
Hedgehog: A Key Signaling in the Development of the Oligodendrocyte Lineage

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
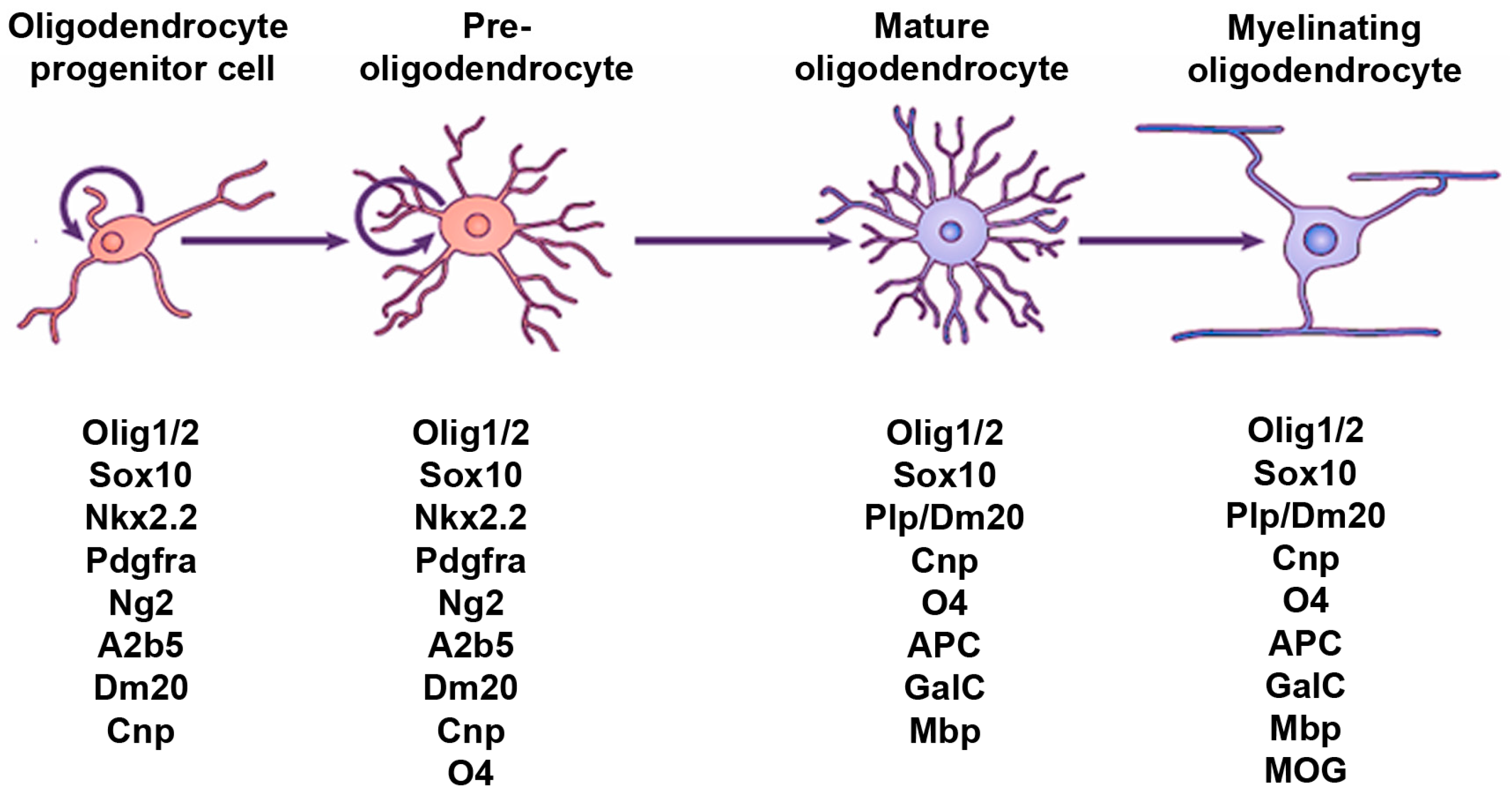
2. Hedgehog Signaling and the OL Lineage in the Spinal Cord
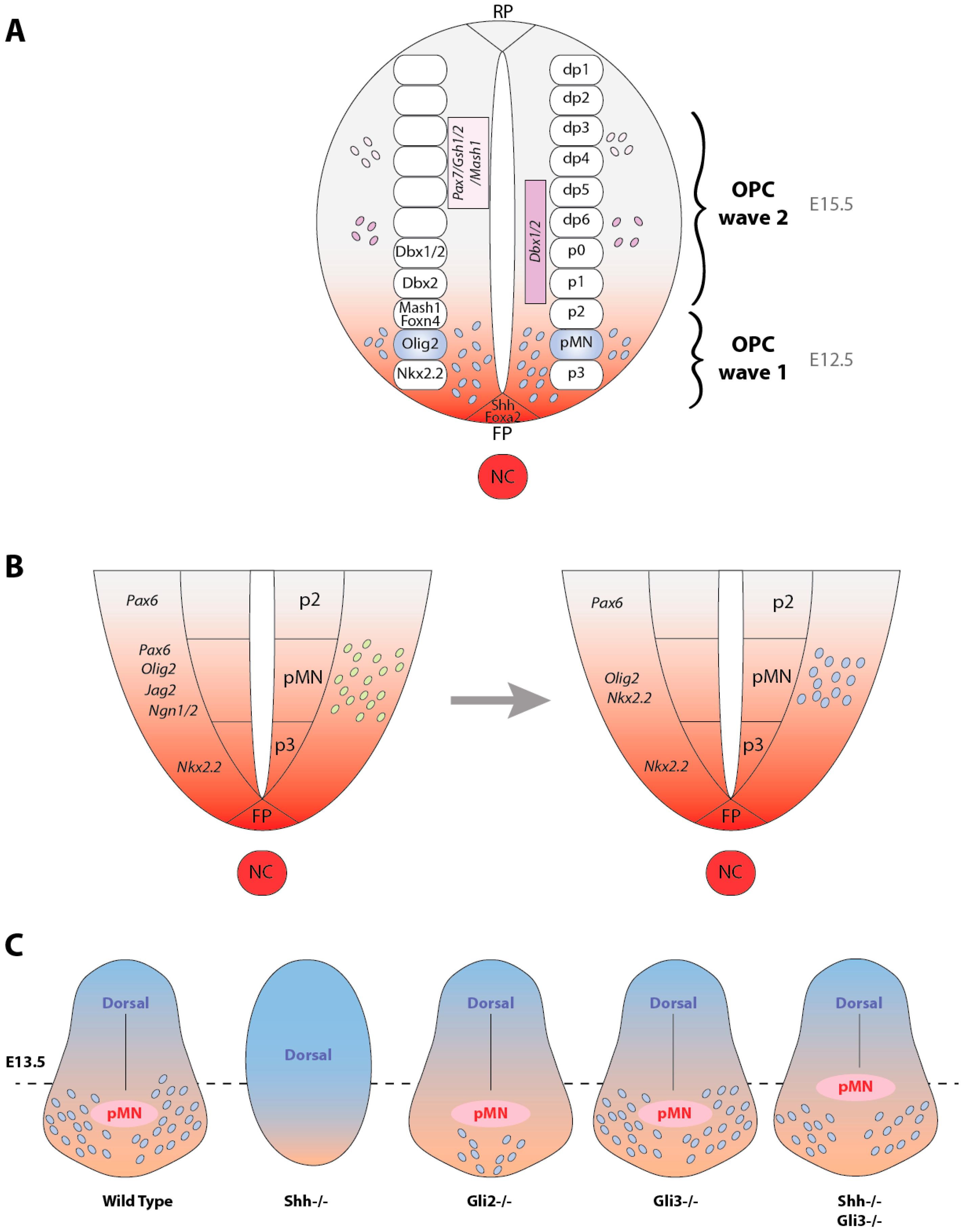
2.1. Hedgehog, a Key Signaling for OPC Specification in the Ventral Spinal Cord
2.2. Hedgehog Transcriptional Targets Leading to OPC Specification in the Ventral Neuroepithelium
2.3. Do MNs and OLs Arise from a Common Population of Progenitor Cells in Response to Hedgehog Signaling?
2.4. Molecular Mechanisms Associated with the Hedgehog-Dependent MN/OL Segregation
2.5. Local Sources of Hedgehog Proteins Contributing to OPC Specification
2.6. Involvement of the Gli Transcription Factors in OPC Specification
2.7. Sources of OPCs Outside the pMN Domain Are Hedgehog-Independent
3. Hedgehog Signaling and the OL Lineage in the Brain
3.1. Hedgehog-Dependent Generation of OPCs in the Hindbrain and Midbrain
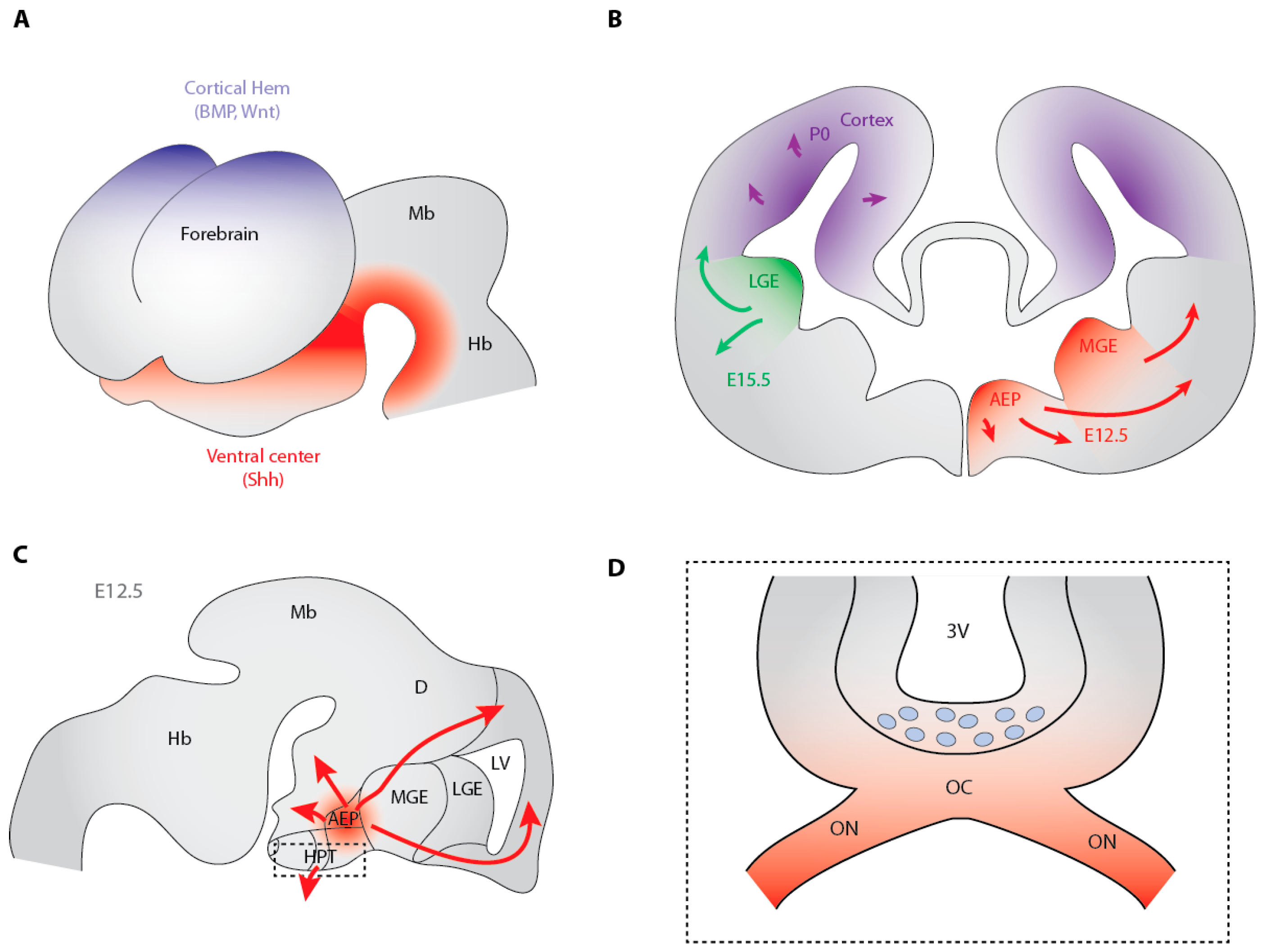
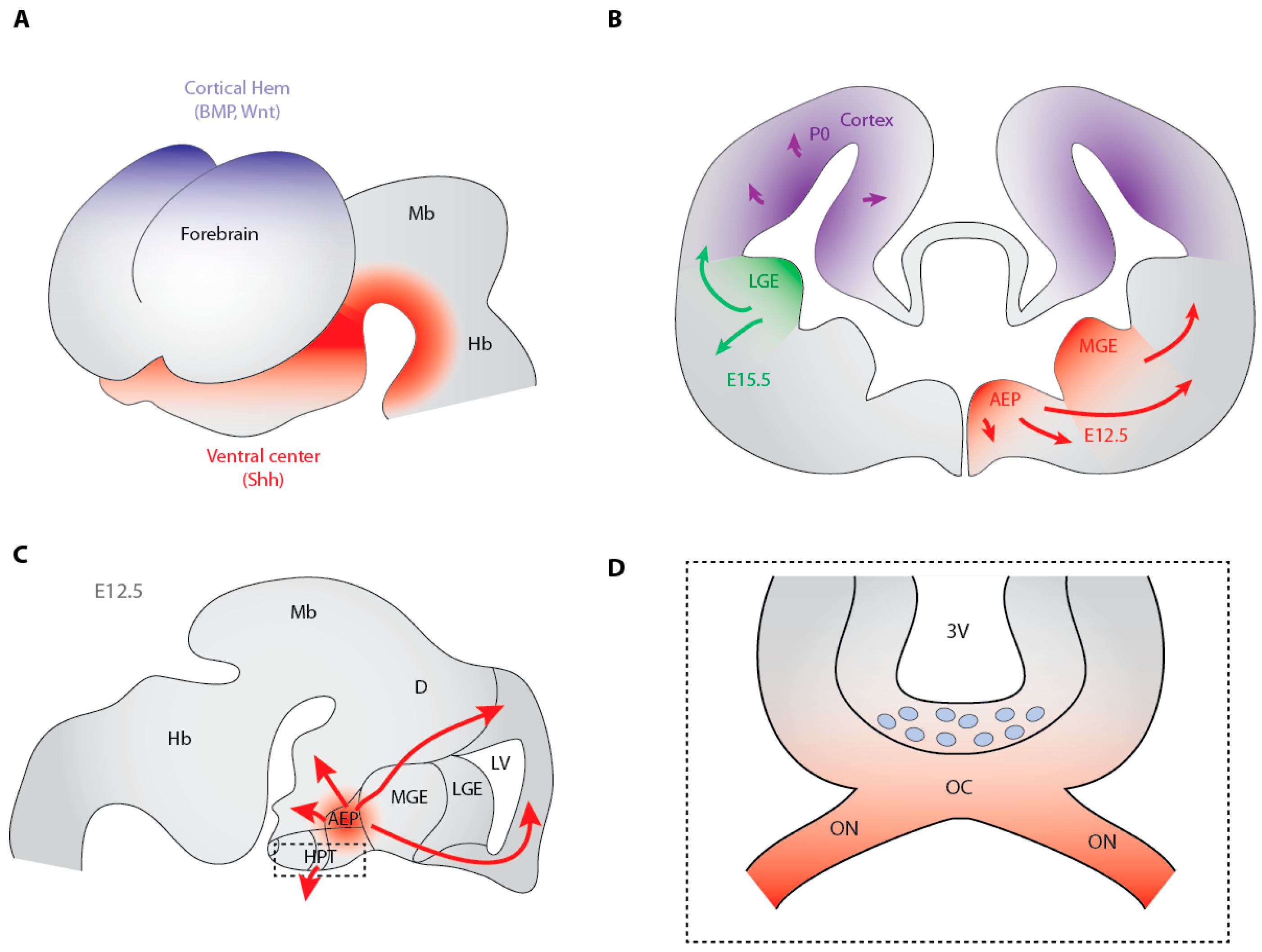
3.2. Hedgehog-Dependent Production of OPCs in the Ventral Telencephalon
3.3. Cooperation between Shh and Fibroblast Growth Factors in the Generation of Ventral Telencephalon OPCs
3.4. OPC Production is Gli2-Independent in the Forebrain
3.5. The Dorsally-Derived Third Wave of OPC Production in the Telencephalon Depends on Shh
3.6. Antagonistic Activities of Shh and BMP on the Transcriptome of Telencephalon OPCs
3.7. Shh-Dependent Generation of OPCs in the Optic Nerve
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hartline, D.K.; Colman, D.R. Rapid conduction and the evolution of giant axons and myelinated fibers. Curr. Biol. 2007, 17, R29–R35. [Google Scholar] [CrossRef] [PubMed]
- Zalc, B.; Goujet, D.; Colman, D. The origin of the myelination program in vertebrates. Curr. Biol. 2008, 18, R511–R512. [Google Scholar] [CrossRef] [PubMed]
- Rowitch, D.H.; Kriegstein, A.R. Developmental genetics of vertebrate glial-cell specification. Nature 2010, 468, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Jessell, T.M. Neuronal specification in the spinal cord: Inductive signals and transcriptional codes. Nat. Rev. Genet. 2000, 1, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Warf, B.C.; Fok-Seang, J.; Miller, R.H. Evidence for the ventral origin of oligodendrocyte precursors in the rat spinal cord. J. Neurosci. 1991, 11, 2477–2488. [Google Scholar] [PubMed]
- Ono, K.; Bansal, R.; Payne, J.; Rutishauser, U.; Miller, R.H. Early development and dispersal of oligodendrocyte precursors in the embryonic chick spinal cord. Development 1995, 121, 1743–1754. [Google Scholar] [PubMed]
- Timsit, S.; Martinez, S.; Allinquant, B.; Peyron, F.; Puelles, L.; Zalc, B. Oligodendrocytes originate in a restricted zone of the embryonic ventral neural tube defined by DM-20 mrna expression. J. Neurosci. 1995, 15, 1012–1024. [Google Scholar] [PubMed]
- Trousse, F.; Giess, M.C.; Soula, C.; Ghandour, S.; Duprat, A.M.; Cochard, P. Notochord and floor plate stimulate oligodendrocyte differentiation in cultures of the chick dorsal neural tube. J. Neurosci. Res. 1995, 41, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Pringle, N.P.; Richardson, W.D. A singularity of pdgf alpha-receptor expression in the dorsoventral axis of the neural tube may define the origin of the oligodendrocyte lineage. Development 1993, 117, 525–533. [Google Scholar] [PubMed]
- Pringle, N.P.; Yu, W.P.; Guthrie, S.; Roelink, H.; Lumsden, A.; Peterson, A.C.; Richardson, W.D. Determination of neuroepithelial cell fate: Induction of the oligodendrocyte lineage by ventral midline cells and sonic hedgehog. Dev. Biol. 1996, 177, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Poncet, C.; Soula, C.; Trousse, F.; Kan, P.; Hirsinger, E.; Pourquie, O.; Duprat, A.M.; Cochard, P. Induction of oligodendrocyte progenitors in the trunk neural tube by ventralizing signals: Effects of notochord and floor plate grafts, and of sonic hedgehog. Mech. Dev. 1996, 60, 13–32. [Google Scholar] [CrossRef]
- Marti, E.; Takada, R.; Bumcrot, D.A.; Sasaki, H.; McMahon, A.P. Distribution of sonic hedgehog peptides in the developing chick and mouse embryo. Development 1995, 121, 2537–2547. [Google Scholar] [PubMed]
- Rowitch, D.H.; S-Jacques, B.; Lee, S.M.; Flax, J.D.; Snyder, E.Y.; McMahon, A.P. Sonic hedgehog regulates proliferation and inhibits differentiation of cns precursor cells. J. Neurosci. 1999, 19, 8954–8965. [Google Scholar] [PubMed]
- Orentas, D.M.; Hayes, J.E.; Dyer, K.L.; Miller, R.H. Sonic hedgehog signaling is required during the appearance of spinal cord oligodendrocyte precursors. Development 1999, 126, 2419–2429. [Google Scholar] [PubMed]
- Soula, C.; Danesin, C.; Kan, P.; Grob, M.; Poncet, C.; Cochard, P. Distinct sites of origin of oligodendrocytes and somatic motoneurons in the chick spinal cord: Oligodendrocytes arise from Nkx2.2-expressing progenitors by a shh-dependent mechanism. Development 2001, 128, 1369–1379. [Google Scholar] [PubMed]
- Lu, Q.R.; Yuk, D.; Alberta, J.A.; Zhu, Z.; Pawlitzky, I.; Chan, J.; McMahon, A.P.; Stiles, C.D.; Rowitch, D.H. Sonic hedgehog—Regulated oligodendrocyte lineage genes encoding BHLH proteins in the mammalian central nervous system. Neuron 2000, 25, 317–329. [Google Scholar] [CrossRef]
- Sussman, C.R.; Davies, J.E.; Miller, R.H. Extracellular and intracellular regulation of oligodendrocyte development: Roles of sonic hedgehog and expression of E proteins. Glia 2002, 40, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Park, H.C.; Mehta, A.; Richardson, J.S.; Appel, B. Olig2 is required for zebrafish primary motor neuron and oligodendrocyte development. Dev. Biol. 2002, 248, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Schebesta, M.; Serluca, F.C. Olig1 expression identifies developing oligodendrocytes in zebrafish and requires hedgehog and notch signaling. Dev. Dyn. 2009, 238, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Takebayashi, H.; Nabeshima, Y.; Yoshida, S.; Chisaka, O.; Ikenaka, K. The basic helix-loop-helix factor Olig2 is essential for the development of motoneuron and oligodendrocyte lineages. Curr. Biol. 2002, 12, 1157–1163. [Google Scholar] [CrossRef]
- Sun, T.; Pringle, N.P.; Hardy, A.P.; Richardson, W.D.; Smith, H.K. Pax6 influences the time and site of origin of Glial precursors in the ventral neural tube. Mol. Cell. Neurosci. 1998, 12, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Qi, Y.; Tan, M.; Cai, J.; Takebayashi, H.; Nakafuku, M.; Richardson, W.; Qiu, M. Dual origin of spinal oligodendrocyte progenitors and evidence for the cooperative role of Olig2 and Nkx2.2 in the control of oligodendrocyte differentiation. Development 2002, 129, 681–693. [Google Scholar] [PubMed]
- Zhou, Q.; Choi, G.; Anderson, D.J. The BHLH transcription factor Olig2 promotes oligodendrocyte differentiation in collaboration with Nkx2.2. Neuron 2001, 31, 791–807. [Google Scholar] [CrossRef]
- Agius, E.; Soukkarieh, C.; Danesin, C.; Kan, P.; Takebayashi, H.; Soula, C.; Cochard, P. Converse control of oligodendrocyte and astrocyte lineage development by sonic hedgehog in the chick spinal cord. Dev. Biol. 2004, 270, 308–321. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Wu, Y.; Capecchi, M.R. Motoneurons and oligodendrocytes are sequentially generated from neural stem cells but do not appear to share common lineage-restricted progenitors in vivo. Development 2006, 133, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Delaunay, D.; Heydon, K.; Cumano, A.; Schwab, M.H.; Thomas, J.L.; Suter, U.; Nave, K.A.; Zalc, B.; Spassky, N. Early neuronal and glial fate restriction of embryonic neural stem cells. J. Neurosci. 2008, 28, 2551–2562. [Google Scholar] [CrossRef] [PubMed]
- Ravanelli, A.M.; Appel, B. Motor neurons and oligodendrocytes arise from distinct cell lineages by progenitor recruitment. Genes Dev. 2015, 29, 2504–2515. [Google Scholar] [CrossRef] [PubMed]
- Park, H.C.; Shin, J.; Appel, B. Spatial and temporal regulation of ventral spinal cord precursor specification by hedgehog signaling. Development 2004, 131, 5959–5969. [Google Scholar] [CrossRef] [PubMed]
- Danesin, C.; Agius, E.; Escalas, N.; Ai, X.; Emerson, C.; Cochard, P.; Soula, C. Ventral neural progenitors switch toward an oligodendroglial fate in response to increased sonic hedgehog (shh) activity: Involvement of sulfatase 1 in modulating shh signaling in the ventral spinal cord. J. Neurosci. 2006, 26, 5037–5048. [Google Scholar] [CrossRef] [PubMed]
- Touahri, Y.; Escalas, N.; Benazeraf, B.; Cochard, P.; Danesin, C.; Soula, C. Sulfatase 1 promotes the motor neuron-to-oligodendrocyte fate switch by activating shh signaling in Olig2 progenitors of the embryonic ventral spinal cord. J. Neurosci. 2012, 32, 18018–18034. [Google Scholar] [CrossRef] [PubMed]
- Al Oustah, A.; Danesin, C.; Khouri-Farah, N.; Farreny, M.A.; Escalas, N.; Cochard, P.; Glise, B.; Soula, C. Dynamics of sonic hedgehog signaling in the ventral spinal cord are controlled by intrinsic changes in source cells requiring sulfatase 1. Development 2014, 141, 1392–1403. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, H.; Ishino, Y.; Jiang, W.; Yoshimura, T.; Takeda-Uchimura, Y.; Uchimura, K.; Kadomatsu, K.; Ikenaka, K. Keratan sulfate regulates the switch from motor neuron to oligodendrocyte generation during development of the mouse spinal cord. Neurochem. Res. 2016, 41, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; McGlynn, S.; Matise, M.P. Floor plate-derived sonic hedgehog regulates glial and ependymal cell fates in the developing spinal cord. Development 2013, 140, 1594–1604. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.Y.; Kim, S.; Kim, E.; Kim, D.; Jeong, I.; Cha, Y.R.; Bae, Y.K.; Park, S.W.; Lee, J.; Park, H.C. Indian hedgehog B function is required for the specification of oligodendrocyte progenitor cells in the zebrafish cns. J. Neurosci. 2013, 33, 1728–1733. [Google Scholar] [CrossRef] [PubMed]
- Ekker, S.C.; Ungar, A.R.; Greenstein, P.; von Kessler, D.P.; Porter, J.A.; Moon, R.T.; Beachy, P.A. Patterning activities of vertebrate hedgehog proteins in the developing eye and brain. Curr. Biol. 1995, 5, 944–955. [Google Scholar] [CrossRef]
- Krauss, S.; Concordet, J.P.; Ingham, P.W. A functionally conserved homolog of the drosophila segment polarity gene hh is expressed in tissues with polarizing activity in zebrafish embryos. Cell 1993, 75, 1431–1444. [Google Scholar] [CrossRef]
- Hajihosseini, M.; Tham, T.N.; Dubois-Dalcq, M. Origin of oligodendrocytes within the human spinal cord. J. Neurosci. 1996, 16, 7981–7994. [Google Scholar] [PubMed]
- Qi, Y.; Tan, M.; Hui, C.C.; Qiu, M. Gli2 is required for normal shh signaling and oligodendrocyte development in the spinal cord. Mol. Cell. Neurosci. 2003, 23, 440–450. [Google Scholar] [CrossRef]
- Tan, M.; Hu, X.; Qi, Y.; Park, J.; Cai, J.; Qiu, M. Gli3 mutation rescues the generation, but not the differentiation, of oligodendrocytes in shh mutants. Brain Res. 2006, 1067, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Huang, X.; Chiang, C. Specific requirements of sonic hedgehog signaling during oligodendrocyte development. Dev. Dyn. 2005, 234, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Cameron-Curry, P.; Le Douarin, N.M. Oligodendrocyte precursors originate from both the dorsal and the ventral parts of the spinal cord. Neuron 1995, 15, 1299–1310. [Google Scholar] [CrossRef]
- Spassky, N.; Goujet-Zalc, C.; Parmantier, E.; Olivier, C.; Martinez, S.; Ivanova, A.; Ikenaka, K.; Macklin, W.; Cerruti, I.; Zalc, B.; et al. Multiple restricted origin of oligodendrocytes. J. Neurosci. 1998, 18, 8331–8343. [Google Scholar] [CrossRef]
- Spassky, N.; Olivier, C.; Perez-Villegas, E.; Goujet-Zalc, C.; Martinez, S.; Thomas, J.; Zalc, B. Single or multiple oligodendroglial lineages: A controversy. Glia 2000, 29, 143–148. [Google Scholar] [CrossRef]
- Chandran, S.; Compston, A.; Jauniaux, E.; Gilson, J.; Blakemore, W.; Svendsen, C. Differential generation of oligodendrocytes from human and rodent embryonic spinal cord neural precursors. Glia 2004, 47, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, M.; Richardson, W.D.; Kessaris, N. A subset of oligodendrocytes generated from radial glia in the dorsal spinal cord. Development 2005, 132, 1951–1959. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Qi, Y.; Hu, X.; Tan, M.; Liu, Z.; Zhang, J.; Li, Q.; Sander, M.; Qiu, M. Generation of oligodendrocyte precursor cells from mouse dorsal spinal cord independent of Nkx6 regulation and shh signaling. Neuron 2005, 45, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Vallstedt, A.; Klos, J.M.; Ericson, J. Multiple dorsoventral origins of oligodendrocyte generation in the spinal cord and hindbrain. Neuron 2005, 45, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Mekki-Dauriac, S.; Agius, E.; Kan, P.; Cochard, P. Bone morphogenetic proteins negatively control oligodendrocyte precursor specification in the chick spinal cord. Development 2002, 129, 5117–5130. [Google Scholar] [PubMed]
- Miller, R.H.; Dinsio, K.; Wang, R.; Geertman, R.; Maier, C.E.; Hall, A.K. Patterning of spinal cord oligodendrocyte development by dorsally derived BMP4. J. Neurosci. Res. 2004, 76, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.B.; Clarke, L.E.; Burzomato, V.; Kessaris, N.; Anderson, P.N.; Attwell, D.; Richardson, W.D. Dorsally and ventrally derived oligodendrocytes have similar electrical properties but myelinate preferred tracts. J. Neurosci. 2011, 31, 6809–6819. [Google Scholar] [CrossRef] [PubMed]
- Timsit, S.G.; Bally-Cuif, L.; Colman, D.R.; Zalc, B. Dm-20 mrna is expressed during the embryonic development of the nervous system of the mouse. J. Neurochem. 1992, 58, 1172–1175. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.E.; Miller, R.H. Local sonic hedgehog signaling regulates oligodendrocyte precursor appearance in multiple ventricular zone domains in the chick metencephalon. Dev. Biol. 2001, 233, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Fujisawa, H.; Hirano, S.; Norita, M.; Tsumori, T.; Yasui, Y. Early development of the oligodendrocyte in the embryonic chick metencephalon. J. Neurosci. Res. 1997, 48, 212–225. [Google Scholar] [CrossRef]
- Alberta, J.A.; Park, S.K.; Mora, J.; Yuk, D.; Pawlitzky, I.; Iannarelli, P.; Vartanian, T.; Stiles, C.D.; Rowitch, D.H. Sonic hedgehog is required during an early phase of oligodendrocyte development in mammalian brain. Mol. Cell. Neurosci. 2001, 18, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Cunliffe, V.T.; Casaccia-Bonnefil, P. Histone deacetylase 1 is essential for oligodendrocyte specification in the zebrafish CNS. Mech. Dev. 2006, 123, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Boyd, P.J.; Cunliffe, V.T.; Roy, S.; Wood, J.D. Sonic hedgehog functions upstream of disrupted-in-schizophrenia 1 (DISC1): Implications for mental illness. Biol. Open 2015, 4, 1336–1343. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Cai, J.; Rutledge, M.; Hu, X.; Qiu, M. Oligodendrocytes can be generated from the local ventricular and subventricular zones of embryonic chicken midbrain. Brain Res. 2003, 143, 161–165. [Google Scholar] [CrossRef]
- Mecklenburg, N.; Garcia-Lopez, R.; Puelles, E.; Sotelo, C.; Martinez, S. Cerebellar oligodendroglial cells have a mesencephalic origin. Glia 2011, 59, 1946–1957. [Google Scholar] [CrossRef] [PubMed]
- Bouslama-Oueghlani, L.; Wehrle, R.; Doulazmi, M.; Chen, X.R.; Jaudon, F.; Lemaigre-Dubreuil, Y.; Rivals, I.; Sotelo, C.; Dusart, I. Purkinje cell maturation participates in the control of oligodendrocyte differentiation: Role of sonic hedgehog and vitronectin. PLoS ONE 2012, 7, e49015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birling, M.C.; Price, J. A study of the potential of the embryonic rat telencephalon to generate oligodendrocytes. Dev. Biol. 1998, 193, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Kalman, M.; Tuba, A. Differences in myelination between spinal cord and corticular tissue transplanted intraocularly in rats. Int. J. Dev. Neurosci. 1998, 16, 115–121. [Google Scholar] [CrossRef]
- Perez Villegas, E.M.; Olivier, C.; Spassky, N.; Poncet, C.; Cochard, P.; Zalc, B.; Thomas, J.L.; Martinez, S. Early specification of oligodendrocytes in the chick embryonic brain. Dev. Biol. 1999, 216, 98–113. [Google Scholar] [CrossRef] [PubMed]
- Nery, S.; Wichterle, H.; Fishell, G. Sonic hedgehog contributes to oligodendrocyte specification in the mammalian forebrain. Development 2001, 128, 527–540. [Google Scholar] [PubMed]
- Qi, Y.; Stapp, D.; Qiu, M. Origin and molecular specification of oligodendrocytes in the telencephalon. Trends Neurosci. 2002, 25, 223–225. [Google Scholar] [CrossRef]
- Tekki-Kessaris, N.; Woodruff, R.; Hall, A.C.; Gaffield, W.; Kimura, S.; Stiles, C.D.; Rowitch, D.H.; Richardson, W.D. Hedgehog-dependent oligodendrocyte lineage specification in the telencephalon. Development 2001, 128, 2545–2554. [Google Scholar] [PubMed]
- Chojnacki, A.; Weiss, S. Isolation of a novel platelet-derived growth factor-responsive precursor from the embryonic ventral forebrain. J. Neurosci. 2004, 24, 10888–10899. [Google Scholar] [CrossRef] [PubMed]
- Kessaris, N.; Jamen, F.; Rubin, L.L.; Richardson, W.D. Cooperation between sonic hedgehog and fibroblast growth factor/MAPK signalling pathways in neocortical precursors. Development 2004, 131, 1289–1298. [Google Scholar] [CrossRef] [PubMed]
- Furusho, M.; Kaga, Y.; Ishii, A.; Hebert, J.M.; Bansal, R. Fibroblast growth factor signaling is required for the generation of oligodendrocyte progenitors from the embryonic forebrain. J. Neurosci. 2011, 31, 5055–5066. [Google Scholar] [CrossRef] [PubMed]
- Miyake, A.; Chitose, T.; Kamei, E.; Murakami, A.; Nakayama, Y.; Konishi, M.; Itoh, N. Fgf16 is required for specification of gabaergic neurons and oligodendrocytes in the zebrafish forebrain. PLoS ONE 2014, 9, e110836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.Y.; Du, Z.W.; Li, X.J.; Ayala, M.; Zhang, S.C. Human oligodendrocytes from embryonic stem cells: Conserved shh signaling networks and divergent fgf effects. Development 2009, 136, 1443–1452. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.B.; Joyner, A.L. Gli1 can rescue the in vivo function of Gli2. Development 2001, 128, 5161–5172. [Google Scholar] [PubMed]
- Kessaris, N.; Fogarty, M.; Iannarelli, P.; Grist, M.; Wegner, M.; Richardson, W.D. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat. Neurosci. 2006, 9, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Olivier, C.; Cobos, I.; Perez Villegas, E.M.; Spassky, N.; Zalc, B.; Martinez, S.; Thomas, J.L. Monofocal origin of telencephalic oligodendrocytes in the anterior entopeduncular area of the chick embryo. Development 2001, 128, 1757–1769. [Google Scholar] [PubMed]
- Machold, R.; Hayashi, S.; Rutlin, M.; Muzumdar, M.D.; Nery, S.; Corbin, J.G.; Gritli-Linde, A.; Dellovade, T.; Porter, J.A.; Rubin, L.L.; et al. Sonic hedgehog is required for progenitor cell maintenance in telencephalic stem cell niches. Neuron 2003, 39, 937–950. [Google Scholar] [CrossRef]
- Lelievre, V.; Ghiani, C.A.; Seksenyan, A.; Gressens, P.; de Vellis, J.; Waschek, J.A. Growth factor-dependent actions of pacap on oligodendrocyte progenitor proliferation. Regul. Pept. 2006, 137, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Tong, C.K.; Fuentealba, L.C.; Shah, J.K.; Lindquist, R.A.; Ihrie, R.A.; Guinto, C.D.; Rodas-Rodriguez, J.L.; Alvarez-Buylla, A. A dorsal shh-dependent domain in the V-SVZ produces large numbers of oligodendroglial lineage cells in the postnatal brain. Stem Cell Rep. 2015, 5, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Samanta, J.; Grund, E.M.; Silva, H.M.; Lafaille, J.J.; Fishell, G.; Salzer, J.L. Inhibition of Gli1 mobilizes endogenous neural stem cells for remyelination. Nature 2015, 526, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Loulier, K.; Ruat, M.; Traiffort, E. Increase of proliferating oligodendroglial progenitors in the adult mouse brain upon sonic hedgehog delivery in the lateral ventricle. J. Neurochem. 2006, 98, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Jakovcevski, I.; Filipovic, R.; Mo, Z.; Rakic, S.; Zecevic, N. Oligodendrocyte development and the onset of myelination in the human fetal brain. Front. Neuroanat. 2009, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Rakic, S.; Zecevic, N. Early oligodendrocyte progenitor cells in the human fetal telencephalon. Glia 2003, 41, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.; Zecevic, N. Human fetal radial glia cells generate oligodendrocytes in vitro. Glia 2009, 57, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Ortega, J.A.; Radonjic, N.V.; Zecevic, N. Sonic hedgehog promotes generation and maintenance of human forebrain Olig2 progenitors. Front. Cell. Neurosci. 2013, 7, 254. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Hernandez, M.; Shen, S.; Sabo, J.K.; Kelkar, D.; Wang, J.; O’Leary, R.; Phillips, G.R.; Cate, H.S.; Casaccia, P. Differential modulation of the oligodendrocyte transcriptome by sonic hedgehog and bone morphogenetic protein 4 via opposing effects on histone acetylation. J. Neurosci. 2012, 32, 6651–6664. [Google Scholar] [CrossRef] [PubMed]
- Small, R.K.; Riddle, P.; Noble, M. Evidence for migration of oligodendrocyte—Type-2 astrocyte progenitor cells into the developing rat optic nerve. Nature 1987, 328, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Miller, R.H. Specification of optic nerve oligodendrocyte precursors by retinal ganglion cell axons. J. Neurosci. 2006, 26, 7619–7628. [Google Scholar] [CrossRef] [PubMed]
- Spassky, N.; de Castro, F.; Le Bras, B.; Heydon, K.; Queraud-LeSaux, F.; Bloch-Gallego, E.; Chedotal, A.; Zalc, B.; Thomas, J.L. Directional guidance of oligodendroglial migration by class 3 semaphorins and netrin-1. J. Neurosci. 2002, 22, 5992–6004. [Google Scholar] [PubMed]
- Dakubo, G.D.; Wang, Y.P.; Mazerolle, C.; Campsall, K.; McMahon, A.P.; Wallace, V.A. Retinal ganglion cell-derived sonic hedgehog signaling is required for optic disc and stalk neuroepithelial cell development. Development 2003, 130, 2967–2980. [Google Scholar] [CrossRef] [PubMed]
- Wallace, V.A.; Raff, M.C. A role for sonic hedgehog in axon-to-astrocyte signalling in the rodent optic nerve. Development 1999, 126, 2901–2909. [Google Scholar] [PubMed]
- Traiffort, E.; Moya, K.L.; Faure, H.; Hassig, R.; Ruat, M. High expression and anterograde axonal transport of aminoterminal sonic hedgehog in the adult hamster brain. Eur. J. Neurosci. 2001, 14, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Macklin, W.; Gerson, J.; Miller, R.H. Intrinsic and extrinsic inhibition of oligodendrocyte development by rat retina. Dev. Biol. 2006, 290, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Merchan, P.; Bribian, A.; Sanchez-Camacho, C.; Lezameta, M.; Bovolenta, P.; de Castro, F. Sonic hedgehog promotes the migration and proliferation of optic nerve oligodendrocyte precursors. Mol. Cell. Neurosci. 2007, 36, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M.C.; Cases, O.; Merchan, P.; Kozyraki, R.; Clemente, D.; de Castro, F. Megalin mediates the influence of sonic hedgehog on oligodendrocyte precursor cell migration and proliferation during development. Glia 2012, 60, 851–866. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Traiffort, E.; Zakaria, M.; Laouarem, Y.; Ferent, J. Hedgehog: A Key Signaling in the Development of the Oligodendrocyte Lineage. J. Dev. Biol. 2016, 4, 28. https://doi.org/10.3390/jdb4030028
Traiffort E, Zakaria M, Laouarem Y, Ferent J. Hedgehog: A Key Signaling in the Development of the Oligodendrocyte Lineage. Journal of Developmental Biology. 2016; 4(3):28. https://doi.org/10.3390/jdb4030028
Chicago/Turabian StyleTraiffort, Elisabeth, Mary Zakaria, Yousra Laouarem, and Julien Ferent. 2016. "Hedgehog: A Key Signaling in the Development of the Oligodendrocyte Lineage" Journal of Developmental Biology 4, no. 3: 28. https://doi.org/10.3390/jdb4030028