Inhibition of Ghrelin Activity by Receptor Antagonist [d-Lys-3] GHRP-6 Attenuates Alcohol-Induced Hepatic Steatosis by Regulating Hepatic Lipid Metabolism


Abstract
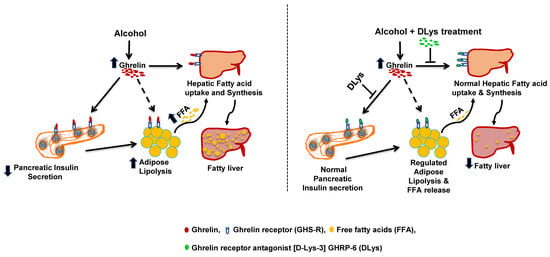
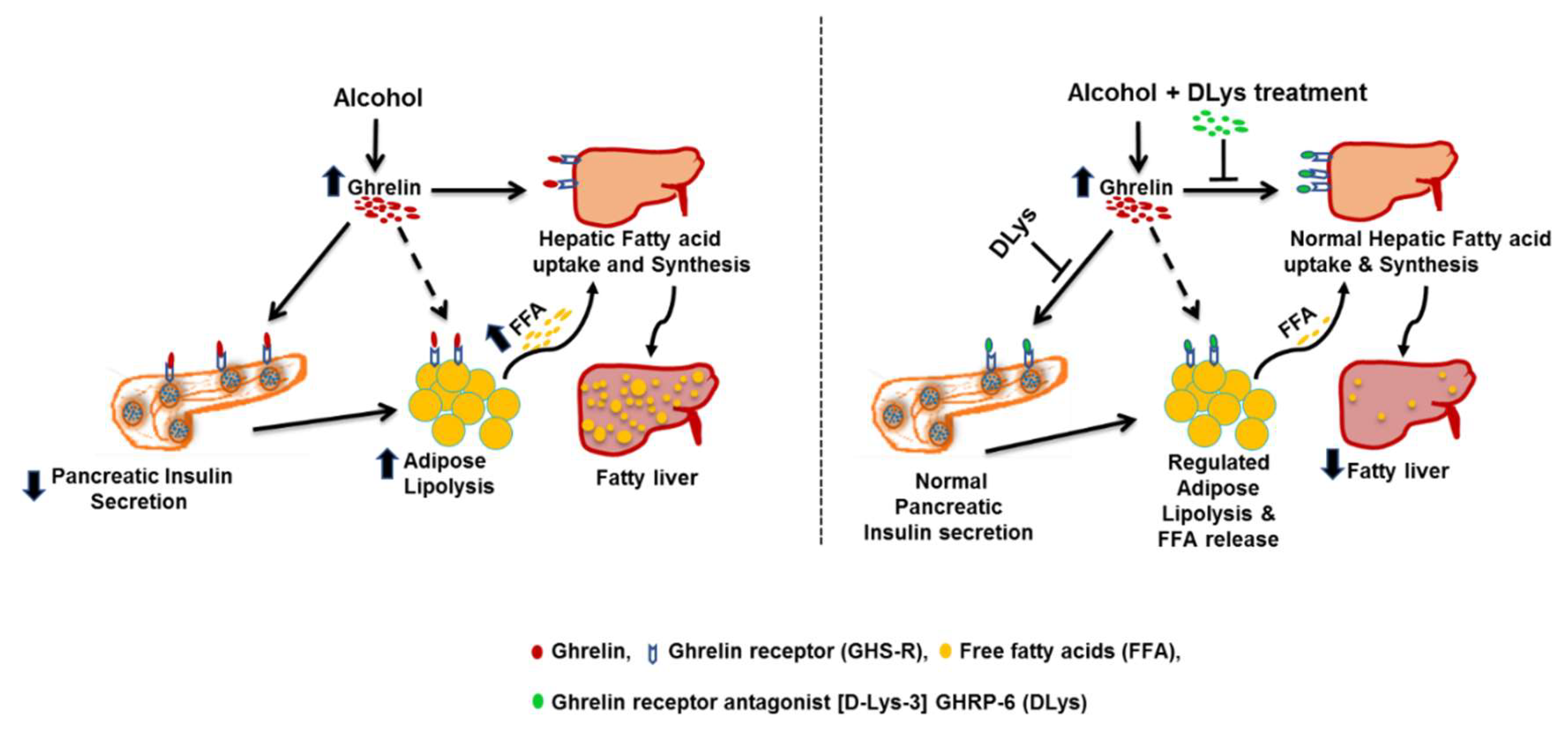
:
1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents
2.2. Animal Maintenance, Treatment, and Tissue Collection
2.3. Liver Triglycerides and Serum Non-Esterified Free Fatty Acids (NEFA)
2.4. Serum Enzymes and Hormones Level
2.5. Gene Expression Analysis
2.6. Immunohistochemistry
2.7. Western Blot
2.8. Statistical Analysis
3. Results
3.1. Food Intake, Liver and Body Weights, and Serum Hepatic Markers
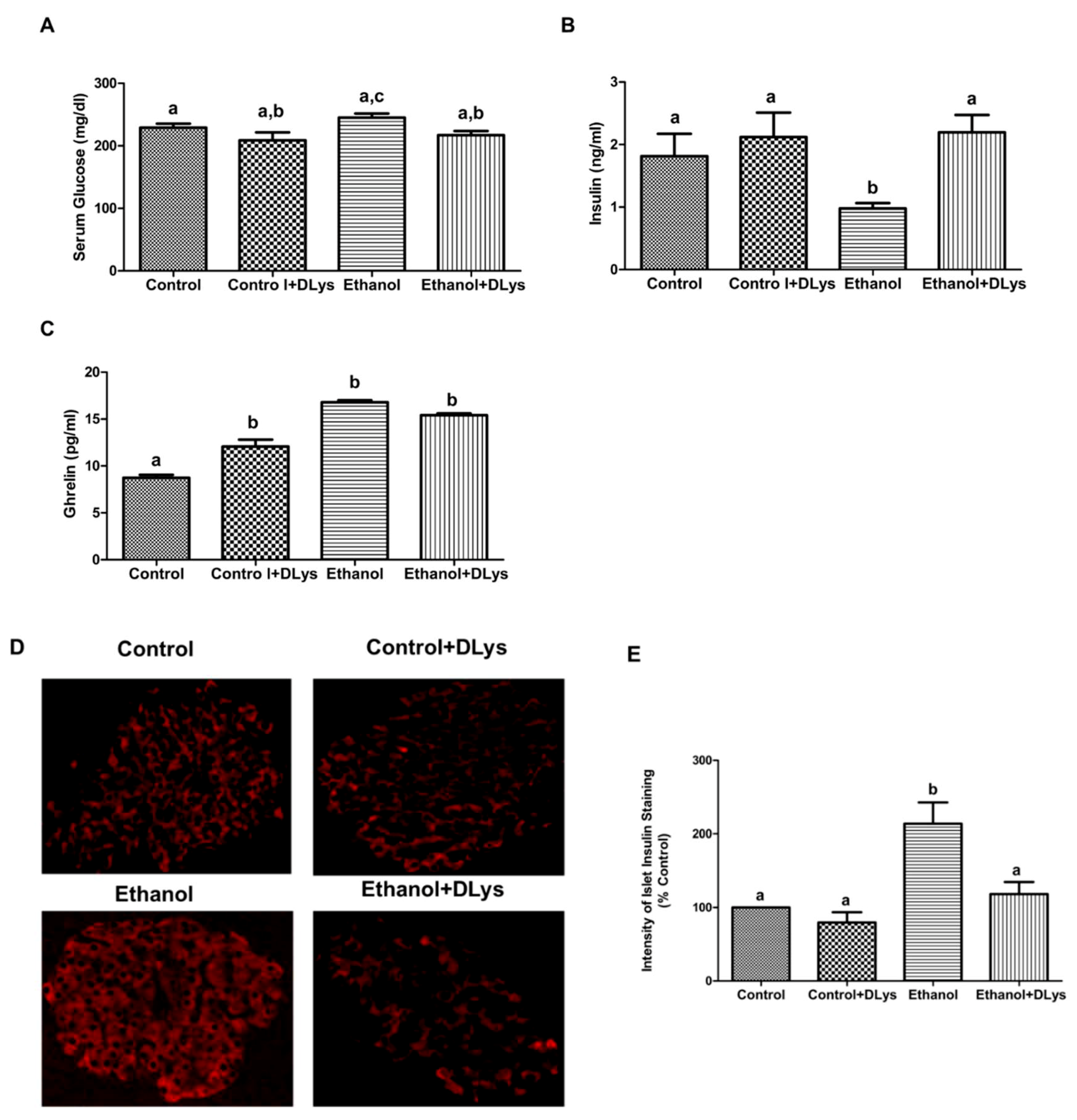
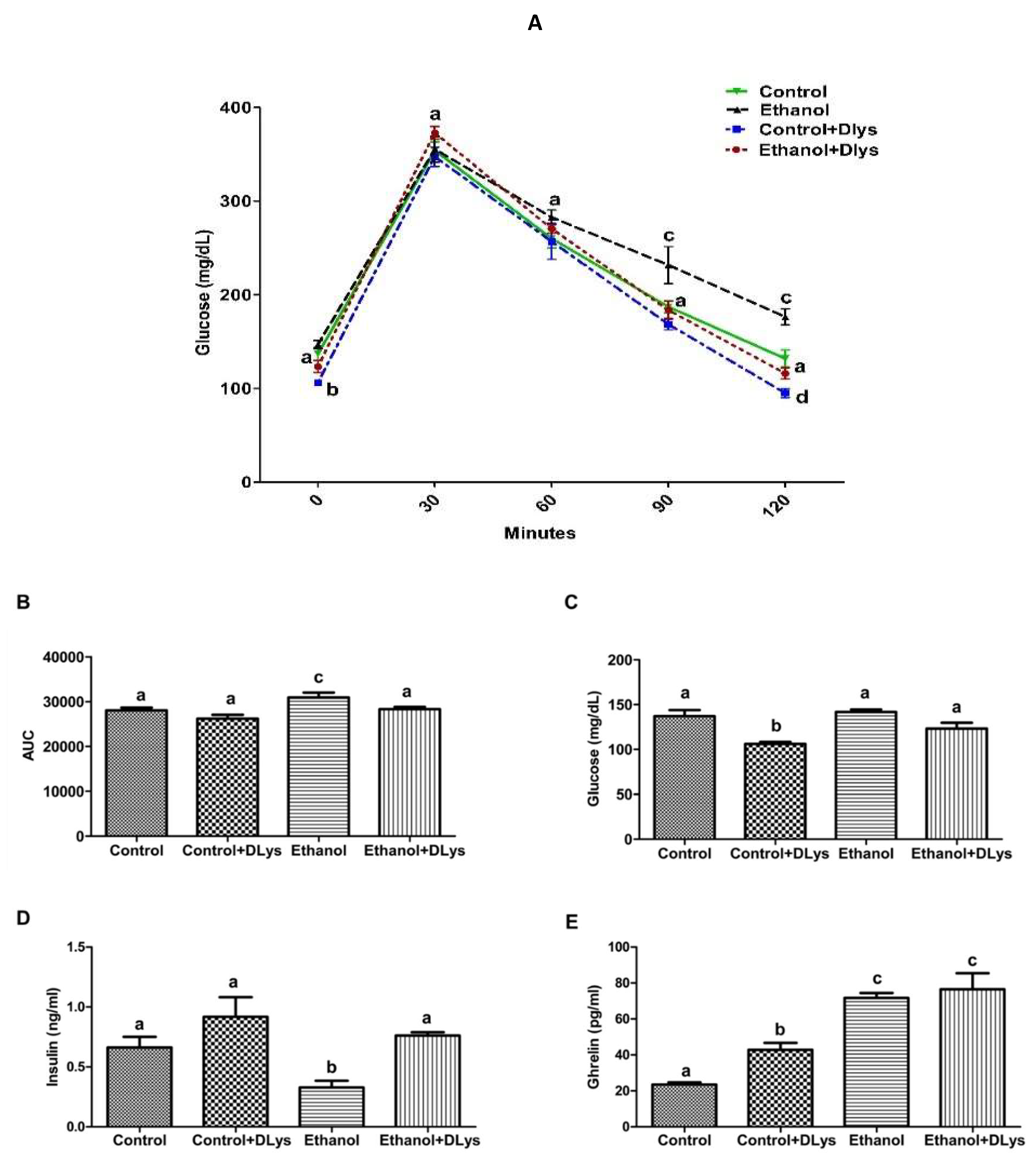
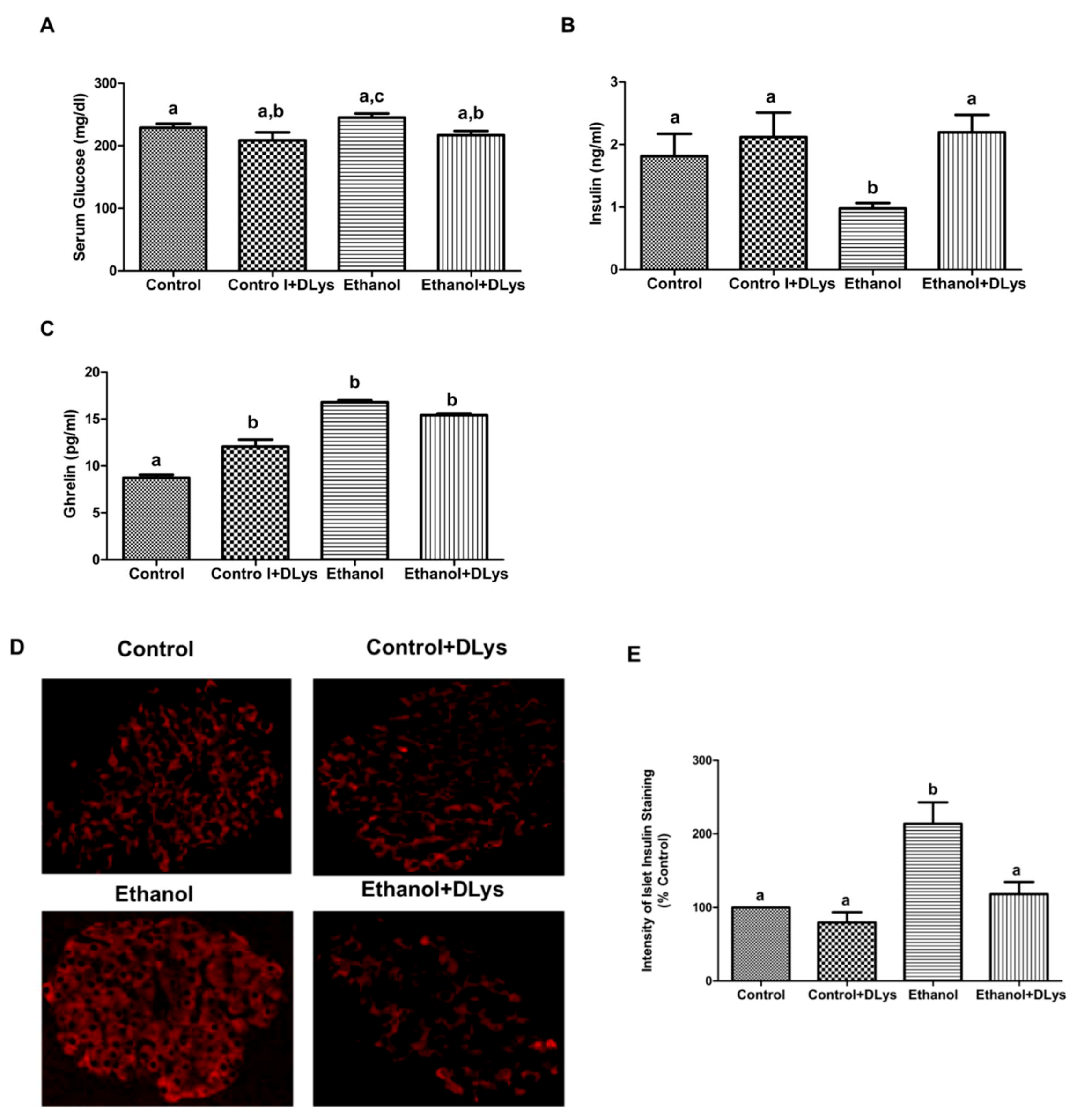
3.2. Ghrelin Receptor Antagonist Abrogated the Ethanol-Induced Impaired Glucose Tolerance by Increasing Serum Insulin Levels via Promotion of Insulin Secretion from the Islets
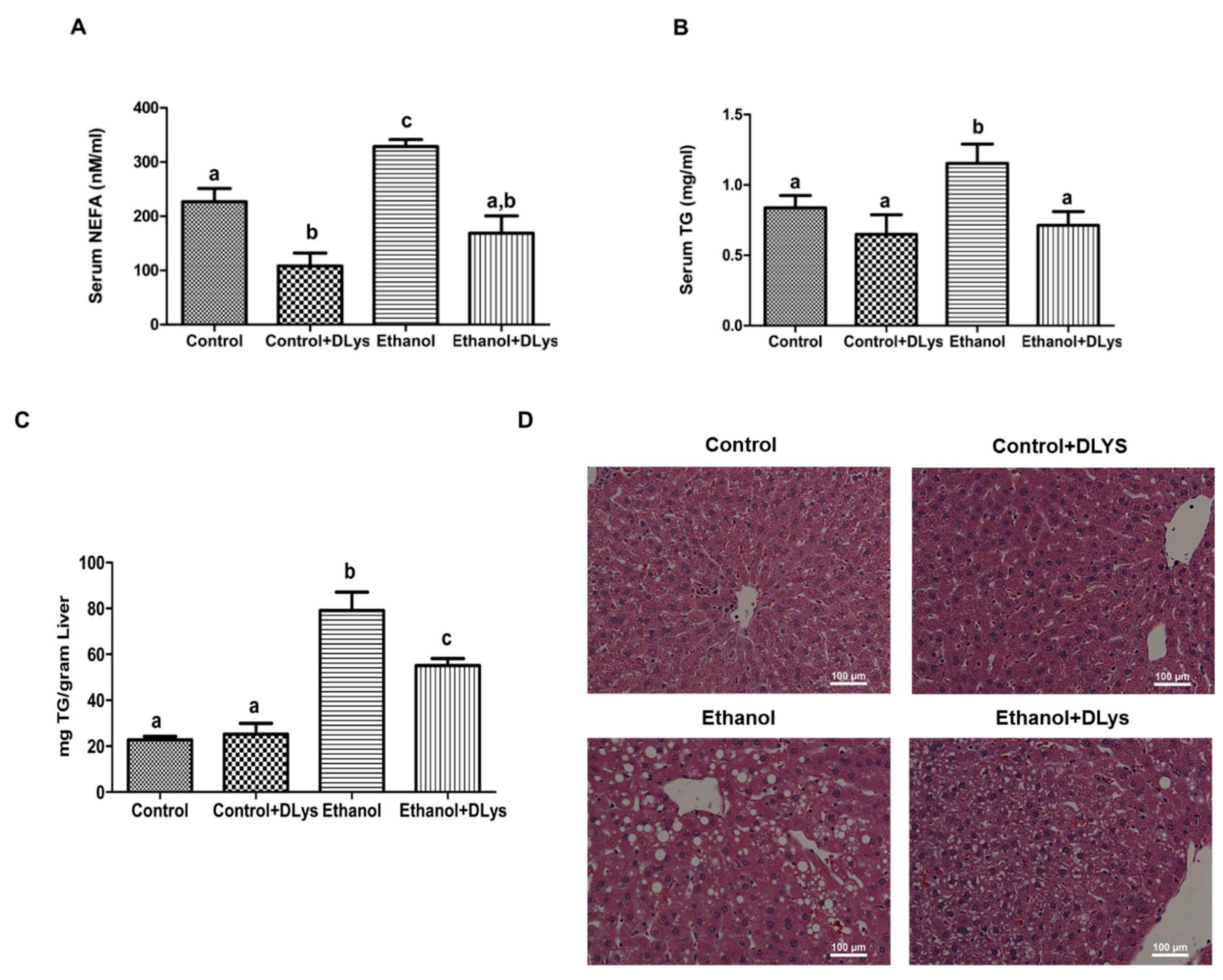
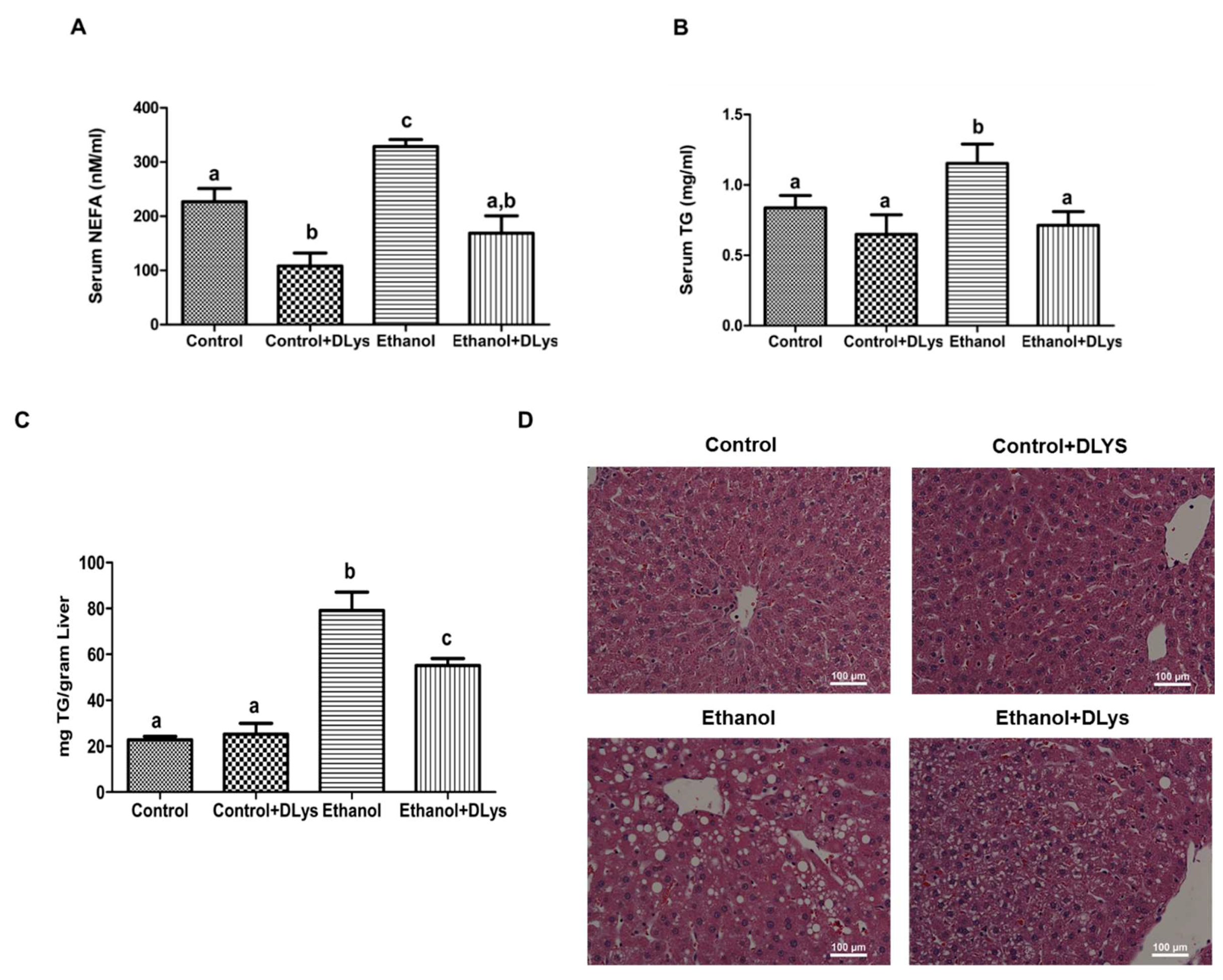
3.3. Inhibition of Ghrelin Activity Attenuates Alcohol-Induced Hepatic Steatosis
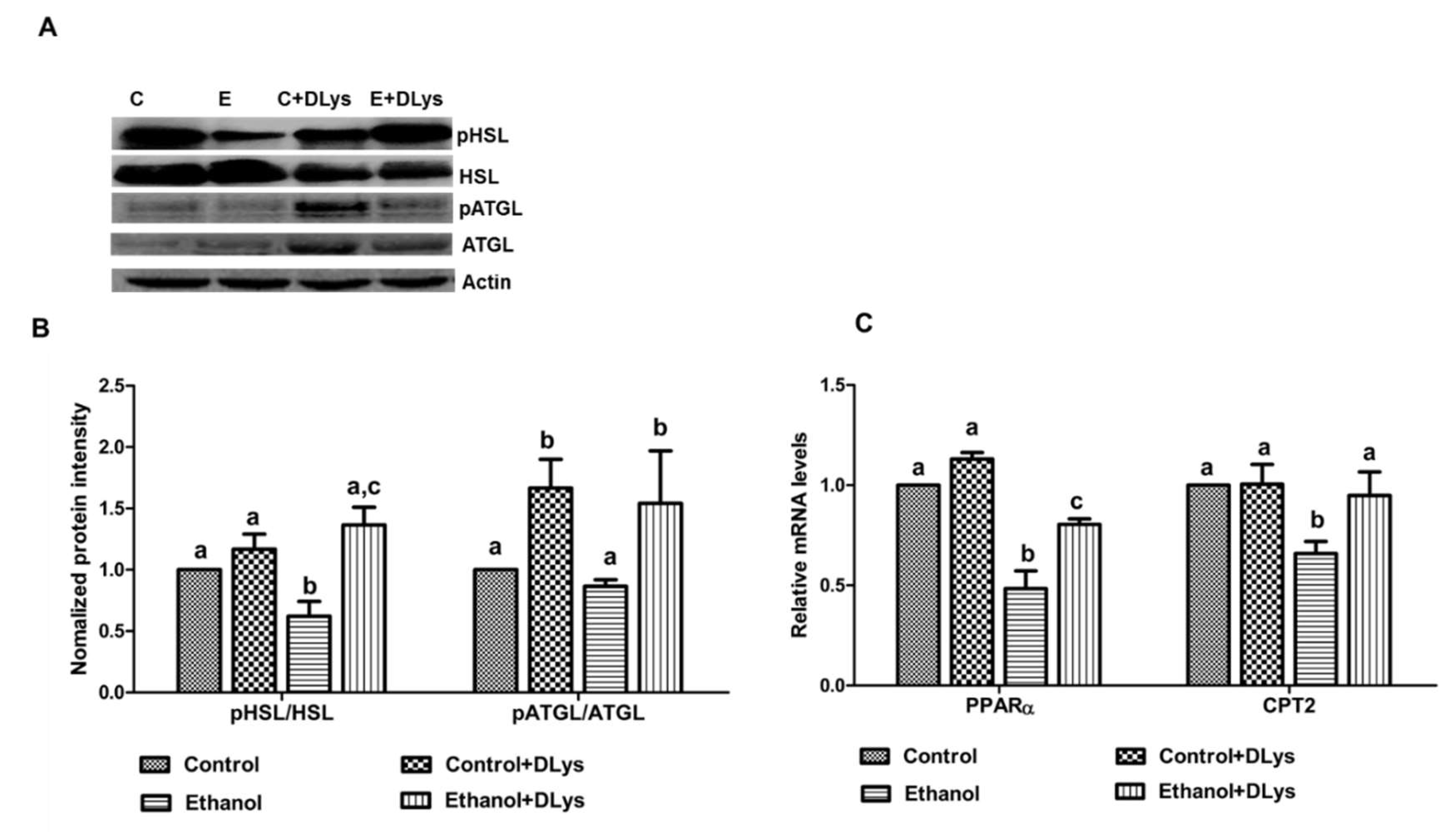
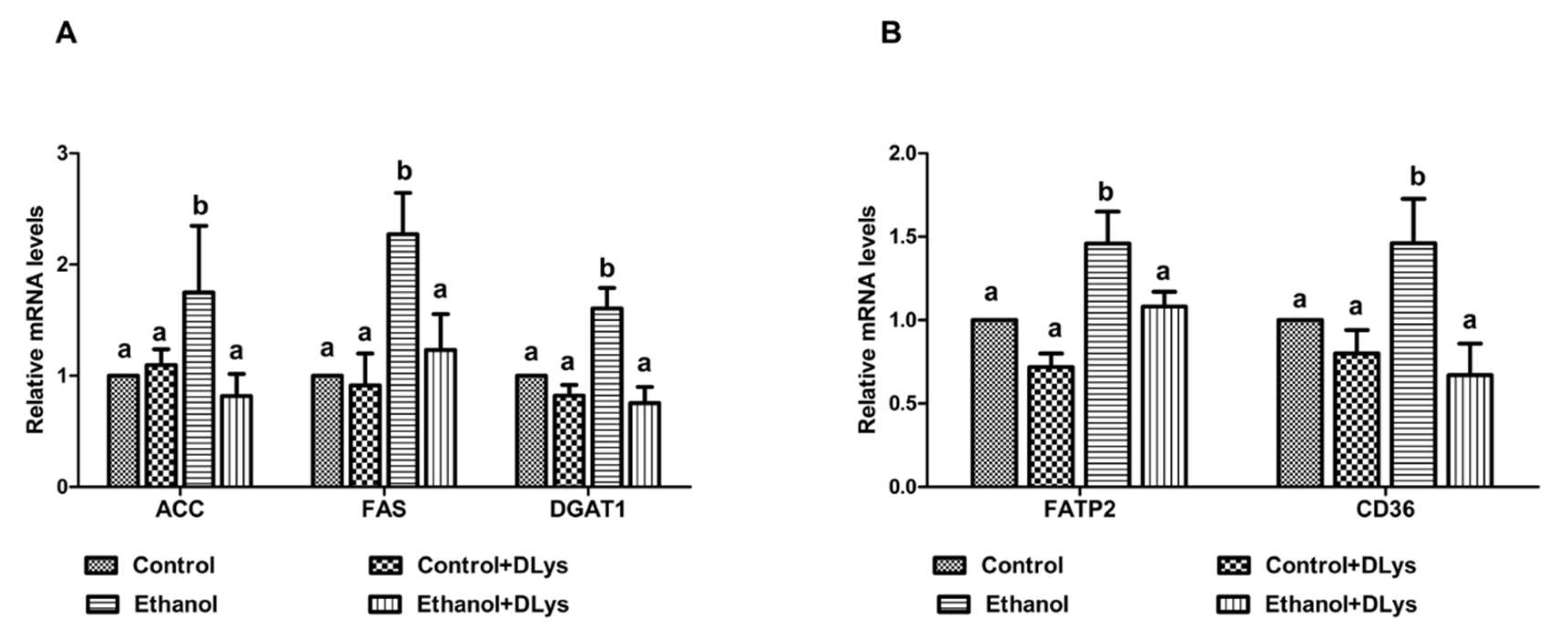
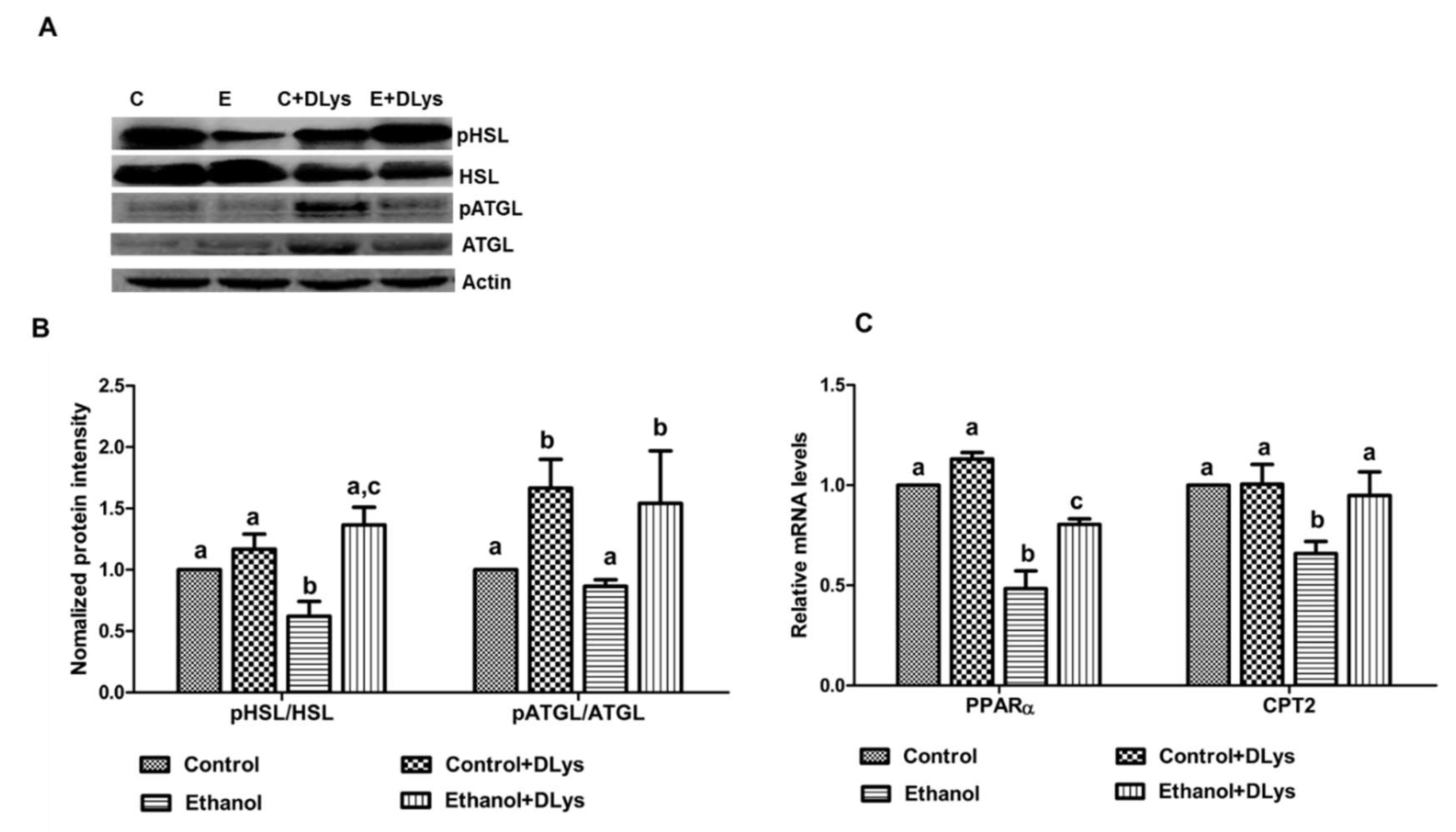
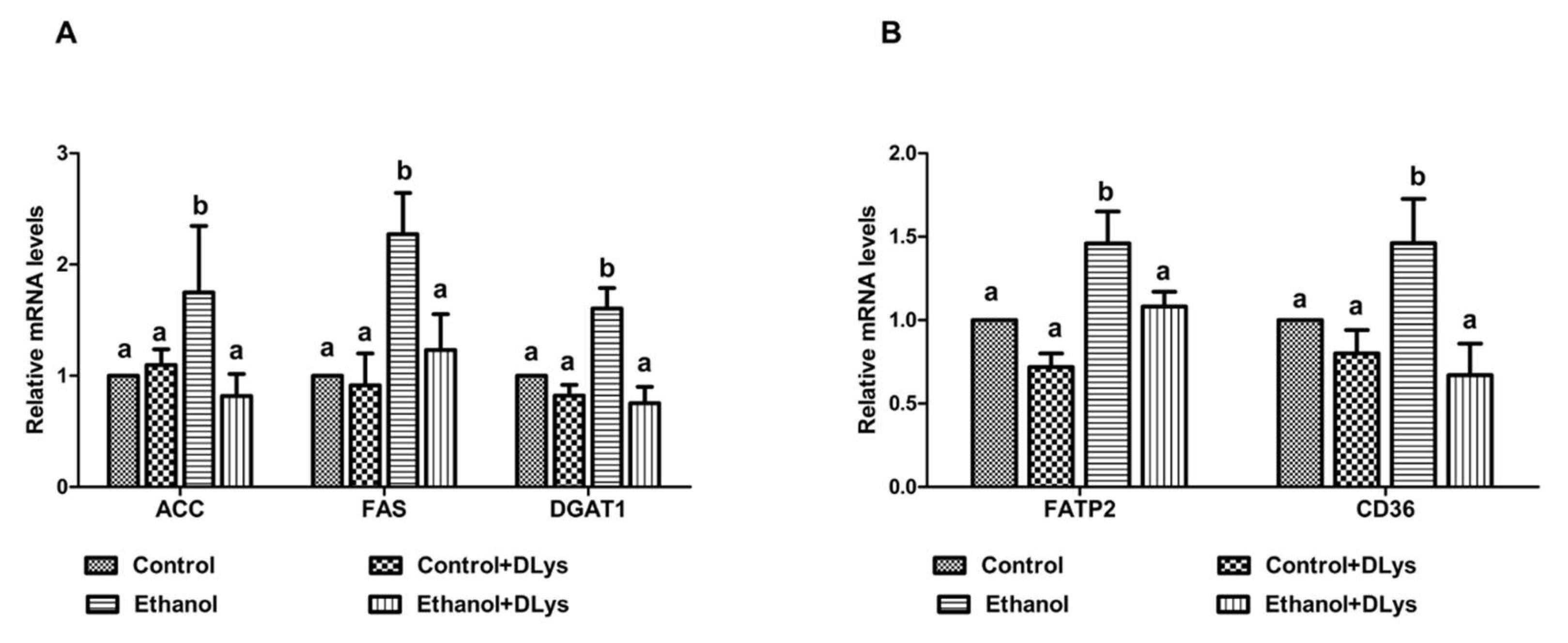
3.4. DLys Treatment Abrogates Ethanol-Induced Fat Accumulation via Increasing Hepatic Lipase Activity, Enhancing Fatty acid Oxidation, and Reducing Fatty Acid Synthesis and Uptake
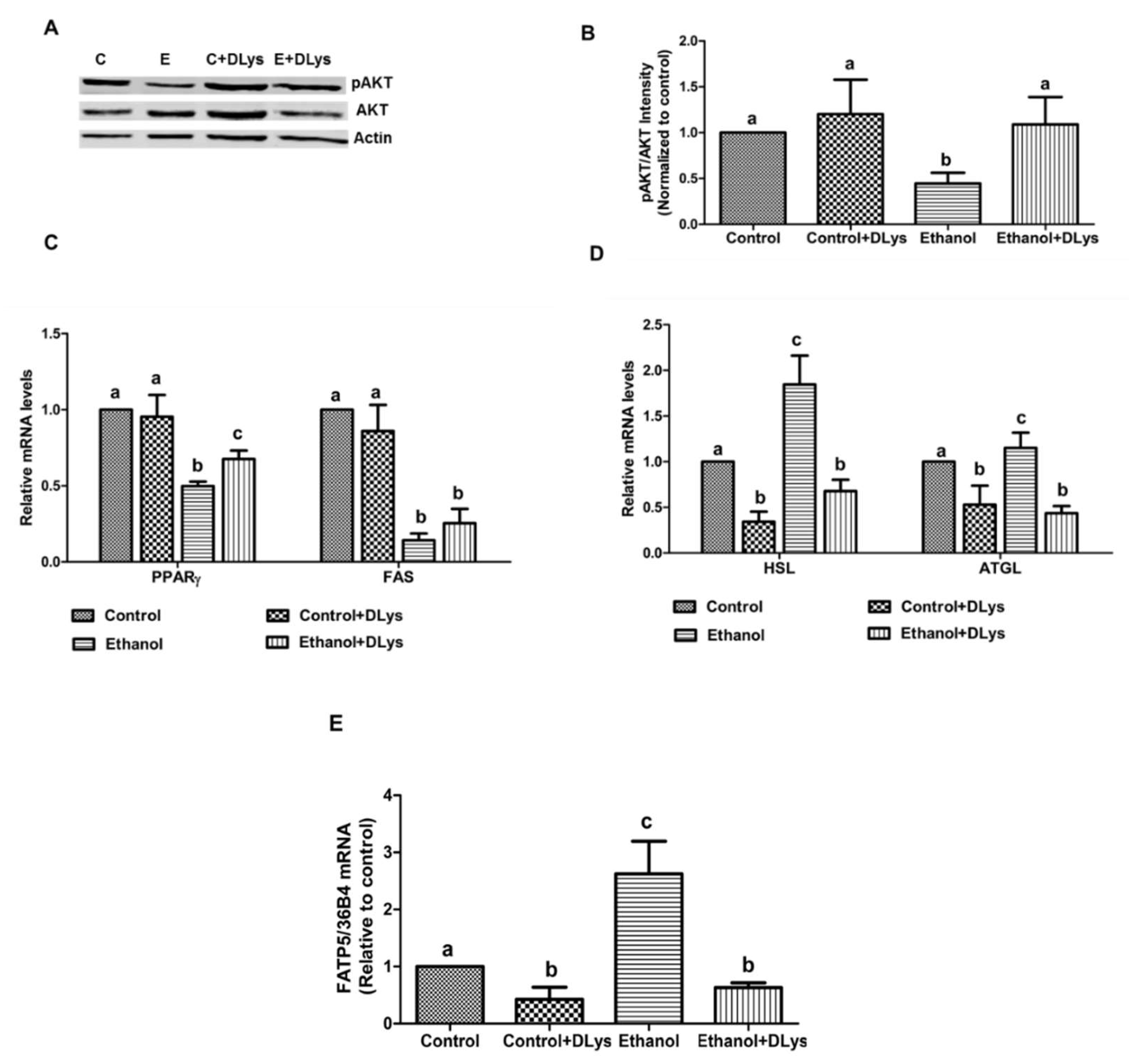
3.5. DLys Treatment Inhibits the Ethanol-Induced Altered Fat Metabolism in the Adipose Tissue
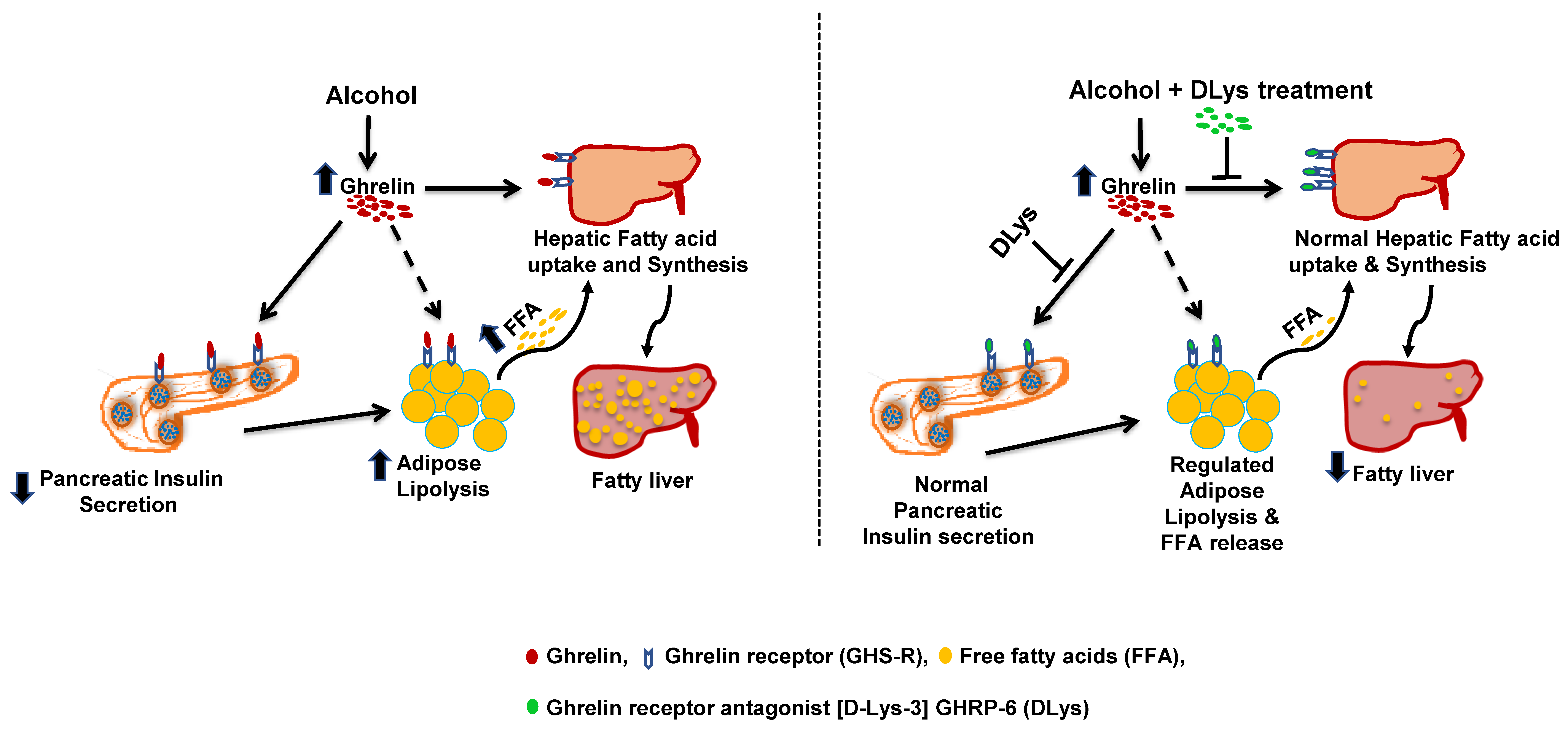
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Singal, A.K.; Anand, B.S. Recent trends in the epidemiology of alcoholic liver disease. Clin. Liver Dis. 2013, 2, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Ishak, K.G.; Zimmerman, H.J.; Ray, M.B. Alcoholic liver disease: Pathologic, pathogenetic and clinical aspects. Alcohol. Clin. Exp. Res. 1991, 15, 45–66. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S. Alcoholic fatty liver: Its pathogenesis and mechanism of progression to inflammation and fibrosis. Alcohol 2004, 34, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; You, M.; Matsumoto, M.; Crabb, D.W. Peroxisome proliferator-activated receptor alpha (PPARalpha) agonist treatment reverses PPARalpha dysfunction and abnormalities in hepatic lipid metabolism in ethanol-fed mice. J. Boil. Chem. 2003, 278, 27997–28004. [Google Scholar] [CrossRef] [PubMed]
- Gearing, K.L.; Gottlicher, M.; Widmark, E.; Banner, C.D.; Tollet, P.; Stromstedt, M.; Rafter, J.J.; Berge, R.K.; Gustafsson, J.A. Fatty acid activation of the peroxisome proliferator activated receptor, a member of the nuclear receptor gene superfamily. J. Nutr. 1994, 124, 1284–1288. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, K.K.; Todero, S.L.; Ward, B.W.; Cannella, J.J.; Tuma, D.J. Betaine administration corrects ethanol-induced defective VLDL secretion. Mol. Cell Biochem. 2009, 327, 75–78. [Google Scholar] [CrossRef] [PubMed]
- McVicker, B.L.; Rasineni, K.; Tuma, D.J.; McNiven, M.A.; Casey, C.A. Lipid droplet accumulation and impaired fat efflux in polarized hepatic cells: Consequences of ethanol metabolism. Int. J. Hepatol. 2012, 2012, 978136. [Google Scholar] [CrossRef]
- Kang, L.; Chen, X.; Sebastian, B.M.; Pratt, B.T.; Bederman, I.R.; Alexander, J.C.; Previs, S.F.; Nagy, L.E. Chronic ethanol and triglyceride turnover in white adipose tissue in rats: Inhibition of the anti-lipolytic action of insulin after chronic ethanol contributes to increased triglyceride degradation. J. Boil. Chem. 2007, 282, 28465–28473. [Google Scholar] [CrossRef]
- Wei, X.; Shi, X.; Zhong, W.; Zhao, Y.; Tang, Y.; Sun, W.; Yin, X.; Bogdanov, B.; Kim, S.; McClain, C.; et al. Chronic alcohol exposure disturbs lipid homeostasis at the adipose tissue-liver axis in mice: Analysis of triacylglycerols using high-resolution mass spectrometry in combination with in vivo metabolite deuterium labeling. PLoS ONE 2013, 8, 55382. [Google Scholar] [CrossRef]
- Kim, J.Y.; Hwang, J.Y.; Lee, D.Y.; Song, E.H.; Park, K.J.; Kim, G.H.; Jeong, E.A.; Lee, Y.J.; Go, M.J.; Kim, D.J.; et al. Chronic ethanol consumption inhibits glucokinase transcriptional activity by Atf3 and triggers metabolic syndrome in vivo. J. Biol. Chem. 2014, 289, 27065–27079. [Google Scholar] [CrossRef]
- Kim, J.Y.; Song, E.H.; Lee, H.J.; Oh, Y.K.; Park, Y.S.; Park, J.W.; Kim, B.J.; Kim, D.J.; Lee, I.; Song, J.; et al. Chronic ethanol consumption-induced pancreatic [2]-cell dysfunction and apoptosis through glucokinase nitration and its down-regulation. J. Biol. Chem. 2010, 285, 37251–37262. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Liang, C.P.; Westerterp, M.; Senokuchi, T.; Welch, C.L.; Wang, Q.; Matsumoto, M.; Accili, D.; Tall, A.R. Hepatic insulin signaling regulates VLDL secretion and atherogenesis in mice. J. Clin. Invest. 2009, 119, 1029–1041. [Google Scholar] [CrossRef] [Green Version]
- Scherer, T.; Lindtner, C.; O’Hare, J.; Hackl, M.; Zielinski, E.; Freudenthaler, A.; Baumgartner-Parzer, S.; Todter, K.; Heeren, J.; Krssak, M.; et al. Insulin Regulates Hepatic Triglyceride Secretion and Lipid Content via Signaling in the Brain. Diabetes 2016, 65, 1511–1520. [Google Scholar] [CrossRef] [Green Version]
- Rasineni, K.; Thomes, P.G.; Kubik, J.L.; Harris, E.N.; Kharbanda, K.K.; Casey, C.A. Chronic alcohol exposure alters circulating insulin and ghrelin levels: Role of ghrelin in hepatic steatosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Laviano, A.; Molfino, A.; Rianda, S.; Rossi Fanelli, F. The growth hormone secretagogue receptor (Ghs-R). Curr. Pharm. Des. 2012, 18, 4749–4754. [Google Scholar] [CrossRef]
- Li, Z.; Xu, G.; Qin, Y.; Zhang, C.; Tang, H.; Yin, Y.; Xiang, X.; Li, Y.; Zhao, J.; Mulholland, M.; et al. Ghrelin promotes hepatic lipogenesis by activation of mTOR-PPARgamma signaling pathway. Proc. Natl. Acad. Sci. USA 2014, 111, 13163–13168. [Google Scholar] [CrossRef]
- Patel, K.; Dixit, V.D.; Lee, J.H.; Kim, J.W.; Schaffer, E.M.; Nguyen, D.; Taub, D.D. Identification of ghrelin receptor blocker, D-[Lys3] GHRP-6 as a CXCR4 receptor antagonist. Int. J. Biol. Sci. 2012, 8, 108–117. [Google Scholar] [CrossRef]
- Gomez, J.L.; Ryabinin, A.E. The Effects of Ghrelin Antagonists [D-Lys(3) ]-GHRP-6 or JMV2959 on Ethanol, Water, and Food Intake in C57BL/6J Mice. Alcohol. Clin. Exp. Res. 2014, 38, 2436–2444. [Google Scholar] [CrossRef]
- Maletinska, L.; Matyskova, R.; Maixnerova, J.; Sykora, D.; Pychova, M.; Spolcova, A.; Blechova, M.; Drapalova, J.; Lacinova, Z.; Haluzik, M.; et al. The Peptidic GHS-R antagonist [D-Lys(3)]GHRP-6 markedly improves adiposity and related metabolic abnormalities in a mouse model of postmenopausal obesity. Mol. Cell Endocrinol. 2011, 343, 55–62. [Google Scholar] [CrossRef]
- Roh, S.-G.; Hong, Y.-H.; Hishikawa, D.; Tsuzuki, H.; Miyahara, H.; Nishimura, Y.; Goto, C.; Suzuki, Y.K.-C.C.; Lee, H.-G.; Sasaki, S.; et al. Inhibition of growth hormone secretagogue receptor antagonist, [D-Lys-3]-GHRP-6, in adipogenesis of ovine and rat adipocytes. Ani. Sci. J. 2005, 76, 381–386. [Google Scholar] [CrossRef]
- Casey, C.A.; McVicker, B.L.; Donohue, T.M., Jr.; McFarland, M.A.; Wiegert, R.L.; Nanji, A.A. Liver asialoglycoprotein receptor levels correlate with severity of alcoholic liver damage in rats. J. Appl. Physiol. 2004, 96, 76–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieber, C.S.; DeCarli, L.M. Liquid diet technique of ethanol administration: 1989 update. Alcohol Alcohol. 1989, 24, 197–211. [Google Scholar] [PubMed]
- Kim, S.J.; Ju, A.; Lim, S.G.; Kim, D.J. Chronic alcohol consumption, type 2 diabetes mellitus, insulin-like growth factor-I (IGF-I), and growth hormone (GH) in ethanol-treated diabetic rats. Life Sci. 2013, 93, 778–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, B.; Richy, S.; Stricker-Krongrad, A. Feeding response to ghrelin agonist and antagonist in lean and obese Zucker rats. Life Sci. 2005, 76, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Shearman, L.P.; Wang, S.P.; Helmling, S.; Stribling, D.S.; Mazur, P.; Ge, L.; Wang, L.; Klussmann, S.; Macintyre, D.E.; Howard, A.D.; et al. Ghrelin neutralization by a ribonucleic acid-SPM ameliorates obesity in diet-induced obese mice. Endocrinology 2006, 147, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.J.; Yao, M.; Yang, J.; Cai, Y.; Zheng, W.J.; Wang, L.; Yao, D.B.; Yao, D.F. Mitochondrial carnitine palmitoyl transferase-II inactivity aggravates lipid accumulation in rat hepatocarcinogenesis. World J. Gastroenterol. 2017, 23, 256–264. [Google Scholar] [CrossRef]
- Kintscher, U.; Law, R.E. PPARgamma-mediated insulin sensitization: The importance of fat versus muscle. Am. J. Physiol. Endocrinol. Metab. 2005, 288, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Rasineni, K.; McVicker, B.L.; Tuma, D.J.; McNiven, M.A.; Casey, C.A. Rab GTPases associate with isolated lipid droplets (LDs) and show altered content after ethanol administration: Potential role in alcohol-impaired LD metabolism. Alcohol. Clin. Exp. Res. 2014, 38, 327–335. [Google Scholar] [CrossRef]
- Rasineni, K.; Penrice, D.D.; Natarajan, S.K.; McNiven, M.A.; McVicker, B.L.; Kharbanda, K.K.; Casey, C.A.; Harris, E.N. Alcoholic vs non-alcoholic fatty liver in rats: Distinct differences in endocytosis and vesicle trafficking despite similar pathology. BMC Gastroenterol. 2016, 16, 27. [Google Scholar] [CrossRef]
- Dezaki, K.; Hosoda, H.; Kakei, M.; Hashiguchi, S.; Watanabe, M.; Kangawa, K.; Yada, T. Endogenous ghrelin in pancreatic islets restricts insulin release by attenuating Ca2+ signaling in beta-cells: Implication in the glycemic control in rodents. Diabetes 2004, 53, 3142–3151. [Google Scholar] [CrossRef]
- Dezaki, K.; Kakei, M.; Yada, T. Ghrelin uses Galphai2 and activates voltage-dependent K+ channels to attenuate glucose-induced Ca2+ signaling and insulin release in islet beta-cells: Novel signal transduction of ghrelin. Diabetes 2007, 56, 2319–2327. [Google Scholar] [CrossRef]
- Dezaki, K.; Sone, H.; Koizumi, M.; Nakata, M.; Kakei, M.; Nagai, H.; Hosoda, H.; Kangawa, K.; Yada, T. Blockade of pancreatic islet-derived ghrelin enhances insulin secretion to prevent high-fat diet-induced glucose intolerance. Diabetes 2006, 55, 3486–3493. [Google Scholar] [CrossRef]
- Addolorato, G.; Capristo, E.; Greco, A.V.; Stefanini, G.F.; Gasbarrini, G. Energy expenditure, substrate oxidation, and body composition in subjects with chronic alcoholism: New findings from metabolic assessment. Alcohol. Clin. Exp. Res. 1997, 21, 962–967. [Google Scholar] [CrossRef]
- Addolorato, G.; Capristo, E.; Greco, A.V.; Stefanini, G.F.; Gasbarrini, G. Influence of chronic alcohol abuse on body weight and energy metabolism: Is excess ethanol consumption a risk factor for obesity or malnutrition? J. Intern. Med. 1998, 244, 387–395. [Google Scholar] [CrossRef]
- Barazzoni, R.; Bosutti, A.; Stebel, M.; Cattin, M.R.; Roder, E.; Visintin, L.; Cattin, L.; Biolo, G.; Zanetti, M.; Guarnieri, G. Ghrelin regulates mitochondrial-lipid metabolism gene expression and tissue fat distribution in liver and skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2005, 288, 228–235. [Google Scholar] [CrossRef]
- Sparks, J.D.; Sparks, C.E. Insulin regulation of triacylglycerol-rich lipoprotein synthesis and secretion. Biochim. Biophys. Acta 1994, 1215, 9–32. [Google Scholar] [CrossRef]
- Flier, J.S.; Underhill, L.H.; Dinneen, S.; Gerich, J.; Rizza, R. Carbohydrate metabolism in non-insulin-dependent diabetes mellitus. N. Engl. J. Med. 1992, 327, 707–713. [Google Scholar]
- Saltiel, A.R.; Kahn, C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef]
- Zhong, W.; Zhao, Y.; Tang, Y.; Wei, X.; Shi, X.; Sun, W.; Sun, X.; Yin, X.; Sun, X.; Kim, S.; et al. Chronic alcohol exposure stimulates adipose tissue lipolysis in mice: Role of reverse triglyceride transport in the pathogenesis of alcoholic steatosis. Am. J. Pathol. 2012, 180, 998–1007. [Google Scholar] [CrossRef]
- Sun, X.; Tang, Y.; Tan, X.; Li, Q.; Zhong, W.; Sun, X.; Jia, W.; McClain, C.J.; Zhou, Z. Activation of peroxisome proliferator-activated receptor-gamma by rosiglitazone improves lipid homeostasis at the adipose tissue-liver axis in ethanol-fed mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, 548–557. [Google Scholar] [CrossRef]
- Wang, N.; Kong, R.; Luo, H.; Xu, X.; Lu, J. Peroxisome Proliferator-Activated Receptors Associated with Nonalcoholic Fatty Liver Disease. PPAR Res. 2017, 2017, 6561701. [Google Scholar] [CrossRef]
- Hougland, J.L. Ghrelin octanoylation by ghrelin O-acyltransferase: Unique protein biochemistry underlying metabolic signaling. Biochem. Soc. Trans. 2019, 47, 169–178. [Google Scholar] [CrossRef]
- Al-Massadi, O.; Muller, T.; Tschop, M.; Dieguez, C.; Nogueiras, R. Ghrelin and LEAP-2: Rivals in Energy Metabolism. Trends Pharmacol. Sci. 2018, 39, 685–694. [Google Scholar] [CrossRef]
- Ge, X.; Yang, H.; Bednarek, M.A.; Galon-Tilleman, H.; Chen, P.; Chen, M.; Lichtman, J.S.; Wang, Y.; Dalmas, O.; Yin, Y.; et al. LEAP2 Is an Endogenous Antagonist of the Ghrelin Receptor. Cell Metab. 2018, 27, 461–469. [Google Scholar] [CrossRef]
- Birdsall, N.J. Class A GPCR heterodimers: Evidence from binding studies. Trends Pharmacol. Sci. 2010, 31, 499–508. [Google Scholar] [CrossRef]
- Gonzalez-Maeso, J. GPCR oligomers in pharmacology and signaling. Mol. Brain 2011, 4, 20. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| CPT2 | CCCTTAGAGATTCAGGCACATC | TAGGCAGAGGCAGAAGACAGCA |
| PPARγ | GAGATCCTCCTGTTGACCCAG | CCACAGAGCTGATTCCGAAGT |
| PPARα | GTCCTCTGGTTGTCCCCTTG | GTCAGTTCACAGGGAAGGCA |
| FAS | TCCCAGGTCTTGCCGTGC | GCGGATGCCTAGGATGTGTGC |
| ATGL | CCCGGTTGTCCCCCAGGAAGA | TCCAGCAGGGCCTCGTTGAGT |
| FATP2 | AGT ACA TCG GTG AAC TGC TTC GGT | TGC CTT CAG TGG AAG CGT AGA ACT |
| FATP5 | TTC AGG GAC CAC TGG ACT TCC AAA | ACC ACA TCA TCA GCT GTT CTC CCA |
| CD36 | AACCCAGAGGAAGTGGCAAAG | GACAGTGAAGGCTCAAAGATGG |
| ACC | TGAGGAGGACCGCATTTATC | GAAGCTTCCTTCGTGACCAG |
| Control | Control + DLys | Ethanol | Ethanol + DLys | |
|---|---|---|---|---|
| Food Intake/Day (ml) | Pair-fed | Pair-fed | 93 ± 2.93 | 85 ± 6.32 |
| Body Weight (g) | 414 ± 23 | 423 ± 16 | 382 ± 18 | 387 ± 11 |
| Relative Liver Weight (g/100 g Body Weight) | 3.03 ± 0.04a | 3.14 ± 0.09a | 3.79 ± 0.32b | 3.54 ± 0.09b |
| Relative Adipose Weight (g/100 g Body Weight) | 2.16 ± 0.05a | 1.93 ± 0.15a | 1.50 ± 0.13b | 1.67 ± 0.13a,b |
| Serum ALT (U/L) | 52.2 ± 3.97a | 53.32 ± 1.99a | 70.76 ± 5.25b | 60.87 ± 3.75b |
| Serum AST (U/L) | 51.2 ± 0.97a | 56.0 ± 4.82a | 81.0 ± 6.15b | 69.8 ± 1.38b |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rasineni, K.; Kubik, J.L.; Casey, C.A.; Kharbanda, K.K. Inhibition of Ghrelin Activity by Receptor Antagonist [d-Lys-3] GHRP-6 Attenuates Alcohol-Induced Hepatic Steatosis by Regulating Hepatic Lipid Metabolism. Biomolecules 2019, 9, 517. https://doi.org/10.3390/biom9100517
Rasineni K, Kubik JL, Casey CA, Kharbanda KK. Inhibition of Ghrelin Activity by Receptor Antagonist [d-Lys-3] GHRP-6 Attenuates Alcohol-Induced Hepatic Steatosis by Regulating Hepatic Lipid Metabolism. Biomolecules. 2019; 9(10):517. https://doi.org/10.3390/biom9100517
Chicago/Turabian StyleRasineni, Karuna, Jacy L. Kubik, Carol A. Casey, and Kusum K. Kharbanda. 2019. "Inhibition of Ghrelin Activity by Receptor Antagonist [d-Lys-3] GHRP-6 Attenuates Alcohol-Induced Hepatic Steatosis by Regulating Hepatic Lipid Metabolism" Biomolecules 9, no. 10: 517. https://doi.org/10.3390/biom9100517