Insights into the Etiology of Mammalian Neural Tube Closure Defects from Developmental, Genetic and Evolutionary Studies

Department of Medical Genetics, University of British Columbia, 2350 Health Sciences Mall, Vancouver, BC V6T 1Z3, Canada
*
Author to whom correspondence should be addressed.
J. Dev. Biol. 2018, 6(3), 22; https://doi.org/10.3390/jdb6030022
Submission received: 15 July 2018
/
Revised: 13 August 2018
/
Accepted: 15 August 2018
/
Published: 21 August 2018
(This article belongs to the Special Issue Development of the Brain in Health and Disease)
Abstract
:The human neural tube defects (NTD), anencephaly, spina bifida and craniorachischisis, originate from a failure of the embryonic neural tube to close. Human NTD are relatively common and both complex and heterogeneous in genetic origin, but the genetic variants and developmental mechanisms are largely unknown. Here we review the numerous studies, mainly in mice, of normal neural tube closure, the mechanisms of failure caused by specific gene mutations, and the evolution of the vertebrate cranial neural tube and its genetic processes, seeking insights into the etiology of human NTD. We find evidence of many regions along the anterior–posterior axis each differing in some aspect of neural tube closure—morphology, cell behavior, specific genes required—and conclude that the etiology of NTD is likely to be partly specific to the anterior–posterior location of the defect and also genetically heterogeneous. We revisit the hypotheses explaining the excess of females among cranial NTD cases in mice and humans and new developments in understanding the role of the folate pathway in NTD. Finally, we demonstrate that evidence from mouse mutants strongly supports the search for digenic or oligogenic etiology in human NTD of all types.
1. Introduction
The mechanisms of closure of the neural tube and the development of neural tube closure defects (NTD) are genetically complex and diverse. With an incidence of one to ten per thousand births, NTD are among the most common severe birth defects [1]. Defects in cranial and spinal neural tube closure (anencephaly and spina bifida) seem to occur at almost equal frequencies in human embryos [1,2]. In the almost 300 mouse mutants with NTD, cranial defects strongly predominate [3,4]. However, much of the molecular genetic understanding of mammalian neural tube development is based on studies of the spinal region in mouse embryos. Many aspects have been comprehensively reviewed previously [4,5,6,7,8,9]. This review gives an overview of the developmental process and the genetic underpinnings of neural tube closure in mammals, and then focuses on some aspects that have potential to bring new insight.
The formation of a neural tube by the mechanism of rolling up a neural plate into a tube evolved before the evolutionary emergence of vertebrates, and some mammalian molecular mechanisms likely reflect this ancient origin. We briefly consider the process of neural tube formation in the animal usually used as a proxy for the ancestral chordate, Amphioxus, and in Ascidian embryos, representative of another ancient pre-vertebrate lineage, the Tunicates. There are extensive differences in the physical process of neural tube closure among modern species and even among mammals; we shall briefly also consider these to point to the need for assessment of these differences before using observations from model animals to predict the etiologies of human NTD.
We give considerable emphasis to the anterior–posterior regional differences in the mechanisms of neural tube closure at the morphological and molecular levels, underlining the concept that defects of closure of the neural tube are mechanistically heterogeneous with distinct or partially overlapping etiologies. This heterogeneity predicts genetic differences in the etiologies of human NTD located at different sites along the anterior–posterior axis, as well as genetic heterogeneity at any given site. We focus on cranial neural tube development, its genetic mechanisms of failure to close, and the evolutionary New Head hypothesis that predicts consequential differences between the cranial and spinal regions in the mechanisms of neural tube closure. These differences should inform genetic studies of human NTD.
We consider recent advances in understanding of the epigenetic mechanism by which the sex chromosome complement of mammalian embryos influences risk of cranial NTD, seeking new insight into the neural tube closure process. Similarly, we focus on the phenomenon of the preventative effect of maternal folic acid supplementation on occurrence of NTD in humans and in mice and recent developments in understanding of folate mechanisms, seeking possible insights into the etiologies of NTD.
That there is a genetic etiology for human NTD is clear, with 30× elevated risk in siblings of NTD cases, but it is complex. Typical recent studies are based on exon sequencing of candidate genes in cohorts of NTD cases, including a variety of locations and types of lesion. This approach for several planar cell polarity (PCP) genes has detected heterozygous rare putatively deleterious variants in a small number of cases [10].
Despite many genetic studies, the etiology is not well understood [11], and likely involves combinations of deleterious versions of genes in affected individuals, with the combinations differing between individuals [12]. We discuss examples of similar genetic complexity in mouse mutant models.
Failure to close the neural tube leads to three principal open neural tube defects: craniorachischisis, in which the entire spinal region and the hindbrain fail to close; spina bifida aperta, in which a limited region of the spinal neural tube fails to close, and anencephaly or exencephaly, in which the midbrain and sometimes also the hindbrain fail to close. There are other defects that arise subsequently from defects in the tissue of the closed neural tube or surrounding mesenchyme, such as spina bifida occulta (abnormal formation of the vertebral arches from mesenchyme) and encephalocele (extrusion of neural tissue through a defect in the formation of the roof of the closed neural tube). Semantically or clinically, they are often included in NTD, but they are not within the scope of the etiology of failure to form a closed neural tube [6,13].
2. The Basics of Neural Tube Formation
2.1. Components
The major physical components of the complex that will comprise the neural tube (Figure 1) include: the anterior to posterior midline neural plate, comprised of neuroectoderm (neuroepithelium) that develops on the dorsal aspect of the embryo, curls from its lateral edges toward the midline and fuses its formerly lateral edges in the midline; the notochord that underlies the neural plate and produces signaling molecules that affect dorsal–ventral cell identity in the incipient neural tube; the lateral ectoderm that initially abuts the lateral edges of the neural plate, covers the lateral surfaces of the elevating neural folds and after the bending of the folds to the midline, meets and fuses to form a contiguous surface ectoderm over the tube; the paraxial mesoderm lateral to, and supporting, the neuroepithelium of the elevating folds; and the neural crest that is induced in the neuroepithelium of the lateral tips of the spinal neural plate or in the dorsal tips of the elevated cranial neural folds. There are several mechanisms affecting these components that lead to failure of neural tube closure (Table 1).
2.2. Convergent Extension and Other Functions of the Planar Cell Polarity Pathway
Immediately preceding the initiation of neural tube formation, the vertebrate embryo undergoes convergent extension, which involves the lateral-to-medial convergence and anterior–posterior extension of axial tissues, including the neural plate. The non-canonical Wnt/PCP (planar cell polarity) pathway is crucial to this process. Cells change position in the tissue, intercalating between surrounding cells to shift toward the midline. A prerequisite is the directional orientation of cells relative to a morphogenic signal. The cells become asymmetrical in the positioning of various subcellular structures, including the core proteins of the PCP pathway [14,15]. Mutations in PCP genes interfere with the directionality of neuroepithelial cell movement in a variety of gene-specific ways [16]. The planar cell polarity (PCP) pathway is complex, comprising two main sets of interacting genes, the “core” pathway, and the Fat-Dachsous pathway, probably connected by Prickle [14]. The “core” pathway is highly conserved and is regulated by gradients of particular Wnt proteins, (e.g., Wnt5a and Wnt11). The “core” proteins include the transmembrane receptors Vangl1/2, Celsr1/2/3, and Fzd (Frizzled) and the cytosolic proteins Dvl, Prickle, Inversin (Invs) and Scrib [14]. The Fat/Dachsous pathway, acting upstream or in parallel with the PCP core pathway, includes the large membrane cadherins, Fat and Dchs, which bind to each other and are regulated by Fjx [5]. An additional assortment of genes serve various functions necessary to the PCP pathways; for example, Ptk7, Sec24b, Intu and Fuz [10].
In addition to its essential role in planar cell polarity required for convergent extension, the PCP pathway affects ciliogenesis, the arrangement of cytoskeletal actin and microtubules, the basal body and the centriole within cells [14,17,18]. Small Rho family-GTPases regulate the localization of actin polymerization. Microtubules are polarized, and their polarity is required for “correct axis establishment of PCP”, then PCP signaling is required for stability, correct localization and remodeling of microtubule networks. It is thought that PCP, actin and microtubules together determine the asymmetrical positioning of the centriole, ciliary basal bodies, cilia and the orientation of the mitotic spindle [14]. There is also interconnection between the convergent extension system and the neural plate bending system based on actomyosin. For example, RhoA and its effector ROCK are involved in both systems [19,20].
2.3. Bending and Hinge Points
Two areas of the neural plate undergo pronounced bending that strongly contributes to forming a tube; these are the medial hinge point (MHP) located at the midline base of the neural tube throughout most of its length, and the dorsolateral hinge point (DLHP) located in the dorsal third of each fold in some zones along the length of the neural tube [21] (Figure 2). In addition, the contraction of actomyosin found in the apices of all neuroepithelial cells has long been thought to contribute to bending the neural folds, and indeed, apical actomyosin is essential for cranial neural tube closure [19], but its specific role is unclear. In contrast, evidence indicates that apical actomyosin is not essential for spinal neural tube closure [19]. Instead, in the neuroepithelium of the spinal region, an orchestrated dynamic of actin turnover and actomyosin disassembly is essential for closure, and has been suggested to function in maintaining a necessary tension in the neuroepithelium (neither too floppy nor too stiff) between the MHP and DLHP sites [19]. The MHP and DLHP in the spinal region differ in their mechanisms that cause neuroepithelial bending. For the MHP to form in mouse embryos, signaling from the notochord is required [22]. At the MHP, bending is attributed to neuroepithelial cell shape. During the cell cycle, in S phase, nuclei move to the base of the cells, causing them to become wedge-shaped, with a narrow apex and wide base. Compared with the rest of the neuroepithelium, the MHP has an increased proportion of cells in S-phase, and also has cell-cycle dependent changes in apical and basolateral junctional proteins that contribute to cell shape [23,24].
The mouse spinal DLHPs form at the dorsoventral location where the neuroepithelium ceases to have basal contact with the paraxial mesoderm and begins basal contact with the surface ectoderm [23]; however, the significance of this relationship is unknown, given that the upper-spinal neural tube, which can form DLHP in the absence of Shh [22], does not have this mesoderm–ectoderm contact transition. Cell wedging related to cell cycle is not seen at the DLHP. Instead, there is cell proliferation and cell movement dorsally in the neuroepithelium, and evidence of a boundary at the DLHP between areas of different cell density. It has been proposed that the neural folds are caused to bend by the cell density difference [23]. There is an important relationship of the formation of the DLHP in the neuroepithelium with Shh signaling molecules from the ventral neural plate, BMP signaling from the surface ectoderm, and Noggin from the neuroepithelium [22]. The dorsal bends that appear to be DLHP in the midbrain neural folds have not been studied and it is possible that, despite appearing similar morphologically, they are driven by different cellular or molecular mechanisms.
2.4. Cilia
Most cells have a primary cilium. In the neural tube, primary cilia can be seen extending from the neuroepithelium into the incipient lumen (see Figure 6 in [25]). Vital components of signaling pathways that are important to development of the neural tube are located in cilia, and if gene mutations cause the structure/function of the cilia to be defective, these signaling pathways are dysregulated [13,26]. The best understood is Sonic hedgehog (Shh) signaling. Other pathways, such as signaling through Wnt, Calcium, Pdgfr, Notch, and Tgfb, may also function via cilia [13,26,27]. Some of the PCP genes affect the position and angle of some types of cilia on cells [28], and some types of PCP genes (the “effectors”) have a role in ciliogenesis in vertebrates [13,29]. There are many components to the cilia, such as intraflagellar transport proteins and parts of the dynein motors, and when they are mutated, they lead to defects in the structure and function of cilia.
2.5. The Sonic Hedgehog Pathway Signaling in NTD
Several components of the Sonic hedgehog (Shh) signaling pathway, including positive and negative regulators, are present in the cilia [13]. Shh is a secreted glycoprotein whose binding with the transmembrane receptor Patched (Ptch1) in the cilia releases Smoothened (Smo) from repression and activates the Shh signaling pathway. The Shh signaling pathway affects the transcription of downstream target genes via the Gli transcription factors; there are many transcriptional targets and complex feedback loops [30]. An active Shh signaling pathway inhibits the processing of a full-length activator form of Gli3 (Gli3FL) into a repressor form (Gli3R) and the ratio of Gli3FL:Gli3R is a measure of functionality of the Shh signaling pathway [13,30].
During neural tube development, Shh is produced by the notochord and prechordal plate extending from the future forebrain area to the caudal area [22], and induces Shh production from the midline of the neural plate with a ventral to dorsal gradient in the developing neural tube [13]. While the notochord is required for formation of the MHP, and Shh signaling is required for development of the floor plate, it is not clear that Shh is required for formation of the MHP; the MHP is not always required for neural tube closure [6,22]. The formation of the DLHP is inhibited by Shh signaling [22]. Absence of Shh signaling in the dorsal neural folds permits a neuroepithelial response to Noggin, which suppresses Bmp signaling from the adjacent surface ectoderm, enabling formation of dorsolateral hinge points (DLHP) [31]. Shh is expressed in a rostral to caudal and temporal gradient in the developing neural tube from the upper spinal region to the caudal spinal region, being expressed in the caudal notochord and floor plate after neural tube closure [31]. In the cranial neural tube, no gradient has been reported and expression is seen from before closure onwards (e.g., Figure 4c in [32] and Figure 1a in [22]). The presence of DLHP in the cranial neural folds, particularly the midbrain, despite possibly robust Shh signaling in the ventral midline seems contradictory to the Shh concentration-dependent inhibition of DLHP in the upper spine. However, in many species, the cranial neural folds are clearly much larger than the spinal neural folds; this is particularly notable in mouse embryos. Therefore, the physical distance for diffusion of signaling molecules from the midline to the dorsal tips is greater than in the spine. We suggest that the large size of the cranial neural folds creates the necessary lack of signaling concentration in the cranial dorsal folds, enabling cranial DLHP formation.
Loss of function of the Shh gene, or loss of function of components of the Shh signaling pathway such as Smo, do not cause NTD; the neural tube forms a midline bend and DLHP and closes. Typically, loss of function of the Shh signaling pathway causes a lack of development of the cranial midline tissues that results in holoprosencephaly and related facial defects [13,33,34]. In contrast, elevated activity of the Shh signaling pathway downstream of Shh due to the loss of negative regulators such as Ptch1, Rab23, Tulp3 or Sufu [13] leads to elevated signaling in the dorsal neural folds, inhibiting development of the DLHP (e.g., Tulp3 [35]) and causing NTD (exencephaly and sometimes caudal spina bifida) in the regions that normally have DLHP (Figure 2).
Mutations in genes that affect the structure or function of cilia fall into two major types with respect to Shh signaling [13]. Some mutations that affect the structure/function of cilia, such as Ttc21b (intraflagellar transport), Kif7 (kinesin motor protein) and Arl13b (cilial structure), seem to increase Shh signaling pathway activity based on expansion of ventral markers, and cause exencephaly, parallel to the other mutants with increased Shh signaling. Others, such as mutants for several intraflagellar transport (Ift) genes, dynein components (Dync2h1, Dync2li1), PCP effector genes (Fuzzy, Inturned), and C2cd3 (which recruits Ift proteins for ciliogenesis) seem to reduce Shh signaling pathway activity, based on reduced ventral markers in the neural tube, but contrary to the mutations with loss of Shh signaling, they also cause exencephaly. Notably, it seems that cilia mutants that cause exencephaly have an increased Gli3FL:Gli3R ratio [13], but there are many other complex changes and regulatory loop alterations specific to each mutant, and possible involvement of the other cilia-based signaling pathways. Shh signaling also functions through a network of non-canonical, Gli-independent pathways, which could be involved [36]. The process of cranial neural fold formation and bending does not seem to have been examined in each mutant with exencephaly and it is unknown whether there is a shared mechanism, such as lack of DLHP formation, or a heterogeneity of mechanisms such as apoptosis in the neuroepithelium or deficient cell adhesion.
2.6. Adhesion and Fusion
To complete neural tube closure, the neuroepithelium and surface ectoderm of the neural fold must make contact with their counterparts in the apposed fold and form de novo adhesions to create a continuous surface ectoderm covering a continuous neuroepithelium. The neuroepithelia and surface ectoderms do not necessarily make contact simultaneously, and the order varies along the anterior–posterior axis. Some details have emerged from recent studies. Filopodia and ruffles project from apposed surface ectoderms into the gap between the apposed folds and are required for successful closure, although their role is unknown [37,38,39]. Various membrane-bound Eph receptor tyrosine kinases and their membrane bound ligands, the Ephrins, which are generally involved in cell–cell attraction and repulsion interactions, are found in the neural folds, including the dorsal tips, just prior to fusion and are possibly involved in initiation of adhesions between apposed folds [38,40].
3. Regional Differences in Closure Mechanisms
Studies of mouse embryos have demonstrated that different regions along the anterior–posterior axis have different major mechanisms for bending the neuroepithelium to meet in the midline and also differ in fusion mechanisms (Table 2). These reflect several factors [5,6] including the early spinal role of planar cell polarity, the timing of anterior–posterior development of the notochord relative to closure, a gradient of Shh signaling along the anterior to posterior spine [22], presence of MHP and DLHP, differences in expression domains along the anterior–posterior axis of various genes important to closure, such as the Grhl gene family [41] and the Ephrin/Eph gene families [40,42,43], types of cell projections, cell types that initiate fusion, and factors in the molecular genetics of closure that are unique to the cranial region, such as the dynamics of the actomyosin cytoskeleton. Given the distinct regional differences in mechanism and genes involved in neurulation, studies to identify causative genetic variants in human NTD would be strengthened by studying pools of cases that share a similar location of lesion.
3.1. Planar Cell Polarity, MHP and DLHP
The upper spine region depends on planar cell polarity for cell movements and convergent extension that narrows the midline gap between the future neural folds, and on cell cycle dynamics in the neural plate midline that cause cell wedging to create a medial hinge point (MHP). The mid-spinal region also depends on the effects of convergent extension, and has the MHP, but additionally has DLHPs. The most caudal area of primary neurulation, the posterior neuropore (PNP), likely also depends on initial convergent extension to narrow the neural plate, but unlike the rest of the neural tube, the notochord is not present before closure [22]. The MHP does not form, but DLHPs bend the folds to the midline; there is also a greater dependence on actomyosin contraction for bending than in the more rostral spinal region [44]. Note: the PNP is sometimes used to refer to an extended region including part of the mid-lower spine to the most caudal end of primary neurulation; in this review we refer to only the most caudal part of this region as the PNP.
Caudal to the PNP, neural folds do not form and a tube is hollowed out from the caudal mesenchyme in a process termed “secondary neurulation”. Defects in secondary neurulation per se would not cause closure defects, but there is evidence that mis-regulation of the development of the junction between the PNP and secondary neurulation may cause some lumbosacral open neural tube defects [45,46].
Overall, given the various overlapping regional differences in molecular genetic mechanisms and closure mechanisms from head to tail, it is likely that NTD in different anterior–posterior regions have region-specific causes that can be predicted. For example, in families with recurrence of NTDs, in about 70% of families, the second cases have the same defect as the first case, but in about 30% of families, one has spina bifida and one has anencephaly [47]. Given that failure of DLHP is a mechanism of NTD shared by anencephaly and lumbosacral spina bifida aperta, we suggest these latter families may be enriched for defects in genes that affect DLHP, which can inform the genetic approaches to unraveling some of the genetic complexity of human NTD.
3.2. Closure Initiation Sites
At several specific locations along the anterior–posterior axis the neural folds achieve close apposition and initiate fusion. Closure then is thought to “zip” along the neural folds until it meets a closed region “zipped” from a different initiation site—an intermittent pattern rather than a single “zipper”. The mechanisms that determine the locations of closure initiation sites are not known. The locations of cranial closure initiation sites differ somewhat between normal mouse strains [48]; the possibility of similar variation within other species should be considered. The locations of cranial closure initiation sites and patterns of closure vary greatly between mammalian species as summarized in Figure 3 (mouse [49,50,51], rat [52,53], hamster [54,55], rabbit [56], pig [57], human [58,59,60,61,62]). The pattern of cranial closure initiation sites differs greatly between humans and mice, rats or hamsters, particularly with respect to the presence of an initiation site near the forebrain–midbrain junction that is not seen in human embryos (Figure 3). The closure process in rabbit and pig seem more similar to human. In rabbit, pig and human, instead of “zipping”, extended regions in the head and spinal region come into close apposition and then fuse simultaneously. Greater axial curvature seems to reduce the rate of closure along the anterior–posterior axis and between species [63]; flat areas tend to close simultaneously rather than “zip”. Otherwise, the location of the Closure 1 initiation site at the future neck (cervical) region and pattern of expansion of closure from it to the posterior neuropore seems generally similar across mammalian species.
The relevance to NTD of species differences in locations of closure initiation sites is debatable. Generally, the locations of open cranial NTD and open spinal NTD in mice and humans appear to be the same, i.e., the same parts of the neural tube have failed to close. As we have said previously “… if the defect in both species is a failure of elevation itself, a species difference in the location of first contact of already elevated folds would be subsequent to, and unrelated to, the cause of NTDs” [64]. Furthermore, the exact positions of open defects may not be interpretable with respect to inferences about locations of closure initiation sites. In mouse models, embryos sharing the same genetic etiology often differ in the exact location of the open neural tube. For example, in the SELH/Bc model, the rostral edge of exencephaly may be located anywhere between the caudal limit of the forebrain and the middle of the midbrain and, independently, the caudal edge of the exencephaly may be located anywhere between the caudal limit of the midbrain and the caudal limit of the hindbrain (Figure 4 in [65]). In the SELH model, we have shown that, in the absence of a midbrain initiation site, the normal closure initiated in the forebrain can compensate by advancing caudally to close the entire midbrain, and that closure of the hindbrain can also compensate by continuing rostrally to close part of the caudal midbrain. The variation in location of exencephaly seems to reflect stochastic variation in the efficiencies of these compensatory processes. We suggest that this type of compensatory mechanism might explain part of the documented variation in locations of human anencephaly [66].
3.3. Cell Type of Initial Contact
The tips of the neural folds expose the border between the neuroepithelium and surface ectoderm. The cell type involved in initial contact between apposed folds varies regionally. In the mouse forebrain region, first contact across the gap is between the apposed neuroepithelia, whereas in the midbrain and hindbrain, the initial contact and apparent adhesion is between cells of the surface ectoderm, which in the midbrain wraps around the neuroectoderm as the folds meet so that surface ectoderm faces across the closing gap [37,67]. In the posterior neuropore, the surface ectoderm may make first contact, based on locations of cellular projections [5].
The fusion of the neuroepithelium and the surface ectoderm may be independent; a closed cerebellar defect arising in an unfused neuroepithelium beneath a fused surface ectoderm has been described in mice [65].
3.4. Cell Projection Types
Fusions between epithelia in various tissues and species are typically preceded by various cellular projections such as ruffles, filopodia and lamellipodia [38]). Dynamic cell behaviors and interactions, such as the interdigitation of ruffles across the gap between the closely apposed neural folds of the closing neural tube, are seen and the projections are required for neural tube closure [38,55,68]. The density and type of cell projections vary along the anterior–posterior axis of the neural folds [38,55]. The forebrain, midbrain and hindbrain differ in the presence of filopodia and ruffles. In the forebrain region, live imaging of surface ectoderm has shown that there are a few small narrow projections close to the area of contact, but the majority of the surface ectoderm does not have projections [39]. In the midbrain region as it approaches closure, there are many surface ectoderm projections that appear to be filopodia [39]. In the closing hindbrain region there are many long round projections from the surface ectoderm, some of which may be filopodia, and others a relatively narrow form of ruffles [39]. The upper spine Closure 1 initiation region has wide membranous ruffles or lamellipodia and narrow bulbous projections, both extending from the surface ectoderm towards the gap at the midline and increasing as the folds move closer together; these projections are the first points of contact across the gap [39]. The caudally advancing closure region of the upper spine region has filopodia [37]. The mid-spinal region has both filopodia and ruffles in the closing area. The most caudal region of primary neurulation has only ruffles [38,55].
The regional differences in the abundance of filopodia and ruffles reflect, at least in part, differences in regulators of the actin cytoskeleton, Rac and Cdc42 [38]. These GTPases are molecular switches that link extracellular signals to specific downstream effectors. In general, Rac1 induces the formation of branched actin networks needed for lamellipodia and ruffles, whereas Cdc42 leads to assembly of unbranched actin bundles that form filopodia [38].
3.5. Regional Differences in Ephrins and Ephrin Receptors
The gene families of membrane-bound Ephrin receptor tyrosine kinases (Ephs) and their membrane-bound ligands, the Ephrins, are involved in cell–cell interactions throughout development. Interaction between Ephrins and their receptors may cause cell adhesion or repulsion. They are involved in the adhesion of apposed neural folds for fusion and may have a role in the etiology of NTD. This area has not been deeply studied and roles of more members of these gene families are likely to be identified in future studies. Expression of some combinations of Ephrins and Ephs may differ between cranial regions and also differ from the caudal region. EphA4 is expressed in the midbrain region in early neural fold stages [69]; EphA2 and EphA4 are expressed in the hindbrain neural folds before closure [70]. EphrinA5 and its receptor EphA7 splice forms are expressed in the dorsal cranial neural folds (midbrain and hindbrain) before and during closure, but not in the spinal neural folds [40]. In the posterior neuropore, EphA1 is expressed in the neural plate and other tissues, EphA2 in the neural plate and tips of the apposed neural folds, EphA4 in the mesoderm and tips of the apposed neural folds, and EphA5 in the neural plate [43]. Further study of EphA2 in the tips of the neural folds located its expression to surface ectoderm cells and their lamellipodia-like protrusions that extend towards the apposed neural fold [43].
3.6. Requirement for Grhl Gene Family Expression
The regionality of the requirement for expression of different members of the Grhl gene family along the anterior–posterior axis of the neural folds in mice is an important demonstration that the locations of NTD reflect the regional requirement for expression of specific genes. Loss of function of Grhl3 causes thoraco-lumbo-sacral spina bifida and curled tail (a microform of spina bifida) in all homozygotes and concomitant exencephaly in a few [71]. Loss of function of Grhl2 causes split face and open cranial neural tube extending through the hindbrain to the cervical region and, depending on the mutant allele and genetic background, spina bifida [37,41]. Heterozygotes at either locus are normal, but some digenic heterozygotes (Grhl2+/−;Grhl3+/−) have lumbo-sacral spina bifida with curled tail or exencephaly and/or curled tail. Grhl2 null and Grhl3 null embryos fail to form DLHP in the regions that will have failed neural tube closure [41].
Further study [41] indicates that Grhl2 is essential to closure of the forebrain and cooperates with Grhl3, whereas Grhl3 is essential to closure posterior to the mid-thoracic region, and cooperates with Grlh2 for closure of the posterior neuropore (PNP). Neither Grhl gene is required for closure of the cervical region to the thoracic region, the rostral part of the Closure 1 zone; this is the only region of the neural tube that does not form a concave shape of neural folds before closure, and may therefore be the only region that does not have DLHPs. DLHPs are lacking in the PNP of both Grhl2 and Grhl3 mutants; other regions were not studied. In summary, the Grhl2 and Grhl3 genes may be respectively essential for DLHP formation in separate zones of the developing neural tube, and may also function cooperatively for DLHP formation in other zones. The Grhl mutants did not alter the expression of some genes known to be important in DLHP formation, such as Bmp2, Noggin, and Zic2 [41]. The failure to form DLHP may be explained by the observation that Grhl2, which is specifically expressed in the surface ectoderm of the cranial neural folds, is required to suppress transformation of ectoderm to mesenchyme [72]. The acquisition of mesenchymal characteristics and loss of integrity of the surface ectoderm likely causes the inability to bend the dorsal neural fold. A similar disruptive effect on the development of DLHPs or the bending to concave shape is suggested for the homolog, Grhl3. In the caudal region Grhl3, is expressed in surface ectoderm during neural tube closure, and lack of Grhl3 seems to cause delay of specification of surface ectoderm [73], a situation somewhat like the findings for Grhl2 [72]. Later, at the most caudal end of primary neurulation, a second mechanism is observed. Grhl3 is expressed in the gut endoderm that underlies the neural tube, and lack of Grhl3 expression there is associated with defective hindgut cell proliferation that causes excessive axial bending that mechanically interferes with closure of the posterior neuropore [73].
3.7. Dynamics of the Actomyosin Cytoskeleton of the Neuroepithelium
Actomyosin “machinery” (actin filaments, phosphorylated myosin light chain (pMLC) and RhoGTPases) is located circumferentially in the apices and apical junctions of all neuroepithelial cells. Its contraction is thought to be responsible for apical constriction, contributing to neural fold bending, and is required for cranial neural tube closure but not spinal neural tube closure. Mice with mutations affecting actin-associated proteins develop cranial, but not spinal, NTDs. Conversely, the caudal spinal region as well as the cranial region requires actin turnover for neural tube closure and abnormal accumulation of apical actomyosin does cause failure of neural fold elevation in both regions [19]. RhoA and its effector ROCK are required in the neuroepithelium, to maintain balanced apical actomyosin accumulation, to regulate actin turnover, as well as for convergent extension [19,20]. A related caudal difference is the contribution of bipotential neuromesodermal progenitors to the caudal neuroectoderm and somatic mesoderm, and the influence of PCP (Vangl2) on their differentiation as well as on neuroepithelial actomyosin contractility [44].
3.8. Other Examples: Hox, Neural Crest and Apoptosis
Other regional differences in the neural tube also have an unknown or uncertain role in closure mechanisms. For example, numerous Hox genes, which convey anterior–posterior positional information, are expressed in many different combinations along the midline axis before neural tube formation, with the earliest and fewest expressed in the hindbrain region and the latest and most numerous expressed in the caudal spinal region; in contrast, none are expressed in the cranial region rostral to the hindbrain [74,75]. Another example is the timing of emigration from the tips of the neural folds by the neural crest, which in mouse emigrates after closure in the spinal region and before closure in the head, and whose role in neural tube closure is uncertain [42,76]. There also may be a requirement for normal apoptosis in the cranial neural folds to enable closure, but having less effect in the spinal region [42,77].
4. Evolutionary Aspects
4.1. Modern Vertebrates
Most of our understanding of neural tube development is based on a small number of species: mouse, chick, frog (Xenopus), rat, and zebrafish. Among these, there are very important differences in the basic mechanisms by which the neural tube is formed. All likely share the basic planar cell polarity/convergent extension mechanism [5]. Details of convergent extension in development of the neural tube have been studied in frog (Xenopus) and zebrafish, but their cellular process is expected to differ from mammals and chick because their organization of neuroepithelial cells is known to differ [16]. For the hinge point mechanism, Xenopus lacks the MHP and DLHP [20]. Overall, the studied species differ in various mechanisms of closure, such as the relationship of the neuroepithelium to the surface ectoderm, the presence of neural folds, the presence of hinge points and the degree to which closure is intermittent or simultaneous along the whole neural tube [5,78,79,80]. Of course, a species that does not normally undergo the cellular/tissue process whose failure causes human NTD cannot model the mechanism of human NTD. Among the well-studied models for neural tube development, mouse, chick and rat appear to be most similar to human, but little is known about human embryos except histological and morphological studies. Based on morphological studies, the little-studied species, rabbit and pig, may be most similar to human (Figure 3).
4.2. Proxies for the Ancestral Pre-Vertebrate
The Cephalochordate, Amphioxus (lancelet), whose lineage diverged from vertebrates 550–600 million years ago, is used as a proxy for the ancestral chordate [81,82]. Like vertebrates, Amphioxus forms a neural plate, with a neural plate border region. Whereas in the representative mammals studied for neural tube development, the border region remains intact and is carried to the midline with the medial folding of the neural folds, in Amphioxus, the neural plate border cells, using lamellipodia, migrate medially to cover the neural plate as continuous surface ectoderm and beneath it, the neuroectoderm bends to form a tube [81,83]. The Amphioxus genome has been sequenced [83]. Noggin/BMP signaling and parts of the Wnt/PCP signaling pathway are present [81] but a detailed analysis of the pathways known to be important in neural tube formation does not seem to have been done.
The lineage of Tunicate chordates, the Ascidians (sea squirts), diverged from the vertebrate lineage around the same time as Cephalochordates [84]. The larval sea squirt forms a neural plate and neural folds in a manner similar to the representative mammals. The neural plate curls towards the midline to form a tube, pulling with it the adjacent surface ectoderm to the midline, which meets and fuses to form the epidermis that covers the neural tube. The neural plate cells elongate and change from columnar to wedge-shaped during this process, with narrow apices. Closure progresses from posterior to anterior [85]. Nodal signaling may be required for neurulation. Convergent extension is present [86]. It is not clear that the notochord has a role in patterning of the neural tube, unlike in vertebrates [87]. As in vertebrate neuroepithelium, cell cycle alterations in both the surface ectoderm and the neural plate facilitate the bending required for closure. Movement of the midline epidermal cells requires Rho/ROCK signaling and medial actin filament accumulation [87].
The similarities of neural tube development in comparisons of mammals and Amphioxus and Ascidian embryos suggests that the rolling up of the neural plate into a tube is the ancient mechanism, and from Ascidian embryos that the formation of neural folds is also the ancient mechanism. The divergences from this pattern in species such as zebrafish are therefore new evolutionary products. Some of the ancient processes, such as migration of the neural plate border cells to form the surface ectoderm over the tube in Amphioxus suggest the possibility of atavistic capabilities lurking in the mammalian genome, potentially available in some circumstances.
5. The Cranial Neural Tube
5.1. The New Head and the Cranial Neural Crest
An important context for understanding cranial neural tube closure is that the vertebrate head and its neural tube are considered by the “New Head” hypothesis to be a new structure that was added by evolution, stepwise, onto the basic chordate ancestor which already had the spinal neural tube [88,89]. The genetic basis used to create the New Head includes conservation of early-expressed genes, co-option of other existing genes, and evolution of novel functions for the newly evolved duplicate orthologs of developmental genes [81,82,90]. Consequently, cranial neural tube developmental processes use a blend of old genes and pathways interacting with more recently evolved genes and pathways; processes in the head that may appear to be similar to those in the spinal neural tube may be different at the molecular genetic level.
The ancestral pre-vertebrate chordate representative, Amphioxus, is a filter-feeder with tentacle-like structures around a small mouth, pharyngeal gill slits, a notochord that extends all the way to the front of the head, a neural tube, and a post-anal tail; it does not have jaws, skull, neural crest or ectodermal placodes (optic, otic, olfactory). It has a very simple tiny brain with rudiments of hindbrain, diencephalon, and perhaps some midbrain, but no telencephalon [81]. Amphioxus has some basic ancient conserved gene networks used in development of the neural plate, neural crest and placodes, but not the more downstream components. It does not have a neural crest per se, because genes required for the early development of neural crest cells, although present, are not expressed at the edges of the neural plate [90].
Evolution of a neural crest set the stage for the development of the New Head [91]. Most of the vertebrate face and jaw complex is derived from cranial neural crest cells [92], which arise from the neuroectoderm at the edge of the neural plate and migrate subectodermally ventrally and rostrally. The sensory placodes, another aspect of the New Head, also arise at the edge of the neural plate, induced in the adjacent non-neural ectoderm [89]. In the mammalian head, the neural crest cells emigrate from the neural folds before the folds have elevated; in the spine, they emigrate after closure [60,76]. The neural crest functions differently in the New Head than in the spinal region. For example, in the New Head, cartilage is derived from neural crest cells; this seems to have been enabled by the acquisition of a new cis regulator on the SoxE gene that causes its expression in cranial neural crest cells [93]. A consideration for cranial neural tube closure is whether there are differences in gene expression in the cranial neural crest versus the spinal neural crest that precede neural tube closure—if so, these differences have potential to affect the process of closure.
Unlike the rest of the vertebrate neural tube, the anterior forebrain neural folds do not generate neural crest cells, and the neural crest cells of the forebrain mesenchyme migrate in from the caudal forebrain and the midbrain [60,94,95,96]. In mouse embryos, the transcription factor Tcf7l1 expressed in the anterior forebrain neural folds acts to repress the Wnt/B-catenin pathway that otherwise would induce neuroectoderm cells to become neural crest cells [97]. Loss of Tcf7l1 leads to conversion of the neuroectoderm of the most rostral neural folds to neural crest cells and a subsequent reduction of the rostral forebrain, which if severe is accompanied by exencephaly [97]. Interestingly, in the images published, the midbrain also appears to be reduced.
5.2. Absence of Hox Gene Expression
Hox genes, some of which are present in Amphioxus, are expressed in the neuroectoderm and paraxial mesoderm of vertebrates and impart anterior–posterior positional identity. Under the regulation of Fgf, Wnt and other genes, various combinations of Hox genes are expressed in the newly formed axial tissues from the primitive streak stage onward [75,98]. In vertebrates, Hox expression extends from the embryonic hindbrain through the tailbud; there is no cranial Hox gene expression rostral of the border between rhombomere 1 and 2 of the hindbrain [98]. An interesting question is whether the absence of Hox gene expression affects the subsequent genetic program determining cranial neural tube closure.
5.3. The Prechordal Plate and Cranial Flexure
The prechordal plate [34,99] underlies only the rostral forebrain (the telencephalon and the diencephalon region that will become the optic primordium). Histologically, the prechordal and notochordal plates appear continuous, but are distinguishable [99]. Located immediately below the midline of the neural plate, the prechordal plate functions in similar ways to the notochord, which underlies the rest of the neural plate, producing essential regulatory factors such as Shh, but it also differs from the notochord, by producing factors such as Goosecoid (Gsc), an inhibitor of convergent extension [100] and by not producing others such as cNot1 [34,101,102]. Thus the basic regulatory mechanism underlying the forebrain neural tube differs from the rest of the neural tube.
Although the prechordal plate is initially located in line with the notochord, it is bent to lie at right angles to the notochord just before and during the time the mesencephalic folds are elevating [34,60,99,103]. Concurrently, and centered on the mid-mesencephalon, the angle of the cranial flexure narrows from 150 degrees to 100 degrees in human, and from 100 degrees to 70 degrees in mouse [60,103]. The increased bend of the mesencephalic neural tube at the time of closure can be hypothesized to be a physical force working in opposition to the elevation of the mesencephalic neural folds, and the relative acuteness of the angle in mouse is notable in this respect. The prechordal plate also takes part in the formation of the oropharygeal membrane, which, beneath the neural tube, temporarily separates the foregut from the stomodeum. The oropharyngeal membrane ruptures around the time the midbrain neural tube is closing [60,61]. It would be interesting to discover whether the rupture has any mechanical effect that offsets the narrowing of the angle of the flexure.
5.4. Optic Sulci
A further complexity in the mechanical context of closure of the cranial neural tube is the development of the optic sulci in the forebrain. These begin as indentations in the neuroepithelium and balloon outward toward the surface ectoderm as the forebrain neural folds elevate toward each other, so that the apposed forebrain folds resemble a pair of castanets [104] (Figure 5c in [105] and Figures 19 and 20 in [106]). This contrasts with the relative simplicity of the shape of the spinal neural folds during elevation and leads to the question whether the process of shaping the neuroepithelium to form optic sulci affects the process of neural fold elevation that occurs concurrently.
5.5. Sonic Hedgehog and Cranial DLHP
A Sonic hedgehog expression gradient in neuroepithelium from upper spine to lower spine accounts for a correlated inverse gradient of dorsal bending of spinal neural folds during closure, as high levels of Shh signaling in the upper spine inhibit formation of DLHP, as discussed in Section 2.5. The levels of Shh in the ventral neural tube of the head do not seem to have been compared to those of the upper spine; in published whole mount in situ hybridization images, the intensity in the cranial region seems as high as the upper spine (Figure 4C in [32]), but this is not a quantitative method. The BMP-Noggin-DLHP relationship and cell behavior at the cranial DLHP does not seem to have been studied, and it is possible that the mechanisms are not entirely the same as in the spine. Conversely, the induction of exencephaly in response to elevated Shh signaling suggests that the relationship of cranial DLHP to Shh is similar to that in the spinal region and modulated by the large size of the cranial neural folds, as discussed in Section 2.5.
5.6. The Actomyosin Cytoskeleton of the Cranial Neuroepithelium
Mammalian cranial neural fold bending for closure requires the actomyosin cytoskeleton, and mutations in its components or chemical disruption of apical actin microfilaments cause exencephaly (summarized in [19]). Thus, mutations in components such as palladin, vinculin, cofilin 1 (Cfl1) and Marcks, or in genes for actin regulatory proteins such as Mena, Vasp and Evl, or in genes for protein kinases with cytoskeletal influence such as Abl1, Abl2, Mapk8 and Mapk9 all cause exencephaly in mice [19]. In contrast, closure of the spinal region does not require these genes. Therefore, these genes important to the actomyosin cytoskeleton are candidate genes specific to human anencephaly etiology.
5.7. Apoptosis in Cranial Neural Folds
Programmed cell death, apoptosis, normally occurs in the neuroepithelium during development of the neural tube [42], with a pattern of areas of particular abundance associated with bending and fusion [107]. Various mouse mutants that demonstrate decreased or increased apoptosis in the neural folds develop exencephaly but not spinal NTD [42]. For example, in mouse embryos that constitutively lack apoptosis, the Casp3 and Apaf1 null mutants, the midbrain and hindbrain fail to close but the rest of the neural tube closes normally. Live imaging of the midbrain–hindbrain neural folds undergoing bending and closure in normal embryos show conventional and unconventional apoptotic cells [77]. Conventional apoptotic cells (having caspase activation, followed by shrinkage and fragmentation) are in the boundary region and surface ectoderm at the tip of the neural folds and at the midline before and after the completion of closure. The unconventional apoptotic cells (having caspase activation, but not shrinkage and fragmentation) emerge from the neuroepithelium of the boundary domain and dorsal neural fold and accumulate as the fold bends; they are round, protrude from the surface, remain attached for a time, and then tumble along the surface of the neuroepithelium into the lumen of the neural tube. In Casp3 and Apaf1 null mutants, neither type of apoptotic cell is found and the apical bending of the folds towards the midline is severely reduced. The mechanism of the effect of apoptosis on bending is not known.
5.8. Mutations in Genes Expressed in the Cranial Neural Crest That Are Associated with NTD
Emigration of neural crest cells from the midbrain and hindbrain open neural folds has been thought to be necessary for cranial neural tube closure [42], but the evidence from mouse mutants is limited. Two genes that cause NTD and are expressed in the cranial neural crest are the transcription factor Tcfap2a and the cell adhesion factor Cdh2 (N-cadherin). In mice on E8.5, Tcfap2a is expressed in the cranial neuroepithelium and cranial neural crest; Cdh2 is expressed in the cranial neural plate. Conditional mutants in which either Tcfap2a or Cdh2 is deleted in the neural crest cells in which Wnt1 is normally expressed, suggest that some aspect of cranial neural crest abnormality causes midbrain exencephaly (15–20% in Tcfap2a, [108]; 100% in Cdh2; [109]). Although for both mutants, it appears that exencephaly is due to the absence of the targeted gene’s expression in the cranial neural crest cells, for neither mutant is the mechanism of interference with neural tube closure understood. In the Tcfap2a mutants, it may involve the positioning of the neural plate border [108]. A complication of these studies is that Wnt1-Cre is expressed in both the lateral non-neural ectoderm of E8.5 cranial neural folds and the neural crest [110]. The potential role of the lateral non-neural ectoderm cells in neural tube closure or its failure has yet to be explored.
It is interesting that whereas 100% of Tcfap2a null embryos have split face and forebrain/midbrain/hindbrain exencephaly [111,112], the cranial neural crest conditional null mutants have failure of closure of only the midbrain. Further evidence for neural crest involvement emerges from study of Cdh2 null mutants rescued from early lethality [113,114], which are 100% exencephalic. On E8.5, the rescued embryos have increased apoptosis in the tips of the cranial neural folds, in what appear to be neural crest cells starting to migrate from the caudal midbrain region [114]. In summary, although abnormality in cranial neural crest cells appears to be the cause of exencephaly in the Wnt1-Cre conditional null Tcfap2a and Cdh2 mutants, the mechanisms are unclear. We have found no definitive evidence from mouse mutants for delayed or abnormal neural crest cell emigration as a cause for exencephaly.
5.9. Role of Cranial Mesoderm in Neural Fold Elevation
The vertebrate cranial mesoderm has an important role in cranial neural fold elevation. The vertebrate cranial mesoderm, created by gastrulation [115], may have accrued differences from spinal mesoderm as part of the evolution of the New Head. Comparisons of mesodermal gene expression between Amphioxus and vertebrates indicate that expression in late gastrula of the mesodermal patterning genes found in both groups is reorganized in vertebrates, limiting expression of some to the head and others to the body, thus creating a novel type of mesoderm in the vertebrate head [116].
The role of cranial mesenchyme (comprised of cranial mesoderm and neural crest cells after their emigration from the neuroectoderm) in the cranial neural tube has been studied in mouse, rat and chick embryos [117]. During cranial neural tube formation, the shape of the folds progresses from biconvex, with the mid-lateral folds cushioned on a large supporting cranial mesenchyme, to biconcave (Figure 4). The neural folds of the spinal region do not have the convex stage. During the process of cranial neural fold elevation, the mesenchyme expands by both increased numbers of cells and increased space between the cells [117,118] and this mesenchymal expansion and reorganization is thought to be important in causing elevation. During the biconvex stage, the neural crest cells leave the neuroectoderm of the lateral tips of the neural folds and begin to migrate subectodermally through the mesenchyme to the first branchial arch and facial prominence areas. As the neural folds elevate, becoming biconcave, and the dorsolateral hinge points form, the mesodermal cells become more widely spaced in the medial region and more closely packed in the lateral region. The extracellular matrix undergoes changes in hyaluronic acid concentration that may provide a mechanism for elevation by mesenchyme expansion, as hydration of hyaluronic acid is known to cause expansion of mesenchyme [117,119].
Several mouse mutants demonstrate the importance of the cranial mesenchyme to cranial neural fold elevation. Loss of function of the transcription factor twist causes severe craniofacial defects, including midbrain exencephaly; the neural folds fail to become concave and remain everted [120]. twist is highly expressed in the cranial mesenchyme just before and during the time of neural tube closure [121,122]. At the normal time of cranial neural fold elevation, mutant embryos have a morphologically normal neuroepithelium, but have abnormal morphology of mesenchyme cells in the forebrain and midbrain, which are rounded, lack normal stellate shape and have greater intercellular space than normal [120]. The exencephaly is attributable to a defective cranial mesenchyme and not the neuroepithelium, based on chimera studies [120] and on conditional mutants. Ablation of twist in the precursors of the cranial mesoderm causes reduced proliferation and abnormal clustering of mesoderm cells in the lateral regions of the head folds and subsequent exencephaly [121]. No evidence of increased cell death has been found [121]. The neural crest seems to form normally [123] and the neural crest cell-based facial abnormalities are considered to be a secondary effect of the abnormal mesenchyme failing to provide essential signals to the neural crest cells as they migrate to the face [121]. Based on studies later in development, the loss of expression of Cart1 (Alx1) and Alx3 in twist null mutant embryos suggests that they are in the same regulatory framework [123], and it is interesting that they also express exencephaly in mutants.
The Cart1 (Alx1) transcription factor is expressed in mesenchyme cells of the forebrain but not the midbrain, and not in neuroepithelium, during neural tube development [124]. Null mutant embryos for Cart1 (Alx1) have increased apoptosis and transient deficiency of mesenchyme in the forebrain, with delayed closure of the forebrain folds; subsequently, the midbrain neural folds fail to elevate, leading to exencephaly [124]. Whereas Cart1 (Alx1) early expression may be limited to the forebrain mesenchyme, a homolog, Alx3, appears to be expressed in paraxial mesoderm of the body and the mesenchyme of the head, but also not in the neuroectoderm, at the time the cranial folds are elevating [125]. Although some authors assume Cart1 (Alx1) and Alx3 expression in the neural fold mesenchyme is in the neural crest cell component, this seems unproven. About 10% of mutants lacking Alx3 develop midbrain exencephaly and/or split face (failure of closure of the forebrain neural folds); the neural folds of these embryos fail to undergo the shift from convex to concave or development of a dorsolateral hinge point and therefore fail to meet in the midline [126]. Increased apoptosis and decreased mesenchymal cell density in mutants are present in later developmental stages [126], but is not known if any abnormalities in apoptosis and mesenchymal cell density are present earlier when they could contribute to the failure of neural tube closure. Interestingly, Alx3 expression is lost during folic acid deficiency at the time of cranial neural tube closure, whereas expression of other neural tube genes such as Cart1 (Alx1), twist, and Cited2 is not affected [126].
Inka1 is first expressed in the cranial mesenchyme prior to and during neural tube formation and then in the cranial neural crest during its migration in the mesenchyme [127]. It is not expressed in the neuroepithelium. The Inka1 protein inhibits the Pak4 kinase that may function in cell–cell contacts and adherent junction formation [128]. Inka1 is regulated by Tcfap2a in non-mammalian vertebrates, but not in mice [127]. Loss of function of Inka1 causes a predisposition to midbrain exencephaly (5–7%); otherwise, individuals are normal and fertile. The mechanism by which the neural tube fails to close has not been demonstrated, and the mechanistic role of the mesoderm or neural crest or both is unknown.
Loss of function of the transcriptional regulator, Ski, [129] which causes exencephaly or split face [130], is sometimes listed as having abnormal cranial mesenchyme [117] but cranial tissues do not seem to have been studied during the period of cranial fold elevation [130,131], and an etiology of exencephaly based on effects on cranial mesenchyme seems unproven.
Null mutant embryos for Smarca4 (Brg1 or SW1/SNF), involved in chromatin remodeling, die before implantation. Heterozygotes often have midbrain exencephaly, and no other defects [132]. Smarca4 is expressed strongly in the cranial mesenchyme before elevation [133]. The mechanism causing the failure of neural tube closure has not been studied.
Mutations causing loss of function of the E3 ubiquitin ligase Hectd1 (opm) lead to midbrain exencephaly in all homozygotes and some heterozygotes [134]. Hectd1 is ubiquitously expressed in embryos, including the head mesenchyme, and the cause of exencephaly is attributable to the effects of Hectd1 deficiency on the cranial mesoderm cells, which are abnormally shaped and packed more densely than normal near the neural tube at the normal time of elevation, and which fail to orient parallel to the neuroepithelium [117,134]. The neural folds lack mesenchyme expansion and remain flat or convex and fail to form the dorsolateral hinge points. The neural crest cells’ migration pattern and differentiation are normal; mesenchymal cell proliferation and apoptosis are normal [134]. Before the normal time of elevation and closure, twist has an expanded area of expression in the midbrain folds in the Hectd1 mutants, and after failure of full elevation, Snail and Pdgfr are abnormally expressed in the dorsal area of the midbrain folds [134]. One of the Hectd1 substrates, heat shock protein 90 (Hsp90), is known to enhance migration in various cell type, is secreted at elevated rates from Hectd1 mutant cranial mesenchyme cells which are abnormally migratory in vitro [135]. It seems that the effects of the lack of Hectd1 in the midbrain neural folds lead to excess Hsp90 secretion, which in turn alters the behavior of the mesoderm cells, leading to their failure to contribute to elevation of the folds.
Loss-of-function mutants for the Cecr2 chromatin-remodeling factor develop exencephaly [136]. Cecr2 is expressed in neuroepithelium and mesenchyme during cranial neural tube development [137]. Studies of gene expression arrays from mutant embryos show reduced expression of Cart1/Alx1 transcription factor [138]. As this mesenchymally-expressed gene is required for cranial neural tube closure [124], it appears that the defect in Cecr2 mutants may be attributable to defective cranial mesenchyme. The observation that in Cecr2 mutants, the cranial neural folds do not elevate sufficiently for contact between the apposed folds, despite forming DLHP [137], is consistent with the hypothesis that the NTD is caused by an insufficient expansion of the cranial mesenchyme to assist elevation.
Exencephaly in mouse mutants is usually caused by a failure of midbrain neural fold elevation and it appears that defects in cranial mesoderm are a common etiology for this defect. Exencephaly becomes anencephaly, owing to degradation of exposed neural tissue during later gestation, and therefore is seen as anencephaly at birth in humans [6]. Interestingly, as in the mesodermal mutants in mice, unelevated everted midbrain neural folds have been observed in some early NTD human embryos [139,140], suggesting that lack of elevation owing to mesodermal defects also could be a common mechanism in human anencephaly.
5.10. Neuroepithelium-Expressed Genes and Mechanisms That Cause Exencephaly
The actomyosin-dependent shaping and bending of the cranial neural folds is based in the neuroepithelium, and it seems predictable that the many actomyosin-related genes that cause NTD would show neuroepithelium-specific expression, but data to demonstrate this is lacking. Many studies report whole mount in situ hybridization in the cranial neural folds without demonstration of the cell type involved (neuroectoderm, mesenchyme, surface ectoderm). A few genes that cause cranial neural tube defects when dysfunctional have been reported to be expressed in the neuroepithelium.
Cfl1 (Cofilin 1, Cofilin-n) is expressed in the pre-closure cranial and caudal neuroepithelium and neural crest [141,142]. Cfl1 enables the dynamic reorganization of the actin cytoskeleton by its function in F-actin, severing and depolymerization and by promoting the recycling of monomeric actin [141]. Mutants have exencephaly and delayed closure of the posterior neuropore (PNP) [19,141]. Originally, the exencephaly was ascribed to a demonstrated lack of neural crest cell migration, but no mechanism for the lack of neural fold elevation was demonstrated [142]. More recent work has shown that the mechanism causing the exencephaly is a lack of activation of apical actomyosin in neuroepithelial cells and abnormal basal accumulation of F-actin [141]. Consistent with the different roles of actomyosin in cranial versus caudal neural folds, the delayed PNP closure of Cfl1 mutants is owing to actomyosin accumulation, lack of F-actin turnover and actomyosin disassembly [19].
Fat1 cadherin, a large cell adhesion molecule, is expressed in the early anterior neural plate in the forebrain and midbrain neural folds during neural fold elevation and in the caudal neural folds [143]. It seems to be located in the neuroepithelium and neural crest. Fat1 is thought to regulate actin cytoskeletal organization at cell peripheries. In cell cultures, it is found in filopodia and cell–cell boundaries, overlapping with dynamic actin structures; it regulates actin polymerization [144]. Therefore, the cause of the NTD is likely a deficiency of neural fold bending. On some strain backgrounds, Fat1 null mutants have exencephaly, but not spina bifida. Although mutations of the family member Fat4 do not cause NTD, and Fat4 binds different actin-regulating and junctional proteins than Fat1, they form heterodimers, and on some strain backgrounds, the combination of homozygous null mutations at both Fat1 and Fat4 cause increased frequencies of exencephaly compared with Fat1 alone [145]; this is an example of the genetic complexity of NTD etiology.
Cited2 (Mrg1) is a transcription regulator that serves as a co-activator for several transcription factors such as Tfcap2, Smad4, Lhx2, Nanog and Tbx3 [146,147,148]. It is expressed during neural fold elevation in the midbrain neuroepithelium and in migrating cranial neural crest [146,149]. Null mutants have exencephaly. The mechanism leading to exencephaly is likely the disrupted integrity of the neuroepithelium, owing to the observed intense localized apoptosis in the midbrain neuroepithelium, particularly at the midbrain–forebrain boundary, at the time of normal midbrain-fold bending [146].
Members of the Sall gene family Sall1, Sall2, and Sall4 are transcription repressors that interact with chromatin remodeling complexes, can form heterodimers [150] and are all expressed in the cranial neuroepithelium during neural fold elevation [151,152]. Loss-of-function mutants for each of these Sall genes cause exencephaly, depending on strain background [150,152,153], and digenic heterozygous mutant combinations of Sall1 with Sall4 or Sall2 with Sall4 also cause exencephaly. Based on embryos with a combination of null Sall1 and hypomorphic Sall4, it appears that the cause of exencephaly is a deficiency of elevation of the cranial neural folds, which do elevate part way but do not reach approximation in order to fuse [152].
Members of the Nuak gene family, Nuak1 and Nuak2, are serine-threonine kinases that have many functions [154]. Nuak1 is expressed in many embryonic tissues including the neuroepithelium, but null mutants do not have NTD; Nuak2 is specifically expressed in the entire neuroepithelium during neural tube closure, and some of the null mutants have midbrain and hindbrain exencephaly; digenic null mutants for both genes all have exencephaly of the forebrain, midbrain and hindbrain [154]. The cranial neural folds in the digenic mutants seem to lack DLHP, and the cause seems to be a significant lack of apical constriction of the cranial neuroepithelial cells during the normal elevation stage [154], which would be expected to cause a lack of bending.
Shroom3 (Shrm), encodes a cytoskeletal protein that binds F-actin and regulates its distribution within the cell. During neural fold elevation, Shroom3 is strongly expressed in the cranial neuroepithelium, rostral to the otic vesicle, but not in the adjacent cranial mesenchyme [155]. Later, it is expressed in the caudal neural folds at the posterior neuropore during elevation (gene k8220b03, Appendix, and Figure 1 in [156]). All Shroom3 mutant homozygotes have midbrain and hindbrain exencephaly, most also have split face (failure of forebrain neural tube closure), and some also have spina bifida aperta. The morphology of the neuroepithelium of the closed spinal neural tube is also abnormal [155]. In Shroom3-mutant embryonic neuroepithelial cells, F-actin is shifted away from the apical surface, whereas in wild-type cells, it is predominantly localized at the apical surface of the cells. This pattern suggests that the mechanism of the neural tube closure defects in Shroom3 mutants is the lack of actomyosin-dependent apical constriction in the neuroepithelium. There is evidence, based on various digenic heterozygous mutants involving Vangl2 (Lp) and Shroom3, that the actomyosin system is linked to the PCP-convergent extension system by Shroom3 [157]. For example, whereas neither Shroom3/+ nor Lp/+ mutants have spina bifida aperta, the digenic mutants (Shroom3/+, Lp/+) have almost 40% spina bifida aperta. At the molecular level, there is evidence that that a PCP factor (Dvl2) determines the location of Shroom3 at mediolateral cell junctions along with F-actin, Rock1 and Myosin IIb, components of the actinomyosin pathway [157], suggesting that Shroom3 and Dvl2 may interact to link PCP signaling to actomyosin contractility. This is an example of how simultaneous reduction in a genetic component of each of two pathways can cause NTD.
5.11. Cranial Ephrins and Ephrin Receptors
An example of the potential molecular complexity underlying neural tube fusion and leading to a cranial-specific etiology of an NTD is an apparent regulatory relationship between the transcription factors Dlx5 and Msx2 with each other and with the downstream membrane-bound cellular adhesion/repulsion factors EphrinA5 and EphA7. Dlx5 is expressed in the dorsal tips of the neural fold before, during, and after neural tube closure, extending from the forebrain to the posterior neuropore [158,159], and null mutants have some (19–28%) exencephaly [159,160]. Similarly, Msx2 is expressed before closure in the dorsal edges of the neural tube, except in the forebrain region [161,162], but null mutants do not have NTD [160,163]. In contrast, double mutant embryos null for Msx2 and Dlx5 have a high frequency (73%) of exencephaly that seems to be caused by their joint regulatory effect on the splicing of EphA7 transcripts [160]. The ligand EphrinA5 and its receptor EphA7 splice forms are expressed in the dorsal cranial neural folds (midbrain and hindbrain) before and during closure [40]. The interaction of EphrinA5 with full length EphA7 causes cellular repulsion, but a co-expressed alternative splice form of truncated EphA7 converts the effect of the interaction to adhesion [40]. EphrinA5 null mutants sometimes develop exencephaly (17%), a few of which have failed forebrain closure as well as midbrain and hindbrain, having failed to fuse despite elevating to become juxtaposed at the midline; some (25%) EphA7 null mutants also have exencephaly [40]. Null mutants for either Msx2 or Dlx5 have fairly normal expression of EphrinA5, full-length EphA7 and truncated EphA7 in the cranial neural tube. In contrast, in Dlx5/Msx2 digenic null mutants during the stage of neural tube closure, the expression of EphrinA5 at the apex of the neural folds is decreased, full length EphA7 expression is unchanged but the truncated form of EphA7 is greatly reduced, a situation that is expected to cause repulsion, rather than adhesion of the apposed midbrain and hindbrain neural folds. It appears, therefore, that the mechanism leading to exencephaly is likely lack of adhesion of neural folds in each of the EphrinA5, EphA7 and digenic null Msx/Dlx5 mutants, and the limitation of the NTD to the midbrain and hindbrain reflects the limited expression domain of EphrinA5 and EphA7.
Demonstrating another kind of complexity, null mutation carriers for the X-linked gene, EphrinB1, express exencephaly at a higher rate in heterozygous females than in homozygotes and null males [164]. Embryos have not been studied during the period of cranial neural tube closure, but the mechanism may be segregation of EphrinB1-positive and -negative cells in the neuroepithelium, affecting its structural integrity [164].
5.12. Cranial Neural Fold Projections
In general, it seems likely that genetic defects whose mechanisms cause cranial neural tube defects through failure of fusion of apposed neural folds, would demonstrate defects in the surface ectoderm projections normally present at the fusion sites. However, it appears that data is lacking in this area.
Loss of function of Myosin-X (Myo10), an unconventional myosin known for its roles in formation of filopodia and localization to the tips of filopodia, causes exencephaly, along with other non-neural-tube defects [165].
5.13. The Phenomenon of Sex Ratio Distortions in Cranial NTD
A striking predominance of females in cranial neural tube closure defects has been recognized for many years; about two-thirds of human anencephalics and mouse exencephalics are female (reviewed in [166]). Human craniorachischisis may also exhibit an excess of females; no sex data are available for mouse mutants with craniorachischisis [166]. Spina bifida as a whole generally does not exhibit female excess in humans or mice [166], although the subtype located in the upper spine may do so [167]. Previously, 12 mouse mutants and strains for which sex had been reported showed an excess of females among exencephalics [166]. There is similar data for two additional mutants: Cecr2 with 66% females among exencephalics [168], and Scarb1 (srb1) with 72% females among exencephalics [169]. The 14 mutants and strains represent a diversity of functions and pathways, suggesting that the increased liability of females to cranial NTD extends across a wide range of causal molecular mechanisms. There may be exceptions. For example, the lack of female predominance in NTD caused by mutations at Gldc [170] and Cited2 [146] requires further confirmation of sex ratios with larger samples. If other exceptional mutants that lack female excess in exencephalics can be found, a comparison of the function of genes with female excess with those without might point to the mechanism responsible for elevated female susceptibility to cranial NTD.
The causes of the female excess among cranial neural tube closure defects in humans are difficult to identify, but in mice, several of the possible explanations have been ruled out by the data. For example, the neural tube develops long before gonadal hormones can be present, the developmental rate of the cranial neural tube during closure does not differ between males and females, and there is no evidence of preferential death of affected males during gestation [166]. In mice, genetic experiments have demonstrated that the higher susceptibility to cranial NTD is caused by the presence of two X chromosomes in females, not the absence of the Y (reviewed in [166]).
There is an intriguing phenomenon of methylation differences correlated to number of X chromosomes in mouse and bovine embryos, during the early developmental period when both X’s are normally active, such as in blastocysts or embryonic stem cells. The global DNA is hypomethylated and the Dnmt3b (DNA methyltransferase 3 beta) transcript and protein levels are reduced in XX embryos relative to embryos with one X [171,172]. Whether this phenomenon has later consequences affecting gene expression in neurulation stage embryos is unknown.
In mammals such as humans and mice, males have one large X chromosome with over a thousand genes and one small Y chromosome, which contains, amongst a few genes, the Sry testis-determining gene; females have two copies of the large X, of which one copy has been mostly silenced beginning from about the time of implantation [173] by a combination of epigenetic changes including DNA methylation, histone modifications and chromatin remodeling [174]. Chromatin (DNA and its attached histones) that has undergone transcriptional silencing and remodeling to become densely packed is termed “heterochromatin”.
A mechanism that has been suggested to explain a variety of disorders that occur at higher frequencies in females than in males is that some genes on the inactive X variably escape from silencing and cause a gene expression imbalance; there is evidence in support of this mechanism for the autoimmune disease lupus erythematosus [174].
Another hypothesis to explain the female excess is that the use of cellular resources to inactivate a large X chromosome in females after every mitosis would competitively reduce the availability of those resources for gene regulation on the autosomes, weakening the process of neural tube closure [64]; updated in [166]. A similar hypothesis has emerged from genetic studies in Drosophila, the “heterochromatin sink” [175]. In Drosophila, a chromosome with a large heterochromatin component can sequester various limited factors required to regulate genes, and this can alter the expression of genes in the rest of the genome [175]. Some observations suggest that the large inactive heterochromatic X chromosome in female mammals can act as a sink that sequesters epigenetic factors that are present in limited amounts (e.g., DNA methyl transferases and histone modifying enzymes), thus altering their availability in the rest of the genome. A mutation at Smchd1, a gene involved in the methylation of DNA (formerly Momme D1) causes reduced methylation and silencing of a reporter transgene and autosomal retrotransposons, but only in females. The female-specific effects led Blewitt et al. [176] to suggest that the inactive X chromosome could be sequestering proteins that are involved in controlling gene expression at autosomal loci. An example of this phenomenon has been demonstrated by the “four core genotypes” system, where the male sex determining factor Sry has been moved to an autosome, comparing XX females, XX males, XY males, and XY females. It shows that the expression level of hundreds of genes in mouse thymus cells is determined by the number of X chromosomes present and not by gender [177]. Genes differ by whether they are sensitive to the number of X chromosome and in degree of sensitivity [177]. The sensitive genes have lower expression in the presence of two X chromosomes [177]. If some of the important genes that have cranial-specific neural tube expression (Section 5) are relatively sensitive, and if there are not sensitive genes among the important spinal-specific neural tube genes, this would explain the female excess in cranial, but not spinal, NTD. Perhaps examination of expression levels of genes in the neural folds of embryos from this “four core genotypes” system would yield insight into the causes of female excess in cranial NTD.
There is an interesting interaction between embryo gender and the preventative effects of folate on human NTD. In at least three populations from Chile, Argentina and Canada, the female excess among anencephalic births disappeared soon after the start of folic acid fortification of wheat flour [167,178]. Prior to folate supplementation, anencephalic births in each country exhibited the typical excess of females, where almost two-thirds of anencephalic births were female. Fortification with folic acid reduced the occurrence of anencephaly in both males and females, but had a greater effect in females; for example, 50% prevention in males versus 70% in females [167]. One way to interpret the pattern would be that the additional folic acid compensates for the “heterochromatin sink” in females and restores availability of essential chromatin regulatory factors, reducing female risk of NTD to the same level as males. Then, through perhaps a different mechanism, the abundant folate interacts with a particular type of genetic weakness present in a subset of cases to reduce NTD risk in that subset equally in both sexes.
6. The Spinal Region, PCP and Craniorachischisis
Much of the understanding of basic mechanisms of neural tube development is based on studies of the spinal region, and spinal neural tube closure has been well reviewed by others [5,6]. We have considered the basic mechanisms (Section 2) and differences between cranial and spinal regions (Section 3). In this section we consider the NTD that is caused by failure of closure of the entire spinal region. The first initiation site for neural tube closure is in the upper spine. There the folds are brought together by, in part, the combined effect of convergent extension and the medial hinge point (MHP, Section 2.3).
The failure of convergent extension (Section 2.2) results in a short body axis and broad neural plate, with no medial hinge point (MHP), placing the lateral neural folds too far apart to meet in the midline and fuse [179,180]. This results in the NTD, craniorachischisis, in which all but the most rostral zone of primary neurulation has failed and the neural tube remains open from the midbrain or hindbrain, along the length of the spine to the lumbar region in humans and to the sacral region in mice. It is interesting to note that the caudal limit of primary neurulation is more rostral in humans than in mice [46]. The constant feature of craniorachischisis, a closed neural tube in the most rostral, forebrain region, is likely explained by Ulmer et al. [100], who show that Goosecoid (Gsc), present in the prechordal plate and not in the notochord, appears to repress convergent extension; i.e., because of Goosecoid, normally there is no convergent extension in the forebrain. The role of a disruption in the planar cell polarity (PCP) pathway and the consequent lack of convergent extension in causing craniorachischisis have been well reviewed recently [5].
In mice, among the numerous mouse gene mutations that cause NTDs, those that produce craniorachischisis are all components of the planar cell polarity pathway, including the PCP core factors, Vangl2, Celsr1, Dvl1/2/3 and Scrib; and the less central factors, Ptk7, Cdx1/2, Sec24b, Sfrp1/2/5 and Smurf1/2 [10].
In humans, craniorachischisis is a relatively rare NTD for which the only known genetic cause is mutations in PCP genes [181]. The developmental morphology and resultant NTD (wide neural plate and wide open neural tube, except for the closed forebrain) is similar in humans and in mice. Histological study of human embryos with craniorachischisis revealed that they have cervical notochord duplication, side by side, beneath the open neural tube, and a broad Sonic hedgehog expression domain; if the neural tube in these embryos appeared closed caudal to the notochord duplication, two parallel neural tubes side by side were found housed in a single dural tube [182]. In the homozygous Lp mouse mutant at the Vangl2 gene (Vangl2-Lp/Lp), the notochord at the time of initiation of neural tube closure is loosely organized and comprised of two or three parallel longitudinal structures [179]. The process of convergent extension has been shown to be cell-autonomously defective in both the notochord and neural plate of these homozygous embryos [180].
It is becoming clear that PCP genes can have roles other than in convergent extension that can influence neural tube closure. One possibility (for spina bifida) is that caudal neural tube closure is delayed in some PCP-mutant heterozygotes, as has been observed in the mouse heterozygous Vangl2-Lp mutant that has 2% spina bifida [183]. A second possibility, which applies more to the cranial region than the spinal region, is a relationship between PCP and cilia that can cause exencephaly in some PCP mutants. Numerous mouse mutants for genes involved in ciliogenesis have exencephaly [13], including mutants for the PCP factor Inturned (Intu) with 30% exencephaly [18]. A third possibility arises from observations of interaction between the PCP pathway and the regulation of actomyosin dynamics. In several mutants, the PCP gene involved has been shown to have a role in both convergent extension and actomyosin dynamics [19]. Conversely, the actin-binding protein, Shroom3, has been found to interact with PCP pathway proteins during neural tube morphogenesis in mice [157]. PCP gene mutations probably contribute to risk of human anencephaly and spina bifida, albeit in a very small proportion of cases [10].
7. Studies of Digenic Mechanisms of NTD Involving PCP Gene Mutations
In mice, a large and growing number of digenic mutants, heterozygous for mutations at two gene loci, have been observed to have NTD. The molecular mechanisms by which these digenic or oligogenic combinations lead to NTD are largely unknown, but likely include both cumulative effects along the same pathway or deficiencies in interacting pathways. The known digenics involve at least twenty different kinds of genes [3,10,12], but the extensive study of the planar cell polarity (PCP) system has generated numerous well-studied examples, which provide insight into mechanisms of the genetic complexity of human NTD. Most of the examples at hand involve two genes in the PCP pathway, many involving interactions between heterozygosity for both the Vangl2 mutant, Lp, and one of several other PCP genes. In addition to the PCP–PCP gene interactions, there are digenic mutant examples of PCP genes interacting with genes in actomyosin dynamics, as in Vangl2;Cfl1 [184] and Vangl2;Shroom3 [44,157].
Mouse PCP-mutant studies focused on the NTD craniorachischisis demonstrate the genetic complexity of the role of PCP genes in craniorachischisis [10]. In some examples, mutants are homozygous at only one PCP gene (e.g., Vangl2, Celsr1, Scrib or Sec24b) and all embryos have craniorachischisis; heterozygotes do not. Although virtually all mouse mutants with craniorachischisis have mutations in genes of the PCP pathway, often mutations at two or more PCP genes are involved. In one type of digenic example, mutants are homozygous at both of two homologous PCP genes with functional redundancy (e.g., Dvl1;Dvl2 or Fzd3;Fzd6) and nearly all embryos have craniorachischisis; homozygotes for any one of these genes do not. In yet another type of digenic cause, high frequencies of craniorachischisis are found in mutants that are double heterozygotes for mutations at two PCP genes of different gene families (e.g., Vangl2;Celsr1, Vangl2;Dvl2, Vangl2;Scrib, or Celsr1;Scrib) [10,185]. It is notable that several of the digenic mutant combinations that cause craniorachischisis also produce some normal, fertile, double heterozygotes.
Craniorachischisis is not the only NTD caused by PCP mutations in mice. Mutations of various single PCP genes or digenic combinations of PCP genes do not cause craniorachischisis, but do cause the more common types of NTD (exencephaly or spina bifida [10]); several others cause craniorachischisis or exencephaly or spina bifida in different individuals. For example, only exencephaly is found in homozygous single mutants at Fzd3, Intu or Fat1, or digenic mutants at Vangl2;Fzd1 (both heterozygous) or Vangl2;Cthrc1 (heterozygous; homozygous). Only spina bifida is found in digenic heterozygotes at Vangl2;Ptk7, Vangl2;Grhl3, Vangl2;Sec24b, and Celsr1;Ptk7 [10]. Finally, different individuals can have craniorachischisis or exencephaly or spina bifida in digenics heterozygous at Vangl2;Scrib or Celsr1;Scrib [185].
The mechanisms by which these non-craniorachischisis defects of neural tube closure are caused by PCP pathway mutations are largely unknown. However, the clear differences known for neural tube formation between anterior–posterior regions (discussed above) raise the possibility that the causal mechanisms involve the ciliogenesis, actomyosin, and microtubule functions of PCP genes, in addition to their convergent extension role (Section 2.2 and Section 6). Effects of the Wnt-PCP pathway on the caudal neural tube that cause spina bifida include effects on cell differentiation, cell shape, and actomyosin contractility [44].
Mouse mutants support the digenic (or oligogenic) hypothesis for human craniorachischisis and, further, the hypothesis that digenic heterozygotes for PCP genes can also cause risk for human exencephaly and spina bifida. Human NTD cases of various types have been screened for rare deleterious variants in several PCP genes. Human craniorachischisis is relatively rare and few studies are of craniorachischisis only. Most variant screening studies include exencephaly, spina bifida and other neural tube abnormalities [10]. Among craniorachischisis cases, about 20% have potentially deleterious variants in single PCP genes (VANGL2, CELSR1, SCRIB or DACT1) [181,186,187]. The mouse digenic PCP mutants support the suggestion that these cases may be undetected double heterozygotes for PCP mutations. Recently, six human NTD cases digenic for PCP variants were reported as follows: three spina bifida and one anencephaly digenic at CELSR1;SCRIB; one anencephaly digenic at CELSR1;DVL3 and one spina bifida digenic at PTK7;SCRIB [188].
Overall, the mouse digenic PCP mutants support the strategy of screening cases with anencephaly, spina bifida or craniorachischisis for PCP gene mutations and, given the usual observation of human heterozygosity for the PCP mutations found, strongly encourage the search for second gene locus mutations in these NTD cases.
8. Folate and NTD
8.1. Human NTD Prevention by Folate
In addition to genetic susceptibility, environmental factors contribute to risk of developing NTD, including various aspects of diet and maternal health [11,189]. One of the important factors is supplementary maternal peri-conceptional intake of folic acid, which has been demonstrated to reduce the occurrence of human NTD by as much as 70% [190,191] and has led to government policies of fortification of various foods with folic acid in some countries [192]. There is a significant residual, perhaps a third, of NTD cases that appear to be “folate unresponsive” [193]. The mechanism of beneficial effects of supplemental folate on human NTD occurrence is unknown. A paradigm of “folate deficiency” is widely assumed, but the data suggest this concept may be an over-simplification [7]. Although in the absence of folic acid supplementation, risk of NTD appears to increase with lower maternal blood folate levels on average [194], the levels are generally in the normal range, not deficient [195]. In animal models, dietary folate deficiency seldom, if ever, causes NTD [189,196,197]. A hypothesis that supplemental folate compensates for a genetic weakness in the folate metabolic pathway is long standing [194,195]. The folate metabolic pathway is well known and frequently reviewed [7,198]. The response to folic acid has directed the search for genes involved in the etiology of human NTD to focus on the genes that affect folate availability to cells and the genes in the known folate metabolic pathways, such as those involved in DNA and protein methylation and in DNA and RNA synthesis [11,199,200]. Despite extensive studies, in general this approach has not discovered genetic variants with major roles in the etiology of human NTD.
8.2. Mouse NTD Prevention by Folate
In parallel to humans, in mice, mutations in most folate metabolic pathway genes that have been tested do not cause NTD [201]. A small proportion of the almost 300 mouse gene mutations that cause NTD have been tested for response to folate supplementation. There are a few mouse mutants whose NTD frequency (usually exencephaly) is reduced by maternal folic acid, and most have no known defect in folate metabolism [201]. Examples are the loss of function mutants for transcription factors Cart1 and Cited2, the histone actetyltransferase Gcn5, and the “Crooked” gain-of-function mutation of the transmembrane co-receptor for Wnt, the Lrp6 gene [202]. Interestingly, the loss-of-function mutation of Lrp6 also causes exencephaly and spina bifida but responds to maternal folate supplementation with increased frequency of these NTDs [202]. These mutants indicate that the NTD preventative effects of folate are not necessarily through compensation for a deficiency in the folate metabolic pathway [201].
The mutant genes in folate-responsive mouse NTD models may have unexpected connections to folate pathways. An example is the Pax3 transcription factor mutations [203,204], which have several effects on neural tube cell functions and whose homozygous “Splotch” mutants have spina bifida, exencephaly, or both. These mutants have a defect in the folate metabolic pathway that leads to deficiency of de novo pyrimidine biosynthesis; this defect seems to be ameliorated by folate supplementation, which reduces the frequency of NTD by about 40% [203,204].
8.3. A Possible Effect of Folate on Cilia
A recent study has suggested that folate deficiency or lack of the folate-dependent methylation process may exert adverse effects on neural tube development through an effect on formation of primary cilia [205]. In Xenopus embryos, mouse embryos and various types of cell cultures, folate deficiency, inhibition of methylation, or loss of the reduced folate carrier Slc19a1 each causes reduced cilia formation, indicating that the folate and methylation pathways are required for cilium formation. The lack of cilia is attributable to the demonstrated deficiency of methylation of Septin2, a cytoskeletal protein whose methylation state affects whether it contributes to cilia formation [205]. Cilia are required for normal functioning of the Hedgehog signaling pathway [13]. An indicator of the functioning of the Hedgehog pathway, the truncated form of Gli3 (Gli3R), is decreased in mouse embryonic fibroblast cells lacking folate or methylation, indicating that the lack of cilia in these cells leads to dysregulation of the Hedgehog pathway [205]. Cilial structural defects often cause over-expression of the Sonic hedgehog signaling pathway in the developing neural tube, which in turn interferes with dorsal bending of the neural folds and causes exencephaly or spina bifida [13] (Section 2.5). In summary, folate status may affect cytoskeletal regulation, cilia formation and Hedgehog signaling.
Folr1 (Fbp1) is a membrane protein that takes folate into cells. Normally, Folr1 in mouse embryos is expressed in the neural folds of the upper spine just before and during closure and expression expands bidirectionally along the anterior–posterior axis with advancing closure; it is also expressed in the cranial neural folds just before and during closure, similarly expanding with the advancing closure; it is not detected in the forebrain region and posterior neuropore [206,207]. The Folr1 null mutant is lethal before neural tube closure, but with maternal folate supplementation, null Folr1 mutant embryos survive to express NTD, particularly exencephaly [208]. FOLR1 gene variants do not seem to be important in human anencephaly [209] nor in spina bifida, although FOLR2 and FOLR3 may have a small role [210]. A functionally related gene that also causes exencephaly in mice is Lrp2, a multi-ligand membrane receptor that seems to mediate uptake of folic acid into cells by Folr1 at the apical surface of the developing neuroepithelium [211]. Maternal injection with folic acid during early gestation reduces the NTD frequency in Lrp2 mutants by half; dietary supplementation with folic acid is not effective [212].
Cellular folate deficiency may not be the mechanism by which mutants for Folr1 and Lrp2 cause exencephaly. There are other mechanisms of uptake of folate into cells that may be more efficient than Folr1, such as the reduced folate carriers [200]. In this light, it is notable that there is evidence that Folr1 can also function as a transcription factor in mammalian cells, with a large number of developmentally important genes as its target [213]. Furthermore, recent observations in Xenopus have pointed to a novel mechanism for the role of Folr1 and folate in neural tube closure [214]. Knockdown of Folr1 during neurulation in Xenopus causes failure to develop a neural tube. In Xenopus, Folr1 is expressed at the apical surface of neural plate cells, and interaction between Folr1 and folate in the medial neural plate is required for these cells’ apical constriction and the bending of the neural plate. Folr1 co-localizes and interacts with C-cadherin and beta-catenin in the medial neural plate cells. Apical constriction requires internalization of apically localized C-cadherin by endocytosis; in Folr1 knockdown neural plates, there is a decrease in C-cadherin–containing endosomes. Overall, the observations indicate that the primary function of Folr1 interacting with folate in Xenopus neural tube development may not be related to its functions in metabolism, but rather as a regulator of the cytoskeleton and cell adhesion remodeling [200,214]. If a similar link of the Folr1-folate interaction to cytoskeletal remodeling can be found in mammals, then a fresh interpretation of the folate-responsiveness of human and mouse NTD occurrences should be undertaken. Perhaps supplemental availability of folate to the Folr1 receptor can maximize its activity, and can also compensate for weaknesses in other unrelated modules that also regulate the cytoskeleton.
8.4. Heterogeneity of Effects of Folate on Neural Tube Closure?
Overall, the mouse NTD mutants that interact with folate present a complex story that suggests that some folate effects on NTD risk may function through connections to the known metabolic roles of folate; other folate effects on NTD may be through previously unknown roles in ciliogenesis and regulation of the cytoskeleton. Given the great variety of cellular functions essential for successful neural tube closure, abundant levels of folic acid may be important via different aspects of its function in multiple aspects of the closure process, such as cell proliferation, regulation of a complex repertoire of developmental gene signaling via DNA and histone methylation, Hedgehog signaling, and cytoskeletal-driven bending of the neural plate. Given the genetic heterogeneity of NTD, individual cases may have different types of weakness in the closure process and interact with the aspect of supplemental folate that matches the functional deficit.
8.5. Mouse NTD Prevention by Formate
A new set of NTD mutants in folate-pathway genes is emerging. The genes are all involved in folate one-carbon metabolism in the mitochondria, the process that supplies one-carbon formate molecules to the cytosol for nucleotide biosynthesis and DNA methylation [6]. The reported mutants, which have between 25% and 100% exencephaly, are in genes Amt, Gldc, Mthfd1l and Slc25a32 [170,215,216,217]. It appears that, whereas most of the non-mitochondrial folate-pathway mutants previously reported [201] do not have NTDs (the exception being Folr1), almost all of the mitochondrial folate-pathway mutants reported to date have exencephaly (with occasional craniorachischisis); none have spina bifida. The exception among the mitochondrial folate-pathway mutants that does not cause NTD is Mthfd2, which has redundancy with Mthfd2l [218].
Interestingly, in three of the mitochondrial mutants (Gldc, Mthfd1l, Slc25a32), the frequency of NTDs is reduced by between 60% and 100% by sodium formate supplementation (in drinking water) to the pregnant dams [170,216,217]; testing of the Amt mutant with formate has not been reported. In none of these mutants has folate/folic acid treatment reduced the NTD frequency.
A further example is the curly tail mouse strain, in which 10–20% of embryos have spina bifida. Folic acid has no preventative effect in curly tail strain embryos, but nucleotide precursors reduce spina bifida by 85% [219]. Recently, expression of the mitochondrial folate-pathway gene, Mthfd1l, was found to be 50% deficient in curly tail strain embryos and, as with the Mthfd1l null mutant (above), treatment of curly tail strain dams with prenatal sodium formate was found to reduce the spina bifida frequency by 75% [220].
Because the main source of one-carbon units for cytoplasmic processes comes from the mitochondria, any failure in a step of mitochondrial folate one-carbon metabolism results in a deficiency of one-carbon components (formate) for essential cellular processes, such as DNA synthesis and methylation. Momb et al. [216] suggest that a metabolic block in the mitochondria creates a “formyl trap”, starving the cytoplasm of formate, even in the presence of plentiful folate. It may be that formate, rather than folate, supplementation could be helpful in a similar category of human NTDs in which folate supplementation has no preventative effect.
9. An Overview of the Genetics of NTD
9.1. Genetic Architecture of Human NTD
Understanding of the genetic architecture of human NTD is largely derived from two approaches: family-based data and population-based data. The family-based approach led to the multifactorial model that explains the pattern of recurrence of NTD in families. Rates of recurrence of NTD in various types of relatives of NTD cases demonstrate that NTD have a strong genetic etiology, with a 30- to 50-fold increased rate of occurrence in siblings of NTD cases, compared to a population rate of about one per 1000 births [11,47]. The pattern of recurrence rates of NTD in other types of relatives does not fit simple single gene locus inheritance, but generally fits predictions for multiple gene locus inheritance, where the effects of variants at the multiple genes add together with each other and with environmental influences. We have summarized the foundational studies previously [12]. Conceptually, the genes involved may be unequal in effect [221,222] and the various genes contributing to risk of NTD in an individual case can be imagined to differ in impact, some having fairly large effects on risk, some having moderate effects, and some, perhaps many common polymorphisms, having small effects. A derivative of quantitative genetics, the multifactorial model assumes that individual cases, even with the same type of NTD, will have different subsets of the risk genes.
Population-based studies aimed at finding statistical associations with single nucleotide polymorphisms (SNPs) in over 200 candidate genes have found a few weak associations often lacking statistical significance [1,223,224]. Association studies likely have been hampered by the heterogeneity of etiology of NTD at different locations along the neural tube, mismatch of candidate genes to the types of NTD in the pool of cases studied, dilution of power by use of pools of multiple types of NTD, and lack of adequate sample size for statistical significance of modest associations.
Studies based on sequencing of coding regions of candidate genes usually are done as a population-based approach, often using pools of various types of NTD cases [10]. This approach does not depend on statistical association, only on the ability to identify sequence changes that affect gene function. As the multifactorial model for NTD predicts that individual cases have different combinations in subsets of the gene variants that cause risk of NTD, sequencing a pool of cases may detect few mutations at any given NTD risk gene sequenced. The pattern of findings fits this prediction. A few putative mutations have been found at each of some of the PCP system loci that have been sequenced in several pools of various types of NTD [10]. When examined, the mutations are usually inherited from a normal parent. The multifactorial concept predicts that the NTD cases have also inherited additional variants or mutations at other genes, probably from the other parent. Sequencing of coding regions of five PCP genes in a pool of 36 human craniorachischisis cases has yielded putative mutations at either of two genes, in about 13% of cases [181]. Similarly, exon sequencing of 191 candidate genes in a pool of 94 cases, most of which were anencephaly, has yielded 21 putative mutations affecting 19 genes [209] in about 22% of cases. In this study, consistent with multifactorial etiology, more of the additional rare/novel sequence variants that might have deleterious effects are found in each of the NTD cases than in the controls. The good yields from these two studies [181,209] perhaps demonstrate that increased effectiveness can be conferred by use of pools of only one type of NTD at a time. It should be noted that the usual sequencing approach cannot detect mutations in the more distant regulatory regions that direct the expression of genes to specific tissues (enhancers), and many such regulatory mutations at previously studied candidate genes may yet be discovered.
A current hypothesis about the genetic architecture of various multifactorial, post-reproductive diseases (“complex traits”) [225] suggests that a large proportion of the genome can have some impact on a “complex trait” disease. This concept is also a long-standing component of genetic selection theory based on empirical data for quantitative traits [226]. The genetic architecture likely differs between NTD and “complex traits” or quantitative traits because of the effects of genetic lethality of NTD on gene frequencies. For NTD, perhaps the important goal is to identify the variants of major effect whose combinations account for most of the risk of NTD, not all of the variants that can add smaller effects. There are probably many variants of major effect.
The common forms of NTD that are explained by the multifactorial model are non-syndromic, meaning the only primary defect is the defect in neural tube closure.
There are some rare human syndromes of multiple unrelated defects that include anencephaly or spina bifida, and they are caused by chromosomal defects or Mendelian mutations [227]. They are not included in studies of common, non-syndromic NTD.
9.2. The Role of Environmental Effects
For a multifactorial trait, it is expected that environmental factors contribute to the trait to some degree. Theoretically, the effects of environmental factors add to the genetic risk factors in individuals. Individual embryos with a load of genetic risk factors but marginal ability to close the neural tube may be hampered enough by the additional effects of environmental factors to fail to close, whereas an exposed embryo with fewer genetic risk factors would still be able to successfully close. Folate supplementation (Section 8) is an environmental factor with a large effect that apparently compensates for the genetic risk factors in some way. Some other environmental factors are deleterious, with much weaker but statistically detectable effects. They include maternal diabetes, maternal obesity, maternal hyperthermia during pregnancy, parental occupations and exposure to chemicals, and a few pharmaceuticals [1].
9.3. The Role of Mouse Mutants in NTD Genetics
The almost 300 mouse NTD mutants cannot model the human genetic architecture of NTD. The mouse mutants are maintained on inbred strain backgrounds, within which there is essentially no genetic variation between individuals. Very few strain backgrounds are used, and the strains themselves are a very limited sample of the variation in the mouse species’ genome. Some mutations show different penetrance or expressivity of NTD on different strain backgrounds, demonstrating their potential for multifactorial genetics. In the studies of digenic effects, to test the combined effects of two different genes on NTD risk, the specific digenic combination of interest is created by crosses between mutant stocks. Unexpected genetic interactions are unlikely to be discovered in this hypothesis-driven system.
The mouse mutations are usually highly penetrant recessive “knockouts” that cause loss of function of the gene, and for many of the genes, these cause syndromes of multiple defects. In general, gene loci that cause syndromic phenotypes from null (knockout) mutations may present non-syndromic phenotypes from hypomorphic (reduced function) mutations. An example in mice is the kidney defect and cleft lip syndrome caused by a null mutation at Wnt9b versus the non-syndromic cleft lip phenotype from a hypomorphic mutation at Wnt9b [228]. Therefore, the mouse mutants serve to identify candidate loci for human NTD despite the severity of the null mutant phenotypes.
The use of candidate genes from mice should be tempered by awareness of the differences in the morphological process between human and mouse neural tube development (Section 4.1), and ideally should be informed by gene expression data from other species whose neural tube closure is morphologically more similar to human, such as pig and rabbit.
Many of the candidate genes that have been tested in human NTD cases are based on various hypotheses, such as roles related to folate metabolism or folate transport, and the lack of finding gene variants associated with human NTD concurs with the mouse mutants; i.e., they do not cause NTD in mouse mutants.
The pools of human cases studied usually focus on spina bifida. The majority of the NTD genes in mouse mutants cause cranial NTD, and based on the regional heterogeneity of etiology discussed in Section 3 and Section 5, these genes would not be expected to participate in human spina bifida. This mismatch may contribute to the disappointing results from some studies.
Two successful sequencing studies are discussed in Section 9.1. Both demonstrate the value of mouse NTD mutants as candidate genes. The study of a few of the PCP genes in craniorachischisis cases [181], based on the observation that nearly all genetic craniorachischisis in mice is caused by PCP gene mutations [12] has been fruitful, yielding putative mutations from 13% of cases. In a similar study [209] based on mostly anencephalic cases, about half of the 19 genes with putative mutations have mouse NTD mutants at the same gene or a member of the same gene family.
10. Concluding Remarks
Various insights emerge from an overview of the many studies of neural tube closure. The mammalian way of forming a neural tube by elevating and bending the lateral edges of a neural plate to meet in the midline is an ancient mechanism that evolved before vertebrates. Several systems contribute to forming the neural tube. Among them are the planar cell polarity/convergent extension system; the medial hinge point and related cell cycle phenomena; the dorsolateral hinge point and the Sonic hedgehog cilia system on which it depends; the actomyosin and cytoskeletal system responsible for some bending of the neuroepithelium; the various types of cell projections and the Ephrin system that facilitate contact and fusion. Each of these systems involves many genes. Despite the understanding of many details of the genetic determination of neural tube development, the genetic causes of human NTD are mostly undiscovered.
The evidence that the planar cell polarity system is involved in various aspects of neural tube closure in addition to convergent extension, such as the cytoskeleton, ciliogenesis, and the actomyosin system, offers an explanation for the observed role of planar cell polarity genes in exencephaly/anencephaly and spina bifida as well as in craniorachischisis.
Although the mouse has been used extensively as the mammalian model for human NTD, the development of the neural tube in other mammals, such as pig and rabbit, is more similar to human in morphology. Gene expression arrays of regions of neural folds from these species during elevation and closure might point to the subset of genes and mechanisms previously identified in mice that are likely to be involved in human neural tube closure.
The vertebrate head evolved as an “add-on” to the pre-vertebrate body plan and its neural tube. The formation of the cranial neural tube consequently differs in several ways from that of the spinal region. For example, the mouse cranial mesoderm has a much greater role in neural tube closure and etiology of NTD. The neuroepithelial actomyosin system is also more important in the cranial region than in the spinal region. The large number of genes involved in these two systems may account for the predominance of genes causing exencephaly among mouse NTD mutants. The genes identified by these mutants may be good candidates for examination in the etiology of human anencephaly.
Cranial NTD occur more frequently in females than in males. This phenomenon is seen in humans and in nearly all mouse NTD mutants examined. Recent advances in the understanding of epigenetics suggest the hypothesis that the second, inactive X chromosome of female embryos is a “heterochromatin sink” that sequesters essential epigenetic factors, altering expression of genes in the rest of the genome and thereby increasing the risk of NTD. The limitation of the effect to cranial NTD suggests that specific genes are involved.
It is well established that peri-conceptional dietary folic acid supplementation reduces the risk of occurrence of NTD in humans and in some mouse mutants, but the genetic risk of folate-responsive NTD does not seem to involve genes in the folate metabolic pathway in either species. There is recent evidence for a function of folate-pathway genes that is separate from the established metabolic functions in DNA methylation and nucleotide synthesis. In Xenopus, the Folr1 membrane protein that takes folate into cells seems to act also as a regulator of the cytoskeleton and of cell adhesion remodeling in the neural plate. Secondly, independent evidence in Xenopus demonstrates a requirement for folate in ciliogenesis, which is known to be involved in NTD. These observations may shift the focus of candidate gene research in folate-responsive human NTD away from the metabolic functions of folate.
There are many examples in mouse mutants of NTD caused by combinations of mutant genes at two or more loci from the same or different genetic pathways. Usually, the molecular mechanism of interaction between the mutant genes is unknown. The mechanism of the interaction effect can be surprising, as in the case of the Msx2/Dlx5 double mutant, where the combined effect of the two mutant genes alters the balance of splice forms of a third gene, EphA7. The extensive study of the planar cell polarity system has generated several examples of digenic combinations of different planar cell polarity genes that cause a variety of types of NTD. The observations in mice suggest that many of the cases of human NTD in which a putative mutation has been detected at one planar cell polarity gene locus may well harbor an undetected mutation at a second planar cell polarity gene locus or other type of gene.
Overall, the cumulative data show that whereas the neural tube appears to be a single anterior–posterior structure, in contradiction, the gene expression, cell behavior, and closure systems occupy a variety of different regions. The neural tube along its length is neither homogeneous nor segmented. Each gene and mechanism is expressed in areas along the length that are different from the expression patterns for many of the other genes or mechanisms so that each linear area is the result of a unique combination. NTD may be a series of genetically distinct defects occurring along a contiguous physical structure. Setting aside the planar cell polarity system mutations, the remainder of human NTD may have genetic etiology that varies by anterior–posterior location of the lesion, a concept that could inform human genetic studies in search of causative variants. For example, families within which there are both anencephalic and spina bifida cases may tend to have mutations in the DLHP system. Perhaps the most important conclusion from this review is recognition of the degree to which the neural tube is regionalized along the anterior–posterior axis.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Au, K.S.; Ashley-Koch, A.; Northrup, H. Epidemiologic and Genetic Aspects of Spina Bifida and Other Neural Tube Defects. Dev. Disabil. Res. Rev. 2010, 16, 6–15. [Google Scholar] [CrossRef] [PubMed]
- McBride, M.L. Sib Risks of Anencephaly and Spina Bifida in British Columbia. Am. J. Med. Genet. 1979, 3, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.J.; Juriloff, D.M. An Update to the List of Mouse Mutants with Neural Tube Closure Defects and Advances toward a Complete Genetic Perspective of Neural Tube Closure. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 653–669. [Google Scholar] [CrossRef] [PubMed]
- Wilde, J.J.; Petersen, J.R.; Niswander, L. Genetic, Epigenetic, and Environmental Contributions to Neural Tube Closure. Annu. Rev. Genet. 2014, 48, 583–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolopoulou, E.; Galea, G.L.; Rolo, A.; Greene, N.D.; Copp, A.J. Neural Tube Closure: Cellular, Molecular and Biomechanical Mechanisms. Development 2017, 144, 552–566. [Google Scholar] [CrossRef] [PubMed]
- Greene, N.D.; Copp, A.J. Neural Tube Defects. Annu. Rev. Neurosci. 2014, 37, 221–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Copp, A.J.; Stanier, P.; Greene, N.D. Neural Tube Defects: Recent Advances, Unsolved Questions, and Controversies. Lancet Neurol. 2013, 12, 799–810. [Google Scholar] [CrossRef]
- Wallingford, J.B.; Niswander, L.A.; Shaw, G.M.; Finnell, R.H. The Continuing Challenge of Understanding, Preventing, and Treating Neural Tube Defects. Science 2013, 339, 1222002. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Miura, M. How to Form and Close the Brain: Insight into the Mechanism of Cranial Neural Tube Closure in Mammals. Cell Mol. Life Sci. 2013, 70, 3171–3186. [Google Scholar] [CrossRef] [PubMed]
- Juriloff, D.M.; Harris, M.J. A Consideration of the Evidence that Genetic Defects in Planar Cell Polarity Contribute to the Etiology of Human Neural Tube Defects. Birth Defects Res. A Clin. Mol. Teratol. 2012, 94, 824–840. [Google Scholar] [CrossRef] [PubMed]
- Au, K.S.; Findley, T.O.; Northrup, H. Finding the Genetic Mechanisms of Folate Deficiency and Neural Tube Defects-Leaving no Stone Unturned. Am. J. Med. Genet. A 2017, 173, 3042–3057. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.J.; Juriloff, D.M. Mouse Mutants with Neural Tube Closure Defects and their Role in Understanding Human Neural Tube Defects. Birth Defects Res. A Clin. Mol. Teratol. 2007, 79, 187–210. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, J.N.; Copp, A.J. The Relationship between Sonic Hedgehog Signaling, Cilia, and Neural Tube Defects. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 633–652. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Gonzalez, J.M.; Mulero-Navarro, S.; Mlodzik, M. Centriole Positioning in Epithelial Cells and its Intimate Relationship with Planar Cell Polarity. Bioessays 2016, 38, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Montcouquiol, M.; Sans, N.; Huss, D.; Kach, J.; Dickman, J.D.; Forge, A.; Rachel, R.A.; Copeland, N.G.; Jenkins, N.A.; Bogani, D.; et al. Asymmetric Localization of Vangl2 and Fz3 Indicate Novel Mechanisms for Planar Cell Polarity in Mammals. J. Neurosci. 2006, 26, 5265–5275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, M.; Yen, W.; Lu, X.; Sutherland, A. Distinct Apical and Basolateral Mechanisms Drive Planar Cell Polarity-Dependent Convergent Extension of the Mouse Neural Plate. Dev. Cell. 2014, 29, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Tissir, F.; Qu, Y.; Montcouquiol, M.; Zhou, L.; Komatsu, K.; Shi, D.; Fujimori, T.; Labeau, J.; Tyteca, D.; Courtoy, P.; et al. Lack of Cadherins Celsr2 and Celsr3 Impairs Ependymal Ciliogenesis, Leading to Fatal Hydrocephalus. Nat. Neurosci. 2010, 13, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Hoover, A.N.; Liu, A. PCP Effector Gene Inturned is an Important Regulator of Cilia Formation and Embryonic Development in Mammals. Dev. Biol. 2010, 339, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Escuin, S.; Vernay, B.; Savery, D.; Gurniak, C.B.; Witke, W.; Greene, N.D.; Copp, A.J. Rho-Kinase-Dependent Actin Turnover and Actomyosin Disassembly are Necessary for Mouse Spinal Neural Tube Closure. J. Cell Sci. 2015, 128, 2468–2481. [Google Scholar] [CrossRef] [PubMed]
- Rolo, A.; Escuin, S.; Greene, N.D.E.; Copp, A.J. Rho GTPases in Mammalian Spinal Neural Tube Closure. Small GTPases 2018, 9, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Shum, A.S.; Copp, A.J. Regional Differences in Morphogenesis of the Neuroepithelium Suggest Multiple Mechanisms of Spinal Neurulation in the Mouse. Anat. Embryol. 1996, 194, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Ybot-Gonzalez, P.; Cogram, P.; Gerrelli, D.; Copp, A.J. Sonic Hedgehog and the Molecular Regulation of Mouse Neural Tube Closure. Development 2002, 129, 2507–2517. [Google Scholar] [PubMed]
- McShane, S.G.; Mole, M.A.; Savery, D.; Greene, N.D.; Tam, P.P.; Copp, A.J. Cellular Basis of Neuroepithelial Bending during Mouse Spinal Neural Tube Closure. Dev. Biol. 2015, 404, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Amarnath, S.; Agarwala, S. Cell-Cycle-Dependent TGFbeta-BMP Antagonism Regulates Neural Tube Closure by Modulating Tight Junctions. J. Cell Sci. 2017, 130, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Chatterjee, B.; Francis, D.; Yu, Q.; SanAgustin, J.T.; Francis, R.; Tansey, T.; Henry, C.; Wang, B.; Lemley, B.; et al. Disruption of Mks1 Localization to the Mother Centriole Causes Cilia Defects and Developmental Malformations in Meckel-Gruber Syndrome. Dis. Model. Mech. 2011, 4, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Wallingford, J.B. Planar Cell Polarity and the Developmental Control of Cell Behavior in Vertebrate Embryos. Annu. Rev. Cell Dev. Biol. 2012, 28, 627–653. [Google Scholar] [CrossRef] [PubMed]
- Pala, R.; Alomari, N.; Nauli, S.M. Primary Cilium-Dependent Signaling Mechanisms. Int. J. Mol. Sci. 2017, 18, 18. [Google Scholar] [CrossRef] [PubMed]
- May-Simera, H.; Kelley, M.W. Planar Cell Polarity in the Inner Ear. Curr. Top. Dev. Biol. 2012, 101, 111–140. [Google Scholar] [PubMed]
- Adler, P.N.; Wallingford, J.B. From Planar Cell Polarity to Ciliogenesis and Back: The Curious Tale of the PPE and CPLANE Proteins. Trends Cell Biol. 2017, 27, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Hui, C.C.; Angers, S. Gli Proteins in Development and Disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 513–537. [Google Scholar] [CrossRef] [PubMed]
- Ybot-Gonzalez, P.; Gaston-Massuet, C.; Girdler, G.; Klingensmith, J.; Arkell, R.; Greene, N.D.; Copp, A.J. Neural Plate Morphogenesis during Mouse Neurulation is Regulated by Antagonism of Bmp Signalling. Development 2007, 134, 3203–3211. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Wang, X.; Li, Z.; Gotoh, N.; Chapman, D.; Skolnik, E.Y. Mesodermal Patterning Defect in Mice Lacking the Ste20 NCK Interacting Kinase (NIK). Development 2001, 128, 1559–1572. [Google Scholar] [PubMed]
- Chiang, C.; Litingtung, Y.; Lee, E.; Young, K.E.; Corden, J.L.; Westphal, H.; Beachy, P.A. Cyclopia and Defective Axial Patterning in Mice Lacking Sonic Hedgehog Gene Function. Nature 1996, 383, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Aoto, K.; Shikata, Y.; Imai, H.; Matsumaru, D.; Tokunaga, T.; Shioda, S.; Yamada, G.; Motoyama, J. Mouse Shh is Required for Prechordal Plate Maintenance during Brain and Craniofacial Morphogenesis. Dev. Biol. 2009, 327, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Patterson, V.L.; Damrau, C.; Paudyal, A.; Reeve, B.; Grimes, D.T.; Stewart, M.E.; Williams, D.J.; Siggers, P.; Greenfield, A.; Murdoch, J.N. Mouse Hitchhiker Mutants have Spina Bifida, Dorso-Ventral Patterning Defects and Polydactyly: Identification of Tulp3 as a Novel Negative Regulator of the Sonic Hedgehog Pathway. Hum. Mol. Genet. 2009, 18, 1719–1739. [Google Scholar] [CrossRef] [PubMed]
- Robbins, D.J.; Fei, D.L.; Riobo, N.A. The Hedgehog Signal Transduction Network. Sci. Signal. 2012, 5, re6. [Google Scholar] [CrossRef] [PubMed]
- Pyrgaki, C.; Trainor, P.; Hadjantonakis, A.K.; Niswander, L. Dynamic Imaging of Mammalian Neural Tube Closure. Dev. Biol. 2010, 344, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Rolo, A.; Savery, D.; Escuin, S.; de Castro, S.C.; Armer, H.E.; Munro, P.M.; Mole, M.A.; Greene, N.D.; Copp, A.J. Regulation of Cell Protrusions by Small GTPases during Fusion of the Neural Folds. Elife 2016, 5, e13273. [Google Scholar] [CrossRef] [PubMed]
- Ray, H.J.; Niswander, L.A. Dynamic Behaviors of the Non-Neural Ectoderm during Mammalian Cranial Neural Tube Closure. Dev. Biol. 2016, 416, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Holmberg, J.; Clarke, D.L.; Frisen, J. Regulation of Repulsion versus Adhesion by Different Splice Forms of an Eph Receptor. Nature 2000, 408, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Rifat, Y.; Parekh, V.; Wilanowski, T.; Hislop, N.R.; Auden, A.; Ting, S.B.; Cunningham, J.M.; Jane, S.M. Regional Neural Tube Closure Defined by the Grainy Head-Like Transcription Factors. Dev. Biol. 2010, 345, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Copp, A.J.; Greene, N.D.; Murdoch, J.N. The Genetic Basis of Mammalian Neurulation. Nat. Rev. Genet. 2003, 4, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Aziz, N.M.; Turmaine, M.; Greene, N.D.; Copp, A.J. EphrinA-EphA Receptor Interactions in Mouse Spinal Neurulation: Implications for Neural Fold Fusion. Int. J. Dev. Biol. 2009, 53, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Escobar, B.; Caro-Vega, J.M.; Vijayraghavan, D.S.; Plageman, T.F.; Sanchez-Alcazar, J.A.; Moreno, R.C.; Savery, D.; Marquez-Rivas, J.; Davidson, L.A.; Ybot-Gonzalez, P. The Non-Canonical Wnt-PCP Pathway Shapes the Mouse Caudal Neural Plate. Development 2018, 145, 145. [Google Scholar] [CrossRef] [PubMed]
- Saitsu, H.; Shiota, K. Involvement of the Axially Condensed Tail Bud Mesenchyme in Normal and Abnormal Human Posterior Neural Tube Development. Congenit. Anom. 2008, 48, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Dady, A.; Havis, E.; Escriou, V.; Catala, M.; Duband, J.L. Junctional Neurulation: A Unique Developmental Program Shaping a Discrete Region of the Spinal Cord Highly Susceptible to Neural Tube Defects. J. Neurosci. 2014, 34, 13208–13221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Detrait, E.R.; George, T.M.; Etchevers, H.C.; Gilbert, J.R.; Vekemans, M.; Speer, M.C. Human Neural Tube Defects: Developmental Biology, Epidemiology, and Genetics. Neurotoxicol. Teratol. 2005, 27, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Juriloff, D.M.; Harris, M.J.; Tom, C.; MacDonald, K.B. Normal Mouse Strains Differ in the Site of Initiation of Closure of the Cranial Neural Tube. Teratology 1991, 44, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, M. Cephalic Neurulation and Optic Vesicle Formation in the Early Mouse Embryo. Am. J. Anat. 1979, 155, 425–443. [Google Scholar] [CrossRef] [PubMed]
- Sakai, Y. Neurulation in the Mouse: Manner and Timing of Neural Tube Closure. Anat. Rec. 1989, 223, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Golden, J.A.; Chernoff, G.F. Intermittent Pattern of Neural Tube Closure in Two Strains of Mice. Teratology 1993, 47, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Morriss, G.M.; Solursh, M. Regional Differences in Mesenchymal Cell Morphology and Glycosaminoglycans in Early Neural-Fold Stage Rat Embryos. J. Embryol. Exp. Morphol. 1978, 46, 37–52. [Google Scholar] [PubMed]
- Christie, G.A. Developmental Stages in Somite and Post-Somite Rat Embryos, Based on External Appearance, and Including some Features of the Macroscopic Development of the Oral Cavity. J. Morphol. 1964, 114, 263–283. [Google Scholar] [CrossRef] [PubMed]
- Keyser, A. The Development of the Diencephalon of the Chinese Hamster. An Investigation of the Validity of the Criteria of Subdivision of the Brain. Acta Anat. Suppl. 1972, 59, 1–178. [Google Scholar] [PubMed]
- Waterman, R.E. Topographical Changes Along the Neural Fold Associated with Neurulation in the Hamster and Mouse. Am. J. Anat. 1976, 146, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Peeters, M.C.; Viebahn, C.; Hekking, J.W.; van Straaten, H.W. Neurulation in the Rabbit Embryo. Anat. Embryol. 1998, 197, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Van Straaten, H.W.; Peeters, M.C.; Hekking, J.W.; van der Lende, T. Neurulation in the Pig Embryo. Anat. Embryol. 2000, 202, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, W.J.; Mossman, H.W. Human Embryology: Prenatal Development of Form and Function, 4th ed.; Williams and Wilkins: Baltimore, MD, USA, 1972; pp. 1–646. [Google Scholar]
- Jirasek, J.E. Atlas of Human Prenatal Morphogenesis, 1st ed.; Springer: Dordrecht, The Netherlands, 1983; p. 168. [Google Scholar]
- Muller, F.; O’Rahilly, R. The First Appearance of the Neural Tube and Optic Primordium in the Human Embryo at Stage 10. Anat. Embryol. 1985, 172, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.; O’Rahilly, R. The Development of the Human Brain and the Closure of the Rostral Neuropore at Stage 11. Anat. Embryol. 1986, 175, 205–222. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, T.; Uwabe, C.; Shiota, K. Neural Tube Closure in Humans Initiates at Multiple Sites; Evidence from Human Embryos and Implications for the Pathogenesis of Neural Tube Defects. Anat. Embryol. 2000, 201, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Peeters, M.C.; Hekking, J.W.; Shiota, K.; Drukker, J.; Van Straaten, H.W. Differences in Axial Curvature Correlate with Species-Specific Rate of Neural Tube Closure in Embryos of Chick, Rabbit, Mouse, Rat and Human. Anat. Embryol. 1998, 198, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Juriloff, D.M.; Harris, M.J. Mouse Models for Neural Tube Closure Defects. Hum. Mol. Genet. 2000, 9, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.J.; Juriloff, D.M.; Gunn, T.M.; Miller, J.E. Development of the Cerebellar Defect in Ataxic SELH/Bc Mice. Teratology 1994, 50, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Golden, J.A.; Chernoff, G.F. Multiple Sites of Anterior Neural Tube Closure in Humans: Evidence from Anterior Neural Tube Defects (Anencephaly). Pediatrics 1995, 95, 506–510. [Google Scholar] [PubMed]
- Geelen, J.A.; Langman, J. Closure of the Neural Tube in the Cephalic Region of the Mouse Embryo. Anat. Rec. 1977, 189, 625–640. [Google Scholar] [CrossRef] [PubMed]
- Geelen, J.A.; Langman, J. Ultrastructural Observations on Closure of the Neural Tube in the Mouse. Anat. Embryol. 1979, 156, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Gilardi-Hebenstreit, P.; Charnay, P.; Wilkinson, D.G. A Receptor Protein Tyrosine Kinase Implicated in the Segmental Patterning of the Hindbrain and Mesoderm. Development 1992, 116, 1137–1150. [Google Scholar] [PubMed]
- Niederreither, K.; Vermot, J.; Schuhbaur, B.; Chambon, P.; Dolle, P. Retinoic Acid Synthesis and Hindbrain Patterning in the Mouse Embryo. Development 2000, 127, 75–85. [Google Scholar] [PubMed]
- Ting, S.B.; Wilanowski, T.; Auden, A.; Hall, M.; Voss, A.K.; Thomas, T.; Parekh, V.; Cunningham, J.M.; Jane, S.M. Inositol- and Folate-Resistant Neural Tube Defects in Mice Lacking the Epithelial-Specific Factor Grhl-3. Nat. Med. 2003, 9, 1513–1519. [Google Scholar] [CrossRef] [PubMed]
- Ray, H.J.; Niswander, L.A. Grainyhead-Like 2 Downstream Targets Act to Suppress Epithelial-to-Mesenchymal Transition during Neural Tube Closure. Development 2016, 143, 1192–1204. [Google Scholar] [CrossRef] [PubMed]
- De Castro, S.C.P.; Hirst, C.S.; Savery, D.; Rolo, A.; Lickert, H.; Andersen, B.; Copp, A.J.; Greene, N.D.E. Neural Tube Closure Depends on Expression of Grainyhead-Like 3 in Multiple Tissues. Dev. Biol. 2018, 435, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Deschamps, J.; van den Akker, E.; Forlani, S.; De Graaff, W.; Oosterveen, T.; Roelen, B.; Roelfsema, J. Initiation, Establishment and Maintenance of Hox Gene Expression Patterns in the Mouse. Int. J. Dev. Biol. 1999, 43, 635–650. [Google Scholar] [PubMed]
- Deschamps, J.; van Nes, J. Developmental Regulation of the Hox Genes during Axial Morphogenesis in the Mouse. Development 2005, 132, 2931–2942. [Google Scholar] [CrossRef] [PubMed]
- Morriss-Kay, G.; Tan, S. Mapping Cranial Neural Crest Cell Migration Pathways in Mammalian Embryos. TIG 1987, 3, 257–261. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Shinotsuka, N.; Nonomura, K.; Takemoto, K.; Kuida, K.; Yosida, H.; Miura, M. Live Imaging of Apoptosis in a Novel Transgenic Mouse Highlights its Role in Neural Tube Closure. J. Cell Biol. 2011, 195, 1047–1060. [Google Scholar] [CrossRef] [PubMed]
- Lowery, L.A.; Sive, H. Strategies of Vertebrate Neurulation and a Re-Evaluation of Teleost Neural Tube Formation. Mech. Dev. 2004, 121, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, T.E. Neurulation in Xenopus Laevis. An Analysis and Model Based upon Light and Electron Microscopy. J. Embryol. Exp. Morphol. 1970, 23, 427–462. [Google Scholar] [PubMed]
- Eagleson, G.W. Developmental Neurobiology of the Anterior Areas in Amphibians: Urodele Perspectives. Int. J. Dev. Biol. 1996, 40, 735–743. [Google Scholar] [PubMed]
- Holland, L.Z. The Origin and Evolution of Chordate Nervous Systems. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 370. [Google Scholar] [CrossRef] [PubMed]
- Shimeld, S.M.; Donoghue, P.C. Evolutionary Crossroads in Developmental Biology: Cyclostomes (Lamprey and Hagfish). Development 2012, 139, 2091–2099. [Google Scholar] [CrossRef] [PubMed]
- Holland, N.D.; Holland, L.Z. The Ups and Downs of Amphioxus Biology: A History. Int. J. Dev. Biol. 2017, 61, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Putnam, N.H.; Butts, T.; Ferrier, D.E.; Furlong, R.F.; Hellsten, U.; Kawashima, T.; Robinson-Rechavi, M.; Shoguchi, E.; Terry, A.; Yu, J.K.; et al. The Amphioxus Genome and the Evolution of the Chordate Karyotype. Nature 2008, 453, 1064–1071. [Google Scholar] [CrossRef] [PubMed]
- Satoh, N. Cellular Morphology and Architecture during Early Morphogenesis of the Ascidian Egg: An SEM Study. Biol. Bull. 1978, 155, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Munro, E.M.; Smith, W.C. Ascidian Prickle Regulates both Mediolateral and Anterior-Posterior Cell Polarity of Notochord Cells. Curr. Biol. 2005, 15, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Sasakura, Y.; Mita, K.; Ogura, Y.; Horie, T. Ascidians as Excellent Chordate Models for Studying the Development of the Nervous System during Embryogenesis and Metamorphosis. Dev. Growth Differ. 2012, 54, 420–437. [Google Scholar] [CrossRef] [PubMed]
- Northcutt, R.G. The New Head Hypothesis Revisited. J. Exp. Zool. B Mol. Dev. Evol. 2005, 304, 274–297. [Google Scholar] [CrossRef] [PubMed]
- Bronner-Fraser, M. On the Trail of the ‘New Head’ in Les Treilles. Development 2008, 135, 2995–2999. [Google Scholar] [CrossRef] [PubMed]
- Holland, L.Z. Chordate Roots of the Vertebrate Nervous System: Expanding the Molecular Toolkit. Nat. Rev. Neurosci. 2009, 10, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Bronner, M.E.; LeDouarin, N.M. Development and Evolution of the Neural Crest: An Overview. Dev. Biol. 2012, 366, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Santagati, F.; Rijli, F.M. Cranial Neural Crest and the Building of the Vertebrate Head. Nat. Rev. Neurosci. 2003, 4, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Jandzik, D.; Garnett, A.T.; Square, T.A.; Cattell, M.V.; Yu, J.K.; Medeiros, D.M. Evolution of the New Vertebrate Head by Co-Option of an Ancient Chordate Skeletal Tissue. Nature 2015, 518, 534–537. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.S.; Morriss-Kay, G. The Development and Distribution of the Cranial Neural Crest in the Rat Embryo. Cell Tissue Res. 1985, 240, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Osumi-Yamashita, N.; Ninomiya, Y.; Doi, H.; Eto, K. The Contribution of both Forebrain and Midbrain Crest Cells to the Mesenchyme in the Frontonasal Mass of Mouse Embryos. Dev. Biol. 1994, 164, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Ezin, M.; Barembaum, M.; Bronner, M.E. Stage-Dependent Plasticity of the Anterior Neural Folds to Form Neural Crest. Differentiation 2014, 88, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Masek, J.; Machon, O.; Korinek, V.; Taketo, M.M.; Kozmik, Z. Tcf7l1 Protects the Anterior Neural Fold from Adopting the Neural Crest Fate. Development 2016, 143, 2206–2216. [Google Scholar] [CrossRef] [PubMed]
- Nolte, C.; Krumlauf, R. Expression of Hox genes in the nervous system of vertebrates. In Hox Gene Expression; Papageorgiou, S., Ed.; Springer: New York, NY, USA, 2007; pp. 14–41. [Google Scholar]
- Muller, F.; O’Rahilly, R. The Prechordal Plate, the Rostral End of the Notochord and Nearby Median Features in Staged Human Embryos. Cells Tissues Organs 2003, 173, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Ulmer, B.; Tingler, M.; Kurz, S.; Maerker, M.; Andre, P.; Monch, D.; Campione, M.; Deissler, K.; Lewandoski, M.; Thumberger, T.; et al. A Novel Role of the Organizer Gene Goosecoid as an Inhibitor of Wnt/PCP-Mediated Convergent Extension in Xenopus and Mouse. Sci. Rep. 2017, 7, 43010. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, K.; Rubenstein, J.L. Inductive Interactions Direct Early Regionalization of the Mouse Forebrain. Development 1997, 124, 2709–2718. [Google Scholar] [PubMed]
- Belo, J.A.; Leyns, L.; Yamada, G.; De Robertis, E.M. The Prechordal Midline of the Chondrocranium is Defective in Goosecoid-1 Mouse Mutants. Mech. Dev. 1998, 72, 15–25. [Google Scholar] [CrossRef]
- Muller, F.; O’Rahilly, R. The First Appearance of the Major Divisions of the Human Brain at Stage 9. Anat. Embryol. 1983, 168, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, A.G.; Tam, P.P. Cephalic Neurulation in the Mouse Embryo Analyzed by SEM and Morphometry. Anat. Rec. 1982, 203, 375–396. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Kojima, M.; Nakajima, K.; Kondo, S. Jumonji Gene is Essential for the Neurulation and Cardiac Development of Mouse Embryos with a C3H/He Background. Mech. Dev. 1999, 86, 29–38. [Google Scholar] [CrossRef]
- Kaufman, M.H. The Role of Embryology in Teratological Research, with Particular Reference to the Development of the Neural Tube and Heart. J. Reprod. Fertil. 1981, 62, 607–623. [Google Scholar] [CrossRef] [PubMed]
- Massa, V.; Savery, D.; Ybot-Gonzalez, P.; Ferraro, E.; Rongvaux, A.; Cecconi, F.; Flavell, R.; Greene, N.D.; Copp, A.J. Apoptosis is Not Required for Mammalian Neural Tube Closure. Proc. Natl. Acad. Sci. USA 2009, 106, 8233–8238. [Google Scholar] [CrossRef] [PubMed]
- Brewer, S.; Feng, W.; Huang, J.; Sullivan, S.; Williams, T. Wnt1-Cre-Mediated Deletion of AP-2alpha Causes Multiple Neural Crest-Related Defects. Dev. Biol. 2004, 267, 135–152. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; High, F.A.; Epstein, J.A.; Radice, G.L. N-Cadherin is Required for Neural Crest Remodeling of the Cardiac Outflow Tract. Dev. Biol. 2006, 299, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Breau, M.A.; Pietri, T.; Stemmler, M.P.; Thiery, J.P.; Weston, J.A. A Nonneural Epithelial Domain of Embryonic Cranial Neural Folds Gives Rise to Ectomesenchyme. Proc. Natl. Acad. Sci. USA 2008, 105, 7750–7755. [Google Scholar] [CrossRef] [PubMed]
- Schorle, H.; Meier, P.; Buchert, M.; Jaenisch, R.; Mitchell, P.J. Transcription Factor AP-2 Essential for Cranial Closure and Craniofacial Development. Nature 1996, 381, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hagopian-Donaldson, S.; Serbedzija, G.; Elsemore, J.; Plehn-Dujowich, D.; McMahon, A.P.; Flavell, R.A.; Williams, T. Neural Tube, Skeletal and Body Wall Defects in Mice Lacking Transcription Factor AP-2. Nature 1996, 381, 238–241. [Google Scholar] [CrossRef] [PubMed]
- Radice, G.L.; Rayburn, H.; Matsunami, H.; Knudsen, K.A.; Takeichi, M.; Hynes, R.O. Developmental Defects in Mouse Embryos Lacking N-Cadherin. Dev. Biol. 1997, 181, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Ferreira-Cornwell, M.; Baldwin, H.; Kostetskii, I.; Lenox, J.; Lieberman, M.; Radice, G. Rescuing the N-Cadherin Knockout by Cardiac-Specific Expression of N- Or E-Cadherin. Development 2001, 128, 459–469. [Google Scholar] [PubMed]
- Kinder, S.J.; Tsang, T.E.; Quinlan, G.A.; Hadjantonakis, A.K.; Nagy, A.; Tam, P.P. The Orderly Allocation of Mesodermal Cells to the Extraembryonic Structures and the Anteroposterior Axis during Gastrulation of the Mouse Embryo. Development 1999, 126, 4691–4701. [Google Scholar] [PubMed]
- Onai, T.; Aramaki, T.; Inomata, H.; Hirai, T.; Kuratani, S. Ancestral Mesodermal Reorganization and Evolution of the Vertebrate Head. Zool. Lett. 2015, 1, 29. [Google Scholar] [CrossRef] [PubMed]
- Zohn, I.E.; Sarkar, A.A. Does the Cranial Mesenchyme Contribute to Neural Fold Elevation during Neurulation? Birth Defects Res. A Clin. Mol. Teratol. 2012, 94, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Copp, A.J. Neurulation in the Cranial Region—Normal and Abnormal. J. Anat. 2005, 207, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Morris-Wiman, J.; Brinkley, L.L. Changes in Mesenchymal Cell and Hyaluronate Distribution Correlate with in vivo Elevation of the Mouse Mesencephalic Neural Folds. Anat. Rec. 1990, 226, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.F.; Behringer, R.R. Twist is Required in Head Mesenchyme for Cranial Neural Tube Morphogenesis. Genes Dev. 1995, 9, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Bildsoe, H.; Loebel, D.A.; Jones, V.J.; Hor, A.C.; Braithwaite, A.W.; Chen, Y.T.; Behringer, R.R.; Tam, P.P. The Mesenchymal Architecture of the Cranial Mesoderm of Mouse Embryos is Disrupted by the Loss of Twist1 Function. Dev. Biol. 2013, 374, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Wolf, C.; Thisse, C.; Stoetzel, C.; Thisse, B.; Gerlinger, P.; Perrin-Schmitt, F. The M-Twist Gene of Mus is Expressed in Subsets of Mesodermal Cells and is Closely Related to the Xenopus X-Twi and the Drosophila Twist Genes. Dev. Biol. 1991, 143, 363–373. [Google Scholar] [CrossRef]
- Soo, K.; O’Rourke, M.P.; Khoo, P.L.; Steiner, K.A.; Wong, N.; Behringer, R.R.; Tam, P.P. Twist Function is Required for the Morphogenesis of the Cephalic Neural Tube and the Differentiation of the Cranial Neural Crest Cells in the Mouse Embryo. Dev. Biol. 2002, 247, 251–270. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Behringer, R.R.; de Crombrugghe, B. Prenatal Folic Acid Treatment Suppresses Acrania and Meroanencephaly in Mice Mutant for the Cart1 Homeobox Gene. Nat. Genet. 1996, 13, 275–283. [Google Scholar] [CrossRef] [PubMed]
- ten Berge, D.; Brouwer, A.; el Bahi, S.; Guenet, J.L.; Robert, B.; Meijlink, F. Mouse Alx3: An Aristaless-Like Homeobox Gene Expressed during Embryogenesis in Ectomesenchyme and Lateral Plate Mesoderm. Dev. Biol. 1998, 199, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Lakhwani, S.; Garcia-Sanz, P.; Vallejo, M. Alx3-Deficient Mice Exhibit Folic Acid-Resistant Craniofacial Midline and Neural Tube Closure Defects. Dev. Biol. 2010, 344, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Reid, B.S.; Sargent, T.D.; Williams, T. Generation and Characterization of a Novel Neural Crest Marker Allele, Inka1-LacZ, Reveals a Role for Inka1 in Mouse Neural Tube Closure. Dev. Dyn. 2010, 239, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Baskaran, Y.; Ang, K.C.; Anekal, P.V.; Chan, W.L.; Grimes, J.M.; Manser, E.; Robinson, R.C. An in Cellulo-Derived Structure of PAK4 in Complex with its Inhibitor Inka1. Nat. Commun. 2015, 6, 8681. [Google Scholar] [CrossRef] [PubMed]
- Baranek, C.; Atanasoski, S. Modulating Epigenetic Mechanisms: The Diverse Functions of Ski during Cortical Development. Epigenetics 2012, 7, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Berk, M.; Desai, S.Y.; Heyman, H.C.; Colmenares, C. Mice Lacking the Ski Proto-Oncogene have Defects in Neurulation, Craniofacial, Patterning, and Skeletal Muscle Development. Genes Dev. 1997, 11, 2029–2039. [Google Scholar] [CrossRef] [PubMed]
- Lyons, G.E.; Micales, B.K.; Herr, M.J.; Horrigan, S.K.; Namciu, S.; Shardy, D.; Stavnezer, E. Protooncogene c-Ski is Expressed in both Proliferating and Postmitotic Neuronal Populations. Dev. Dyn. 1994, 201, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Bultman, S.; Gebuhr, T.; Yee, D.; La Mantia, C.; Nicholson, J.; Gilliam, A.; Randazzo, F.; Metzger, D.; Chambon, P.; Crabtree, G.; et al. A Brg1 Null Mutation in the Mouse Reveals Functional Differences among Mammalian SWI/SNF Complexes. Mol. Cell 2000, 6, 1287–1295. [Google Scholar] [CrossRef]
- Randazzo, F.M.; Khavari, P.; Crabtree, G.; Tamkun, J.; Rossant, J. Brg1: A Putative Murine Homologue of the Drosophila Brahma Gene, a Homeotic Gene Regulator. Dev. Biol. 1994, 161, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Zohn, I.E.; Anderson, K.V.; Niswander, L. The Hectd1 Ubiquitin Ligase is Required for Development of the Head Mesenchyme and Neural Tube Closure. Dev. Biol. 2007, 306, 208–221. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.A.; Zohn, I.E. Hectd1 Regulates Intracellular Localization and Secretion of Hsp90 to Control Cellular Behavior of the Cranial Mesenchyme. J. Cell Biol. 2012, 196, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Banting, G.S.; Barak, O.; Ames, T.M.; Burnham, A.C.; Kardel, M.D.; Cooch, N.S.; Davidson, C.E.; Godbout, R.; McDermid, H.E.; Shiekhattar, R. CECR2, a Protein Involved in Neurulation, Forms a Novel Chromatin Remodeling Complex with SNF2L. Hum. Mol. Genet. 2005, 14, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Dawe, C.E.; Kooistra, M.K.; Fairbridge, N.A.; Pisio, A.C.; McDermid, H.E. Role of Chromatin Remodeling Gene Cecr2 in Neurulation and Inner Ear Development. Dev. Dyn. 2011, 240, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Fairbridge, N.A.; Dawe, C.E.; Niri, F.H.; Kooistra, M.K.; King-Jones, K.; McDermid, H.E. Cecr2 Mutations Causing Exencephaly Trigger Misregulation of mesenchymal/ectodermal Transcription Factors. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Nishi, T.; Nakano, R. First-Trimester Diagnosis of Exencephaly by Transvaginal Ultrasonography. J. Ultrasound Med. 1994, 13, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Chatzipapas, I.K.; Whitlow, B.J.; Economides, D.L. The ‘Mickey Mouse’ Sign and the Diagnosis of Anencephaly in Early Pregnancy. Ultrasound Obstet. Gynecol. 1999, 13, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Grego-Bessa, J.; Hildebrand, J.; Anderson, K.V. Morphogenesis of the Mouse Neural Plate Depends on Distinct Roles of Cofilin 1 in Apical and Basal Epithelial Domains. Development 2015, 142, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Gurniak, C.B.; Perlas, E.; Witke, W. The Actin Depolymerizing Factor n-Cofilin is Essential for Neural Tube Morphogenesis and Neural Crest Cell Migration. Dev. Biol. 2005, 278, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Cox, B.; Hadjantonakis, A.K.; Collins, J.E.; Magee, A.I. Cloning and Expression Throughout Mouse Development of mfat1, a Homologue of the Drosophila Tumour Suppressor Gene Fat. Dev. Dyn. 2000, 217, 233–240. [Google Scholar] [CrossRef]
- Tanoue, T.; Takeichi, M. Mammalian Fat1 Cadherin Regulates Actin Dynamics and Cell-Cell Contact. J. Cell Biol. 2004, 165, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Badouel, C.; Zander, M.A.; Liscio, N.; Bagherie-Lachidan, M.; Sopko, R.; Coyaud, E.; Raught, B.; Miller, F.D.; McNeill, H. Fat1 Interacts with Fat4 to Regulate Neural Tube Closure, Neural Progenitor Proliferation and Apical Constriction during Mouse Brain Development. Development 2015, 142, 2781–2791. [Google Scholar] [CrossRef] [PubMed]
- Barbera, J.P.; Rodriguez, T.A.; Greene, N.D.; Weninger, W.J.; Simeone, A.; Copp, A.J.; Beddington, R.S.; Dunwoodie, S. Folic Acid Prevents Exencephaly in Cited2 Deficient Mice. Hum. Mol. Genet. 2002, 11, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Bamforth, S.D.; Braganca, J.; Eloranta, J.J.; Murdoch, J.N.; Marques, F.I.; Kranc, K.R.; Farza, H.; Henderson, D.J.; Hurst, H.C.; Bhattacharya, S. Cardiac Malformations, Adrenal Agenesis, Neural Crest Defects and Exencephaly in Mice Lacking Cited2, a New Tfap2 Co-Activator. Nat. Genet. 2001, 29, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Kranc, K.R.; Oliveira, D.V.; Armesilla-Diaz, A.; Pacheco-Leyva, I.; Catarina Matias, A.; Luisa Escapa, A.; Subramani, C.; Wheadon, H.; Trindade, M.; Nichols, J.; et al. Acute Loss of Cited2 Impairs Nanog Expression and Decreases Self-Renewal of Mouse Embryonic Stem Cells. Stem Cells 2015, 33, 699–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunwoodie, S.L.; Rodriguez, T.A.; Beddington, R.S. Msg1 and Mrg1, Founding Members of a Gene Family, show Distinct Patterns of Gene Expression during Mouse Embryogenesis. Mech. Dev. 1998, 72, 27–40. [Google Scholar] [CrossRef]
- Sakaki-Yumoto, M.; Kobayashi, C.; Sato, A.; Fujimura, S.; Matsumoto, Y.; Takasato, M.; Kodama, T.; Aburatani, H.; Asashima, M.; Yoshida, N.; et al. The Murine Homolog of SALL4, a Causative Gene in Okihiro Syndrome, is Essential for Embryonic Stem Cell Proliferation, and Cooperates with Sall1 in Anorectal, Heart, Brain and Kidney Development. Development 2006, 133, 3005–3013. [Google Scholar] [CrossRef] [PubMed]
- Buck, A.; Kispert, A.; Kohlhase, J. Embryonic Expression of the Murine Homologue of SALL1, the Gene Mutated in Townes–Brocks Syndrome. Mech. Dev. 2001, 104, 143–146. [Google Scholar] [CrossRef]
- Bohm, J.; Buck, A.; Borozdin, W.; Mannan, A.U.; Matysiak-Scholze, U.; Adham, I.; Schulz-Schaeffer, W.; Floss, T.; Wurst, W.; Kohlhase, J.; et al. Sall1, sall2, and sall4 are Required for Neural Tube Closure in Mice. Am. J. Pathol. 2008, 173, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, S.M.; Ohlemiller, K.K.; Yang, J.; McDill, B.W.; Kohlhase, J.; Rauchman, M. Expression of a Truncated Sall1 Transcriptional Repressor is Responsible for Townes-Brocks Syndrome Birth Defects. Hum. Mol. Genet. 2003, 12, 2221–2227. [Google Scholar] [CrossRef] [PubMed]
- Ohmura, T.; Shioi, G.; Hirano, M.; Aizawa, S. Neural Tube Defects by NUAK1 and NUAK2 Double Mutation. Dev. Dyn. 2012, 241, 1350–1364. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, J.D.; Soriano, P. Shroom, a PDZ Domain-Containing Actin-Binding Protein, is Required for Neural Tube Morphogenesis in Mice. Cell 1999, 99, 485–497. [Google Scholar] [CrossRef]
- Sousa-Nunes, R.; Rana, A.A.; Kettleborough, R.; Brickman, J.M.; Clements, M.; Forrest, A.; Grimmond, S.; Avner, P.; Smith, J.C.; Dunwoodie, S.L.; et al. Characterizing Embryonic Gene Expression Patterns in the Mouse using Nonredundant Sequence-Based Selection. Genome Res. 2003, 13, 2609–2620. [Google Scholar] [CrossRef] [PubMed]
- McGreevy, E.M.; Vijayraghavan, D.; Davidson, L.A.; Hildebrand, J.D. Shroom3 Functions Downstream of Planar Cell Polarity to Regulate Myosin II Distribution and Cellular Organization during Neural Tube Closure. Biol. Open 2015, 4, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, H.; Hu, G.; Wang, H.; Abate-Shen, C.; Shen, M.M. An Early Phase of Embryonic Dlx5 Expression Defines the Rostral Boundary of the Neural Plate. J. Neurosci. 1998, 18, 8322–8330. [Google Scholar] [CrossRef] [PubMed]
- Depew, M.J.; Liu, J.K.; Long, J.E.; Presley, R.; Meneses, J.J.; Pedersen, R.A.; Rubenstein, J.L. Dlx5 Regulates Regional Development of the Branchial Arches and Sensory Capsules. Development 1999, 126, 3831–3846. [Google Scholar] [PubMed]
- Lee, J.; Corcoran, A.; Han, M.; Gardiner, D.M.; Muneoka, K. Dlx5 and Msx2 Regulate Mouse Anterior Neural Tube Closure through ephrinA5-EphA7. Dev. Growth Differ. 2013, 55, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Ishii, M.; Han, J.; Yen, H.Y.; Sucov, H.M.; Chai, Y.; Maxson, R.E., Jr. Combined Deficiencies of Msx1 and Msx2 Cause Impaired Patterning and Survival of the Cranial Neural Crest. Development 2005, 132, 4937–4950. [Google Scholar] [CrossRef] [PubMed]
- Trainor, P.A.; Sobieszczuk, D.; Wilkinson, D.; Krumlauf, R. Signalling between the Hindbrain and Paraxial Tissues Dictates Neural Crest Migration Pathways. Development 2002, 129, 433–442. [Google Scholar] [PubMed]
- Han, J.; Ishii, M.; Bringas, P., Jr.; Maas, R.L.; Maxson, R.E., Jr.; Chai, Y. Concerted Action of Msx1 and Msx2 in Regulating Cranial Neural Crest Cell Differentiation during Frontal Bone Development. Mech. Dev. 2007, 124, 729–745. [Google Scholar] [CrossRef] [PubMed]
- Arvanitis, D.N.; Behar, A.; Tryoen-Toth, P.; Bush, J.O.; Jungas, T.; Vitale, N.; Davy, A. Ephrin B1 Maintains Apical Adhesion of Neural Progenitors. Development 2013, 140, 2082–2092. [Google Scholar] [CrossRef] [PubMed]
- Heimsath, E.G., Jr.; Yim, Y.I.; Mustapha, M.; Hammer, J.A.; Cheney, R.E. Myosin-X Knockout is Semi-Lethal and Demonstrates that Myosin-X Functions in Neural Tube Closure, Pigmentation, Hyaloid Vasculature Regression, and Filopodia Formation. Sci. Rep. 2017, 7, 17354. [Google Scholar] [CrossRef] [PubMed]
- Juriloff, D.M.; Harris, M.J. Hypothesis: The Female Excess in Cranial Neural Tube Defects Reflects an Epigenetic Drag of the Inactivating X Chromosome on the Molecular Mechanisms of Neural Fold Elevation. Birth Defects Res. A Clin. Mol. Teratol. 2012, 94, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Poletta, F.A.; Rittler, M.; Saleme, C.; Campana, H.; Gili, J.A.; Pawluk, M.S.; Gimenez, L.G.; Cosentino, V.R.; Castilla, E.E.; Lopez-Camelo, J.S. Neural Tube Defects: Sex Ratio Changes After Fortification with Folic Acid. PLoS ONE 2018, 13, e0193127. [Google Scholar] [CrossRef] [PubMed]
- Davidson, C.E.; Li, Q.; Churchill, G.A.; Osborne, L.R.; McDermid, H.E. Modifier Locus for Exencephaly in Cecr2 Mutant Mice is Syntenic to the 10q25.3 Region Associated with Neural Tube Defects in Humans. Physiol. Genom. 2007, 31, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Santander, N.G.; Contreras-Duarte, S.; Awad, M.F.; Lizama, C.; Passalacqua, I.; Rigotti, A.; Busso, D. Developmental Abnormalities in Mouse Embryos Lacking the HDL Receptor SR-BI. Hum. Mol. Genet. 2013, 22, 1086–1096. [Google Scholar] [CrossRef] [PubMed]
- Pai, Y.J.; Leung, K.Y.; Savery, D.; Hutchin, T.; Prunty, H.; Heales, S.; Brosnan, M.E.; Brosnan, J.T.; Copp, A.J.; Greene, N.D. Glycine Decarboxylase Deficiency Causes Neural Tube Defects and Features of Non-Ketotic Hyperglycinemia in Mice. Nat. Commun. 2015, 6, 6388. [Google Scholar] [CrossRef] [PubMed]
- Zvetkova, I.; Apedaile, A.; Ramsahoye, B.; Mermoud, J.E.; Crompton, L.A.; John, R.; Feil, R.; Brockdorff, N. Global Hypomethylation of the Genome in XX Embryonic Stem Cells. Nat. Genet. 2005, 37, 1274–1279. [Google Scholar] [CrossRef] [PubMed]
- Dobbs, K.B.; Rodriguez, M.; Sudano, M.J.; Ortega, M.S.; Hansen, P.J. Dynamics of DNA Methylation during Early Development of the Preimplantation Bovine Embryo. PLoS ONE 2013, 8, e66230. [Google Scholar] [CrossRef] [PubMed]
- Vallot, C.; Ouimette, J.F.; Rougeulle, C. Establishment of X Chromosome Inactivation and Epigenomic Features of the Inactive X Depend on Cellular Contexts. Bioessays 2016, 38, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Carrel, L.; Brown, C.J. When the Lyon(Ized Chromosome) Roars: Ongoing Expression from an Inactive X Chromosome. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 372. [Google Scholar] [CrossRef] [PubMed]
- Wijchers, P.J.; Festenstein, R.J. Epigenetic Regulation of Autosomal Gene Expression by Sex Chromosomes. Trends Genet. 2011, 27, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Blewitt, M.E.; Vickaryous, N.K.; Hemley, S.J.; Ashe, A.; Bruxner, T.J.; Preis, J.I.; Arkell, R.; Whitelaw, E. An N-Ethyl-N-Nitrosourea Screen for Genes Involved in Variegation in the Mouse. Proc. Natl. Acad. Sci. USA 2005, 102, 7629–7634. [Google Scholar] [CrossRef] [PubMed]
- Wijchers, P.J.; Yandim, C.; Panousopoulou, E.; Ahmad, M.; Harker, N.; Saveliev, A.; Burgoyne, P.S.; Festenstein, R. Sexual Dimorphism in Mammalian Autosomal Gene Regulation is Determined Not Only by Sry but by Sex Chromosome Complement as Well. Dev. Cell. 2010, 19, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.A. Comment on Changes in Sex Ratio in Neural Tube Defects since Food Fortification with Folic Acid: Re “Hypothesis: The Female Excess in Cranial Neural Tube Defects Reflects an Epigenetic Drag of the Inactivating X Chromosome on the Molecular Mechanisms of Neural Tube Fold Elevation”. Birth Defects Res. A Clin. Mol. Teratol. 2012, 94, 958. [Google Scholar] [CrossRef] [PubMed]
- Greene, N.D.; Gerrelli, D.; van Straaten, H.W.; Copp, A.J. Abnormalities of Floor Plate, Notochord and Somite Differentiation in the Loop-Tail (Lp) Mouse: A Model of Severe Neural Tube Defects. Mech. Dev. 1998, 73, 59–72. [Google Scholar] [CrossRef]
- Ybot-Gonzalez, P.; Savery, D.; Gerrelli, D.; Signore, M.; Mitchell, C.E.; Faux, C.H.; Greene, N.D.; Copp, A.J. Convergent Extension, Planar-Cell-Polarity Signalling and Initiation of Mouse Neural Tube Closure. Development 2007, 134, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.; Escuin, S.; Doudney, K.; Vekemans, M.; Stevenson, R.E.; Greene, N.D.; Copp, A.J.; Stanier, P. Mutations in the Planar Cell Polarity Genes CELSR1 and SCRIB are Associated with the Severe Neural Tube Defect Craniorachischisis. Hum. Mutat. 2012, 33, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Kirillova, I.; Novikova, I.; Auge, J.; Audollent, S.; Esnault, D.; Encha-Razavi, F.; Lazjuk, G.; Attie-Bitach, T.; Vekemans, M. Expression of the Sonic Hedgehog Gene in Human Embryos with Neural Tube Defects. Teratology 2000, 61, 347–354. [Google Scholar] [CrossRef]
- Murdoch, J.N.; Doudney, K.; Paternotte, C.; Copp, A.J.; Stanier, P. Severe Neural Tube Defects in the Loop-Tail Mouse Result from Mutation of Lpp1, a Novel Gene Involved in Floor Plate Specification. Hum. Mol. Genet. 2001, 10, 2593–2601. [Google Scholar] [CrossRef] [PubMed]
- Mahaffey, J.P.; Grego-Bessa, J.; Liem, K.F., Jr.; Anderson, K.V. Cofilin and Vangl2 Cooperate in the Initiation of Planar Cell Polarity in the Mouse Embryo. Development 2013, 140, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, J.N.; Damrau, C.; Paudyal, A.; Bogani, D.; Wells, S.; Greene, N.D.; Stanier, P.; Copp, A.J. Genetic Interactions between Planar Cell Polarity Genes Cause Diverse Neural Tube Defects in Mice. Dis. Model. Mech. 2014, 7, 1153–1163. [Google Scholar] [CrossRef] [PubMed]
- Doudney, K.; Ybot-Gonzalez, P.; Paternotte, C.; Stevenson, R.E.; Greene, N.D.; Moore, G.E.; Copp, A.J.; Stanier, P. Analysis of the Planar Cell Polarity Gene Vangl2 and its Co-Expressed Paralogue Vangl1 in Neural Tube Defect Patients. Am. J. Med. Genet. A 2005, 136, 90–92. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Ding, Y.; Lei, Y.P.; Yang, X.Y.; Xie, G.M.; Wen, J.; Cai, C.Q.; Li, H.; Chen, Y.; Zhang, T.; et al. Identification of Novel Rare Mutations of DACT1 in Human Neural Tube Defects. Hum. Mutat. 2012, 33, 1450–1455. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xiao, Y.; Tian, T.; Jin, L.; Lei, Y.; Finnell, R.H.; Ren, A. Digenic Variants of Planar Cell Polarity Genes in Human Neural Tube Defect Patients. Mol. Genet. Metab. 2018, 124, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.M.; Kirke, P.N.; Weir, D.G. The Role of Nutrition in Neural Tube Defects. Annu. Rev. Nutr. 1990, 10, 277–295. [Google Scholar] [CrossRef] [PubMed]
- MRC Vitamin Study Research Group. Prevention of Neural Tube Defects: Results of the Medical Research Council Vitamin Study. MRC Vitamin Study Research Group. Lancet 1991, 338, 131–137. [Google Scholar] [CrossRef]
- Czeizel, A.E.; Dudas, I. Prevention of the First Occurrence of Neural-Tube Defects by Periconceptional Vitamin Supplementation. N. Engl. J. Med. 1992, 327, 1832–1835. [Google Scholar] [CrossRef] [PubMed]
- Garrett, G.S.; Bailey, L.B. A Public Health Approach for Preventing Neural Tube Defects: Folic Acid Fortification and Beyond. Ann. N. Y. Acad. Sci. 2018, 1414, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Greene, N.D.; Leung, K.Y.; Copp, A.J. Inositol, Neural Tube Closure and the Prevention of Neural Tube Defects. Birth Defects Res. 2017, 109, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Kirke, P.N.; Molloy, A.M.; Daly, L.E.; Burke, H.; Weir, D.G.; Scott, J.M. Maternal Plasma Folate and Vitamin B12 are Independent Risk Factors for Neural Tube Defects. Q. J. Med. 1993, 86, 703–708. [Google Scholar] [PubMed]
- Scott, J.M. Folate and Vitamin B12. Proc. Nutr. Soc. 1999, 58, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Heid, M.K.; Bills, N.D.; Hinrichs, S.H.; Clifford, A.J. Folate Deficiency Alone does Not Produce Neural Tube Defects in Mice. J. Nutr. 1992, 122, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Burgoon, J.M.; Selhub, J.; Nadeau, M.; Sadler, T.W. Investigation of the Effects of Folate Deficiency on Embryonic Development through the Establishment of a Folate Deficient Mouse Model. Teratology 2002, 65, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Blom, H.J.; Shaw, G.M.; den Heijer, M.; Finnell, R.H. Neural Tube Defects and Folate: Case Far from Closed. Nat. Rev. Neurosci. 2006, 7, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Doudney, K.; Grinham, J.; Whittaker, J.; Lynch, S.A.; Thompson, D.; Moore, G.E.; Copp, A.J.; Greene, N.D.; Stanier, P. Evaluation of Folate Metabolism Gene Polymorphisms as Risk Factors for Open and Closed Neural Tube Defects. Am. J. Med. Genet. A 2009, 149A, 1585–1589. [Google Scholar] [CrossRef] [PubMed]
- Balashova, O.A.; Visina, O.; Borodinsky, L.N. Folate Action in Nervous System Development and Disease. Dev. Neurobiol. 2018, 78, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.J. Insights into Prevention of Human Neural Tube Defects by Folic Acid Arising from Consideration of Mouse Mutants. Birth Defects Res. A Clin. Mol. Teratol. 2009, 85, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.D.; Nakouzi, G.; Slowinska-Castaldo, B.; Dazard, J.E.; Rao, J.S.; Nadeau, J.H.; Ross, M.E. Functional Interactions between the LRP6 WNT Co-Receptor and Folate Supplementation. Hum. Mol. Genet. 2010, 19, 4560–4572. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.; Copp, A.J. Embryonic Folate Metabolism and Mouse Neural Tube Defects. Science 1998, 280, 2107–2109. [Google Scholar] [CrossRef] [PubMed]
- Greene, N.D.; Massa, V.; Copp, A.J. Understanding the Causes and Prevention of Neural Tube Defects: Insights from the Splotch Mouse Model. Birth Defects Res. A Clin. Mol. Teratol. 2009, 85, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Toriyama, M.; Toriyama, M.; Wallingford, J.B.; Finnell, R.H. Folate-Dependent Methylation of Septins Governs Ciliogenesis during Neural Tube Closure. FASEB J. 2017, 31, 3622–3635. [Google Scholar] [CrossRef] [PubMed]
- Saitsu, H.; Ishibashi, M.; Nakano, H.; Shiota, K. Spatial and Temporal Expression of Folate-Binding Protein 1 (Fbp1) is Closely Associated with Anterior Neural Tube Closure in Mice. Dev. Dyn. 2003, 226, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Saitsu, H. Folate Receptors and Neural Tube Closure. Congenit. Anom. 2017, 57, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.S.; Finnell, R.H. Neural and Orofacial Defects in Folp1 Knockout Mice [Corrected]. Birth Defects Res. A Clin. Mol. Teratol. 2003, 67, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Ishida, M.; Cullup, T.; Boustred, C.; James, C.; Docker, J.; English, C.; GOSgene; Lench, N.; Copp, A.J.; Moore, G.E.; et al. A Targeted Sequencing Panel Identifies Rare Damaging Variants in Multiple Genes in the Cranial Neural Tube Defect, Anencephaly. Clin. Genet. 2018, 93, 870–879. [Google Scholar] [CrossRef] [PubMed]
- O’Byrne, M.R.; Au, K.S.; Morrison, A.C.; Lin, J.I.; Fletcher, J.M.; Ostermaier, K.K.; Tyerman, G.H.; Doebel, S.; Northrup, H. Association of Folate Receptor (FOLR1, FOLR2, FOLR3) and Reduced Folate Carrier (SLC19A1) Genes with Meningomyelocele. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Kur, E.; Mecklenburg, N.; Cabrera, R.M.; Willnow, T.E.; Hammes, A. LRP2 Mediates Folate Uptake in the Developing Neural Tube. J. Cell Sci. 2014, 127, 2261–2268. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, J.A.; Stokes, B.A.; Zohn, I.E. Prevention of Neural Tube Defects in Lrp2 Mutant Mouse Embryos by Folic Acid Supplementation. Birth Defects Res. 2017, 109, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Boshnjaku, V.; Shim, K.W.; Tsurubuchi, T.; Ichi, S.; Szany, E.V.; Xi, G.; Mania-Farnell, B.; McLone, D.G.; Tomita, T.; Mayanil, C.S. Nuclear Localization of Folate Receptor Alpha: A New Role as a Transcription Factor. Sci. Rep. 2012, 2, 980. [Google Scholar] [CrossRef] [PubMed]
- Balashova, O.A.; Visina, O.; Borodinsky, L.N. Folate Receptor 1 is Necessary for Neural Plate Cell Apical Constriction during Xenopus Neural Tube Formation. Development 2017, 144, 1518–1530. [Google Scholar] [CrossRef] [PubMed]
- Narisawa, A.; Komatsuzaki, S.; Kikuchi, A.; Niihori, T.; Aoki, Y.; Fujiwara, K.; Tanemura, M.; Hata, A.; Suzuki, Y.; Relton, C.L.; et al. Mutations in Genes Encoding the Glycine Cleavage System Predispose to Neural Tube Defects in Mice and Humans. Hum. Mol. Genet. 2012, 21, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Momb, J.; Lewandowski, J.P.; Bryant, J.D.; Fitch, R.; Surman, D.R.; Vokes, S.A.; Appling, D.R. Deletion of Mthfd1l Causes Embryonic Lethality and Neural Tube and Craniofacial Defects in Mice. Proc. Natl. Acad. Sci. USA 2013, 110, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lei, Y.; Guo, J.; Kim, S.E.; Wlodarczyk, B.J.; Cabrera, R.M.; Lin, Y.L.; Nilsson, T.K.; Zhang, T.; Ren, A.; et al. Formate Rescues Neural Tube Defects Caused by Mutations in Slc25a32. Proc. Natl. Acad. Sci. USA 2018, 115, 4690–4695. [Google Scholar] [PubMed]
- Di Pietro, E.; Sirois, J.; Tremblay, M.L.; MacKenzie, R.E. Mitochondrial NAD-Dependent Methylenetetrahydrofolate Dehydrogenase-Methenyltetrahydrofolate Cyclohydrolase is Essential for Embryonic Development. Mol. Cell. Biol. 2002, 22, 4158–4166. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.Y.; De Castro, S.C.; Savery, D.; Copp, A.J.; Greene, N.D. Nucleotide Precursors Prevent Folic Acid-Resistant Neural Tube Defects in the Mouse. Brain 2013, 136, 2836–2841. [Google Scholar] [CrossRef] [PubMed]
- Sudiwala, S.; De Castro, S.C.; Leung, K.Y.; Brosnan, J.T.; Brosnan, M.E.; Mills, K.; Copp, A.J.; Greene, N.D. Formate Supplementation Enhances Folate-Dependent Nucleotide Biosynthesis and Prevents Spina Bifida in a Mouse Model of Folic Acid-Resistant Neural Tube Defects. Biochimie 2016, 126, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Lalouel, J.M.; Rao, D.C.; Morton, N.E.; Elston, R.C. A Unified Model for Complex Segregation Analysis. Am. J. Hum. Genet. 1983, 35, 816–826. [Google Scholar] [PubMed]
- Morton, N.E.; MacLean, C.J. Analysis of Family Resemblance. III. Complex Segregation of Quantitative Traits. Am. J. Hum. Genet. 1974, 26, 489–503. [Google Scholar] [PubMed]
- Greene, N.D.E.; Stanier, P.; Copp, A.J. Genetics of Human Neural Tube Defects. Hum. Mol. Genet. 2009, 18, R113–R129. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Guzman, A.R.; Yang, W.; Chapa, C.J.; Shaw, G.M.; Greene, R.M.; Pisano, M.M.; Lammer, E.J.; Finnell, R.H.; Zhu, H. Genes Encoding Critical Transcriptional Activators for Murine Neural Tube Development and Human Spina Bifida: A Case-Control Study. BMC Med. Genet. 2010, 11, 141. [Google Scholar] [CrossRef] [PubMed]
- Boyle, E.A.; Li, Y.; Pritchard, J.K. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 2017, 169, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Hill, W.G. Understanding and Using Quantitative Genetic Variation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.A. Non-multifactorial Neural Tube Defects. Am. J. Med. Genet. C Semin. Med. Genet. 2005, 135C, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Juriloff, D.M.; Harris, M.J.; McMahon, A.P.; Carroll, T.J.; Lidral, A.C. Wnt9b is the Mutated Gene Involved in Multifactorial Nonsyndromic Cleft Lip with or without Cleft Palate in A/WySn Mice, as Confirmed by a Genetic Complementation Test. Birth Defects Res. A Clin. Mol. Teratol. 2006, 76, 574–579. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Diagram of a transverse section of upper spinal neural folds of an E8 mouse embryo before neural tube closure. Somites not shown.
Figure 1.
Diagram of a transverse section of upper spinal neural folds of an E8 mouse embryo before neural tube closure. Somites not shown.

Figure 2.
Conceptual views of the shape of neuroepithelium in transverse sections, at various anterior–posterior locations, of a mouse neural tube before closure, demonstrating differences in bending mechanisms. Shading represents the concentration of Shh signaling. DLHP, dorsolateral hinge point. MHP, medial hinge point. Modified from Figure 3 in [13].
Figure 2.
Conceptual views of the shape of neuroepithelium in transverse sections, at various anterior–posterior locations, of a mouse neural tube before closure, demonstrating differences in bending mechanisms. Shading represents the concentration of Shh signaling. DLHP, dorsolateral hinge point. MHP, medial hinge point. Modified from Figure 3 in [13].

Figure 3.
An interpretation of the patterns of neural tube closure in various mammalian species based on published images and studies. Colors denote future anterior–posterior fates of the neural tube. F, forebrain; M, midbrain; H, hindbrain; S, spine. Short arrows indicate direction of zipping. Long stems on arrows and lines lacking arrowheads denote areas that appose and then fuse simultaneously. Triangles and circled numbers indicate closure initiation sites. The forebrain oblong denotes a region that appears to close from all sides, rather than apposition or zipping. 2°, region of secondary neurulation.
Figure 3.
An interpretation of the patterns of neural tube closure in various mammalian species based on published images and studies. Colors denote future anterior–posterior fates of the neural tube. F, forebrain; M, midbrain; H, hindbrain; S, spine. Short arrows indicate direction of zipping. Long stems on arrows and lines lacking arrowheads denote areas that appose and then fuse simultaneously. Triangles and circled numbers indicate closure initiation sites. The forebrain oblong denotes a region that appears to close from all sides, rather than apposition or zipping. 2°, region of secondary neurulation.

Figure 4.
Diagrams of transverse sections of midbrain neural folds during their elevation, showing the morphological change from convex to concave shape. Based on Figure 1 in [117].
Figure 4.
Diagrams of transverse sections of midbrain neural folds during their elevation, showing the morphological change from convex to concave shape. Based on Figure 1 in [117].


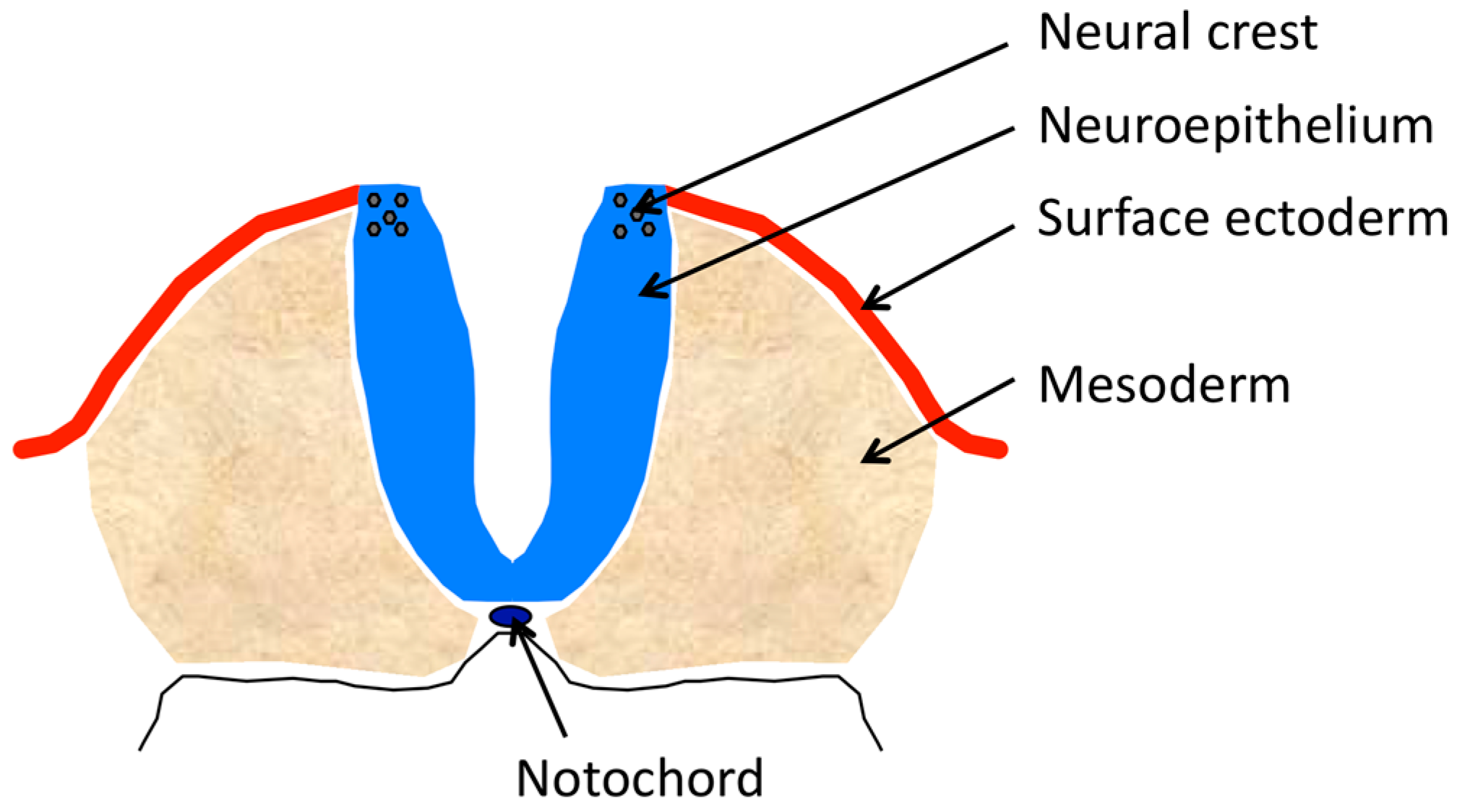{kind=link}
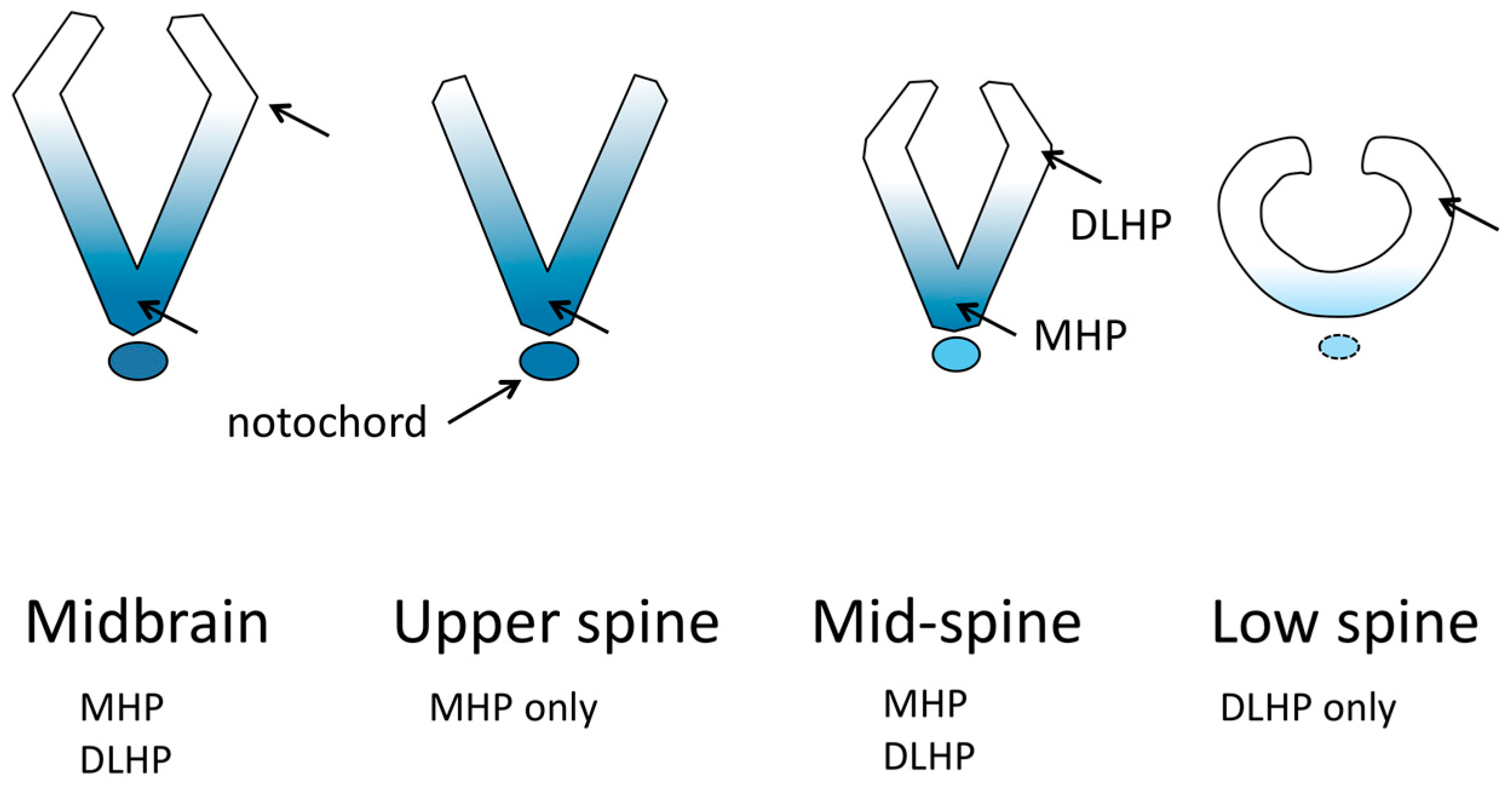{kind=link}
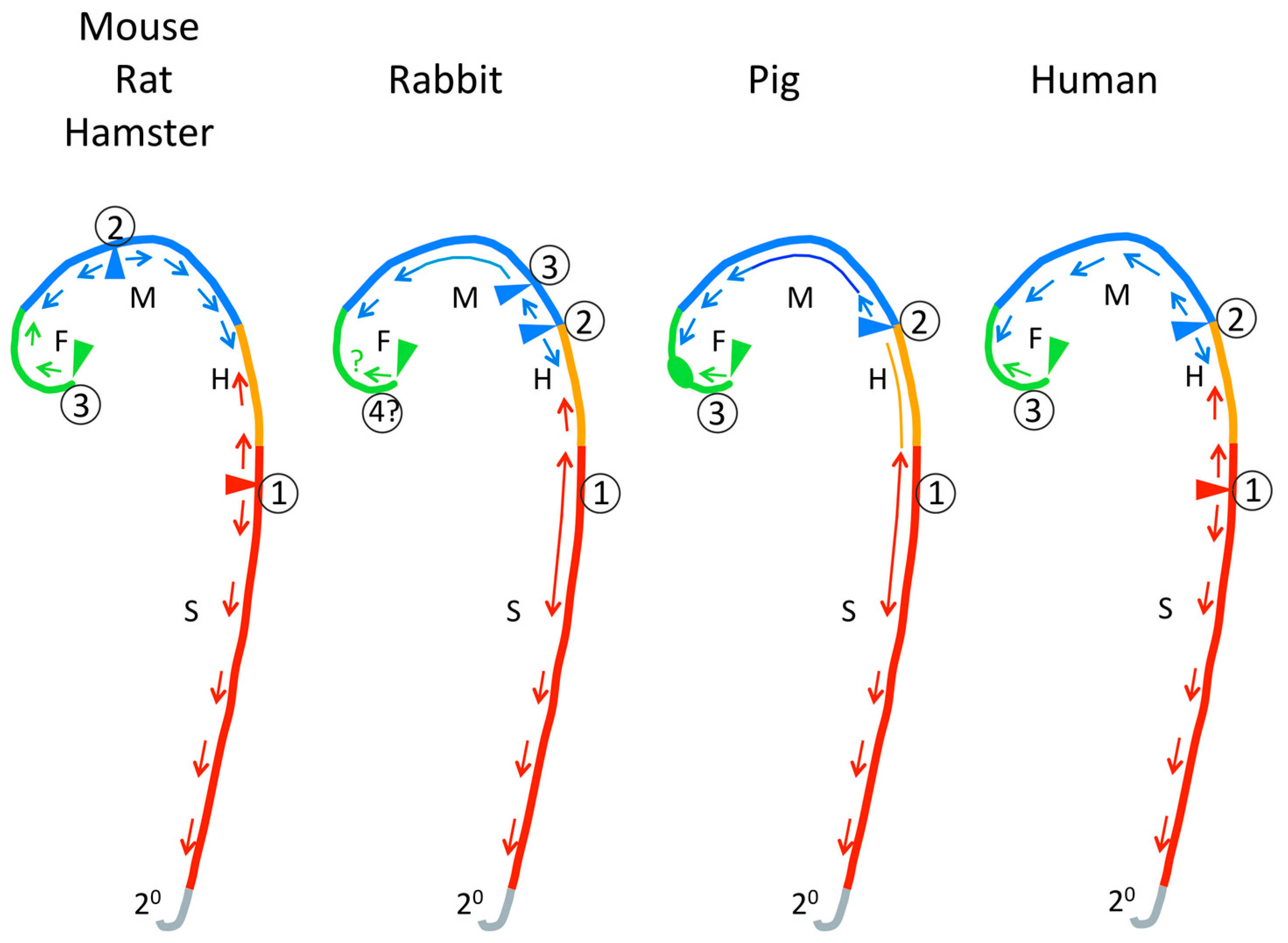{kind=link}
{kind=link}
Table 1.
Ways in which the neural tube can fail to form.
| Lack of convergent extension |
| Lack of medial hinge point (MHP) 1 |
| Lack of dorsolateral hinge point (DLHP) 1 |
| Lack of neuroepithelial bending by apical constriction (cranial region) |
| Lack of structural integrity of the neuroepithelium |
| Lack of support from surrounding mesenchyme Lack of midline fusion of neuroepithelium and/or surface ectoderm Interference by excessive anterior-posterior curvature of the neural plate |
1 Shown in Figure 2.
Table 2.
Regional differences in mechanisms for neural tube closure.
| Mechanism | Difference between Regions of Neural Tube |
|---|---|
| Convergent extension | Absent in forebrain |
| Notochord in head | Absent in forebrain |
| Notochord in spinal region | Absent at posterior neuropore during closure |
| Level of Shh expression | Gradient; lowest in posterior neuropore |
| Medial Hinge Point | Absent in posterior neuropore |
| Dorsolateral Hinge Point | Absent in upper spinal region |
| Convex mesenchymal expansion | Midbrain only |
| Grhl2, Grhl3 expression | Cranial, caudal and upper spine differ |
| Ephrins and Ephrin receptors | Different members of gene family regionally |
| Hox gene expression | Absent in midbrain and forebrain regions |
| Closure initiation site spacing | Caudal closure furthest from an initiation site |
| “Zipping” vs. simultaneous closure | Regional differences in axial bending |
| Meeting of neural folds in midline | Contact of neuroepithelium vs. surface ectoderm |
| Ruffles versus filopodia | Forebrain, midbrain, hindbrain, spine differ |
| Apical actomyosin contractility | Required for cranial closure, not spinal |
| Neural crest emigration | Spinal after closure; cranial before closure |
| Apoptosis | Required for cranial closure, not caudal |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Juriloff, D.M.; Harris, M.J. Insights into the Etiology of Mammalian Neural Tube Closure Defects from Developmental, Genetic and Evolutionary Studies. J. Dev. Biol. 2018, 6, 22. https://doi.org/10.3390/jdb6030022
AMA Style
Juriloff DM, Harris MJ. Insights into the Etiology of Mammalian Neural Tube Closure Defects from Developmental, Genetic and Evolutionary Studies. Journal of Developmental Biology. 2018; 6(3):22. https://doi.org/10.3390/jdb6030022
Chicago/Turabian StyleJuriloff, Diana M., and Muriel J. Harris. 2018. "Insights into the Etiology of Mammalian Neural Tube Closure Defects from Developmental, Genetic and Evolutionary Studies" Journal of Developmental Biology 6, no. 3: 22. https://doi.org/10.3390/jdb6030022
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.