ERK/CREB Activation. Front. Neurosci. 2017, 11, 87. [Google Scholar] [CrossRef] [PubMed]
- Hane, F.T.; Lee, B.Y.; Leonenko, Z. Recent progress in Alzheimer’s disease research, Part 1: Pathology. J. Alzheimers Dis. 2017, 57, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Nicol, C.J.B.; Cheng, Y.C.; Chen, S.J.; Yen, C.H.; Huang, R.N.; Chiang, M.C. Rosiglitazone rescues human neural stem cells from amyloid-β induced ER stress via PPARgamma dependent signaling. Exp. Cell Res. 2018, 370, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Huang, L.P.; Li, Y.; Liu, C.; Wang, S.; Meng, W.; Wei, S.; Liu, X.P.; Gong, Y.; Yao, L.H. Neuroprotective effects of cordycepin inhibit Aβ-induced apoptosis in hippocampal neurons. Neurotoxicology 2018, 68, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Zhang, B.; Luan, P.; Gu, B.; Wan, Q.; Huang, X.; Liao, W.; Liu, J. PI3K/AKT/mTOR/p70S6K pathway is involved in Aβ25-35-Induced autophagy. BioMed Res. Int. 2015, 2015, 161020. [Google Scholar] [CrossRef] [PubMed]
- Coronel, R.; Bernabeu-Zornoza, A.; Palmer, C.; Muniz-Moreno, M.; Zambrano, A.; Cano, E.; Liste, I. Role of amyloid precursor protein (APP) and its derivatives in the biology and cell fate specification of neural stem cells. Mol. Neurobiol. 2018, 55, 7107–7117. [Google Scholar] [CrossRef] [PubMed]
- Bernabeu-Zornoza, A.; Coronel, R.; Palmer, C.; Calero, M.; Martinez-Serrano, A.; Cano, E.; Zambrano, A.; Liste, I. Aβ42 peptide promotes proliferation and gliogenesis in human neural stem cells. Mol. Neurobiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, M.K.; Abdollah, N.A.; Shafie, N.H.; Yusof, N.M.; Razak, S.R.A. Dual-specificity phosphatase 6 (DUSP6): A review of its molecular characteristics and clinical relevance in cancer. Cancer Biol. Med. 2018, 15, 14–28. [Google Scholar] [PubMed]
- Wang, Z.; Chen, Y.; Li, X.; Sultana, P.; Yin, M.; Wang, Z. Amyloid-β1-42 dynamically regulates the migration of neural stem/progenitor cells via MAPK-ERK pathway. Chem.-Biol. Interact. 2018, 298, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Harada, T.; Morooka, T.; Ogawa, S.; Nishida, E. ERK induces p35, a neuron-specific activator of Cdk5, through induction of Egr1. Nat. Cell Biol. 2001, 3, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Giusti-Rodriguez, P.; Gao, J.; Graff, J.; Rei, D.; Soda, T.; Tsai, L.H. Synaptic deficits are rescued in the p25/Cdk5 model of neurodegeneration by the reduction of β-secretase (BACE1). J. Neurosci. 2011, 31, 15751–15756. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Rice, A.E.; Denu, J.M. Intramolecular dephosphorylation of ERK by MKP3. Biochemistry 2003, 42, 15197–15207. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Kobayashi, S.; Borczuk, A.C.; Leidner, R.S.; Laframboise, T.; Levine, A.D.; Halmos, B. Dual specificity phosphatase 6 (DUSP6) is an ETS-regulated negative feedback mediator of oncogenic ERK signaling in lung cancer cells. Carcinogenesis 2010, 31, 577–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen Brain Atlas. Available online: http://mouse.brain-map.org/experiment/show/79632277 (accessed on 1 December 2018).
- Jung, S.; Nah, J.; Han, J.; Choi, S.G.; Kim, H.; Park, J.; Pyo, H.K.; Jung, Y.K. Dual-specificity phosphatase 26 (DUSP26) stimulates Aβ42 generation by promoting amyloid precursor protein axonal transport during hypoxia. J. Neurochem. 2016, 137, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mut, J.V.; Aso, E.; Heyn, H.; Matsuda, T.; Bock, C.; Ferrer, I.; Esteller, M. Promoter hypermethylation of the phosphatase DUSP22 mediates PKA-dependent TAU phosphorylation and CREB activation in Alzheimer’s disease. Hippocampus 2014, 24, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lin, J.; Chen, Q.Z.; Zhu, N.; Jiang, D.Q.; Li, M.X.; Wang, Y. Overexpression of mitochondrial Hsp75 protects neural stem cells against microglia-derived soluble factor-induced neurotoxicity by regulating mitochondrial permeability transition pore opening in vitro. Int. J. Mol. Med. 2015, 36, 1487–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes-Xavier, C.E.; Tarrega, C.; Cejudo-Marin, R.; Frijhoff, J.; Sandin, A.; Ostman, A.; Pulido, R. Differential up-regulation of MAP kinase phosphatases MKP3/DUSP6 and DUSP5 by Ets2 and c-Jun converge in the control of the growth arrest versus proliferation response of MCF-7 breast cancer cells to phorbol ester. J. Biol. Chem. 2010, 285, 26417–26430. [Google Scholar] [CrossRef] [PubMed]
- Wolvetang, E.J.; Wilson, T.J.; Sanij, E.; Busciglio, J.; Hatzistavrou, T.; Seth, A.; Hertzog, P.J.; Kola, I. ETS2 overexpression in transgenic models and in Down syndrome predisposes to apoptosis via the p53 pathway. Hum. Mol. Genet. 2003, 12, 247–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Um, J.W.; Kaufman, A.C.; Kostylev, M.; Heiss, J.K.; Stagi, M.; Takahashi, H.; Kerrisk, M.E.; Vortmeyer, A.; Wisniewski, T.; Koleske, A.J.; et al. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer aβ oligomer bound to cellular prion protein. Neuron 2013, 79, 887–902. [Google Scholar] [CrossRef] [PubMed]
- Luan, P.; Zhou, H.H.; Zhang, B.; Liu, A.M.; Yang, L.H.; Weng, X.L.; Tao, E.X.; Liu, J. Basic fibroblast growth factor protects C17.2 cells from radiation-induced injury through ERK1/2. CNS Neurosci. Ther. 2012, 18, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Oyarce, K.; Silva-Alvarez, C.; Ferrada, L.; Martinez, F.; Salazar, K.; Nualart, F. SVCT2 is expressed by cerebellar precursor cells, which differentiate into neurons in response to ascorbic acid. Mol. Neurobiol. 2018, 55, 1136–1149. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Li, J.; Zheng, B.; Hua, L.; Zuo, Z. Enriched environment attenuates surgery-induced impairment of learning, memory, and neurogenesis possibly by preserving BDNF expression. Mol. Neurobiol. 2016, 53, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.Y.; Luan, P.; Huang, S.X.; Xiao, S.H.; Zhao, J.; Zhang, B.; Gu, B.B.; Pi, R.B.; Liu, J. Edaravone protects HT22 neurons from H2O2-induced apoptosis by inhibiting the MAPK signaling pathway. CNS Neurosci. Ther. 2013, 19, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.L.; Zhao, D.Q.; Wang, F.; Li, M.; Fan, S.N.; Liao, W.; Zheng, Y.Q.; Liao, S.W.; Xiao, S.H.; Luan, P.; et al. Neurotropin (R) alleviates hippocampal neuron damage through a HIF-1/MAPK pathway. Cns Neurosci. Ther. 2017, 23, 428–437. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, W.; Zheng, Y.; Fang, W.; Liao, S.; Xiong, Y.; Li, Y.; Xiao, S.; Zhang, X.; Liu, J. Dual Specificity Phosphatase 6 Protects Neural Stem Cells from β-Amyloid-Induced Cytotoxicity through ERK1/2 Inactivation. Biomolecules 2018, 8, 181. https://doi.org/10.3390/biom8040181
Liao W, Zheng Y, Fang W, Liao S, Xiong Y, Li Y, Xiao S, Zhang X, Liu J. Dual Specificity Phosphatase 6 Protects Neural Stem Cells from β-Amyloid-Induced Cytotoxicity through ERK1/2 Inactivation. Biomolecules. 2018; 8(4):181. https://doi.org/10.3390/biom8040181
Chicago/Turabian StyleLiao, Wang, Yuqiu Zheng, Wenli Fang, Shaowei Liao, Ying Xiong, Yi Li, Songhua Xiao, Xingcai Zhang, and Jun Liu. 2018. "Dual Specificity Phosphatase 6 Protects Neural Stem Cells from β-Amyloid-Induced Cytotoxicity through ERK1/2 Inactivation" Biomolecules 8, no. 4: 181. https://doi.org/10.3390/biom8040181