Macrophage Migration Inhibitory Factor (MIF) Expression Increases during Myocardial Infarction and Supports Pro-Inflammatory Signaling in Cardiac Fibroblasts

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Model
2.2. Cell Culture
2.3. Simulated Ischemia, Mechanical Stretch and Cytokine Stimulation in Cell Culture Experiments
2.4. Histology and Immunofluorescence
2.5. RNA Isolation
2.6. Reverse Transcription and Gene Expression Analysis
2.7. Statistics
3. Results
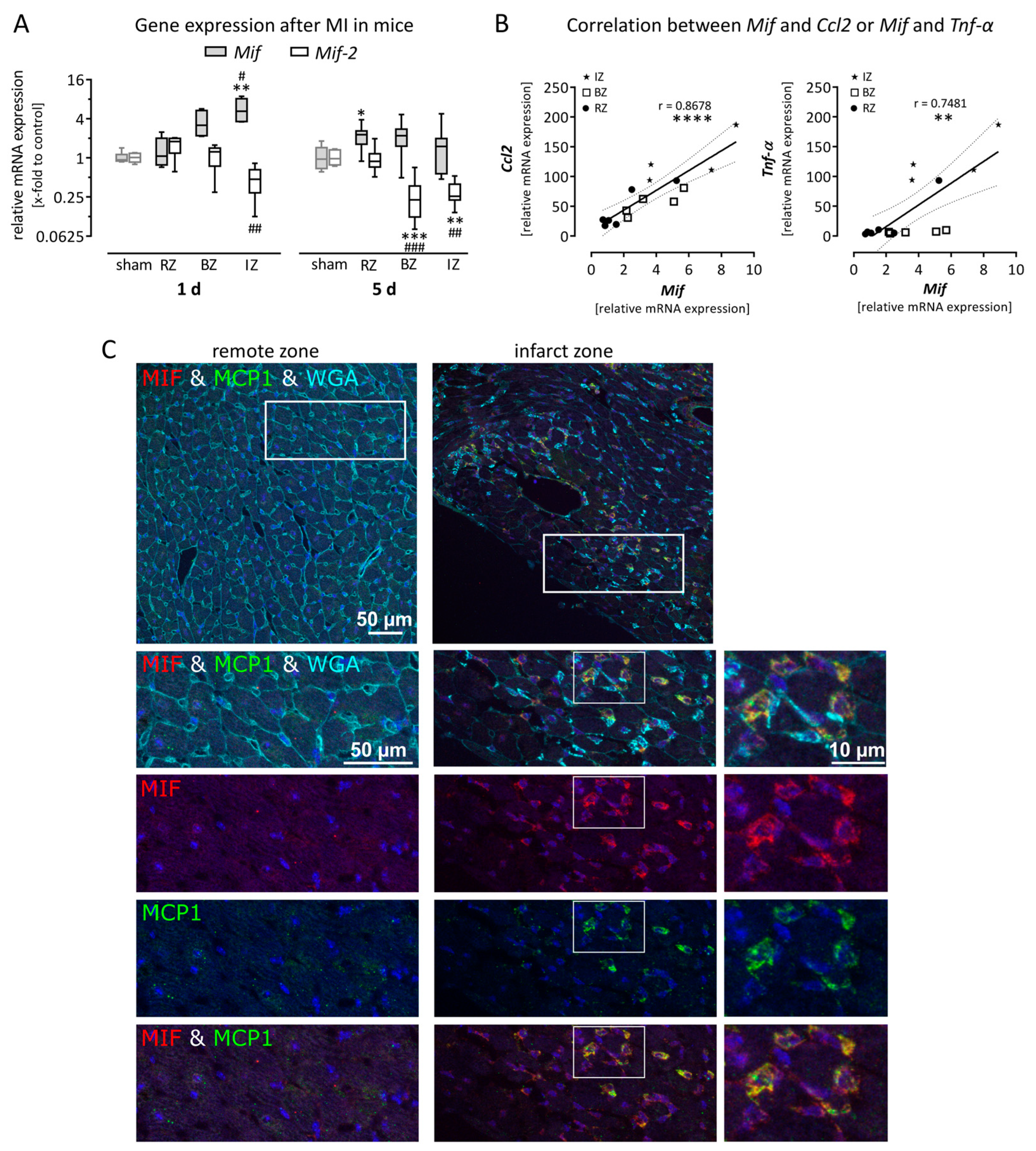
3.1. Expression of Macrophage Migration Inhibitory Factor is Upregulated in Mice After Myocardial Infarction
3.2. Gene Expression of Mif Receptors Cxcr4 as Well as Cd74/Cd44 Was Increased after Myocardial Infarction as a Result of Recruited Leukocytes
3.3. Macrophage Migration Inhibitory Factor is Expressed in Murine Left Ventricular Tissue and Upregulated in Activated Cardiac Cells
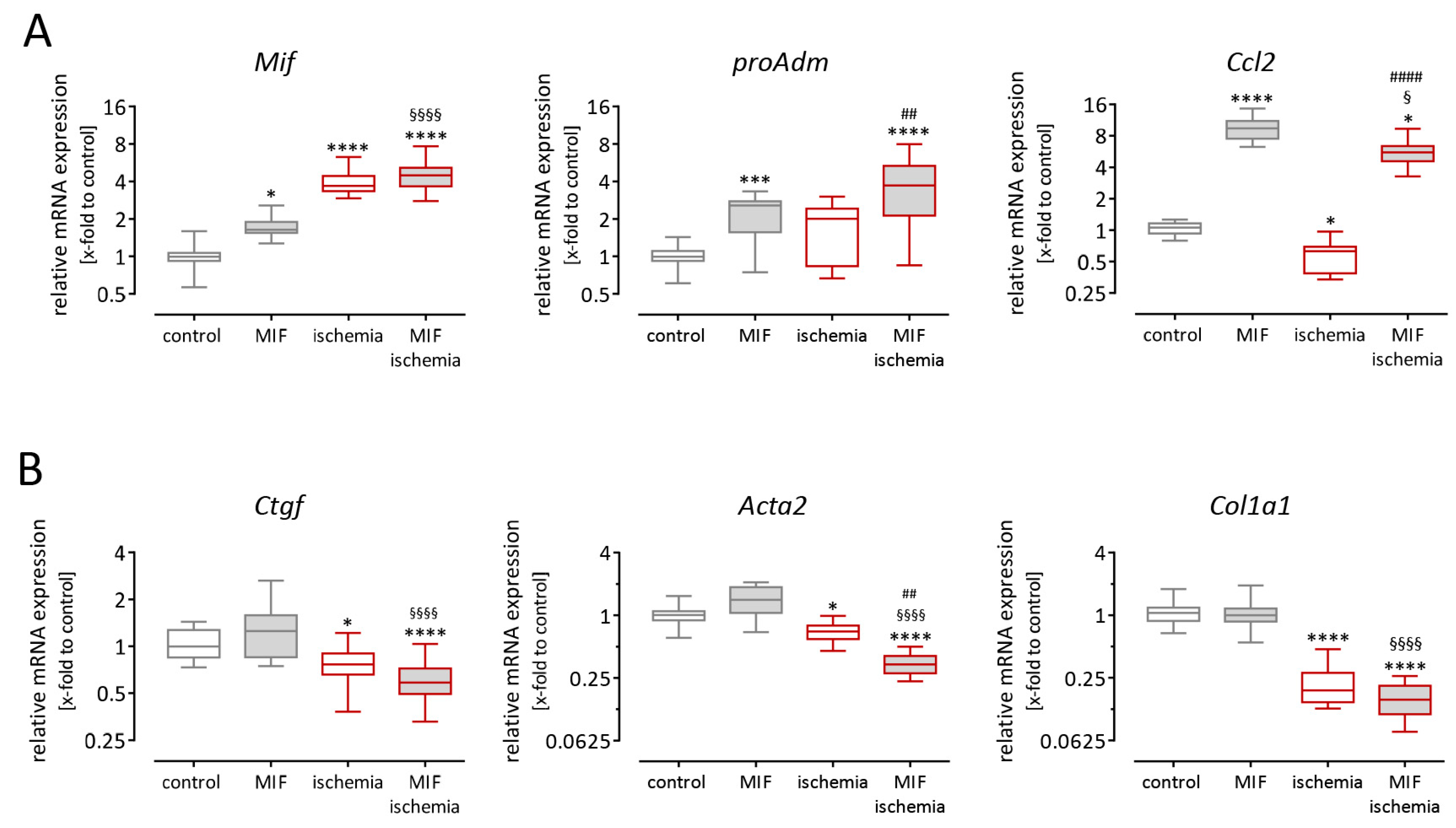
3.4. Recombinant Macrophage Migration Inhibitory Factor Induced Paracrine Effects on Cardiac Fibroblasts but Not on Cardiomyocytes
3.5. Gene Expression in Cardiac Fibroblasts is Altered after Stimulation with Macrophage Migration Inhibitory Factor and Simulated Ischemia
4. Discussion
4.1. Cardio-Protective Function of Macrophage Migration Inhibitory Factor
4.2. Cellular Origin of Macrophage Migration Inhibitory Factor after Myocardial Infarction
4.3. Macrophage Migration Inhibitory Factor and Cardiac Inflammation after Myocardial Infarction
4.4. Macrophage Migration Inhibitory Factor—Utilizing Its Cardio-Protective Function but Treating the Pro-Inflammatory Function?
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Simoons, M.L.; Chaitman, B.R.; White, H.D.; Thygesen, K.; Alpert, J.S.; White, H.D.; Jaffe, A.S.; et al. Third universal definition of myocardial infarction. Eur. Heart J. 2012, 33, 2551–2567. [Google Scholar] [CrossRef] [PubMed]
- Lopez, A.D.; Mathers, C.D.; Ezzati, M.; Jamison, D.T.; Murray, C.J. Global and regional burden of disease and risk factors, 2001: Systematic analysis of population health data. Lancet 2006, 367, 1747–1757. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Hinrichs, S.; Scherschel, K.; Kruger, S.; Neumann, J.T.; Schwarzl, M.; Yan, I.; Warnke, S.; Ojeda, F.M.; Zeller, T.; Karakas, M.; et al. Precursor proadrenomedullin influences cardiomyocyte survival and local inflammation related to myocardial infarction. Proc. Natl. Acad. Sci. USA 2018, 115, E8727–E8736. [Google Scholar] [CrossRef] [PubMed]
- White, D.A.; Fang, L.; Chan, W.; Morand, E.F.; Kiriazis, H.; Duffy, S.J.; Taylor, A.J.; Dart, A.M.; Du, X.J.; Gao, X.M. Pro-inflammatory action of MIF in acute myocardial infarction via activation of peripheral blood mononuclear cells. PLoS ONE 2013, 8, e76206. [Google Scholar] [CrossRef]
- Lindner, D.; Zietsch, C.; Tank, J.; Sossalla, S.; Fluschnik, N.; Hinrichs, S.; Maier, L.; Poller, W.; Blankenberg, S.; Schultheiss, H.P.; et al. Cardiac fibroblasts support cardiac inflammation in heart failure. Basic Res. Cardiol. 2014, 109, 428. [Google Scholar] [CrossRef] [PubMed]
- Westermann, D.; Lindner, D.; Kasner, M.; Zietsch, C.; Savvatis, K.; Escher, F.; von Schlippenbach, J.; Skurk, C.; Steendijk, P.; Riad, A.; et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ. Heart Fail. 2011, 4, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Tilstam, P.V.; Qi, D.; Leng, L.; Young, L.; Bucala, R. MIF family cytokines in cardiovascular diseases and prospects for precision-based therapeutics. Expert Opin. Ther. Targets 2017, 21, 671–683. [Google Scholar] [CrossRef]
- van Zuylen, V.L.; den Haan, M.C.; Geutskens, S.B.; Roelofs, H.; Fibbe, W.E.; Schalij, M.J.; Atsma, D.E. Post-myocardial infarct inflammation and the potential role of cell therapy. Cardiovasc. Drugs Ther. 2015, 29, 59–73. [Google Scholar] [CrossRef]
- Lolis, E.; Bucala, R. Crystal structure of macrophage migration inhibitory factor (MIF), a glucocorticoid-induced regulator of cytokine production, reveals a unique architecture. Proc. Assoc. Am. Physicians 1996, 108, 415–419. [Google Scholar]
- Muhlhahn, P.; Bernhagen, J.; Czisch, M.; Georgescu, J.; Renner, C.; Ross, A.; Bucala, R.; Holak, T.A. NMR characterization of structure, backbone dynamics, and glutathione binding of the human macrophage migration inhibitory factor (MIF). Protein Sci. 1996, 5, 2095–2103. [Google Scholar] [CrossRef] [PubMed]
- Baugh, J.A.; Chitnis, S.; Donnelly, S.C.; Monteiro, J.; Lin, X.; Plant, B.J.; Wolfe, F.; Gregersen, P.K.; Bucala, R. A functional promoter polymorphism in the macrophage migration inhibitory factor (MIF) gene associated with disease severity in rheumatoid arthritis. Genes Immun. 2002, 3, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Calandra, T.; Echtenacher, B.; Roy, D.L.; Pugin, J.; Metz, C.N.; Hultner, L.; Heumann, D.; Mannel, D.; Bucala, R.; Glauser, M.P. Protection from septic shock by neutralization of macrophage migration inhibitory factor. Nat. Med. 2000, 6, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Morand, E.F.; Bucala, R.; Leech, M. Macrophage migration inhibitory factor: An emerging therapeutic target in rheumatoid arthritis. Arthritis Rheum. 2003, 48, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Morand, E.F.; Leech, M.; Bernhagen, J. MIF: A new cytokine link between rheumatoid arthritis and atherosclerosis. Nat. Rev. Drug Discov. 2006, 5, 399–410. [Google Scholar] [CrossRef] [PubMed]
- David, J.R. Delayed hypersensitivity in vitro: Its mediation by cell-free substances formed by lymphoid cell-antigen interaction. Proc. Natl. Acad. Sci. USA 1966, 56, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Bloom, B.R.; Bennett, B. Mechanism of a reaction in vitro associated with delayed-type hypersensitivity. Science 1966, 153, 80–82. [Google Scholar] [CrossRef]
- Takahashi, M.; Nishihira, J.; Shimpo, M.; Mizue, Y.; Ueno, S.; Mano, H.; Kobayashi, E.; Ikeda, U.; Shimada, K. Macrophage migration inhibitory factor as a redox-sensitive cytokine in cardiac myocytes. Cardiovasc. Res. 2001, 52, 438–445. [Google Scholar] [CrossRef]
- Roger, T.; David, J.; Glauser, M.P.; Calandra, T. MIF regulates innate immune responses through modulation of Toll-like receptor 4. Nature 2001, 414, 920–924. [Google Scholar] [CrossRef]
- Jian, Z.; Li, J.B.; Ma, R.Y.; Chen, L.; Zhong, Q.J.; Wang, X.F.; Wang, W.; Hong, Y.; Xiao, Y.B. Increase of macrophage migration inhibitory factor (MIF) expression in cardiomyocytes during chronic hypoxia. Clin. Chim. Acta 2009, 405, 132–138. [Google Scholar] [CrossRef]
- Gregory, J.L.; Leech, M.T.; David, J.R.; Yang, Y.H.; Dacumos, A.; Hickey, M.J. Reduced leukocyte-endothelial cell interactions in the inflamed microcirculation of macrophage migration inhibitory factor-deficient mice. Arthritis Rheum. 2004, 50, 3023–3034. [Google Scholar] [CrossRef] [PubMed]
- Lan, H.Y.; Bacher, M.; Yang, N.; Mu, W.; Nikolic-Paterson, D.J.; Metz, C.; Meinhardt, A.; Bucala, R.; Atkins, R.C. The pathogenic role of macrophage migration inhibitory factor in immunologically induced kidney disease in the rat. J. Exp. Med. 1997, 185, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Bernhagen, J.; Krohn, R.; Lue, H.; Gregory, J.L.; Zernecke, A.; Koenen, R.R.; Dewor, M.; Georgiev, I.; Schober, A.; Leng, L. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat. Med. 2007, 13, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Leng, L.; Metz, C.N.; Fang, Y.; Xu, J.; Donnelly, S.; Baugh, J.; Delohery, T.; Chen, Y.; Mitchell, R.A.; Bucala, R. MIF signal transduction initiated by binding to CD74. J. Exp. Med. 2003, 197, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Leng, L.; Wang, T.; Wang, W.; Du, X.; Li, J.; McDonald, C.; Chen, Z.; Murphy, J.W.; Lolis, E.; et al. CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex. Immunity 2006, 25, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Merk, M.; Mitchell, R.A.; Endres, S.; Bucala, R. D-dopachrome tautomerase (D-DT or MIF-2): Doubling the MIF cytokine family. Cytokine 2012, 59, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Merk, M.; Zierow, S.; Leng, L.; Das, R.; Du, X.; Schulte, W.; Fan, J.; Lue, H.; Chen, Y.; Xiong, H.; et al. The D-dopachrome tautomerase (DDT) gene product is a cytokine and functional homolog of macrophage migration inhibitory factor (MIF). Proc. Natl. Acad. Sci. USA 2011, 108, E577–E585. [Google Scholar] [CrossRef]
- Takahashi, M.; Nishihira, J.; Katsuki, T.; Kobayashi, E.; Ikeda, U.; Shimada, K. Elevation of plasma levels of macrophage migration inhibitory factor in patients with acute myocardial infarction. Am. J. Cardiol. 2002, 89, 248–249. [Google Scholar] [CrossRef]
- Yu, C.M.; Lau, C.P.; Lai, K.W.; Huang, X.R.; Chen, W.H.; Lan, H.Y. Elevation of plasma level of macrophage migration inhibitory factor in patients with acute myocardial infarction. Am. J. Cardiol. 2001, 88, 774–777. [Google Scholar] [CrossRef]
- Chan, W.; White, D.A.; Wang, X.Y.; Bai, R.F.; Liu, Y.; Yu, H.Y.; Zhang, Y.Y.; Fan, F.; Schneider, H.G.; Duffy, S.J.; et al. Macrophage migration inhibitory factor for the early prediction of infarct size. J. Am. Heart Assoc. 2013, 2, e000226. [Google Scholar] [CrossRef]
- Miller, E.J.; Li, J.; Leng, L.; McDonald, C.; Atsumi, T.; Bucala, R.; Young, L.H. Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature 2008, 451, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.M.; Liu, Y.; White, D.; Su, Y.; Drew, B.G.; Bruce, C.R.; Kiriazis, H.; Xu, Q.; Jennings, N.; Bobik, A.; et al. Deletion of macrophage migration inhibitory factor protects the heart from severe ischemia-reperfusion injury: A predominant role of anti-inflammation. J. Mol. Cell. Cardiol. 2011, 50, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Rassaf, T.; Weber, C.; Bernhagen, J. Macrophage migration inhibitory factor in myocardial ischaemia/reperfusion injury. Cardiovasc. Res. 2014, 102, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Westermann, D.; Riad, A.; Lettau, O.; Roks, A.; Savvatis, K.; Becher, P.M.; Escher, F.; Jan Danser, A.H.; Schultheiss, H.P.; Tschope, C. Renin inhibition improves cardiac function and remodeling after myocardial infarction independent of blood pressure. Hypertension 2008, 52, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Claycomb, W.C.; Lanson, N.A., Jr.; Stallworth, B.S.; Egeland, D.B.; Delcarpio, J.B.; Bahinski, A.; Izzo, N.J., Jr. Hl-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. USA 1998, 95, 2979–2984. [Google Scholar] [CrossRef] [PubMed]
- Jungen, C.; Scherschel, K.; Eickholt, C.; Kuklik, P.; Klatt, N.; Bork, N.; Salzbrunn, T.; Alken, F.; Angendohr, S.; Klene, C.; et al. Disruption of cardiac cholinergic neurons enhances susceptibility to ventricular arrhythmias. Nat. Commun. 2017, 8, 14155. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔCTmethod. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Dayawansa, N.H.; Gao, X.M.; White, D.A.; Dart, A.M.; Du, X.J. Role of MIF in myocardial ischaemia and infarction: Insight from recent clinical and experimental findings. Clin. Sci. 2014, 127, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Qi, D.; Hu, X.; Wu, X.; Merk, M.; Leng, L.; Bucala, R.; Young, L.H. Cardiac macrophage migration inhibitory factor inhibits JNK pathway activation and injury during ischemia/reperfusion. J. Clin. Investig. 2009, 119, 3807–3816. [Google Scholar] [CrossRef] [PubMed]
- Koga, K.; Kenessey, A.; Powell, S.R.; Sison, C.P.; Miller, E.J.; Ojamaa, K. Macrophage migration inhibitory factor provides cardioprotection during ischemia/reperfusion by reducing oxidative stress. Antioxid. Redox Signal. 2011, 14, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Hua, Y.; Nair, S.; Bucala, R.; Ren, J. Macrophage migration inhibitory factor deletion exacerbates pressure overload-induced cardiac hypertrophy through mitigating autophagy. Hypertension 2014, 63, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Tong, C.; Morrison, A.; Yan, X.; Zhao, P.; Yeung, E.D.; Wang, J.; Xie, J.; Li, J. Macrophage migration inhibitory factor deficiency augments cardiac dysfunction in type 1 diabetic murine cardiomyocytes. J. Diabetes 2010, 2, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Bucala, R.; Ren, J. Macrophage migration inhibitory factor deficiency augments doxorubicin-induced cardiomyopathy. J. Am. Heart Assoc. 2013, 2, e000439. [Google Scholar] [CrossRef] [PubMed]
- White, D.A.; Su, Y.; Kanellakis, P.; Kiriazis, H.; Morand, E.F.; Bucala, R.; Dart, A.M.; Gao, X.M.; Du, X.J. Differential roles of cardiac and leukocyte derived macrophage migration inhibitory factor in inflammatory responses and cardiac remodelling post myocardial infarction. J. Mol. Cell. Cardiol. 2014, 69, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.S.; Tilstam, P.V.; Hwang, S.S.; Simons, D.; Schulte, W.; Leng, L.; Sauler, M.; Ganse, B.; Averdunk, L.; Kopp, R.; et al. D-dopachrome tautomerase in adipose tissue inflammation and wound repair. J. Cell. Mol. Med. 2017, 21, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.S.; Rongisch, R.; Hager, S.; Grieb, G.; Nourbakhsh, M.; Rennekampff, H.O.; Bucala, R.; Bernhagen, J.; Pallua, N. Macrophage migration inhibitory factor in acute adipose tissue inflammation. PLoS ONE 2015, 10, e0137366. [Google Scholar] [CrossRef] [PubMed]
- Kerschbaumer, R.J.; Rieger, M.; Volkel, D.; Le Roy, D.; Roger, T.; Garbaraviciene, J.; Boehncke, W.H.; Mullberg, J.; Hoet, R.M.; Wood, C.R.; et al. Neutralization of macrophage migration inhibitory factor (MIF) by fully human antibodies correlates with their specificity for the beta-sheet structure of MIF. J. Biol. Chem. 2012, 287, 7446–7455. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Species | Dilution | Company | Catalogue No.# |
|---|---|---|---|---|
| Primary antibodies | ||||
| anti-MIF (FL-115) | Rabbit | 1:100 | Santa Cruz | sc-20121 |
| anti-MCP-1 (ECE.2) | Rat | 1:50 | abcam | ab8101 |
| Secondary antibodies | ||||
| anti-rabbit IgG (H + L) Alexa-568 | Donkey | 1:500 | Life technologies | A10042 |
| anti-rat IgG (H + L) Alexa-488 | Donkey | 1:500 | Life technologies | A21208 |
| Gene Symbol | Gene Name | Assay ID |
|---|---|---|
| mActa2 | α smooth muscle actin | Mm00725412_s1 |
| mCcl2 | chemokine (C-C motif) ligand 2 | Mm99999056_m1 |
| mCd44 | cluster of differentiation 44 | Mm01277161_m1 |
| mCd74 | cluster of differentiation 74 | Mm00658576_m1 |
| mCdkn1b | cyclin-dependent kinase inhibitor 1B | Mm00438167_g1 |
| mCol1a1 | collagen type I, α 1 | Mm01302043_g1 |
| mCtgf | connective tissue growth factor | Mm00515790_g1 |
| mCxcr4 | chemokine (C-X-C motif) receptor 4 | Mm01996749_s1 |
| mDdt (Mif-2) | D-dopachrome tautomerase | Mm00515641_m1 |
| mMif | macrophage migration inhibitory factor | Mm01611157_gH |
| mproAdm | pro-adrenomedullin | Mm00437438_g1 |
| mTnfα | tumor necrosis factor α | Mm00443258_m1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voss, S.; Krüger, S.; Scherschel, K.; Warnke, S.; Schwarzl, M.; Schrage, B.; Girdauskas, E.; Meyer, C.; Blankenberg, S.; Westermann, D.; et al. Macrophage Migration Inhibitory Factor (MIF) Expression Increases during Myocardial Infarction and Supports Pro-Inflammatory Signaling in Cardiac Fibroblasts. Biomolecules 2019, 9, 38. https://doi.org/10.3390/biom9020038
Voss S, Krüger S, Scherschel K, Warnke S, Schwarzl M, Schrage B, Girdauskas E, Meyer C, Blankenberg S, Westermann D, et al. Macrophage Migration Inhibitory Factor (MIF) Expression Increases during Myocardial Infarction and Supports Pro-Inflammatory Signaling in Cardiac Fibroblasts. Biomolecules. 2019; 9(2):38. https://doi.org/10.3390/biom9020038
Chicago/Turabian StyleVoss, Svenja, Saskia Krüger, Katharina Scherschel, Svenja Warnke, Michael Schwarzl, Benedikt Schrage, Evaldas Girdauskas, Christian Meyer, Stefan Blankenberg, Dirk Westermann, and et al. 2019. "Macrophage Migration Inhibitory Factor (MIF) Expression Increases during Myocardial Infarction and Supports Pro-Inflammatory Signaling in Cardiac Fibroblasts" Biomolecules 9, no. 2: 38. https://doi.org/10.3390/biom9020038