
Primary Cilia Dysfunction in Neurodevelopmental Disorders beyond Ciliopathies

1
The Rosamund Stone Zander Translational Neuroscience Center, Department of Neurology Boston Children’s Hospital, Harvard Medical School, Boston, MA 02115, USA
2
FM Kirby Neurobiology Center, Boston Children’s Hospital, Boston, MA 02115, USA
*
Author to whom correspondence should be addressed.
J. Dev. Biol. 2022, 10(4), 54; https://doi.org/10.3390/jdb10040054
Submission received: 4 November 2022
/
Revised: 4 December 2022
/
Accepted: 9 December 2022
/
Published: 13 December 2022
(This article belongs to the Special Issue Cilia in Development)
Abstract
:Primary cilia are specialized, microtubule-based structures projecting from the surface of most mammalian cells. These organelles are thought to primarily act as signaling hubs and sensors, receiving and integrating extracellular cues. Several important signaling pathways are regulated through the primary cilium including Sonic Hedgehog (Shh) and Wnt signaling. Therefore, it is no surprise that mutated genes encoding defective proteins that affect primary cilia function or structure are responsible for a group of disorders collectively termed ciliopathies. The severe neurologic abnormalities observed in several ciliopathies have prompted examination of primary cilia structure and function in other brain disorders. Recently, neuronal primary cilia defects were observed in monogenic neurodevelopmental disorders that were not traditionally considered ciliopathies. The molecular mechanisms of how these genetic mutations cause primary cilia defects and how these defects contribute to the neurologic manifestations of these disorders remain poorly understood. In this review we will discuss monogenic neurodevelopmental disorders that exhibit cilia deficits and summarize findings from studies exploring the role of primary cilia in the brain to shed light into how these deficits could contribute to neurologic abnormalities.
1. Introduction
The first observation of cilia was reported in 1674–1675, when Antoni van Leeuwenhoek was studying pond protozoa and noticed small hairlike structures that he described as “incredibly thin feet, which moved very nimbly” [1]. Later, these structures were identified as cilia, a highly conserved organelle that is currently classified into two groups: motile and non-motile cilia, with the latter group being much less understood. Both motile and non-motile cilia contain an axoneme, a structure made of nine peripheral microtubule doublets. In motile cilia, there is a central pair of microtubules (referred to as 9 + 2 arrangement), which is lacking in non-motile cilia (referred to as 9 + 0 arrangement). However, not all non-motile cilia conform to this 9 + 0 arrangement and among the ones that do, some exhibit motility, suggesting that there might be structural variability within these groups [2,3,4]. Non-motile cilia, today called primary cilia, are present in almost all types of cells in the body and are thought to serve mainly as ‘the cell’s antennae’, receiving and integrating extracellular chemical and mechanical signals.
Although primary cilia were observed as early as 1898 [5], their study was delayed for almost 50 years due to a lack of tools [4]. Only after the advent of transmission electron microscopy (TEM) and its subsequent commercialization did the primary cilia field gain minor traction in the 1950s and 60s [6,7,8]. For most of the 20th century, primary cilia were considered “rudimentary” or “vestigial” organelles [9,10]. However, in 1992, the ciliary intraflagellar transport (IFT) system was discovered [11] and associated with polycystic kidney disease [12], indicating that these organelles had a more important role than previously thought. These discoveries catalyzed our current ‘Golden Age’ of primary cilia research, in which much has been elucidated about primary cilia’s biological relevance (Figure 1).
The primary cilium’s function is closely related to the cell cycle. Specifically, primary cilia formation occurs during the G1/G0 phase, followed by resorption upon cell cycle re-entry [13]. Ciliogenesis is initiated upon formation of the basal body from the centrosome (the mother and daughter centriole pair). The basal body docks at the apical plasma membrane, and microtubules nucleate to begin axoneme formation [14], with the axoneme projecting outward beneath the plasma membrane [15]. The region where the basal body and axoneme meet is called the transition zone, and it is required for cilium compartmentalization. In the transition zone, Y-shaped protein link the axoneme to the plasma membrane, restricting the movement of proteins and lipids into and out of the ciliary compartment [16]. To elongate the cilium, proteins are selectively imported and transported to the ciliary tip via anterograde transport by the IFT system. Anterograde and retrograde ciliary transport are regulated by distinct IFT complex/motor protein pairs: IFT-B with homotrimeric Kinesin-2, and IFT-A with cytoplasmic dynein 2, respectively [15,17]. Proteins which enter the cilium via anterograde transport can be removed through retrograde transport or secreted within ciliary ectosomes to the extracellular space [18,19].
The primary cilium actively imports receptors and signaling molecules [20,21] making it uniquely suited to sense and integrate signals. Several signaling pathways are coordinated through the primary cilium, including Sonic Hedgehog (Shh), G-protein-coupled receptors (GPCR), platelet-derived growth factor receptor α (PDGFα), fibroblast growth factor (FGF), transforming growth factor β (TGF-β), Wnt, Hippo, Notch, and mechanistic target of rapamycin (mTOR) signaling. These pathways have been extensively reviewed elsewhere [21,22].
The first evidence for the presence of primary cilia on neurons was reported in 1969 [23]. However, their contribution to brain development and function was not investigated until the early 21st century [24,25,26] (Figure 1). Today it is known that the majority of mature neurons and astrocytes in the central nervous system (CNS) have a primary cilium, but so far primary cilia have not been detected on mature oligodendrocytes or microglia [27]. Mutations in genes that affect primary cilia have major neurologic consequences and are currently an active field of research. Interestingly, recent studies identified impaired neuronal primary cilia in monogenic neurodevelopmental disorders in which the disease-causing mutations do not have clear links to the primary cilium. In this review we discuss (1) monogenic neurodevelopmental disorders that were shown to exhibit impaired ciliation and (2) evidence that could shed light on the mechanisms via which defective cilia could contribute to these disorders’ neurologic manifestations.
2. Primary Cilia Deficits in CNS Disorders: Ciliopathies and Beyond
Primary cilia host some of the most important signaling pathways for proper brain development and function. Several of these signaling pathways function exclusively through cilia, making these organelles vital cellular compartments [21,22]. Therefore, it is not surprising that defects in cilia underlie several disorders collectively termed “ciliopathies” [28].
Ciliopathies are a group of inherited genetic disorders caused by mutations in genes encoding proteins essential for the structure and function of cilia [29]. Since cilia are components of nearly all human cells, pathological manifestations of ciliopathies are multisystemic, and include retinopathy, obesity, diabetes, skeletal malformations, and hepatic disease [29,30]. Interestingly, certain ciliopathies specifically affect only a subset of organs such as polycystic kidney disease (PKD), which is characterized by multiple cysts in the kidney and liver [29,30].
The importance of cilia function in the brain is underscored by the presence of a wide range of neurologic manifestations in ciliopathies such as Joubert and Bardet–Biedl syndromes. These disorders exhibit brain malformations, ataxia, and cognitive deficits [31,32]. In addition to the role of cilia dysfunction in ciliopathies, cilia deficits have been observed in a broad range of CNS disorders suggesting that these organelles might contribute to the pathology of disorders beyond ciliopathies [33,34,35]. Specifically, it has been proposed that abnormal primary cilia function may be involved in neuropsychiatric conditions, such as schizophrenia and bipolar disorder [33]. In addition, recent reports implicate cilia in neurodegenerative disorders such as Alzheimer’s and Parkinson’s disease [34,35]. These data emphasize the importance of primary cilia in the proper development and function of the brain across the lifespan.
While cilia dysfunction plays a clear role in the development of neurological manifestations in ciliopathies, there is increasing evidence that other monogenetic neurodevelopmental syndromes indirectly impinge on primary cilia function. This has provided the intriguing hypothesis that ciliary dysfunction may be an integral part of the pathological mechanisms underlying these disorders. Therefore, these disorders provide a further framework to investigate (1) novel key players in the molecular mechanisms that regulate primary cilia structure and function and (2) the roles of primary cilia in normal and diseased brain.
3. Primary Cilia Defects in Monogenic Neurodevelopmental Disorders
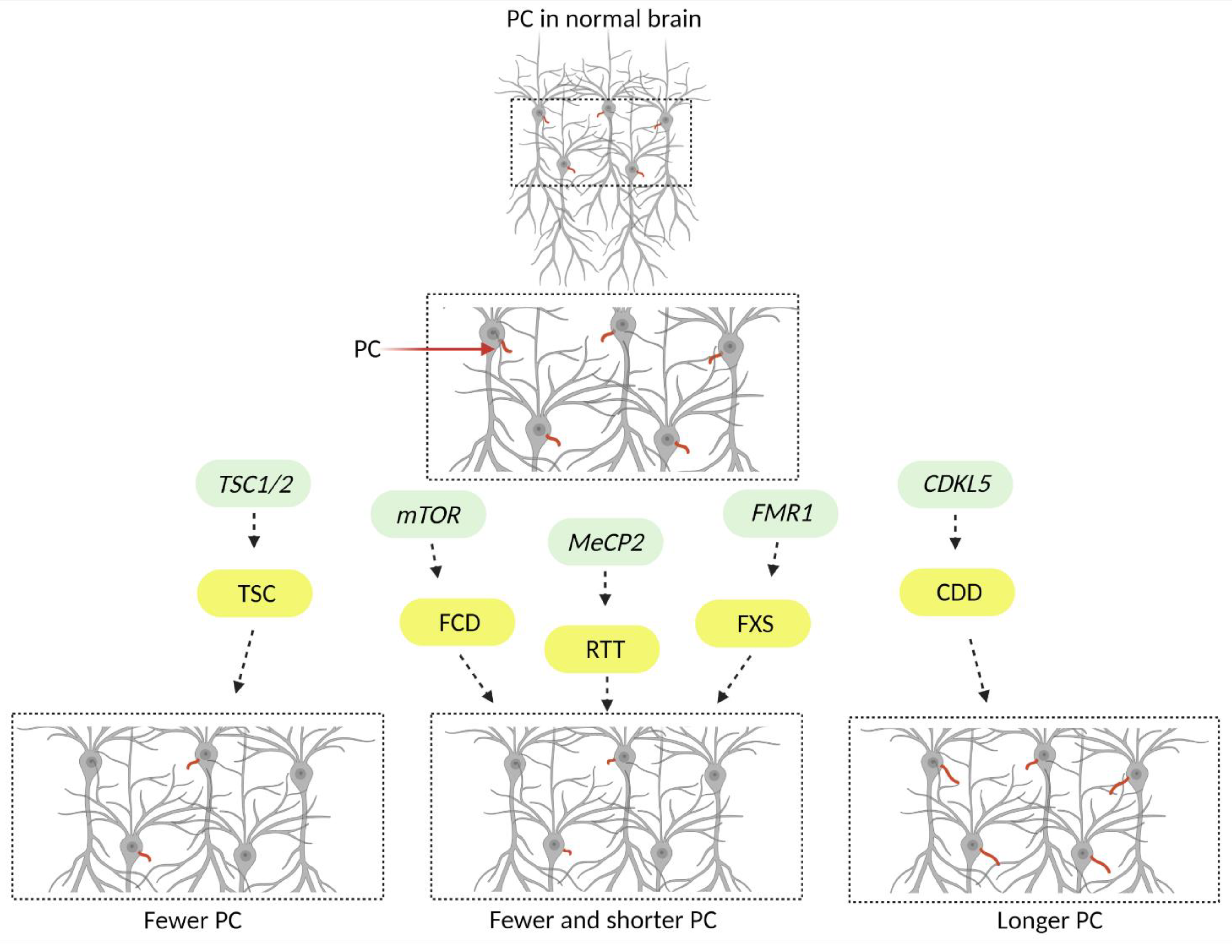
Due to the significant role of neuronal primary cilia in the brain, studies have investigated whether and how these organelles are impacted in developmental disorders with severe neuropsychiatric manifestations. A group of monogenic neurodevelopmental disorders that exhibit several common neurologic manifestations and were found to have defects in neuronal primary cilia are discussed below. Figure 2 and Table 1 summarize the findings of the studies, focusing on neuronal primary cilia.
3.1. Fragile X Syndrome
Fragile X syndrome (FXS) is caused by loss of function of the fragile X messenger ribonucleoprotein (FMRP), which is typically caused by epigenetic silencing of the FMR1 gene due to an expanded CGG repeat in the promoter [50]. FMRP is a widely expressed RNA-binding protein and a translational repressor that has been linked to proper development and maintenance of neuronal synapses. Patients with FXS present with a range of neurological symptoms, including intellectual disability, autism spectrum disorder (ASD), and seizures [36,38].
A recent study by Lee et al. showed that Fmr1-KO mice exhibit reduced primary cilia number and length, within mature granule neurons of the dentate gyrus (DG) after postnatal day 14 (Figure 2, Table 1). Interestingly, these primary cilia abnormalities were not observed in other hippocampal regions [37]. In addition, when the authors examined the two populations that give rise to the granule neuronal population of the DG, they found that cilia were affected only in the subgranular zone (SGZ) newborn neurons and not in neurons originating from the dentate neuroepithelium (DNe) [37]. Notably, while astrocytes of the DG in Fmr1-KO mice did not exhibit ciliary deficits [37], another study from the same group, showed that cerebellar Fmr1-KO Bergmann glia in the Purkinje cell layer of the posterior cerebellum had downregulated Shh signaling and fewer primary cilia after postnatal day 10 [51]. These studies suggest that loss of FMRP may have a cell type- and age-dependent effect on primary cilia, but the mechanism by which FMRP impacts cilia remains unknown.
3.2. Tuberous Sclerosis Complex
Tuberous Sclerosis Complex (TSC) is a developmental multisystem Mendelian disorder that is caused by loss of either the TSC1 or TSC2 genes [40,41,52]. The products of these genes, TSC1 and TSC2 proteins, together with the protein TBC1D7, form a heterotrimeric complex that negatively regulates mTOR signaling pathway [40,41,52]. TSC affects multiple organs, including the brain, and patients often present with various neurological and neuropsychiatric conditions including focal malformations called tubers, ASD, intellectual disability, and early onset epilepsy [40,41,52].
Several studies in different models have showed that loss of Tsc1 or Tsc2 genes from kidney cells and embryonic fibroblasts leads to impaired ciliary structure [53,54,55]. Tsc1-KO or Tsc2-KO mouse embryonic fibroblasts (MEFs) were more likely to be ciliated with elongated cilia in comparison to wild-type control MEFs [53]. Notably, a separate study in Tsc1-KO or Tsc2-KO MEFs observed that while cilia in Tsc1-KO MEFs were elongated, cilia in Tsc2-KO MEFs were shorter when compared to control [54]. Additionally, loss of Tsc1 from mouse in the distal convoluted tubules cells led to elongated cilia [55]. Collectively, these studies indicate that TSC1 and TSC2 proteins are important for cilia function, and further studies are necessary to resolve the discrepancies observed in the cilia phenotypes.
A recent study, by Di Nardo et al., explored how loss of TSC1 and TSC2 genes affects primary cilia in surgically resected patient brain samples and rodent models of TSC [39]. Loss of either gene in patient samples and in vitro or in vivo rodent models led to a reduction in the number of neuronal primary cilia (Figure 2, Table 1). Notably, the authors showed that shRNA mediated knockdown of Tsc2 in rat primary hippocampal neurons resulted in loss of primary cilia over time suggesting that the TSC2 protein function and consequent mTOR suppression may be important for cilia stability and maintenance. Furthermore, deciliation could be prevented by knockdown of the heat shock protein 27 (Hsp27), which was upregulated upon loss of Tsc2 [39].
Interestingly, while rapamycin treatment was able to prevent loss of cilia, it was not sufficient to reverse this phenotype [39] suggesting that there is a critical temporal window for mTOR inhibition to be effective in preventing deciliation. Rapamycin and other related compounds (rapalogs) are FDA approved for treatment of multiple manifestations of TSC. While these drugs are effective at treating some neurologic abnormalities, they are not effective in treating TSC-related cognitive deficits [56,57,58]. Of note, these patients were all treated in late childhood or adolescence, which may be ineffective in preventing loss of cilia. Hence, it would be worthwhile to explore whether development of therapeutics aiming to restore or prevent primary cilia deficits could be a more effective therapeutic strategy for cognitive deficits in TSC and potentially other disorders.
3.3. Focal Cortical Dysplasia
Focal cortical dysplasia (FCD) belongs to a group of disorders that are collectively referred to as focal malformations of cortical development (FMCD) [42,43]. FCD is characterized by the presence of hypertrophic “balloon-like” cells, dysplastic neurons with abnormal orientation and processes, disorganized cortical lamination, and gliosis. FCD is one of the most common underlying causes of refractory pediatric epilepsy and intellectual disability [42,43].
A recent study by Park et al., examined the role of primary cilia in the pathogenesis of FMCDs caused by somatic mutations in the mTOR gene. The authors examined surgically resected tissue from patients and found a reduction in the number and the length of neuronal primary cilia [44] (Figure 2, Table 1). To explore the molecular mechanisms underlying these defects, the authors utilized several models, including in utero electroporated mice that expressed mTOR harboring a somatic mutation identified in FCD patients. Notably, they found that mTOR-related autophagy defects led to accumulation of oral-facial-digital syndrome 1 protein (OFD1) at the centriolar satellites [44], which was previously shown to inhibit ciliogenesis [59]. They showed that Ofd1 suppression was sufficient to restore primary cilia in mTOR mutant cells. They proposed that OFD1 accumulation and impaired ciliogenesis disrupt Wnt signaling, leading to the cortical dyslamination phenotypes observed in their models [44].
Cortical malformations are also present in multiple developmental disorders, including TSC and phosphatase and tensin homolog (PTEN) hamartoma tumor syndrome (PHTS) [60]. Thus, it would be very interesting to examine whether the mechanism identified in this study is also implicated in the cortical malformation phenotypes in these disorders.
3.4. Cyclin-Dependent Kinase-like 5 Deficiency Disorder
CDKL5 (cyclin-dependent kinase-like 5) Deficiency Disorder (CDD) is a rare genetic disorder characterized by severe neurologic manifestations, including infantile epileptic encephalopathy and cognitive disabilities [46,47,61] and has been also associated with ASD [62,63]. The CDKL5 gene is an X-linked gene that encodes a serine/threonine kinase, which is highly expressed in the brain and known to be critical for proper dendritic arborization, axonal growth, and synaptic plasticity [64].
The importance of CDKL5 in cilia function and structure has been proposed by several studies [65,66,67]. Specifically, it has been shown that CDKL5 localizes to the centrosome, controls ciliary length, and is a key component of ciliogenesis in several systems, including C. elegans, Chlamydomonas, and proliferating cells lines such as HeLa [65,66,67]. While there are not many studies describing the role of CDKL5 in neurons, one recent study, using in vitro and in vivo rodent models of CDD showed that Cdkl5 null neurons exhibit increased primary cilia length [45] (Figure 2, Table 1). Even though the relationship between CDKL5 and primary cilia has been explored, the molecular mechanisms underlying this relationship, and the consequences of the neuronal cilia structural changes remain elusive.
3.5. Rett Syndrome
Rett syndrome (RTT) is a monogenic X-linked rare neurodevelopmental disorder. RTT patients exhibit significant developmental regression and intellectual disability [49]. Mutations in the methyl-CpG-binding protein 2 (MeCP2) gene is the leading cause of Rett syndrome [49,68]. MeCP2 is a multifunctional epigenetic regulator that controls the expression of several genes. Notably, MeCP2 recognizes histone methylation marks and can act either as a transcriptional activator or repressor, depending on the presence of various cofactors [69,70,71]. Studies in rodent models explored the effects of MeCP2 loss from the brain and found that this protein is important for energy metabolism and proteostasis [72].
MeCP2 has been shown to localize at the centrosome, affecting the cell cycle and the cytoskeleton stability [73], suggesting that this protein might be important for ciliogenesis. A recent study examined loss of Mecp2 in several mouse models including primary cortical neuronal and astrocytic cultures as well as fibroblasts from RTT patients. The authors found that loss of MeCP2 led to fewer and shorter primary cilia and associated reduction in the Shh signaling pathway activity (Figure 2, Table 1). Interestingly, this phenotype could be rescued with a histone deacetylase 6 (HDAC6) inhibitor suggesting that microtubule instability contributes to the primary cilia deficits observed in these models [48]. Notably, while this study provides strong evidence that support a relationship between MeCP2 function and primary cilia, another study showed that of loss of Mecp2 in mouse retina cells did not affect cilia formation [74]. Taken together, these studies suggest that MeCP2 effect on primary cilia might be cell-type dependent.
4. Ciliary Defects as a Convergent Mechanism for Neurologic Phenotypes
Several of the aforementioned monogenic neurodevelopmental disorders exhibit common neuropsychiatric manifestations including early onset epilepsy, intellectual disability, and ASD. In addition, the majority of these disorders exhibit similar neuronal primary cilia defects, namely fewer and shorter cilia (Table 1). Given the critical role of primary cilia in the brain, it is possible that defects in these organelles contribute to the neuropsychiatric manifestations. It is worth noting however, that primary cilia dysfunction might cause different changes on the molecular level depending on the cell type and the specific genetic perturbation. Hence, while the phenotypic manifestations are shared, different mechanisms could be responsible in each disorder. Therefore, it is important to examine (1) whether and how the genetic perturbations underlying these disorders lead to abnormal cilia structure and function, and (2) if and to what extent, primary cilia dysfunction is involved in this wide range of neurologic abnormalities in each of these disorders. Such studies could open the road for discovery of novel therapeutic targets and development of new treatment strategies.
Crosstalk between Signaling Pathways in Monogenic Neurodevelopmental Disorders
Most studies identifying deficits in primary cilia in monogenic neurodevelopmental disorders have not characterized the mechanisms involved. Some of these studies, however, have suggested that alterations in major cellular functions, such as autophagy, are involved [44,75]. It is unclear whether there is a convergent mechanism or independent mechanisms underlying cilia deficits in each of these diseases. While each disorder likely has unique features, several lines of evidence support bidirectional interactions among key components of the molecular cascades involved in these disorders. Interestingly, many potential shared mechanisms center around direct or indirect mTOR dysregulation [76].
Genetic mutations underlying FCD and TSC affect mTOR signaling directly by altering the balance between activation and inhibition [40,43]. However, there are also proposed interactions between the RTT related protein, MeCP2, and mTOR protein. Specifically, aberrant mTOR signaling has been shown in patients with RTT syndrome [77]. In addition, Mecp2 mutations in mouse models of RTT lead to downregulation of mTOR signaling activity and reduced neuronal size [78], a known phenotype controlled by the mTOR pathway. Interestingly, in Mecp2 null or heterozygous mice, downregulation of the phosphorylated form of ribosomal protein S6 (p-rpS6), a well-established mTOR target, is detectable prior to the appearance of obvious RTT-related neurologic manifestations [79]. mTOR signaling was also found to be altered in FXS [75,80]. For example, Sharma et al. showed increased mTORC1 activity in the hippocampal region of a mouse model of fragile X [80]. Additionally, Yan et al., showed that mTOR-dependent decreased autophagy is responsible for several of the phenotypes observed in Fmr1-KO mice, including spine and synaptic plasticity defects as well as impaired cognition [75]. MeCP2 was also shown to interact with the FMRP. Specifically, a study showed reciprocal regulation between the expression levels of these two proteins both in vitro and in vivo [81].
CDKL5 has also been shown to affect the mTOR pathway [82,83,84]. Studies in Cdkl5 mutant mouse models reveal downregulation of Akt and mTORC1 activity, hence disruption of the Akt/mTOR signaling cascade [83,84]. Notably, one of these studies showed that by boosting phosphorylation of GSK-3b, an Akt downstream target, in Cdkl5 null neuronal precursor cells, several developmental alterations including neuronal survival and maturation were rescued [84]. Another study examined how loss of Cdkl5 affected the mTOR signaling cascade by examining components of the mTOR pathway in different neuronal types. The authors examined cortical excitatory and inhibitory neurons, as well as striatal inhibitory neurons, and observed differential perturbation of the mTOR signaling cascade, suggesting that Cdkl5 affects mTOR in a cell type-dependent manner [82].
The mTOR dysregulation seen in FCD, TSC, RTT, FXS and CDD is noteworthy given that several lines of evidence propose that that mTOR and primary cilia regulate each other [85]. Specifically, primary cilia inhibit mTORC1 activity via several proposed mechanisms involving proteins such as Lkb1, Folliculin, AMPK and polycystin-1 [86,87,88]. Reciprocally, several studies have shown that mTORC1 activity affects cilia formation and length [44,89,90].
Outside of mTOR, MeCP2 and CDKL5 have been shown to interact in various systems. Specifically, it has been shown that MeCP2 can be phosphorylated in a Cdkl5-dependent manner [91,92] and that Cdkl5 is a MeCP2-repressed target gene in the rat brain [93]. In addition, patient stem cells that express mutated MeCP2 or CDKL5 exhibit common phenotypes such as upregulation of glutamate D1 receptor (GluD1) [94].
Taken together these data suggest that there is some crosstalk between components of these signaling pathways and support the hypothesis of a convergent mechanism that could act independently or synergistically with other mechanisms to underlie cilia defects. One of the most intriguing signaling cascades that appears dysregulated in all these disorders is the Akt/mTOR signaling pathway. In FCD, TSC and FXS models Akt/mTOR appears upregulated and the number of primary neuronal cilia is reduced [37,39,40,43,44,75,80]. Additionally, in FCD and FXS, remaining primary cilia are also shorter in length [37,44]. On the other hand, a few studies have shown that in CDD, Akt/mTOR is downregulated and primary neuronal cilia length is increased [45,82,83,84]. Taken together these data suggest that Akt/mTOR activity can bidirectionally affect the number and length of primary neuronal cilia. However, RTT syndrome appears to be an exception to this Akt/mTOR activity–cilia phenotype pattern as mTOR in RTT is downregulated and primary cilia are reduced both in number and length [48,78,79]. One explanation could be that different mechanisms in each genetic perturbations could underlie and/or contribute to the primary cilia phenotypes. Further studies are warranted to elucidate the molecular mechanisms leading to impaired ciliation in these disorders.
5. Impact of Primary Cilia Loss in Neurons and Neuronal Networks
While primary cilia defects have been identified in the neurodevelopmental disorders discussed above, the extent of their contribution to the neurologic abnormalities remains elusive. Neuronal primary cilia regulate a plethora of signaling cascades and extensive studies have shown that their proper function is crucial both in the developing and the adult brain [34,35,95]. These studies, a few of which will be discussed here, can shed light into the potential contributions of primary cilia into brain-wide disease phenotypes such as structural deficits, seizures, and cognitive disabilities. (For a more comprehensive list of studies focusing on mouse models see Table 2 and Reviews [96,97,98]).
Numerous studies have examined the roles of primary cilia in the developing brain [32], since these organelles regulate signaling pathways essential for brain patterning, neuronal migration and differentiation (reviewed elsewhere) [95,116,117]. These studies have provided a significant framework to explore the contribution of cilia in disease phenotypes. Interestingly, impaired neuronal migration is observed in some of the monogenic neurodevelopmental disorders discussed above such as in TSC, RTT and FMCDs [40,43,118]. Notably, one study examining models of FCD, showed that aberrant Wnt signaling due to cilia loss was the underlying cause for impaired neuronal migration and cortical lamination deficits [44], supporting the key role of primary cilia in proper brain development.
Abnormal neuronal and network activity is involved in seizures and cognitive deficits such as ASD which are common phenotypes in several neurodevelopmental disorders [119,120]. Recent studies have revealed that primary cilia are important for proper neuronal structure, function, and network connectivity [108,110,114,121], suggesting that cilia deficits might play a role in these neurologic manifestations. Specifically, a study by Kumamoto at al. showed that primary cilia are essential for the integration of hippocampal neurons into existing neuronal circuits [108]. In this study the authors manipulated primary cilia in mouse adult-born neurons and showed that newborn dentate granule cells (DGCs) lacking primary cilia, exhibit glutamatergic synapse formation defects and dendritic refinement deficits. The authors also noted that loss of primary cilia enhanced Wnt/β-catenin signaling activity and expression of a constitutively active form of β-catenin in newborn DGCs was sufficient to recapitulate the dendritic defects [108]. Another study by Rhee et al., explored the effects of primary cilia depletion from mouse mature DGCs and found hippocampus-dependent memory and synaptic plasticity defects [110]. Specifically, the authors showed that these mice exhibited impairment in contextual fear and spatial recognition memory. Further examination of brain slice preparations revealed increased long-term potentiation (LTP) in the CA3 region, which could potentially account for the observed behavioral changes [110]. Bowie et al. deleted the tau tubulin kinase 2 (Ttbk2) gene which encodes for an essential regulator of ciliogenesis in young adult mice, using a tamoxifen inducible line and examined the effects of cilia loss in the cerebellum [114]. When they examined Purkinje cells, the authors found significant defects including altered intracellular Ca2+ concentrations, loss of excitatory synapses from climbing fibers, and Purkinje cell death [114]. This study signifies the importance of cilia in the maintenance of connectivity between neurons, as well as neuronal survival [114]. Interestingly, another study from Tereshko et al., showed that acute disruption of ciliary signaling in rat cortical cultured neurons lead to strengthening of glutamatergic synapses and increased spontaneous firing, while there was no effect on dendritic morphology, passive neuronal properties, or intrinsic excitability [121].
Besides their role as sensory “antennae”, neuronal primary cilia were recently showed to be sites of synaptic contacts [122]. The axo-ciliary synapse was first showed by Sheu et al., using enhanced focused ion beam-scanning electron microscopy [122]. The authors discovered functional synapses between brainstem serotonergic axons and primary cilia from CA1 hippocampal neurons that express serotonin (5-hydroxytryptamine, 5-HT) receptor type 6 (5-HTR6) [122]. They also found that stimulation of this type of receptor is linked to nuclear actin modifications in post-mitotic and post-migratory CA1 neurons [122]. Taken together these studies highlight the importance of primary cilia in proper neuronal morphology, function, and connectivity.
Primary Cilia Signaling Pathways in Brain Pathology
Several models of the neurodevelopmental disorders discussed in this review show that ablation of primary cilia occurs only in a minority of neurons, sparing most neuronal primary cilia. For example, Di Nardo et al., showed that in the hippocampus of Tsc2-KO mice there is on average only a ~20% decrease in the number of ciliated neurons within the CA1 region in comparison to WT mice [39]. A similar magnitude of decrease in ciliation was observed in an Fmr1-KO mouse model where the authors noted that only a subset of DG neurons had lost their cilia [37]. Surprisingly, most neurons in these models have seemingly structurally intact cilia. However, it is still unknown whether the remaining neuronal cilia retain normal function, and if not, whether their impaired function contributes to brain pathology.
Primary neuronal cilia in the brain express components of numerous signaling pathways, with several receptors being localized either primarily or exclusively in the primary cilium [21,22,123]. In order to elucidate the roles of specific pathways some studies selectively manipulated receptors and key components affecting the activity of these pathways while retaining the cilium structure intact. For example, Einstein et al., examined the effects of somatostatin receptor type 3 (SSTR3) loss in mice [103]. SSTR3 is a GPCR, located exclusively in primary neuronal cilia in the brain [124,125]. The authors examined Sstr3-KO mice and found that there is no apparent disruption of cilia structure nor changes in the number of primary cilia. Interestingly, this study revealed that SSTR3 signaling is critical for hippocampal synaptic plasticity and object recognition memory [103]. A more recent study by Tereshko et al., further showed that SSTR3 almost exclusively localizes to primary cilia of excitatory neurons in the cerebral cortex in rodent models and modulates excitatory synaptic properties [121]. Melanin concentrating hormone receptor 1 (MCHR1) is another GPCR abundantly expressed in neuronal primary cilia within several brain structures including cerebral cortex, hippocampus, and amygdala [126]. Adamantidis et al. showed that MCHR1 knock out mice exhibited reduced NMDA receptor 1 subunit in the CA1 region and deficits in learning and memory [99]. Another study exploring the role of 5-HTR6 signaling reported that 5-Htr6-KO mice exhibited cognitive impairments and had altered gene expression, impaired morphology and physiology in hippocampal neurons [115].
Type 3 adenylyl cyclase (AC3) belongs to the cAMP signal transduction pathway predominantly localizes to primary cilia throughout the brain [127]. Several studies examining AC3 knock out mice have reported phenotypes such as obesity, major depression, and learning and memory deficits [106,127]. Small GTPase ADP-ribosylation factors (ARF), are essential for membrane trafficking [128]. Arl13b, a small GTPase that belongs to the ARF family, is highly enriched in primary cilia. Loss of Arl13b from mouse cortical progenitor cells leads to abnormal neuronal placement in the developing cerebral cortex [109]. Another study examined how conditional deletion of Arl13b from cortical projection neurons and interneurons affects their migration. Notably the authors found that Arl13b is specifically required for proper interneuron cortical migration but not for cortical projection neurons [107]. Another study found that loss of Arl13b from postnatal interneurons in the striatum caused reduction of dendritic and axonal complexity, synaptic connectivity deficits, and altered Ca2+ signaling and ciliary localization of GPCRs [113]. Interestingly, chemogenetic activation of GPCR signaling or expression of Sstr3 in the Arl13b deficient interneurons was sufficient to rescue morphology and connectivity abnormalities [113]. Changes in interneuron migration or morphology can result in changes in brain’s network activity and connectivity. Such alterations could lead to imbalanced excitation/inhibition (E/I) ratio which has been implicated in several neuropsychiatric conditions including epilepsy and autism [120].
Overall, these studies suggest that aberrant ciliary signaling can be sufficient to drive neurologic abnormalities. Therefore, it is important to develop tools to measure functional changes of primary cilia that appear structurally intact in neurodevelopmental and other brain disorders.
6. Conclusions and Future Directions
The field of neuronal primary cilia has been gaining significant attention in recent years. These organelles, which were once thought to be vestigial structures, now appear to be key players not only for proper brain development but also for proper function of the adult brain, and they are implicated in a wide range of neurologic and neuropsychiatric disorders [32,34,35]. However, the functions of neuronal primary cilia remain enigmatic due to the substantial complexity and breadth of actions they exhibit which can vary significantly based on the cell-type, age, and brain region. The implications of primary neuronal cilia in neurologic and neuropsychiatric presentations, however, have prompted efforts to generate tools and assays to characterize their function.
Major efforts are currently focused on dissecting the proteomic composition of primary cilia in different cell types [129,130,131,132]. Such studies have already revealed novel key components of cilia signaling pathways [133]. Identifying proteins unique to cilia subtypes will pave the way for our understanding of how primary cilia contents change to support the function of specific neuronal and glial cell types. Furthermore, development of tools that will enable the study of cilia function and structure are also currently in progress. A group recently developed a novel approach to study ciliary signaling and function using a nanobody-based targeting approach combined with optogenetics tools and biosensors [134]. Moreover, another exciting area currently under development is the functional and structural assessment of primary cilia via application of new imaging methods [122,135]. Imaging techniques including expansion microscopy [135] and enhanced focused ion beam-scanning electron microscopy [122] have already uncovered novel roles for neuronal cilia such as the fact that they can be sites of synaptic contact [122].
In summary, while the primary cilia field is still at its infancy, there have been increasing efforts to understand the role of these organelles in the healthy and diseased brain. Advancing our knowledge in neuronal primary cilia biology and shedding light on the contributions of these organelles to brain pathologies will potentially facilitate the development of novel therapeutic targets and treatment strategies for neurologic abnormalities.
Author Contributions
V.K., K.E.D. and M.S. outlined, drafted, and edited the review text. V.K. and K.E.D. prepared the tables and the figure. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the NIH (R01NS113591 to M.S., NRSA T32 NS007473 to V.K.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank Kellen Winden for critical reading of this manuscript.
Conflicts of Interest
Mustafa Sahin reports grant support from Novartis, Biogen, Astellas, Aeovian, Bridgebio, and Aucta. He has served on Scientific Advisory Boards for Novartis, Roche, Regenxbio, SpringWorks Therapeutics, Jaguar Therapeutics and Alkermes.
Abbreviations
| AC3 | Type 3 adenyl cyclase |
| ARF | ADP-ribosylation factors |
| ASD | Autism spectrum disorder |
| CDD | CDKL5 deficiency disorder |
| CDKL5 | Cyclin-dependent kinase-like 5 |
| CNS | Central nervous system |
| DG | Dentate gyrus |
| DGC | Dentate granule cell |
| DNe | Dentate neuroepithelium |
| E/I | Excitation/inhibition |
| FCD | Focal cortical dysplasia |
| FGF | Fibroblast growth factor |
| FMCD | Focal malformations of cortical development |
| FMRP | Fragile X messenger ribonucleoprotein |
| FXS | Fragile X syndrome |
| GluD1 | Glutamate D1 receptor |
| GPCR | G-protein-coupled receptor |
| HDAC6 | Histone deacetylase 6 |
| Hsp27 | Heat shock protein 27 |
| ID | Intellectual disability |
| IFT | Intraflagellar transport |
| KD | Knockdown |
| KO | Knockout |
| LTP | Long term potentiation |
| MCHR1 | Melanin concentrating hormone receptor 1 |
| MeCP2 | Methyl-CpG-binding protein 2 |
| MEF | Mouse embryonic fibroblast |
| mTOR | Mechanistic target of rapamycin |
| OFD1 | Oral-facial-digital syndrome 1 protein |
| PDGFα | Platelet-derived growth factor α |
| PHTS | PTEN hamartoma tumor syndrome |
| PKD | Polycystic kidney disease |
| p-rpS6 | Phosphorylated ribosomal protein S6 |
| PTEN | Phosphatase and tensin homolog |
| RTT | Rett syndrome |
| Shh | Sonic hedgehog |
| SGZ | Subgranular zone |
| SSTR3 | Somatostatin receptor type 3 |
| TEM | Transmission electron microscopy |
| TGF-β | Transforming growth factor β |
| TSC | Tuberous sclerosis complex |
| Ttbk2 | Tau tubulin kinase 2 |
| 5-HT | 5-hydroxytryptamine |
| 5-HTR6 | 5-HT receptor type 6 |
References
- Leeuwenhoek, A. Concerning little animals observed in rain-, well-, sea- and snow-water; as also in water wherein pepper had lain infused. Philos. Trans. 1677, 12, 821–831. [Google Scholar]
- Kiesel, P.; Viar, G.A.; Tsoy, N.; Maraspini, R.; Gorilak, P.; Varga, V.; Honigmann, A.; Pigino, G. The molecular structure of mammalian primary cilia revealed by cryo-electron tomography. Nat. Struct. Mol. Biol. 2020, 27, 1115–1124. [Google Scholar] [CrossRef]
- Satir, P.; Christensen, S.T. Overview of Structure and Function of Mammalian Cilia. Annu. Rev. Physiol. 2007, 69, 377–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloodgood, R.A. From Central to Rudimentary to Primary: The History of an Underappreciated Organelle Whose Time Has Come. The Primary Cilium. Methods Cell Biol. 2009, 94, 2–52. [Google Scholar]
- Zimmermann, K.W. Beiträge zur Kenntniss einiger Drüsen und Epithelien. Arch. Für Mikrosk. Anat. 1898, 52, 552–706. [Google Scholar] [CrossRef]
- Sjostrand, F.S. The ultrastructure of the innersegments of the retinal rods of the guinea pig eye as revealed by electron microscopy. J. Cell Comp. Physiol. 1953, 42, 45–70. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, D.W.; Porter, K.R. A study of the fine structure of ciliated epithelia. J. Morphol. 1954, 94, 221–281. [Google Scholar] [CrossRef]
- De Robertis, E. Electron microscope observations on the submicroscopic organization of the retinal rods. J. Biophys. Biochem. Cytol. 1956, 2, 319–330. [Google Scholar] [CrossRef]
- Sorokin, S. Centrioles and The Formation of Rudimentary Cilia by Fibroblasts and Smooth Muscle Cells. J. Cell Biol. 1962, 15, 363–377. [Google Scholar] [CrossRef] [Green Version]
- Flood, P.R.; Totland, G.K. Substructure of solitary cilia in mouse kidney. Cell Tissue Res. 1977, 183, 281–290. [Google Scholar] [CrossRef]
- Kozminski, K.G.; Johnson, K.A.; Forscher, P.; Rosenbaum, J.L. A motility in the eukaryotic flagellum unrelated to flagellar beating. Proc. Natl. Acad. Sci. USA 1993, 90, 5519–5523. [Google Scholar] [CrossRef] [Green Version]
- Pazour, G.J.; Dickert, B.L.; Vucica, Y.; Seeley, E.S.; Rosenbaum, J.L.; Witman, G.B.; Cole, D.G. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene Tg737, are required for assembly of cilia and flagella. J. Cell Biol. 2000, 151, 709–718. [Google Scholar] [CrossRef]
- Izawa, I.; Goto, H.; Kasahara, K.; Inagaki, M. Current topics of functional links between primary cilia and cell cycle. Cilia 2015, 4, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.E.L.; Lake, A.V.R.; Johnson, C.A. Primary Cilia, Ciliogenesis and the Actin Cytoskeleton: A Little Less Resorption, A Little More Actin Please. Front. Cell Dev. Biol. 2020, 8, 622822. [Google Scholar] [CrossRef]
- Ishikawa, H.; Marshall, W.F. Ciliogenesis: Building the cell’s antenna. Nat. Rev. Mol. Cell Biol. 2011, 12, 222–234. [Google Scholar] [CrossRef]
- Goncalves, J.; Pelletier, L. The Ciliary Transition Zone: Finding the Pieces and Assembling the Gate. Mol. Cells 2017, 40, 243–253. [Google Scholar] [CrossRef] [Green Version]
- Klena, N.; Pigino, G. Structural Biology of Cilia and Intraflagellar Transport. Annu. Rev. Cell Dev. Biol. 2022, 38, 103–123. [Google Scholar] [CrossRef]
- Wood, C.R.; Rosenbaum, J.L. Ciliary ectosomes: Transmissions from the cell’s antenna. Trends Cell Biol. 2015, 25, 276–285. [Google Scholar] [CrossRef] [Green Version]
- Long, H.; Huang, K. Transport of Ciliary Membrane Proteins. Front. Cell Dev. Biol. 2019, 7, 381. [Google Scholar] [CrossRef] [Green Version]
- Nachury, M.V.; Mick, D.U. Establishing and regulating the composition of cilia for signal transduction. Nat. Rev. Mol. Cell Biol. 2019, 20, 389–405. [Google Scholar] [CrossRef]
- Anvarian, Z.; Mykytyn, K.; Mukhopadhyay, S.; Pedersen, L.B.; Christensen, S.T. Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol. 2019, 15, 199–219. [Google Scholar] [CrossRef]
- Wheway, G.; Nazlamova, L.; Hancock, J.T. Signaling through the Primary Cilium. Front. Cell Dev. Biol. 2018, 6, 8. [Google Scholar] [CrossRef]
- Del Cerro, M.P.; Snider, R.S. The Purkinje cell cilium. Anat. Rec. 1969, 165, 127–130. [Google Scholar] [CrossRef]
- Banizs, B.; Pike, M.M.; Millican, C.L.; Ferguson, W.B.; Komlosi, P.; Sheetz, J.; Bell, P.D.; Schwiebert, E.M.; Yoder, B.K. Dysfunctional cilia lead to altered ependyma and choroid plexus function, and result in the formation of hydrocephalus. Development 2005, 132, 5329–5339. [Google Scholar] [CrossRef] [Green Version]
- Spassky, N.; Han, Y.-G.; Aguilar, A.; Strehl, L.; Besse, L.; Laclef, C.; Ros, M.R.; Garcia-Verdugo, J.; Alvarez-Buylla, A. Primary cilia are required for cerebellar development and Shh-dependent expansion of progenitor pool. Dev. Biol. 2008, 317, 246–259. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.-G.; Spassky, N.; Romaguera-Ros, M.; Garcia-Verdugo, J.-M.; Aguilar, A.; Schneider-Maunoury, S.; Alvarez-Buylla, A. Hedgehog signaling and primary cilia are required for the formation of adult neural stem cells. Nat. Neurosci. 2008, 11, 277–284. [Google Scholar] [CrossRef]
- Sipos, É.; Komoly, S.; Ács, P. Quantitative Comparison of Primary Cilia Marker Expression and Length in the Mouse Brain. J. Mol. Neurosci. 2018, 64, 397–409. [Google Scholar] [CrossRef]
- Badano, J.L.; Mitsuma, N.; Beales, P.L.; Katsanis, N. The Ciliopathies: An Emerging Class of Human Genetic Disorders. Annu. Rev. Genom. Hum. Genet. 2006, 7, 125–148. [Google Scholar] [CrossRef] [Green Version]
- Waters, A.M.; Beales, P.L. Ciliopathies: An expanding disease spectrum. Pediatr. Nephrol. 2011, 26, 1039–1056. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.E.; Gleeson, J.G. A systems-biology approach to understanding the ciliopathy disorders. Genome Med. 2011, 3, 59. [Google Scholar] [CrossRef] [Green Version]
- Louvi, A.; Grove, E.A. Cilia in the CNS: The Quiet Organelle Claims Center Stage. Neuron 2011, 69, 1046–1060. [Google Scholar] [CrossRef] [Green Version]
- Valente, E.M.; Rosti, R.O.; Gibbs, E.; Gleeson, J.G. Primary cilia in neurodevelopmental disorders. Nat. Rev. Neurol. 2014, 10, 27–36. [Google Scholar] [CrossRef]
- Muñoz-Estrada, J.; Lora-Castellanos, A.; Meza, I.; Elizalde, S.A.; Benítez-King, G. Primary cilia formation is diminished in schizophrenia and bipolar disorder: A possible marker for these psychiatric diseases. Schizophr. Res. 2018, 195, 412–420. [Google Scholar] [CrossRef]
- Ma, R.; Kutchy, N.A.; Chen, L.; Meigs, D.D.; Hu, G. Primary cilia and ciliary signaling pathways in aging and age-related brain disorders. Neurobiol. Dis. 2022, 163, 105607. [Google Scholar] [CrossRef]
- Ki, S.M.; Jeong, H.S.; Lee, J.E. Primary Cilia in Glial Cells: An Oasis in the Journey to Overcoming Neurodegenerative Diseases. Front. Neurosci. 2021, 15, 736888. [Google Scholar] [CrossRef]
- Sandoval, G.M.; Shim, S.; Hong, D.S.; Garrett, A.S.; Quintin, E.-M.; Marzelli, M.J.; Patnaik, S.; Lightbody, A.A.; Reiss, A.L. Neuroanatomical abnormalities in fragile X syndrome during the adolescent and young adult years. J. Psychiatr. Res. 2018, 107, 138–144. [Google Scholar] [CrossRef]
- Lee, B.; Panda, S.; Lee, H.Y. Primary Ciliary Deficits in the Dentate Gyrus of Fragile X Syndrome. Stem Cell Rep. 2020, 15, 454–466. [Google Scholar] [CrossRef]
- Razak, K.A.; Dominick, K.C.; Erickson, C.A. Developmental studies in fragile X syndrome. J. Neurodev. Disord. 2020, 12, 13–15. [Google Scholar] [CrossRef]
- Di Nardo, A.; Lenoel, I.; Winden, K.D.; Ruhmkorf, A.; Modi, M.E.; Barrett, L.; Marzelli, A. Phenotypic Screen with TSC-Deficient Neurons Reveals Heat-Shock Machinery as a Druggable Pathway for mTORC1 and Reduced Cilia. Cell Rep. 2020, 31, 107780. [Google Scholar] [CrossRef]
- Crino, P.B. Evolving neurobiology of tuberous sclerosis complex. Acta Neuropathol. 2013, 125, 317–332. [Google Scholar] [CrossRef]
- Di Mario, F.J., Jr.; Sahin, M.; Ebrahimi-Fakhari, D. Tuberous sclerosis complex. Pediatr. Clin. N. Am. 2015, 62, 633–648. [Google Scholar] [CrossRef]
- Kabat, J.; Krol, P. Focal cortical dysplasia-review. Pol. J. Radiol. 2012, 77, 35–43. [Google Scholar] [CrossRef]
- Lim, K.-C.; Crino, P. Focal malformations of cortical development: New vistas for molecular pathogenesis. Neuroscience 2013, 252, 262–276. [Google Scholar] [CrossRef]
- Park, S.M.; Lim, J.S.; Ramakrishina, S.; Kim, S.H.; Kim, W.K.; Lee, J.; Kang, H.-C.; Reiter, J.F.; Kim, D.S.; Kim, H.H.; et al. Brain Somatic Mutations in MTOR Disrupt Neuronal Ciliogenesis, Leading to Focal Cortical Dyslamination. Neuron 2018, 99, 83–97.e7. [Google Scholar] [CrossRef] [Green Version]
- Di Nardo, A.; Ruhmkorf, A.; Award, P.; Brennecke, A.; Fagiolini, M.; Sahin, M. Phenotypic characterization of Cdkl5-knockdown neurons establishes elongated cilia as a functional assay for CDKL5 Deficiency Disorder. Neurosci. Res. 2022, 176, 73–78. [Google Scholar] [CrossRef]
- Olson, H.E.; Demarest, S.T.; Pestana-Knight, E.M.; Swanson, L.C.; Iqbal, S.; Lal, D.; Leonard, H.; Cross, H.; Devinsky, O.; Benke, T.A. Cyclin-Dependent Kinase-Like 5 Deficiency Disorder: Clinical Review. Pediatr. Neurol. 2019, 97, 18–25. [Google Scholar] [CrossRef]
- Bahi-Buisson, N.; Nectoux, J.; Rosas-Vargas, H.; Milh, M.; Boddaert, N.; Girard, B.; Cances, C.; Ville, D.; Afenjar, A.; Rio, M.; et al. Key clinical features to identify girls with CDKL5 mutations. Brain 2008, 131 Pt 10, 2647–2661. [Google Scholar] [CrossRef] [Green Version]
- Frasca, A.; Spiombi, E.; Palmieri, M.; Albizzati, E.; Valente, M.M.; Bergo, A.; Leva, B.; Kilstrup-Nielsen, C.; Bianchi, F.; Di Carlo, V.; et al. MECP2 mutations affect ciliogenesis: A novel perspective for Rett syndrome and related disorders. EMBO Mol. Med. 2020, 12, e10270. [Google Scholar] [CrossRef]
- Smeets, E.E.; Pelc, K.; Dan, B. Rett Syndrome. Mol. Syndromol. 2012, 2, 113–127. [Google Scholar] [CrossRef]
- Yu, S.; Pritchard, M.; Kremer, E.; Lynch, M.; Nancarrow, J.; Baker, E.; Holman, K.; Mulley, J.C.; Warren, S.T.; Schlessinger, D.; et al. Fragile X Genotype Characterized by an Unstable Region of DNA. Science 1991, 252, 1179–1181. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.; Beuhler, L.; Lee, H.Y. The Primary Ciliary Deficits in Cerebellar Bergmann Glia of the Mouse Model of Fragile X Syndrome. Cerebellum 2022, 21, 801–813. [Google Scholar] [CrossRef]
- Uysal, S.P.; Şahin, M. Tuberous sclerosis: A review of the past, present, and future. Turk. J. Med Sci. 2020, 50, 1665–1676. [Google Scholar] [CrossRef]
- Hartman, T.R.; Liu, D.; Zilfou, J.T.; Robb, V.; Morrison, T.; Watnick, T.; Henske, E.P. The tuberous sclerosis proteins regulate formation of the primary cilium via a rapamycin-insensitive and polycystin 1-independent pathway. Hum. Mol. Genet. 2009, 18, 151–163. [Google Scholar] [CrossRef] [Green Version]
- Rosengren, T.; Larsen, L.J.; Pedersen, L.B.; Christensen, S.T.; Møller, L.B. TSC1 and TSC2 regulate cilia length and canonical Hedgehog signaling via different mechanisms. Cell. Mol. Life Sci. 2018, 75, 2663–2680. [Google Scholar] [CrossRef] [Green Version]
- Armour, E.A.; Carson, R.P.; Ess, K.C. Cystogenesis and elongated primary cilia in Tsc1-deficient distal convoluted tubules. Am. J. Physiol. Physiol. 2012, 303, F584–F592. [Google Scholar] [CrossRef] [Green Version]
- Krueger, D.A.; Sadhwani, A.; Byars, A.W.; De Vries, P.J.; Franz, D.N.; Whittemore, V.H.; Filip-Dhima, R.; Murray, D.; Kapur, K.; Sahin, M. Everolimus for treatment of tuberous sclerosis complex-associated neuropsychiatric disorders. Ann. Clin. Transl. Neurol. 2017, 4, 877–887. [Google Scholar] [CrossRef]
- Overwater, I.E.; Rietman, A.B.; Mous, S.E.; Heus, K.B.-D.; Rizopoulos, D.; Hoopen, L.W.T.; Van Der Vaart, T.; Jansen, F.E.; Elgersma, Y.; Moll, H.A.; et al. A randomized controlled trial with everolimus for IQ and autism in tuberous sclerosis complex. Neurology 2019, 93, e200–e209. [Google Scholar] [CrossRef] [Green Version]
- Karalis, V.; Bateup, H.S. Current Approaches and Future Directions for the Treatment of mTORopathies. Dev. Neurosci. 2021, 43, 143–158. [Google Scholar] [CrossRef]
- Tang, Z.; Lin, M.G.; Stowe, T.R.; Chen, S.; Zhu, M.; Stearns, T.; Franco, B.; Zhong, Q. Autophagy promotes primary ciliogenesis by removing OFD1 from centriolar satellites. Nature 2013, 502, 254–257. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.D.; Achkar, C.M.; Lai, A.; Srivastava, S.; Doan, R.N.; Rodan, L.H.; Chen, A.Y.; Poduri, A.; Yang, E.; Walsh, C.A.; et al. Polymicrogyria is Associated With Pathogenic Variants in PTEN. Ann. Neurol. 2020, 88, 1153–1164. [Google Scholar] [CrossRef]
- Nabbout, R.; Dulac, O. Epilepsy. Genetics of early-onset epilepsy with encephalopathy. Nat. Rev. Neurol. 2012, 8, 129–130. [Google Scholar] [CrossRef] [PubMed]
- Sakai, Y.; Shaw, C.A.; Dawson, B.C.; Dugas, D.V.; Al-Mohtaseb, Z.; Hill, D.E.; Zoghbi, H.Y. Protein Interactome Reveals Converging Molecular Pathways Among Autism Disorders. Sci. Transl. Med. 2011, 3, 86ra49. [Google Scholar] [CrossRef] [PubMed]
- Talkowski, M.E.; Rosenfeld, J.A.; Blumenthal, I.; Pillalamarri, V.; Chiang, C.; Heilbut, A.; Ernst, C.; Hanscom, C.; Rossin, E.; Lindgren, A.M.; et al. Sequencing Chromosomal Abnormalities Reveals Neurodevelopmental Loci that Confer Risk across Diagnostic Boundaries. Cell 2012, 149, 525–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rusconi, L.; Salvatoni, L.; Giudici, L.; Bertani, I.; Kilstrup-Nielsen, C.; Broccoli, V.; Landsberger, N. CDKL5 Expression Is Modulated during Neuronal Development and Its Subcellular Distribution Is Tightly Regulated by the C-terminal Tail. J. Biol. Chem. 2008, 283, 30101–30111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, L.-W.; Ranum, P.T.; Lefebvre, P.A. CDKL5 regulates flagellar length and localizes to the base of the flagella in Chlamydomonas. Mol. Biol. Cell 2013, 24, 588–600. [Google Scholar] [CrossRef] [PubMed]
- Barbiero, I.; Valente, D.; Chandola, C.; Magi, F.; Bergo, A.; Monteonofrio, L.; Tramarin, M.; Fazzari, M.; Soddu, S.; Landsberger, N.; et al. CDKL5 localizes at the centrosome and midbody and is required for faithful cell division. Sci. Rep. 2017, 7, 6228. [Google Scholar] [CrossRef] [Green Version]
- Canning, P.; Park, K.; Gonçalves, J.; Li, C.; Howard, C.; Sharpe, T.; Holt, L.J.; Pelletier, L.; Bullock, A.N.; Leroux, M.R. CDKL Family Kinases Have Evolved Distinct Structural Features and Ciliary Function. Cell Rep. 2018, 22, 885–894. [Google Scholar] [CrossRef] [Green Version]
- Good, K.V.; Vincent, J.B.; Ausió, J. MeCP2: The Genetic Driver of Rett Syndrome Epigenetics. Front. Genet. 2021, 12, 620859. [Google Scholar] [CrossRef]
- Banerjee, A.; Castro, J.; Sur, M. Rett syndrome: Genes, synapses, circuits, and therapeutics. Front. Psychiatry 2012, 3, 34. [Google Scholar] [CrossRef] [Green Version]
- Yasui, D.H.; Peddada, S.; Bieda, M.C.; Vallero, R.O.; Hogart, A.; Nagarajan, R.P.; Thatcher, K.N.; Farnham, P.J.; LaSalle, J.M. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc. Natl. Acad. Sci. USA 2007, 104, 19416–19421. [Google Scholar] [CrossRef] [Green Version]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.C.; Qin, J.; Zoghbi, H.Y. MeCP2, a Key Contributor to Neurological Disease, Activates and Represses Transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortelazzo, A.; De Felice, C.; Guy, J.; Timperio, A.M.; Zolla, L.; Guerranti, R.; Leoncini, S.; Signorini, C.; Durand, T.; Hayek, J. Brain protein changes in Mecp2 mouse mutant models: Effects on disease progression of Mecp2 brain specific gene reactivation. J. Proteom. 2019, 210, 103537. [Google Scholar] [CrossRef]
- Bergo, A.; Strollo, M.; Gai, M.; Barbiero, I.; Stefanelli, G.; Sertic, S.; Bergo, M. Methyl-CpG binding protein 2 (MeCP2) localizes at the centrosome and is required for proper mitotic spindle organization. J. Biol. Chem. 2015, 290, 3223–3237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.; Mariappan, R.; De, K.; Ohn, T. Loss of MeCP2 causes subtle alteration in dendritic arborization of retinal ganglion cells. Anim. Cells Syst. 2021, 25, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Porch, M.W.; Court-Vazquez, B.; Bennett, M.V.L.; Zukin, R.S. Activation of autophagy rescues synaptic and cognitive deficits in fragile X mice. Proc. Natl. Acad. Sci. USA 2018, 115, E9707–E9716. [Google Scholar] [CrossRef] [Green Version]
- Winden, K.D.; Ebrahimi-Fakhari, D.; Sahin, M. Abnormal mTOR Activation in Autism. Annu. Rev. Neurosci. 2018, 41, 1–23. [Google Scholar] [CrossRef]
- Olson, C.O.; Pejhan, S.; Kroft, D.; Sheikholeslami, K.; Fuss, D.; Buist, M.; Vallero, A. MECP2 Mutation Interrupts Nucleolin-mTOR-P70S6K Signaling in Rett Syndrome Patients. Front. Genet. 2018, 9, 635. [Google Scholar] [CrossRef] [Green Version]
- Rangasamy, S.; Olfers, S.; Gerald, B.; Hilbert, A.; Svejda, S.; Narayanan, V. Reduced neuronal size and mTOR pathway activity in the Mecp2 A140V Rett syndrome mouse model. F1000Research 2016, 5, 2269. [Google Scholar] [CrossRef] [Green Version]
- Ricciardi, S.; Boggio, E.M.; Grosso, S.; Lonetti, G.; Forlani, G.; Stefanelli, G.; Calcagno, E.; Morello, N.; Landsberger, N.; Biffo, S.; et al. Reduced AKT/mTOR signaling and protein synthesis dysregulation in a Rett syndrome animal model. Hum. Mol. Genet. 2011, 20, 1182–1196. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Hoeffer, C.A.; Takayasu, Y.; Miyawaki, T.; McBride, S.M.; Klann, E.; Zukin, R.S. Dysregulation of mTOR Signaling in Fragile X Syndrome. J. Neurosci. 2010, 30, 694–702. [Google Scholar] [CrossRef] [Green Version]
- Arsenault, J.; Hooper, A.W.M.; Gholizadeh, S.; Kong, T.; Pacey, L.K.; Koxhioni, E.; Niibori, Y.; Eubanks, J.H.; Wang, L.-Y.; Hampson, D.R. Interregulation between fragile X mental retardation protein and methyl CpG binding protein 2 in the mouse posterior cerebral cortex. Hum. Mol. Genet. 2020, 29, 3744–3756. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, E.; Yuan, L.; Seong, E.; Ligon, C.; DeKorver, N.; Gurumurthy, C.; Arikkath, J. Neuron-Type Specific Loss of CDKL5 Leads to Alterations in mTOR Signaling and Synaptic Markers. Mol. Neurobiol. 2018, 56, 4151–4162. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.-T.J.; Allen, M.; Goffin, D.; Zhu, X.; Fairless, A.H.; Brodkin, E.S.; Siegel, S.J.; Marsh, E.D.; Blendy, J.A.; Zhou, Z. Loss of CDKL5 disrupts kinome profile and event-related potentials leading to autistic-like phenotypes in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 21516–21521. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, C.; Trazzi, S.; Torricella, R.; Viggiano, R.; De Franceschi, M.; Amendola, E. Loss of CDKL5 impairs survival and dendritic growth of newborn neurons by altering AKT/GSK-3beta signaling. Neurobiol. Dis. 2014, 70, 53–68. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.; Jiang, Y. Reciprocal Regulation between Primary Cilia and mTORC1. Genes 2020, 11, 711. [Google Scholar] [CrossRef] [PubMed]
- Boehlke, C.; Kotsis, F.; Patel, V.; Braeg, S.; Voelker, H.; Bredt, S.; Beyer, T.; Janusch, H.; Hamann, C.; Gödel, M.; et al. Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat. Cell Biol. 2010, 12, 1115–1122. [Google Scholar] [CrossRef] [Green Version]
- Zhong, M.; Zhao, X.; Li, J.; Yuan, W.; Yan, G.; Tong, M.; Guo, S.; Zhu, Y.; Jiang, Y.; Liu, Y.; et al. Tumor Suppressor Folliculin Regulates mTORC1 through Primary Cilia. J. Biol. Chem. 2016, 291, 11689–11697. [Google Scholar] [CrossRef] [Green Version]
- Shillingford, J.M.; Murcia, N.S.; Larson, C.H.; Low, S.H.; Hedgepeth, R.; Brown, N.; Flask, C.A.; Novick, A.C.; Goldfarb, D.A.; Kramer-Zucker, A.; et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5466–5471. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Nagai, T.; Chiba, S.; Nakayama, K.; Mizuno, K. Glucose deprivation induces primary cilium formation through mTORC1 inactivation. J. Cell Sci. 2018, 131, jcs208769. [Google Scholar] [CrossRef] [Green Version]
- Sherpa, R.T.; Atkinson, K.F.; Ferreira, V.P.; Nauli, S.M. Rapamycin Increases Length and Mechanosensory Function Of Primary Cilia In Renal Epithelial And Vascular Endothelial Cells. IERJ 2016, 2, 91–97. [Google Scholar]
- Mari, F.; Azimonti, S.; Bertani, I.; Bolognese, F.; Colombo, E.; Caselli, R.; Scala, E.; Longo, I.; Grosso, S.; Pescucci, C.; et al. CDKL5 belongs to the same molecular pathway of MeCP2 and it is responsible for the early-onset seizure variant of Rett syndrome. Hum. Mol. Genet. 2005, 14, 1935–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertani, I.; Rusconi, L.; Bolognese, F.; Forlani, G.; Conca, B.; De Monte, L.; Larson, L. Functional consequences of mutations in CDKL5, an X-linked gene involved in infantile spasms and mental retardation. J. Biol. Chem. 2006, 281, 32048–32056. [Google Scholar] [CrossRef] [PubMed]
- Carouge, D.; Host, L.; Aunis, D.; Zwiller, J.; Anglard, P. CDKL5 is a brain MeCP2 target gene regulated by DNA methylation. Neurobiol. Dis. 2010, 38, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Livide, G.; Patriarchi, T.; Amenduni, M.; Amabile, S.; Yasui, D.; Calcagno, E.; Rizzo, C.L.; De Falco, G.; Ulivieri, C.; Ariani, F.; et al. GluD1 is a common altered player in neuronal differentiation from both MECP2-mutated and CDKL5-mutated iPS cells. Eur. J. Hum. Genet. 2015, 23, 195–201. [Google Scholar] [CrossRef]
- Park, S.M.; Jang, H.J.; Lee, J.H. Roles of Primary Cilia in the Developing Brain. Front. Cell. Neurosci. 2019, 13, 218. [Google Scholar] [CrossRef] [Green Version]
- Hasenpusch-Theil, K.; Theil, T. The Multifaceted Roles of Primary Cilia in the Development of the Cerebral Cortex. Front. Cell Dev. Biol. 2021, 9, 630161. [Google Scholar] [CrossRef]
- Guemez-Gamboa, A.; Coufal, N.G.; Gleeson, J.G. Primary Cilia in the Developing and Mature Brain. Neuron 2014, 82, 511–521. [Google Scholar] [CrossRef] [Green Version]
- Suciu, S.K.; Caspary, T. Cilia, neural development and disease. Semin. Cell Dev. Biol. 2021, 110, 34–42. [Google Scholar] [CrossRef]
- Adamantidis, A.; Thomas, E.; Foidart, A.; Tyhon, A.; Coumans, B.; Minet, A.; Tirelli, E.; Seutin, V.; Grisar, T.; Lakaye, B. Disrupting the melanin-concentrating hormone receptor 1 in mice leads to cognitive deficits and alterations of NMDA receptor function. Eur. J. Neurosci. 2005, 21, 2837–2844. [Google Scholar] [CrossRef]
- May, S.R.; Ashique, A.M.; Karlen, M.; Wang, B.; Shen, Y.; Zarbalis, K.; Gleeson, M. Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev. Biol. 2005, 287, 378–389. [Google Scholar] [CrossRef] [Green Version]
- Komada, M.; Saitsu, H.; Kinboshi, M.; Miura, T.; Shiota, K.; Ishibashi, M. Hedgehog signaling is involved in development of the neocortex. Development 2008, 135, 2717–2727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willaredt, M.A.; Hasenpusch-Theil, K.; Gardner, H.A.R.; Kitanovic, I.; Hirschfeld-Warneken, V.C.; Gojak, C.P.; Gorgas, K.; Bradford, C.L.; Spatz, J.; Wölfl, S.; et al. A Crucial Role for Primary Cilia in Cortical Morphogenesis. J. Neurosci. 2008, 28, 12887–12900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Einstein, E.B.; Patterson, C.A.; Hon, B.J.; Regan, K.A.; Reddi, J.; Melnikoff, D.E.; Mariani, P. Somatostatin signaling in neuronal cilia is critical for object recognition memory. J. Neurosci. 2010, 30, 4306–4314. [Google Scholar] [CrossRef]
- Amador-Arjona, A.; Elliott, J.; Miller, A.; Ginbey, A.; Pazour, G.J.; Enikolopov, G.; Roberts, A.J.; Terskikh, A.V. Primary Cilia Regulate Proliferation of Amplifying Progenitors in Adult Hippocampus: Implications for Learning and Memory. J. Neurosci. 2011, 31, 9933–9944. [Google Scholar] [CrossRef] [Green Version]
- Besse, L.; Neti, M.; Anselme, I.; Gerhardt, C.; Rüther, U.; Laclef, C.; Schneider-Maunoury, S. Primary cilia control telencephalic patterning and morphogenesis via Gli3 proteolytic processing. Development 2011, 138, 2079–2088. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Phan, T.; Storm, D.R. The Type 3 Adenylyl Cyclase Is Required for Novel Object Learning and Extinction of Contextual Memory: Role of cAMP Signaling in Primary Cilia. J. Neurosci. 2011, 31, 5557–5561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higginbotham, H.; Eom, T.-Y.; Mariani, L.E.; Bachleda, A.; Hirt, J.; Gukassyan, V.; Cusack, C.L.; Lai, C.; Caspary, T.; Anton, E. Arl13b in Primary Cilia Regulates the Migration and Placement of Interneurons in the Developing Cerebral Cortex. Dev. Cell 2012, 23, 925–938. [Google Scholar] [CrossRef] [Green Version]
- Kumamoto, N.; Gu, Y.; Wang, J.; Janoschka, S.; Takemaru, K.-I.; Levine, J.; Ge, S. A role for primary cilia in glutamatergic synaptic integration of adult-born neurons. Nat. Neurosci. 2012, 15, 399–405. [Google Scholar] [CrossRef] [Green Version]
- Higginbotham, H.; Guo, J.; Yokota, Y.; Umberger, N.L.; Su, C.-Y.; Li, J.; Verma, N.; Hirt, J.; Ghukasyan, V.; Caspary, T.; et al. Arl13b-regulated cilia activities are essential for polarized radial glial scaffold formation. Nat. Neurosci. 2013, 16, 1000–1007. [Google Scholar] [CrossRef]
- Rhee, S.; Kirschen, G.W.; Gu, Y.; Ge, S. Depletion of primary cilia from mature dentate granule cells impairs hippocampus-dependent contextual memory. Sci. Rep. 2016, 6, 34370. [Google Scholar] [CrossRef]
- Gazea, M.; Tasouri, E.; Tolve, M.; Bosch, V.; Kabanova, A.; Gojak, C.; Kurtulmus, B.; Novikov, O.; Spatz, J.; Pereira, G.; et al. Primary cilia are critical for Sonic hedgehog-mediated dopaminergic neurogenesis in the embryonic midbrain. Dev. Biol. 2016, 409, 55–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foerster, P.; Daclin, M.; Asm, S.; Faucourt, M.; Boletta, A.; Genovesio, A.; Spassky, N. mTORC1 signaling and primary cilia are required for brain ventricle morphogenesis. Development 2017, 144, 201–210. [Google Scholar] [PubMed] [Green Version]
- Guo, J.; Otis, J.M.; Higginbotham, H.; Monckton, C.; Cheng, J.; Asokan, A.; Mykytyn, K.; Caspary, T.; Stuber, G.D.; Anton, E. Primary Cilia Signaling Shapes the Development of Interneuronal Connectivity. Dev. Cell 2017, 42, 286–300.e4. [Google Scholar] [CrossRef] [PubMed]
- Bowie, E.; Goetz, S.C. TTBK2 and primary cilia are essential for the connectivity and survival of cerebellar Purkinje neurons. eLife 2020, 9, e51166. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Wang, B.; Chen, C.; Li, C.; Zhang, Y. 5-HT6R null mutatrion induces synaptic and cognitive defects. Aging Cell 2021, 20, e13369. [Google Scholar] [CrossRef]
- Han, Y.-G.; Alvarez-Buylla, A. Role of primary cilia in brain development and cancer. Curr. Opin. Neurobiol. 2010, 20, 58–67. [Google Scholar] [CrossRef] [Green Version]
- Youn, Y.H.; Han, Y.-G. Primary Cilia in Brain Development and Diseases. Am. J. Pathol. 2018, 188, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Feldman, D.; Banerjee, A.; Sur, M. Developmental Dynamics of Rett Syndrome. Neural Plast. 2016, 2016, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Stefan, H.; Lopes da Silva, F.H. Epileptic neuronal networks: Methods of identification and clinical relevance. Front. Neurol. 2013, 4, 8. [Google Scholar] [CrossRef] [Green Version]
- Sohal, V.S.; Rubenstein, J.L.R. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol. Psychiatry 2019, 24, 1248–1257. [Google Scholar] [CrossRef]
- Tereshko, L.; Gao, Y.; Cary, B.A.; Turrigiano, G.G.; Sengupta, P. Ciliary neuropeptidergic signaling dynamically regulates excitatory synapses in postnatal neocortical pyramidal neurons. eLife 2021, 10, e65427. [Google Scholar] [CrossRef] [PubMed]
- Sheu, S.-H.; Upadhyayula, S.; Dupuy, V.; Pang, S.; Deng, F.; Wan, J.; Walpita, D.; Pasolli, H.A.; Houser, J.; Sanchez-Martinez, S.; et al. A serotonergic axon-cilium synapse drives nuclear signaling to alter chromatin accessibility. Cell 2022, 185, 3390–3407.e18. [Google Scholar] [CrossRef] [PubMed]
- Wachten, D.; Mick, D.U. Signal transduction in primary cilia–analyzing and manipulating GPCR and second messenger signaling. Pharmacol. Ther. 2021, 224, 107836. [Google Scholar] [CrossRef]
- Barbeito, P.; Garcia-Gonzalo, F.R. HTR6 and SSTR3 targeting to primary cilia. Biochem. Soc. Trans. 2021, 49, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Händel, M.; Schulz, S.; Stanarius, A.; Schreff, M.; Erdtmann-Vourliotis, M.; Schmidt, H.; Wolf, G.; Höllt, V. Selective targeting of somatostatin receptor 3 to neuronal cilia. Neuroscience 1999, 89, 909–926. [Google Scholar] [CrossRef]
- Diniz, G.B.; Battagello, D.S.; Klein, M.O.; Bono, B.S.M.; Ferreira, J.G.P.; Motta-Teixeira, L.C.; Gygi, D. Ciliary melanin-concentrating hormone receptor 1 (MCHR1) is widely distributed in the murine CNS in a sex-independent manner. J. Neurosci. Res. 2020, 98, 2045–2071. [Google Scholar] [CrossRef]
- Qiu, L.; LeBel, R.P.; Storm, D.R.; Chen, X. Type 3 adenylyl cyclase: A key enzyme mediating the cAMP signaling in neuronal cilia. Int. J. Physiol. Pathophysiol. Pharmacol. 2016, 8, 95–108. [Google Scholar]
- D’Souza-Schorey, C.; Chavrier, P. ARF proteins: Roles in membrane traffic and beyond. Nat. Rev. Mol. Cell Biol. 2006, 7, 347–358. [Google Scholar] [CrossRef]
- Kohli, P.; Höhne, M.; Jüngst, C.; Bertsch, S.; Ebert, L.K.; Schauss, A.C.; Benzing, T.; Rinschen, M.M.; Schermer, B. The ciliary membrane-associated proteome reveals actin-binding proteins as key components of cilia. EMBO Rep. 2017, 18, 1521–1535. [Google Scholar] [CrossRef]
- Mick, D.U.; Rodrigues, R.B.; Leib, R.D.; Adams, C.M.; Chien, A.S.; Gygi, S.P.; Nachury, M.V. Proteomics of Primary Cilia by Proximity Labeling. Dev. Cell 2015, 35, 497–512. [Google Scholar] [CrossRef] [Green Version]
- May, E.A.; Kalocsay, M.; D’Auriac, I.G.; Schuster, P.S.; Gygi, S.P.; Nachury, M.V.; Mick, D.U. Time-resolved proteomics profiling of the ciliary Hedgehog response. J. Cell Biol. 2021, 220, e202007207. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shi, Z.; Yang, F.; Zhou, T.; Xie, S. Deciphering cilia and ciliopathies using proteomic approaches. FEBS J. 2022; online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yam, P.T.; Chen, W.; Schlienger, S.; Gutierrez, O.T.; Cai, E.; Klein, G. Cilium proteomics reveals Numb as a positive regulator of the Hedgehog signaling pathway. bioRxiv 2022. [Google Scholar] [CrossRef]
- Hansen, J.N.; Kaiser, F.; Klausen, C.; Stüven, B.; Chong, R.; Bönigk, W.; Mick, D.U.; Möglich, A.; Jurisch-Yaksi, N.; Schmidt, F.I.; et al. Nanobody-directed targeting of optogenetic tools to study signaling in the primary cilium. eLife 2020, 9, e57907. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Chiba, S.; Nakayama, K. Practical method for superresolution imaging of primary cilia and centrioles by expansion microscopy using an amplibody for fluorescence signal amplification. Mol. Biol. Cell 2020, 31, 2195–2206. [Google Scholar] [CrossRef]
Figure 1.
Key events in primary cilia research history. Schematic illustration of the timeline of the major discoveries in the field of primary cilia. (TEM) Transmission electron microscopy, (IFT) Intraflagellar transport, (PKD) Polycystic kidney disease.
Figure 1.
Key events in primary cilia research history. Schematic illustration of the timeline of the major discoveries in the field of primary cilia. (TEM) Transmission electron microscopy, (IFT) Intraflagellar transport, (PKD) Polycystic kidney disease.
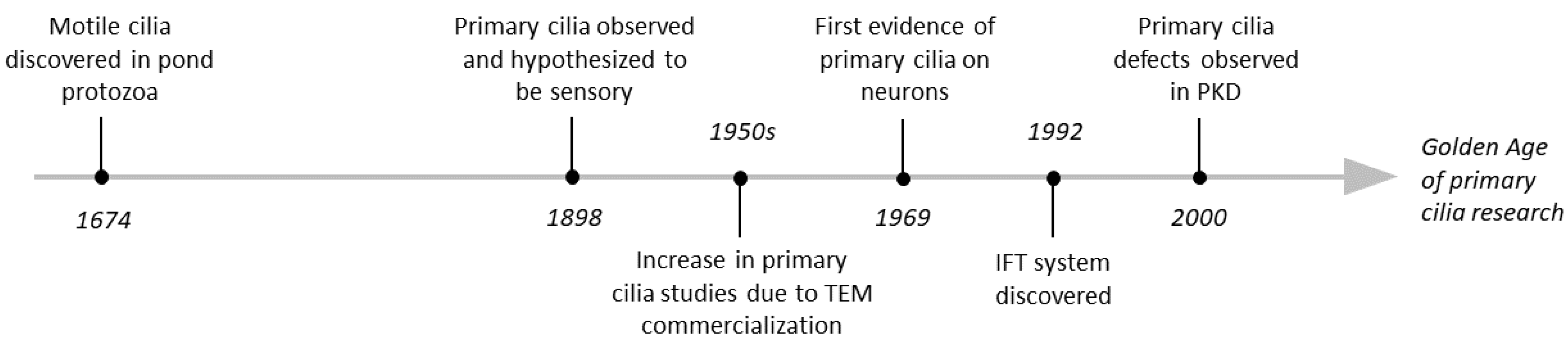
Figure 2.
Primary cilia phenotypes in monogenic neurodevelopmental disorders. Mutations in genes (denoted in green) underly a group of neurodevelopmental disorders (denoted in yellow) that exhibit severe neurologic and neuropsychiatric manifestations including cortical malformation, epilepsy, ASD, and ID. Some of the neurological phenotypes are unique to certain disorders while others are shared (see Table 1). Recently several studies discovered that these disorders exhibit neuronal primary cilia (colored red in the illustration) deficits raising the question of whether these organelles contribute to the neurologic manifestations that patients exhibit. (TSC) Tuberous sclerosis complex, (FCD) Focal cortical dysplasia, (RTT) Rett syndrome, (FXS) Fragile-X syndrome, (CDD) CDKL5 deficiency disorder, (ASD) Autism spectrum disorder, (ID) Intellectual disabilities. Created with BioRender.com (www.biorender.com, accessed on 2 December 2022).
Figure 2.
Primary cilia phenotypes in monogenic neurodevelopmental disorders. Mutations in genes (denoted in green) underly a group of neurodevelopmental disorders (denoted in yellow) that exhibit severe neurologic and neuropsychiatric manifestations including cortical malformation, epilepsy, ASD, and ID. Some of the neurological phenotypes are unique to certain disorders while others are shared (see Table 1). Recently several studies discovered that these disorders exhibit neuronal primary cilia (colored red in the illustration) deficits raising the question of whether these organelles contribute to the neurologic manifestations that patients exhibit. (TSC) Tuberous sclerosis complex, (FCD) Focal cortical dysplasia, (RTT) Rett syndrome, (FXS) Fragile-X syndrome, (CDD) CDKL5 deficiency disorder, (ASD) Autism spectrum disorder, (ID) Intellectual disabilities. Created with BioRender.com (www.biorender.com, accessed on 2 December 2022).


{kind=link}
{kind=link}
Table 1.
Primary cilia phenotypes in neurodevelopmental disorders discussed in this review.
| Mutated Gene | Disease | Key Neurologic Features * | Experimental Systems + | Neuronal PC Phenotypes + | Ref |
|---|---|---|---|---|---|
| FMR1 | Fragile X Syndrome (FXS) | ASD, ID, seizures, ADHD, neuroanatomical abnormalities, i.e., larger volume of lateral ventricles |
| ↓ Number ↓ Length | [36,37,38] |
| TSC1/2 | Tuberous Sclerosis Complex (TSC) | Tubers, SENs, SEGAs, epilepsy, disorganized WM, ID, ASD, ADHD |
| ↓ Number | [39,40,41] |
| mTOR | Focal Cortical Dysplasia (FCD) | Epilepsy, ID, ASD, altered cortical architecture |
| ↓ Number ↓ Length | [42,43,44] |
| CDKL5 | CDKL5 Deficiency Disorder (CDD) | Infantile spasms, ASD, epilepsy, ID |
| ↑ Length | [45,46,47] |
| MeCP2 | Rett Syndrome (RTT) | ID, ASD, seizures |
| ↓ Number ↓ Length | [48,49] |
* The primary neurologic features of these disorders are listed based on the references noted, as well as in the OMIM database and the NIH Genetic and Rare Diseases Information Center. Other neurologic phenotypes may be present in these disorders and not all patients may present the ones listed here. + Listed here are the primary cilia phenotypes and the experimental systems from the studies discussed in this review. # k allele is full KO while c allele is a conditional mutation leading to ~7% expression of Tsc2 protein. (ASD)Autism spectrum disorder, (ID) Intellectual disability, (ADHD) Attention deficit hyperactivity disorder, (SENs) subependymal nodules, (SEGAs) subependymal giant cell astrocytomas, (WM) White matter, (KD) Knockdown, (FMCD) Focal malformations of cortical development. Arrows indicate either reduction or increase.
Table 2.
Ciliary, neurological, and behavioral phenotypes observed in primary cilia deficit mouse models.
Table 2.
Ciliary, neurological, and behavioral phenotypes observed in primary cilia deficit mouse models.
| Mouse Model | Primary Cilia Phenotypes * | Neurological Phenotypes + | Behavioral Phenotypes + | Ref |
|---|---|---|---|---|
| Mchr1Neo/Neo | n/a |
| Impaired learning and memory | [99] |
| Dnchc2 mutant | Structurally impaired in neuroectoderm |
| n/a | [100] |
| hGFAP-Cre; Kif3afl/fl | Loss from granule neuron precursors in DG |
| n/a | [26] |
| Emx1-Cre; Shhfl/- Emx1-Cre; Smofl/- | n/a |
| n/a | [101] |
| Cobblestone (hypomorphic Ift88) | Normal morphology in ventricles |
| n/a | [102] |
| Sst3 knockout | Normal in CA1 |
| Impaired recognition memory | [103] |
| Ift20fl/fl::mGFAP-Cre | Loss from radial neural stem cells in the SGZ |
| Delay in learning and enhanced cue-based fear responses | [104] |
| Ftm mutant |
Loss from telencephalic neuroepithelial cells |
| n/a | [105] |
| Ac3 mutant | Structurally intact in hippocampus | n/a | Impaired learning and memory | [106] |
| Arl13bfl/fl; Nex-Cre | n/a |
| n/a | [107] |
| Arl13bfl/fl; Dlx5/6-CIE | Shorter on interneurons |
| n/a | [107] |
| Inducible dominant negative Kif3a expressed in the hilus region of DG of adult mice | Loss from newborn DGCs |
| n/a | [108] |
| Arl13bhnn/hnn (null allele) | Shorter with disrupted morphological plasticity on radial progenitors in cortex |
| n/a | [109] |
| Ift20fl/fl AAV-CaMKII-eGFP-Cre injected in DG of adult mice | Loss from majority of GFP+ cells in DG |
| Impaired memory | [110] |
| Cobblestone (hypomorphic Ift88) | Normal morphology in VZ of ventral midbrain |
| n/a | [111] |
| Nestin-Kif3afl/fl | Loss from cortical ventricular surface and somatosensory cortex |
| n/a | [112] |
| Nkx2.1Cre; Arl13bfl/fl | Defective intraciliary Ca2+ signaling in striatal interneurons |
| n/a | [113] |
| Ttbk2fl/fl; Ubc-Cre-ERT2+ (Tamoxifen on P21) | Loss from cerebellum, brainstem, hippocampus, and cortex |
| Locomotor deficiencies | [114] |
| 5-ht6r mutant | Normal morphology in hippocampus |
| Anxiety and cognitive impairments | [115] |
This table contains information from studies which investigated primary cilia deficits in mouse models. A subset of these studies is discussed in the review. Arrows indicate either reduction or increase. * Listed are the primary cilia phenotypes within the brain regions investigated in the referenced study. + Listed are the key neurological and behavioral phenotypes reported in the reference study. Some studies might have additional phenotypes not listed in this table. (Nmdar1) N-methyl-D-aspartate receptor subunit 1, (DG) Dentate gyrus, (Shh) Sonic Hedgehog, (DT) Dorsal telencephalon, (LTP) Long-term potentiation, (SGZ) Subgranular zone, (OB) Olfactory bulb, (IC) Internal capsule, (DGCs) Dentate granule cells, (MF) Mossy fibers, (RGCs) Radial glia cells, (VZ) Ventricular zone, (mDA) Midbrain dopaminergic, (BP) Basal progenitors, (LVs) lateral ventricles, (VGLUT2) vesicular-glutamate transporter 2.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Karalis, V.; Donovan, K.E.; Sahin, M. Primary Cilia Dysfunction in Neurodevelopmental Disorders beyond Ciliopathies. J. Dev. Biol. 2022, 10, 54. https://doi.org/10.3390/jdb10040054
AMA Style
Karalis V, Donovan KE, Sahin M. Primary Cilia Dysfunction in Neurodevelopmental Disorders beyond Ciliopathies. Journal of Developmental Biology. 2022; 10(4):54. https://doi.org/10.3390/jdb10040054
Chicago/Turabian StyleKaralis, Vasiliki, Kathleen E. Donovan, and Mustafa Sahin. 2022. "Primary Cilia Dysfunction in Neurodevelopmental Disorders beyond Ciliopathies" Journal of Developmental Biology 10, no. 4: 54. https://doi.org/10.3390/jdb10040054
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.