Identification of the Genetic Variation and Gene Exchange between Citrus Trifoliata and Citrus Clementina

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. DNA Isolation and Genome Resequencing
2.3. Detection of Single Nucleotide Polymorphisms and Insertion/Deletion Polymorphism
2.4. Annotation of Single Nucleotide Polymorphisms and Insertion/Deletion Polymorphism
2.5. RNA Sequencing Analyses
2.6. Detection of Mobile Genes between Scions and Rootstocks from Grafted Plants
2.7. Real-Time Polymerase Chain Reaction
3. Results
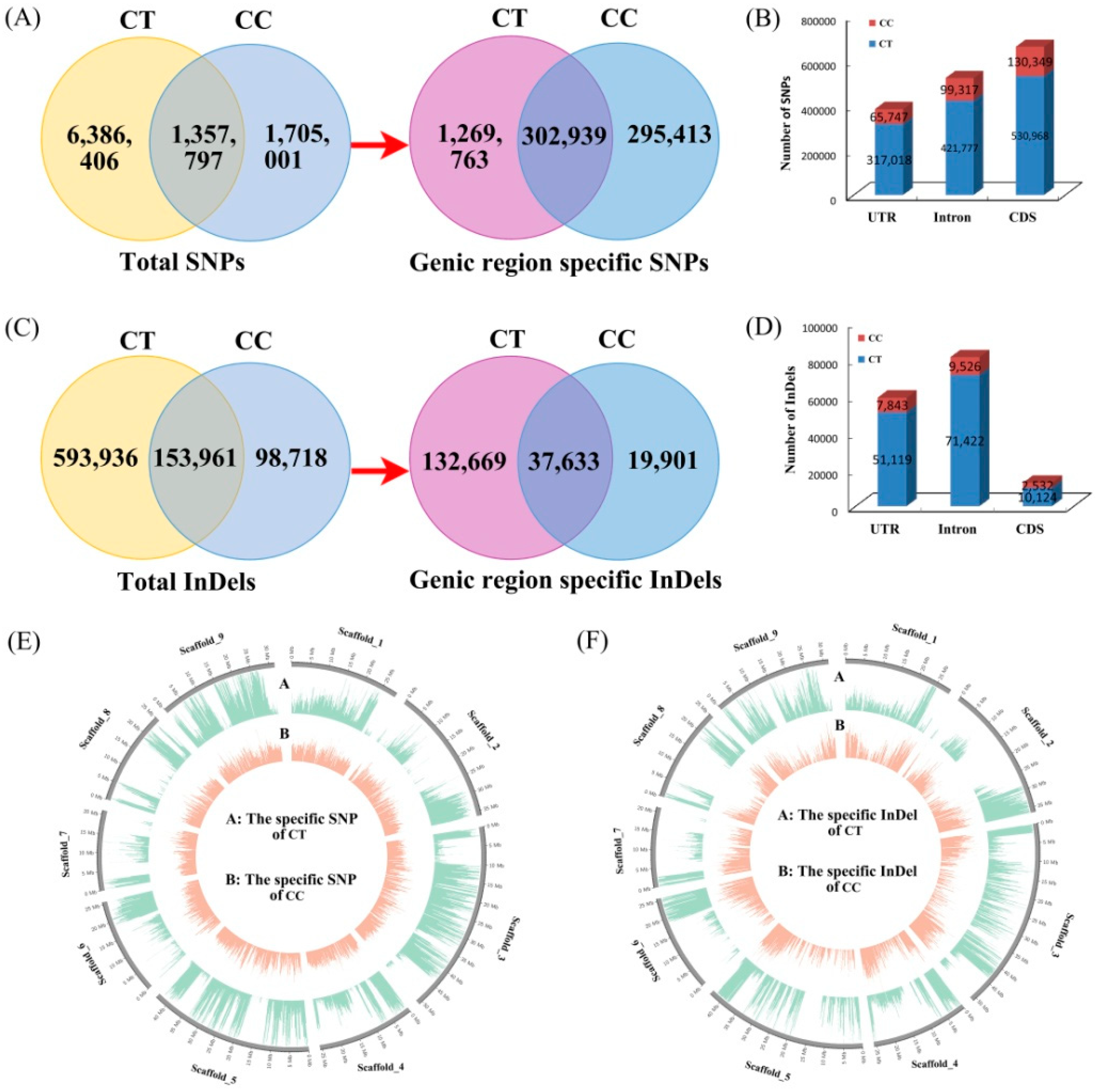
3.1. Genome Resequencing, Detection, and Characteristics of Genetic Variations
3.2. Analysis of Genetic Variation between C. Trifoliata and C. Clementina
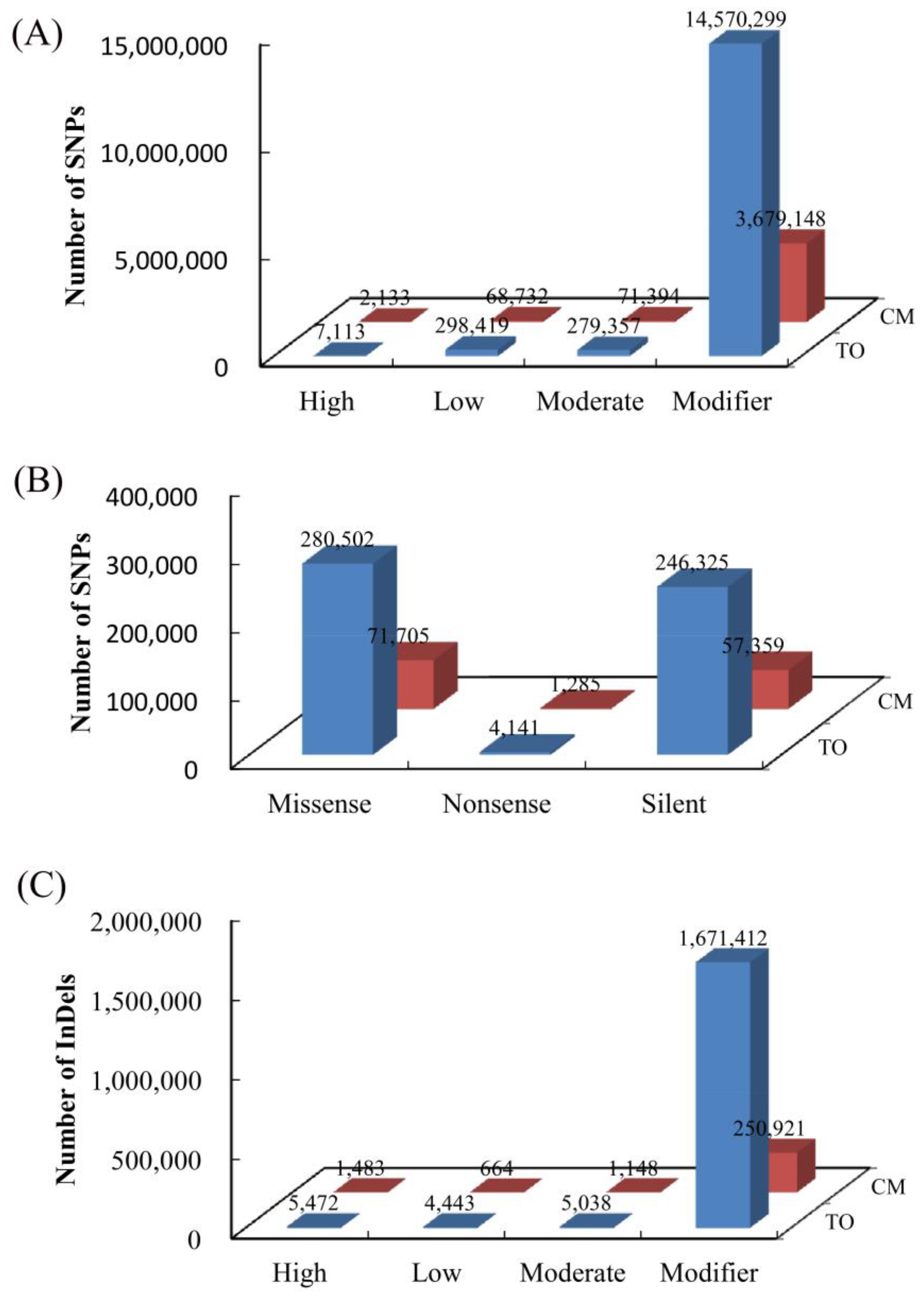
3.3. Annotation of Species-Specific Single Nucleotide Polymorphisms and Insertion/Deletion Polymorphism
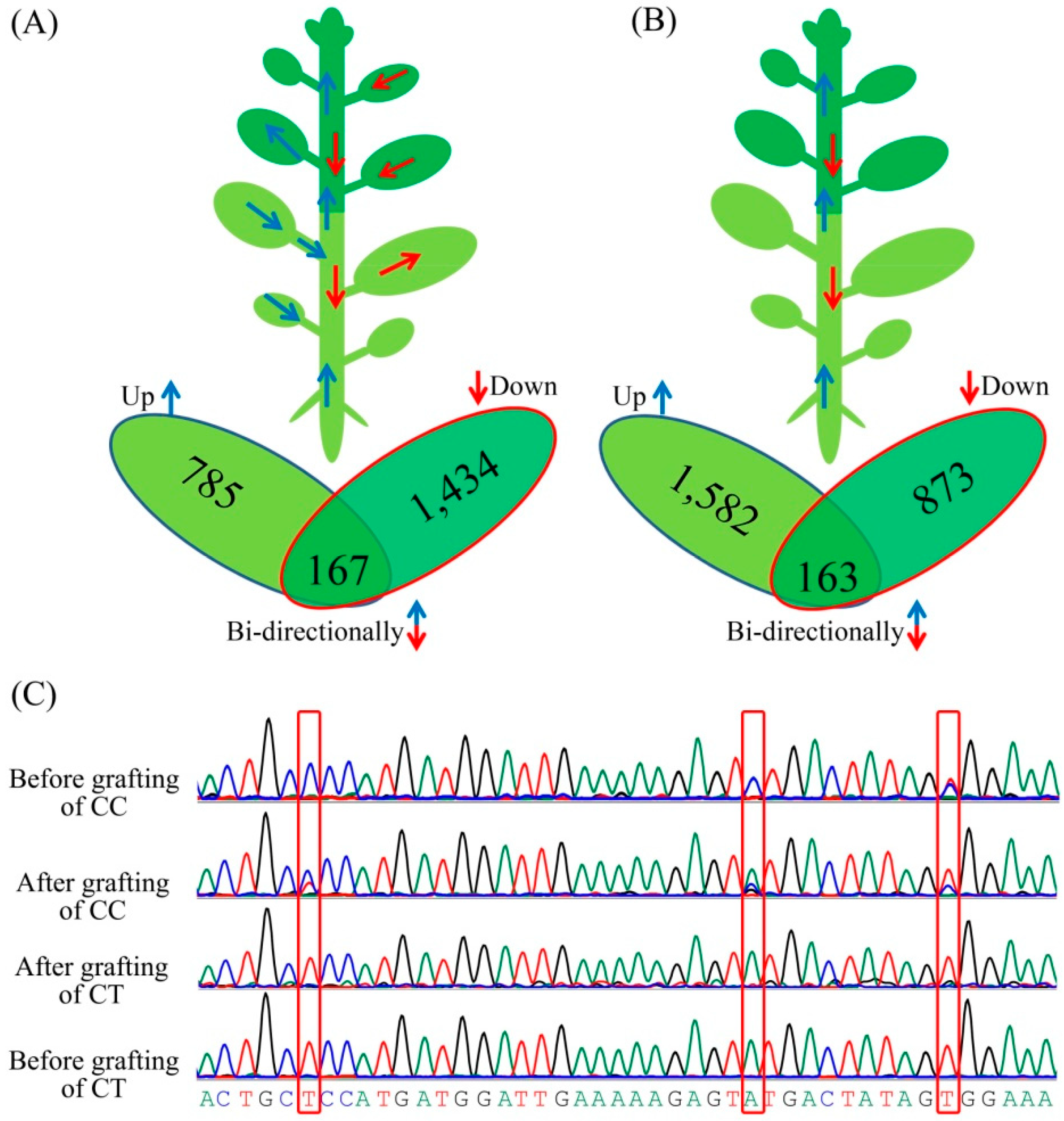
3.4. Messenger RNA Movement between Citrus Trifoliata and Citrus Clementina
3.5. Classification of the Biological Functions of the Transmitting Genes
3.6. Identification of Differentially Expressed Genes (DEGs) during Grafting Process
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Albacete, A.; Martinez-Andujar, C.; Martinez-Perez, A.; Thompson, A.J.; Dodd, I.C.; Perez-Alfocea, F. Unravelling rootstockxscion interactions to improve food security. J. Exp. Bot. 2015, 66, 2211–2226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.M.; Oda, M. Grafting of herbaceous vegetable and ornamental crops. Hortic. Rev. 2003, 28, 61–124. [Google Scholar]
- Nawaz, M.A.; Imtiaz, M.; Kong, Q.S.; Cheng, F.; Ahmed, W.; Huang, Y.; Bie, Z.L. Grafting: A technique to modify Ion accumulation in horticultural crops. Front. Plant Sci. 2016, 7, 1457. [Google Scholar] [CrossRef] [PubMed]
- Mudge, K.; Janick, J.; Scofield, S.; Goldschmidt, E.E. A history of grafting. Hortic. Rev. 2009, 35, 437–493. [Google Scholar]
- Warschefsky, E.J.; Klein, L.L.; Frank, M.H.; Chitwood, D.H.; Londo, J.P.; von Wettberg, E.J.; Miller, A.J. Rootstocks: Diversity, domestication, and Impacts on shoot phenotypes. Trends Plant. Sci. 2016, 21, 418–437. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Marguerit, E.; Rossdeutsch, L.; Ollat, N.; Gambetta, G.A. The influence of grapevine rootstocks on scion growth and drought resistance. Theor. Exp. Plant Physiol. 2016, 28, 143–157. [Google Scholar]
- Corso, M.; Vannozzi, A.; Ziliotto, F.; Zouine, M.; Maza, E.; Nicolato, T.; Vitulo, N.; Meggio, F.; Valle, G.; Bouzayen, M. Grapevine rootstocks differentially affect the rate of ripening and modulate auxin-related genes in cabernet sauvignon berries. Front. Plant Sci. 2016, 7, 69. [Google Scholar] [CrossRef]
- Liu, N.; Yang, J.; Fu, X.; Zhang, L.; Tang, K.; Guy, K.M.; Hu, Z.; Guo, S.; Xu, Y.; Zhang, M. Genome-wide identification and comparative analysis of grafting-responsive mRNA in watermelon grafted onto bottle gourd and squash rootstocks by high-throughput sequencing. Mol. Genet. Genom. 2016, 291, 621–633. [Google Scholar] [CrossRef]
- Chitarra, W.; Perrone, I.; Avanzato, C.G.; Minio, A.; Boccacci, P.; Santini, D.; Gilardi, G.; Siciliano, I.; Gullino, M.L.; Delledonne, M. Grapevine Grafting: Scion Transcript Profiling and Defense-Related Metabolites Induced by Rootstocks. Front. Plant Sci. 2017, 8, 654. [Google Scholar] [CrossRef]
- Cookson, S.J.; Moreno, M.J.C.; Hevin, C.; Mendome, L.Z.N.; Delrot, S.; Trossat-Magnin, C.; Ollat, N. Graft union formation in grapevine induces transcriptional changes related to cell wall modification, wounding, hormone signalling, and secondary metabolism. J. Exp. Bot. 2013, 64, 2997–3008. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wang, L.; Tian, J.; Li, J.; Sun, J.; He, L.; Guo, S.; Tezuka, T. Proteomic study participating the enhancement of growth and salt tolerance of bottle gourd rootstock-grafted watermelon seedlings. Plant Physiol. Biochem. 2012, 58, 54–65. [Google Scholar] [CrossRef]
- Xu, D.; Yuan, H.; Tong, Y.; Zhao, L.; Qiu, L.; Guo, W.; Shen, C.; Liu, H.; Yan, D.; Zheng, B. Comparative proteomic analysis of the graft unions in hickory (Carya cathayensis) provides insights into response mechanisms to grafting process. Front. Plant Sci. 2017, 8, 676. [Google Scholar] [CrossRef]
- Oka-Kira, E.; Kawaguchi, M. Long-distance signaling to control root nodule number. Curr. Opin. Plant Biol. 2006, 9, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Lifschitz, E.; Eshed, Y. Universal florigenic signals triggered by FT homologues regulate growth and flowering cycles in perennial day-neutral tomato. J. Exp. Bot. 2006, 57, 3405–3414. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.; Adam, H.; Diaz-Mendoza, M.; Zurczak, M.; Gonzalez-Schain, N.D.; Suarez-Lopez, P. Graft-transmissible induction of potato tuberization by the microRNA miR172. Development 2009, 136, 2873–2881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haywood, V.; Yu, T.S.; Huang, N.C.; Lucas, W.J. Phloem long-distance trafficking of Gibberellic acid-insensitive RNA regulates leaf development. Plant J. 2005, 42, 49–68. [Google Scholar] [CrossRef] [PubMed]
- Beveridge, C.A. Axillary bud outgrowth: Sending a message. Curr. Opin. Plant Biol. 2006, 9, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q. Plant grafting and its application in biological research. Chin. Sci. Bull. 2011, 56, 3511–3517. [Google Scholar] [CrossRef]
- Harada, T. Grafting and RNA transport via phloem tissue in horticultural plants. Sci. Hortic. Amst. 2010, 125, 545–550. [Google Scholar] [CrossRef]
- Kehr, J.; Buhtz, A. Long distance transport and movement of RNA through the phloem. J. Exp. Bot. 2008, 59, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.E.; Bauerle, T.L. Long-distance plant signaling pathways in response to multiple stressors: The gap in knowledge. J. Exp. Bot. 2016, 67, 2063–2079. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Y.; Li, J.; Liu, M.M.; Yao, Q.; Chen, J.Z. Transcriptome profiling to understand the effect of Citrus rootstocks on the growth of ‘Shatangju’ Mandarin. PLoS ONE 2017, 12, e0169897. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Wang, Y.; Chen, Q.; Sun, B.; Tang, H.R.; Pan, D.M.; Wang, X.R. Dissection of the mechanism for compatible and incompatible graft combinations of Citrus grandis (L.) Osbeck (‘Hongmian Miyou’). Int. J. Mol. Sci. 2018, 19, 505. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.A.; Prochnik, S.; Jenkins, J.; Salse, J.; Hellsten, U.; Murat, F.; Perrier, X.; Ruiz, M.; Scalabrin, S.; Terol, J. Sequencing of diverse mandarin, pummelo and orange genomes reveals complex history of admixture during citrus domestication. Nat. Biotechnol. 2014, 32, 656–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.-J.; Guo, W.-W.; Yi, H.-L.; Pang, X.-M.; Deng, X. An efficient protocol for genomic DNA extraction from Citrus species. Plant Mol. Biol. Report. 2003, 21, 177–178. [Google Scholar] [CrossRef]
- Datta, S.; Datta, S.; Kim, S.; Chakraborty, S.; Gill, R.S. Statistical analyses of next generation sequence data: A partial overview. J. Proteom. Bioinform. 2010, 3, 183–190. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Le, L.W.; Coon, M.; Nguyen, T.; Luan, W.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Blankenberg, D.; Gordon, A.; Von Kuster, G.; Coraor, N.; Taylor, J.; Nekrutenko, A. Manipulation of FASTQ data with Galaxy. Bioinformatics 2010, 26, 1783–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; Mccue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Tarazona, S.; Garcíaalcalde, F.; Dopazo, J.; Ferrer, A.; Conesa, A. Differential expression in RNA-seq: A matter of depth. Genome Res. 2011, 21, 2213–2223. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Yang, Y.; Mao, L.; Jittayasothorn, Y.; Kang, Y.; Jiao, C.; Fei, Z.; Zhong, G.Y. Messenger RNA exchange between scions and rootstocks in grafted grapevines. BMC Plant Biol. 2015, 15, 251. [Google Scholar] [CrossRef]
- Thieme, C.J.; Rojastriana, M.; Stecyk, E.; Schudoma, C.; Zhang, W.; Yang, L.; Miñambres, M.; Walther, D.; Schulze, W.X.; Pazares, J. Endogenous Arabidopsis messenger RNAs transported to distant tissues. Nat. Plants 2015, 1, 15025. [Google Scholar] [CrossRef]
- Zhang, J.Z.; Li, Z.M.; Yao, J.L.; Hu, C.G. Identification of flowering-related genes between early flowering trifoliate orange mutant and wild-type trifoliate orange (Poncirus trifoliata L. Raf.) by suppression subtraction hybridization (SSH) and macroarray. Gene 2009, 430, 95–104. [Google Scholar] [CrossRef]
- Wang, X.; Xu, Y.; Zhang, S.; Cao, L.; Huang, Y.; Cheng, J.; Wu, G.; Tian, S.; Chen, C.; Liu, Y.; et al. Genomic analyses of primitive, wild and cultivated citrus provide insights into asexual reproduction. Nat. Genet. 2017, 49, 765. [Google Scholar] [CrossRef]
- Natarajan, S.; Kim, H.T.; Thamilarasan, S.K.; Veerappan, K.; Park, J.I.; Nou, I.S. Whole Genome Re-Sequencing and Characterization of Powdery Mildew Disease-Associated Allelic Variation in Melon. PLoS ONE 2016, 11, e0157524. [Google Scholar] [CrossRef]
- Wu, G.A.; Terol, J.; Ibanez, V.; Lopez-Garcia, A.; Perez-Roman, E.; Borreda, C.; Domingo, C.; Tadeo, F.R.; Carbonell-Caballero, J.; Alonso, R.; et al. Genomics of the origin and evolution of Citrus. Nature 2018, 554, 311–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, W.B.; Huang, D.; Xing, F.; Hu, Y.; Deng, X.X.; Xu, Q.; Chen, L.L. Genome-wide characterization and expression analysis of genetic variants in sweet orange. Plant J. 2013, 75, 954–964. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073. [Google Scholar] [CrossRef] [PubMed]
- Shabala, S.; White, R.G.; Djordjevic, M.A.; Ruan, Y.-L.; Mathesius, U. Root-to-shoot signalling: Integration of diverse molecules, pathways and functions. Funct. Plant Biol. 2016, 43, 87–104. [Google Scholar] [CrossRef]
- Kim, G.; Leblanc, M.L.; Wafula, E.K.; Depamphilis, C.W.; Westwood, J.H. Genomic-scale exchange of mRNA between a parasitic plant and its hosts. Science 2014, 345, 808–811. [Google Scholar] [CrossRef] [PubMed]
- Brosnan, C.A.; Mitter, N.; Christie, M.; Smith, N.A.; Waterhouse, P.M.; Carroll, B.J. Nuclear gene silencing directs reception of long-distance mRNA silencing in Arabidopsis. Proc. Natl. Acad. Sci. USA 2007, 104, 14741–14746. [Google Scholar] [CrossRef] [Green Version]
- Ayre, B.G.; Turgeon, R. Graft transmission of a floral stimulant derived from CONSTANS. Plant Physiol. 2004, 135, 2271–2278. [Google Scholar] [CrossRef]
- Gómez-Ariza, J.; Campo, S.; Rufat, M.; Estopà, M.; Messeguer, J.; San, S.B.; Coca, M. Sucrose-mediated priming of plant defense responses and broad-spectrum disease resistance by overexpression of the maize pathogenesis-related PRms protein in rice plants. Mol. Plant Microbe Interact. 2007, 20, 832–842. [Google Scholar] [CrossRef]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, T.-J.; Zhou, J.-J.; Chen, F.-Y.; Gan, Z.-M.; Li, Y.-P.; Zhang, J.-Z.; Hu, C.-G. Identification of the Genetic Variation and Gene Exchange between Citrus Trifoliata and Citrus Clementina. Biomolecules 2018, 8, 182. https://doi.org/10.3390/biom8040182
Liu T-J, Zhou J-J, Chen F-Y, Gan Z-M, Li Y-P, Zhang J-Z, Hu C-G. Identification of the Genetic Variation and Gene Exchange between Citrus Trifoliata and Citrus Clementina. Biomolecules. 2018; 8(4):182. https://doi.org/10.3390/biom8040182
Chicago/Turabian StyleLiu, Tian-Jia, Jing-Jing Zhou, Fa-Yi Chen, Zhi-Meng Gan, Yong-Ping Li, Jin-Zhi Zhang, and Chun-Gen Hu. 2018. "Identification of the Genetic Variation and Gene Exchange between Citrus Trifoliata and Citrus Clementina" Biomolecules 8, no. 4: 182. https://doi.org/10.3390/biom8040182