Prenatal Neuropathologies in Autism Spectrum Disorder and Intellectual Disability: The Gestation of a Comprehensive Zebrafish Model

Department of Biology, University of Miami, Coral Gables, FL 33315, USA
J. Dev. Biol. 2018, 6(4), 29; https://doi.org/10.3390/jdb6040029
Submission received: 12 September 2018
/
Revised: 20 November 2018
/
Accepted: 27 November 2018
/
Published: 30 November 2018
(This article belongs to the Special Issue Zebrafish - A Model System for Developmental Biology Study)
Abstract
:Autism spectrum disorder (ASD) and intellectual disability (ID) are neurodevelopmental disorders with overlapping diagnostic behaviors and risk factors. These include embryonic exposure to teratogens and mutations in genes that have important functions prenatally. Animal models, including rodents and zebrafish, have been essential in delineating mechanisms of neuropathology and identifying developmental critical periods, when those mechanisms are most sensitive to disruption. This review focuses on how the developmentally accessible zebrafish is contributing to our understanding of prenatal pathologies that set the stage for later ASD-ID behavioral deficits. We discuss the known factors that contribute prenatally to ASD-ID and the recent use of zebrafish to model deficits in brain morphogenesis and circuit development. We conclude by suggesting that a future challenge in zebrafish ASD-ID modeling will be to bridge prenatal anatomical and physiological pathologies to behavioral deficits later in life.
1. Introduction
Autism spectrum disorder (ASD) and intellectual disability (ID) share diagnostic features, such as deficits in social behavior, and problems with communication and stereotypies [1,2]. As such, individuals with ASD are commonly diagnosed with ID (50–70%) and vis-versa (28–70%) [1,3,4]. These common co-diagnoses are likely a product of environmental and genetic factors that converge on similar molecular pathways [1,5]. Due to the relative diagnostic age (Median ASD diagnosis = 4.5 years; ID = 1–18 years) and enrichment of synaptic gene mutations ASD-ID, animal models initially focused on postnatal time periods [6,7,8,9]. However, studies have recently shown that disruptions in fetal development can manifest as childhood behavioral deficits [10,11]. These studies include zebrafish models that are being used to pinpoint critical periods and to provide a more complete picture of the mechanisms that underlie symptoms [12,13].
Zebrafish are increasingly used as a prenatal animal model for neurodevelopmental disorders, due to several anatomical and behavioral characteristics. First, zebrafish are small and transparent during early life stages which facilitates visualization of the nervous system, both anatomically and physiologically [14,15,16,17]. Therefore, developmental disruptions that might impact circuits can be investigated morphologically using transgenics to label specific neuronal populations [18,19], or physiologically using genetically encoded calcium indicators to assay neuronal activity [20,21,22]. Stereotyped swimming behaviors also emerge embryonically and rapidly transition to evasive and predatory behavior [23,24]. Disruptions in critical periods of development can therefore be identified by analyzing these behavioral milestones, such as embryonic tail coiling [25] and larval swimming [26]. Finally, one week old larval zebrafish have a small brain that simplifies studying how circuit-level disruptions affect behaviors at the whole-brain level [22]. Together these attributes produce a cellular- to circuit-level model for establishing functional relationships between prenatal developmental deficits and behavioral phenotypes.
In this paper, we look at how zebrafish are used to study mechanisms by which causal genetic mutations for ASD-ID impact embryonic development. We review recent studies of prenatal factors affecting ASD-ID, that detail critical time periods and the cellular processes responsible for later behavioral deficits. Then, we look at zebrafish proof-of-concept ASD-ID models that investigated embryonic and larval phenotypes related to ASD-ID. Finally, we conclude by discussing how recent advances in methodology are now being employed to investigate how developmental changes in zebrafish ASD-ID models contribute to behavioral phenotypes.
2. What Prenatal Factors Contribute to ASD-ID
The relationship between prenatal development and symptoms of ASD-ID was first established by investigating maternal teratogen exposure and gestationally derived deficits in ASD-ID syndromes. Teratogens are agents, chemical or infectious, that cause malformations to an embryo or fetus [27]. Many teratogenic effects depend on the time of exposure and can be traced back via hospital records or estimated by linking developmental milestones to somatic defects [27,28]. In addition to teratogens, many genes are expressed prenatally and several well-studied ASD-ID syndromes, such as Tuberous Sclerosis, are known to include gestationally derived malformations [10]. Recent research has expanded these prenatal factors to include maternal cytokines, genetic networks and non-coding genetic contributions [29,30,31,32]. However many individuals with ASD-ID have a non-etiological diagnosis, with neuropathological roots that may stem from prenatal time periods. In fact, some anatomical features arising from prenatal deficits have been described in adults with ASD-ID, including ectopic superior olivary nucleus neurons and loss of cerebellar Purkinje Cells with intact inferior olivary nuclei [33,34,35,36]. These features highlight the need for prenatal animal models to determine whether environmental and genetic risk factors for ASD-ID influence later behavioral deficits.
2.1. Environmental Risk Factors
Prenatal critical periods for ASD-ID were initially identified by unintentional population scale exposures to teratogens, that were initially prescribed as medications during pregnancy [37,38,39]. These studies showed that children exposed to teratogens in utero, such as thalidomide or valproic acid (VPA), developed symptoms of ASD-ID later in life [27]. To narrow down critical periods, researchers matched somatic defects with an accompanying developmental window, such as neural tube defects with neural tube closure [27]. For example, some children of mothers exposed to thalidomide developed autistic symptoms and cranial nerve defects consistent with exposure around 20 to 24 days post conception [27,28]. Despite these temporal signatures exposure can occur throughout the entire pregnancy, making it difficult to ascribe prenatal disruptions to later behavioral deficits. To identify whether these critical periods affect post-natal behavior, animal models have been screened for behavioral deficits, following exposure to teratogens at different time intervals. For example, following exposure to VPA during embryogenesis (E10–E13), rats exhibited learning and social deficits as adults [40,41]. Several zebrafish labs have also modeled the teratogenic effects of thalidomide [42] and VPA [43]. A recent study exposed zebrafish to VPA from gastrulation to neural tube formation, mimicking the window of exposure used in mouse VPA models [44]. VPA exposed larval fish showed decreases in histaminergic, noradrenergic and dopaminergic systems; accompanied by changes to larval sensory behavior and adult social behavior [44]. These models highlight the usefulness of testing teratogenic hypotheses from clinical data, to determine critical embryonic time periods that underlie later ASD-ID symptoms.
Another environmental factor that can increase the risk of ASD-ID are maternal immune responses [32,37,45]. In the 1970’s, Chess and colleagues found that mothers exposed to rubella during pregnancy had children with a high incidence of ASD-ID [37]. Recent studies have identified several immune responses, such as higher concentrations of intergerons in ASD-ID versus the general population, or ASD alone [32,45]. Additionally, researchers have shown that inflammatory molecules influence fetal gene expression, including dysregulation of ASD-ID genes associated with mTOR and EIF4E dependent signaling [30]. A recent ASD-ID zebrafish model looked at larval immunological function to determine whether immune responses were affected in a Methyl-CpG-Binding Protein 2 (MECP2) null background [46]. MECP2 mutations cause Rett Syndrome, which includes symptoms associated with inflammation, such as gastrointestinal disturbances and oxidative stress [47]. Mecp2 mutants exhibited decreased expression of tumor necrosis factor a (tnfa) during early embryogenesis and increased expression of interleukin-1b and interleukin-10 throughout larval stages [46]. Inflammatory molecules from these genes are needed to stimulate the bodies response to a disturbance and promote tissue repair [46]. Therefore, these changes in gene expression may be a response to tissue disturbances or oxidative stress, including a dysregulated inflammatory response of the gastrointestinal tract. These studies provide evidence for an interaction between the environment, inflammation and genetic risk factors in ASD-ID.
2.2. Genetic Risk Factors
Syndromes associated with ASD-ID, including Rett, Fragile X Syndrome (FXS) and Tuberous Sclerosis (TSC) identified the first genetic mutations of large-effect to cause ASD-ID [48]. Causal mutations for all of these conditions occur in genes normally expressed during prenatal periods. The Fragile X Mental Retardation (FMRP) gene is expressed throughout development and produces many alternatively spliced variants. Because different protein isoforms are expressed at different times and in different cell types [49,50], phenotypes vary depending on the nature and position of FMRP mutations. For example, while deletion of FMRP in humans and mice is linked to cortical defects, a common FMRP repeat mutation in FXS does not cause obvious gross anatomical deficits [50]. None-the-less, pluripotent stem-cells derived from individuals with FXS exhibit developmental delay in neuronal differentiation during embryogenesis [51]. Likewise TSC, caused by mutations in TSC1/2, is a ASD-ID syndrome and characterized by large benign cortical tubers and giant cells [1,7]. Individuals with TSC have been shown to have gross anatomical defects during mid-fetal development, including focal lesions [52,53] and giant cells [54]. Finally, recent ASD-ID mutational models have functionally linked deficits in prenatal cellular processes, such as neurogenesis, with postnatal ASD-ID behavioral deficits [11,55,56]. These embryonically expressed single gene mutations point to prenatal time periods as being critical to symptom development in ASD-ID.
In addition to these syndromic causes of ASD-ID, the vast majority of individuals have non-syndromic rare genetic variants, or are diagnosed with idiopathic ASD-ID. Fortunately, more high-confidence ASD-ID genes are being identified, as a result of world-wide consortia, and whole-genome and whole-exome sequencing [57,58,59,60]. These studies have shown that ASD-ID gene mutations are heterogenous and rare, implicating hundreds of ASD-ID associated genes. This recent increase in gene discovery, along with transcriptomic and proteomic analysis, has enriched our understanding of molecular networks associated with ASD-ID (recently reviewed in References [9,61,62]. The focus has thus shifted from defining genetic causes of ASD-ID to understanding how biologically relevant networks contribute to ASD-ID symptoms [63,64,65]. These biologically relevant networks are constructed by analyzing gene expression profiles, including gene ontology terms and protein-protein interactions [31,66,67]. These integrated models continue to identify prenatal ASD-ID clusters with greater temporal and spatial specificity, highlighting commonly impacted structures, like the fetal amygdala and mid-fetal cortical projection neurons [31] and molecular pathways, such as Notch and Wnt signaling [66]. In addition to rare genetic variants, common genetic variation of small-effect size also contribute to the risk and development of ASD-ID [61,68]. Although a single variant confers a small increase in ASD-ID risk, cumulative variation across the genome could account for large contributions (15–50%) towards the genetic etiology of ASD-ID [68]. These common genetic variants may also impact the penetrance of rare genetic variants, influencing the development and severity of ASD-ID phenotypes [69]. The ASD-ID genetic landscape continues to expand, with genetic variants and biological networks explaining a large percentage of the ASD-ID etiology.
Maternal genetics can also contribute to symptoms of ASD-ID via maternal effects. Maternal effects are maternally derived material that can contribute to the development of an offspring [70,71]. Maternal effects include embryonic translation of maternally loaded mRNA transcripts, genetic imprinting and the influence of intrauterine factors [70]. While maternally derived mechanisms including 15q duplications and imprinting in Angelman syndrome are well known, recent studies have discovered novel maternal effects. For example, a recent genome-wide association study linked a parent-of-origin effect with the transfer of a maternal ASD-ID gene SH3 and multiple ankyrin repeat domain protein 3 (SHANK3) variant, suggesting that this parent-of-origin variant increased the likelihood of developing ASD [72]. Finally, maternally loaded genetic variants can also affect developing oocytes. Following long-term storage a Drosophila FXS model exhibited dysregulated neuronal development, due to translational deficiencies of transcripts encoding large proteins [73]. This finding supports previous research that identified deficits in translation of transcripts encoding large proteins as a mechanism plaquing local translation at synapses [73,74]. These studies indicate that mutations in genes linked to ASD-ID can influence neuropathology even before the embryo begins to develop.
3. Comparative Neurodevelopment and Critical Periods: Can Embryonic and Larval Periods in Zebrafish be Compared to Prenatal Development in Humans?
The use of animal models to study human neurodevelopmental disorders requires the ability to translate critical periods across species. However, this can be problematic due to varying rates of development and dramatically different life histories. For instance, the human prenatal period is defined by prolonged in utero gestation and infants are born dependent on parental care [75,76]. In contrast, zebrafish develop externally in a protective chorion and hatch after a few days as fully independent larvae [14]. Moreover, morphogenesis of the zebrafish brain is complete in three days, compared to twenty one days in mice and eight weeks in humans [14,77]. Therefore, internal neurodevelopmental milestones should be used to define critical periods, from the completion of embryogenesis to the maturation of neural circuits essential for complex behaviors.
Currently, a comprehensive guide for comparing zebrafish and mammalian neuronal development is lacking. In mammals a group recently made a resource for comparing critical periods, by using internal and external developmental events. These events included the onset and offset of neurogenesis, cellular processes like myelination and the appearance of behaviors [76,78]. This comparative mammalian study revealed that the order of these events is largely conserved across species, despite differences in gestational time and alternative life histories (e.g., precocial versus altricial development). This order of events may also be conserved in zebrafish; however, a major knowledge gap exists concerning periods of extensive brain growth, between late larval and juvenile stages. Fortunately, research in zebrafish embryos have confirmed that major brain regions, gene expression patterns and cell types remain homologous with mammals (Table 1) [79,80,81]. This section focuses on comparing zebrafish and mammalian neural development and a strategy for translating prenatal regional or circuit specific critical periods.
3.1. Conserved Neuronal Development with Differences in Morphogenesis
Although neuronal development is largely conserved among zebrafish and mammals, several differences exist in morphogenesis and neurogenesis [14,81,96]. Zebrafish and mammals utilize different strategies during the formation of the neural tube: while zebrafish develop a neural thickening or keel that is hollowed by cavitation, mammals create a neural tube by enfolding the neural epithelial sheet [14,97]. Although these mechanisms differ, the underlying genes and signaling pathways responsible are largely conserved [97,98]. Morphogenesis of the telencephalon also differs: the mammalian neural tube evaginates and ventricles develop internally (medial), while the zebrafish telencephalon everts and ventricles develop on the surface of the brain [99,100]. This eversion process rearranges the topographic layout of functionally homologous brain regions, that results in a dorsal-medial amygdala and lateral hippocampus. Furthermore, there is still debate in the fish community over the severity of eversion and the final placement of regions within the forebrain [101,102]. Despite morphogenic differences, regions similar to the amygdala [103,104] and hippocampus [105,106] have been identified by conserved gene expression and function [102]. This provides a rationale for using zebrafish to study neuropsychiatric disorders in relation to emotive, social and memory-based behaviors [107,108].
3.2. Integrated Critical Periods: Gene Expression, Morphogenesis and Function
Currently zebrafish researchers must utilize molecular markers, cellular processes and circuit function to identify translatable critical periods. Some brain regions such as the cerebellum maintain remarkable genetic, cytoarchitectural and functional homology [80,109,110,111]. As an example of regional developmental staging, milestones of cerebellar development can be used to translate developmentally similar critical periods between zebrafish and mammals (Table 2). First the primordial cerebellum is defined by the expression of engrailed1/2 in rhombomere 1 of zebrafish (1–2 dpf), mouse (10–12 dpf) and humans (28 dpf) [109,112]. Cerebellar progenitor pools are then specified and can be identified by their expression of the transcription factors Math1/Atoh1a and Ptf1a [109]. Math1/Atoh1a labels the upper rhombic lip that will give rise to excitatory granule cells, while Ptf1a labels the ventral cerebellar proliferation zone that gives rise to inhibitory purkinje cells [113,114]. Following neuronal proliferation, the granule, purkinje and deep cerebellar neurons (eurydendroid cells in fish) migrate to assemble a trilaminar structure [113,114]. Critical periods can also be identified as cerebellar regions become compartmentalized for specific functions, similarly to mammals [115,116]. For example, the caudal purkinje cell layer in zebrafish has conserved efferent projections and functions with the dorsal cerebellar vermis in mammals [111,117]. The caudal vermis has indeed been implicated in ASD-ID, including loss of purkinje cells and deficits in saccade adaptation [35,118]. This specific example shows that several criteria can be used to define regional critical periods, including the appearance of a structure, neurogenesis of specific cell types, formation of a functional circuit and emergence of function related to behavior (Table 2).
4. Zebrafish ASD-ID Models: Critical Periods and Developmental Mechanisms
Recently there have been several proof-of-concept studies that have used zebrafish to model inherited neuropsychological disorders [108,121,122,123,124,125,126,127,128]. The discovery of high-confidence ASD-ID genes was initially slow, due to genome-wide studies with small sample sizes and an ever evolving set of diagnostic criteria (Figure 1) [59,129,130]. Many of the original ASD-ID zebrafish models represent genetic variants that, with high-confidence, were deemed causal for ASD-ID (16p11.2 CNV, SHANK3, FMRP, MET, TSC1) [127,128,131,132,133,134]. Additionally, the majority of ASD-ID zebrafish models were made using morpholino knockdown technology, predating the widespread availability of gene editing techniques to generate site specific knockouts. Interpretations of knockdown phenotypes are limited due to off-targeting concerns and the transient nature of knockdown technologies, restricting analyses to early stages [135]. Comparisons between knockdowns and knockouts of the same gene are complicated by off-target effects induced by morpholinos and genetic compensation seen in mutants ([136] discussed in Section 5.2). Given the recent rate of discovery of causal genetic mutations [9,64], future studies should produce a rich understanding of critical developmental time periods and mechanisms. None-the-less, past ASD-ID zebrafish models illustrate the importance of neuronal critical periods and developmental check points; when initiation of gene expression, activity dependent growth and synaptic refinement are required to build behavioral circuits [137].
4.1. Disruptions in Morphogenesis and Circuit Development of the Embryonic Brain
Early in development the embryo must undergo major morphological changes to specify neural ectoderm, establish the divisions of the nervous system and grow a brain anteriorly. During gastrulation cells of the blastodisc migrate to engulf the yolk, while cells at the leading edge involute to establish the epiblast (ectoderm) and the hypoblast (endo-mesoderm) [163,164,165]. As gastrulation and neurulation progress, molecules such as noggin and chordin are expressed to establish anterior brain regions [166]. Due to embryonic accessibility, seminal work in zebrafish helped to further elucidate morphogenic mechanisms that lead to the specification and formation of the forebrain, midbrain and hindbrain [167,168,169]. These studies illustrate how transcription factors and signaling molecules are expressed to pattern the neural tube, including feedback loops to restrict brain regions and produce a myriad of neuronal subtypes necessary to build diverse neural circuits [79,81]. For instance, the mid-hindbrain boundary (MHB) is demarcated by midbrain Orthodenticle Homeobox 2 (Otx2) and hindbrain Gastrulation Brain Homeobox 2 (Gbx2) expression. This MHB is further maintained by the mid-hindbrain program consisting of Fibroblast growth factor 8 (Fgf8), Wingless (Wnt1), Engrailed 1/2 (En1/2) and Paired Box (Pax) genes [81]. These genes form a complex network of regional signaling that differentially restrict gene expression and promote the generation of the midbrain optic tectum and the hindbrain cerebellum [81]. Many early studies focused on genetic mutants that exhibited the loss of a brain region, such as no isthmus (noi) and acerebellar (ace), that lacked the mid-hindbrain boundary and cerebellum, respectively [170]. These studies exemplify the importance of tightly controlled cellular signaling to establish boundaries and provide lineage restricted differential growth of brain regions.
4.1.1. The Dysregulation of Morphogenic Genes and Conserved Signaling Pathways Influences Brain Size
Some ASD-ID genes are expressed during embryogenesis, when loss-of-function has the potential to affect the spatial and temporal growth of neuronal tissue [12,72,73]. These changes in growth can be caused by dysregulated expression of genes involved in conserved signaling pathways, many of which are responsible for delineating the boundaries of tissues and brain regions. The morphogenic gene chordin encodes a BMP antagonist necessary for neurulation, that is instrumental in determining the size of anterior brain regions [171]. Zebrafish ASD-ID gene models have shown disrupted chordin expression during gastrulation that greatly influences the size of brain regions. For instance, knocking-down or knocking-out the ASD gene chromodomain helicase DNA binding protein 8 (chd8) in zebrafish causes an expanded expression of chordin and the forebrain marker otx2 [160]. These changes in expression increase the tissue that will give rise to the forebrain, that the authors suggest may underlie macrocephaly and increased brain size seen in a subset of individuals carrying CHD8 mutations [160]. In contrast to chordin expansion seen in chd8 models, knocking-down Delta Catenin-2 (CTNND2), another gene linked to ASD-ID, causes a shortening and widening of chordin expression during epiboly and a reduction in presumptive forebrain tissue [172]. Direct interactions between CHD8 and CTNND2 with Wnt signaling components may therefore explain changes in expression of chordin and other brain region specific markers [173]. For instance, CHD8 indirectly influences embryonic cortical progenitors through chromatin remodeling, affecting the expression of several Wnt signaling genes [55]. Additionally, inactivation of beta catenin is known to cause dlx2 upregulation and dlx2 expression was increased in the prethalamus of chd8 morphants [160,174]. Furthermore, upregulated dlx2 expression coincides with an enlarged midbrain that, in chd8 morphants, is likely due to an earlier increase in neuronal proliferation [175]. Finally, Ctnnd1 also interacts directly with beta-catenin and ctnnd1 morphants have reduced axin expression that may underlie the changes seen in chordin expression [57]. These studies demonstrate how gene mutations can affect gastrulation and neurulation through convergent developmental mechanisms, producing diametrically opposite phenotypes.
Zebrafish knockdown and knockout models of autism susceptibility candidate 2 (auts2) and shank3 also suggest ASD-ID genes play essential roles in brain morphogenesis. Auts2 morphants are microcephalic, have increased apoptosis and a decreased number of neurons in the forebrain [158,176]. Morphant and mutant models of shank3 are also microcephalic and appear developmentally delayed [126,162]. Shank3 microcephaly is likely due to increased cell death or decreased neuronal proliferation. Mid-hindbrain apoptosis was increased in shank3a morphants, while shank3b homozygous mutants exhibited brain-wide decreases in HuC:RFP fluorescence [126,162]. Furthermore, olfactory placodal neurons derived from humans with SHANK3 microdeletions exhibited developmental phenotypes [177]. These young post-mitotic neurons had smaller cell bodies, more extensively branched neurites and motility deficits, that likely impact early morphogenesis [177]. While these studies provide evidence that ASD-ID genes play important roles during brain morphogenesis, more research is needed to identify the molecular mechanisms underlying these early developmental phenotypes.
Zebrafish knockdown and knockout strategies have also been used to model the ASD associated 16p11.2 deletion syndrome. These 16p11.2 models have identified several genes that may contribute to changes in brain size. With 29 core genes affected, a 16p11.2 knockdown screen identified multiple genes that may contribute to brain growth [127]. A follow up study used genetic knockouts to further investigate developmental phenotypes and found that homozygote fam57ba mutations contribute specifically to macrocephaly [178]. In contrast, two 16p11.2 models have focused on potassium channel tetramerization domain containing 13 (kctd13), with morphant and mutant models producing different results [134,179]. A knockdown and overexpression study found that Kctd13 caused brain morphogenesis phenotypes in a dosage-dependent manner [134]. Kctd13 morphants exhibited increased proliferation and macrocephaly, while overexpression of kctd13 mRNA caused increased apoptosis, decreased proliferation and microcephaly [134]. In contrast, a more recent kctd13 gene deletion model found no phenotypes associated with head size or neuronal proliferation [179]. These researchers suggest that the discordance in knockdown and knockout phenotypes could either be due to off-target effects that enhance phenotypes in morphants or genetic compensation that masks phenotypes in kctd13 mutants [179]. These studies highlight the challenges in comparing mutant and morphant phenotypes (see Section 5.2).
4.1.2. Deficits in Neurogenesis Negatively Impact Axonogenesis and Circuit Connectivity
During neurogenesis primary neurons extend axons throughout the nervous system that act as a foundation for future circuits. Primary neurons are born within the first 24 h after fertilization, repeat serially and have large cell bodies with long axons [180,181]. Because later-born neurons navigate to their targets along the primary neuron axons, changes in primary neuron axonogenesis can have a significant impact on the function of mature circuits. As these axons grow, synchronized developmental steps establish a delicate balance of circuit components, eventually supporting a flexible repertoire of behaviors. These steps include the differential expression of signaling pathway genes that guide them to correct targets, activity dependent neurite growth and synaptogenesis, and both neuronal and synaptic refinement [137,182,183]. These events are especially important for maintaining balance of excitatory and inhibitory (E/I) inputs, whose homeostasis is necessary to coordinate complex behaviors [184]. Although circuits are resilient, disruptions to E/I balance are common in ASD-ID, underlying comorbid symptoms such as epilepsy and hypotonia [184,185].
ASD-ID mutations have been shown to impact axonogenesis, including several zebrafish models investigating embryonic axonogenesis. The X-linked intellectual disability gene FGF13 was found to play a role in stabilizing microtubules to control proper extension of growth cones [186]. When FGF13 is mutated, growth cones overextend past their targets, which likely explains defects in FGF13 null cortical layering. This axonogenic deficit may also underlie learning and memory deficits seen in mice carrying Fgf13 LOF mutations [186]. Several zebrafish ASD-ID gene models show similar deficits, for example, the Disrupted In Schizophrenia 1 (DISC1) gene is a negative regulator of Wnt signaling that influences cellular migration, axonal transport and axonogenesis [187]. Both mutant and morphant zebrafish disc1 models exhibit axonogenesis phenotypes, including the absence of supraoptic tracts and deficits in rhombomeric axon development [122,187]. Importantly, these zdisc1 morphant phenotypes could be rescued by full-length human DISC1 mRNA but not by mRNA encoding Bipolar/Schizophrenia causing DISC1 variants [122]. Axon tract phenotypes have also phenocopied anatomical deficits seen in ASD-ID patients. The RAC1 pathway has been implicated as a common molecular pathway affecting individuals with ASD-ID and RAC1 gene mutations have been implicated in several developmental disorders [188]. Cerebellar hypoplasia is common in individuals with RAC1 mutations, while over-expression of patient RAC1 mutations in zebrafish caused disruptions in cerebellar axonogenesis [188]. As mentioned previously, a recent study knocking-down 16p11.2 genes found that 80% of genes tested showed deficits in axonogenesis, including reduced fasciculation, disorganized morphology and supernumerary axon tracts that could be rescued by the corresponding human mRNA [127]. These results suggest that several 16p11.2 CNV genes are necessary for early brain development and contribute to neurological phenotypes. Furthermore, because axon tracts are shaped by environmental cues, disruptions to morphogenesis and axonogenesis are likely mechanistically linked.
Developing circuits can also be impacted by disruptions in the balance of excitatory and inhibitory (E/I) inputs [184]. Evidence for increased or decreased excitatory (glutamatergic) and inhibitory (GABAergic) neurons have been reported in different zebrafish ASD-ID gene models. Recent zebrafish ASD-ID studies have used markers that specify E/I neuronal populations to assess changes in the number and distribution of E/I components. The ASD-ID genes SHANK3 and SYNGAP1 encode post-synaptic density proteins, that are essential for maintaining synaptic strength of excitatory glutamatergic neurons via dendritic spine growth and AMPA receptor expression [189,190]. Morphant models of shank3a and syngap1b exhibited severe behavioral deficits, including seizure-like bouts, unproductive body undulations and decreased sensitivity to touch [126]. Shank3a and syngap1b morphants showed broad decreases in inhibitory GABAergic neurons, suggesting a shift towards an increased ratio of excitatory neurons [126]. A zebrafish mutant loss-of-function Contactin Associated Protein 2 (CNTNAP2) ASD-ID model also quantified E/I balance [125]. In contrast to SHANK3 and SYNGAP1, CNTNAP2 is expressed in inhibitory GABAergic neurons, localizes voltage-gated potassium channels and plays a role in migration of GABAergic neurons [125]. Cntnap2ab mutant zebrafish exhibited a decrease in forebrain GABAergic neurons, night-time hyperactivity and increased seizure susceptibility [125]. To determine the possible neurological basis for these behavioral deficits, a drug screen was performed confirming that cntnap2ab mutants were sensitive to GABAergic agonists. Surprisingly, NMDA receptor antagonists in wild-type larvae strongly correlated with mutant phenotypes suggesting that cntnap2ab mutations also affect behaviorally relevant excitatory components. This combination of systems level neuronal deficits, behavioral phenotypes and drug screening provide a compelling model for future ASD-ID studies.
Future studies investing changes in E/I balance will require the identification of ASD-ID relevant neuronal subtypes in zebrafish. Many ASD-ID genes are expressed in glutamatergic excitatory or inhibitory GABAergic neurons, including Neurexin (NRX) and SHANK3, or Sodium Voltage-Gated Channels Alpha Subunit 1 (SCN1A) and CNTNAP2, respectively [185,191]. Several of these genes have been selectively knocked-out in specific neuronal subtypes to demonstrate that genetic mutations in those neuronal subtypes are sufficient to cause ASD-ID behavioral deficits [185,192]. For example, knocking-out Scn1a in forebrain GABAergic neurons recapitulated several Dravet syndrome phenotypes, including seizure susceptibility [192]. These phenotypes are likely due to Scn1a loss-of-function in fast-spiking Parvalbumin positive cell types [192]. Zebrafish scn1lLab mutants were recently used to discover novel anti-convulsant molecules. This study provided evidence for a GABAergic component, however cellular level and subtype specific neuropathology was not investigated [193]. Identifying similar Parvalbumin positive GABAergic neurons in zebrafish forebrain could provide a better understanding of whether scn1Lab mutations affect the same cell types, while determining how those neurons relate to seizure susceptibility and deficits in behavior. Studies looking at peptidergic cell types in the hypothalamus [194,195] and dopaminergic neurons [196,197] have been well characterized by specific molecular markers. Further studies are needed to define other neuronal subtypes, including a thorough molecular census of glutamatergic and GABAergic sub-populations, to further validate zebrafish ASD-ID models.
5. Current Limitations for Using Zebrafish to Study Human Disease and Disorder Genes
Although zebrafish provide many advantages for modeling human genetic disorders, the retention of many duplicate genes after a whole genome duplication and the differences in morphant and mutant phenotypes provide challenges for researchers. For example, duplicate genes in zebrafish require researchers to study two genes in parallel, due to the likelihood that subfunctionalization has split ancestral functions between duplicate pairs [198]. In addition due to off targeting and toxicity with morpholinos and genetic compensation in mutants, knockdown and knockout models of the same gene can present different phenotypes [135,199]. Fortunately these issues are not barriers to using zebrafish for disease models and can be tackled via careful consideration while planning projects.
5.1. The Teleost Whole Genome Duplication
A whole genome duplication occurred during the evolution of teleosts (zebrafish infraclass) and resulted in the retention of many disorder related genes in zebrafish [200]. When a duplicate is retained, the duplicate pair either share ancestral functions (functional redundancy), split functions (subfunctionalization), or develop new functions (neofunctionalization) [201]. In an effort to resolve the ancestral state of zebrafish genes, recent studies have compared genes in the spotted gar (diverged before the Teleostei duplication) with zebrafish, to determine which gene within a pair is most similar to the ancestral orthologue [198,202]. This method can also be combined with gene expression data (temporal and spatial patterns) and genetic manipulation, to determine whether a gene duplicate is relevant to a human disorder [202]. A recent study showed that a majority of duplicate pairs experience subfunctionalization, requiring double knockouts for disorder modeling [203]. Although only a minority of zebrafish duplicates have been retained (~13%), [203] developmental disorder duplicates have been retained at a much higher rate (~60%) [12]. Therefore, the majority of ASD-ID gene orthologues are likely duplicated and will require studying two genes. Although studying two genes becomes a daunting task, clear advantageous can be garnered from studying a duplicate pair. For example, subfunctionalization could provide a means for teasing out the relationship between phenotypes. One gene may retain a role in ASD-ID social behavior while the duplicate retains a role important for a comorbid phenotype, such as intestinal distress. While subfunctionalization may be advantageous for studying certain genotype-phenotype relationship, gene duplicates remain challenging to study, and researchers need to utilize improved methodology for evaluating ancestral functions when starting a project.
5.2. Gene Knockdowns Versus Knockouts
Genetic knockdown and knockout technologies have strengths and weaknesses when modeling human disorders [199]. Gene knockdown techniques rely on anti-sense morpholinos or short-hairpin RNA molecules, to either inhibit mRNA translation or disrupt pre-mRNA splicing [135]. Gene knockdown technologies have been used extensively to functionally validate human disease genes [204,205,206,207,208]. However, in some cases, morpholinos cause severe phenotypes due to off-target effects, activation of p53 cell death pathways and toxicity [135,209,210]. Alternatively, some morpholinos faithfully phenocopy stable mutant phenotypes that can be rescued by injecting mRNA encoding the targeted gene [170,211,212]. In these cases morpholinos can complement mutant studies, allowing loss-of-function phenotypes to be studied rapidly in multiple transgenic zebrafish lines. Because gene knockdowns are transient, phenotypes must be studied in the embryo [135,213]. Therefore, gene knockdowns could be useful for future embryonic models of ASD-ID and researchers should follow the guidelines for using morpholino knockdown technology. These guidelines detail the proper control experiments needed for morphant studies and suggest using morphants as a complement to genetic mutants [213].
More recently, CRISPR-Cas9, TALENS and zinc finger endonucleases have provided a powerful tool, not only to produce gene knockouts but to induce site-specific mutations and knock-ins [214,215]. In some cases, phenotypes are subtler than expected due to genetic compensation, though the pervasiveness of such compensation has yet to be determined [136,216]. None-the-less, targeted knockouts and tailored mutants provide a better representation of the genetic state in human disorders, in comparison to morpholinos [209]. Additionally, when mutagenesis efficiencies are high, phenotypes can be assessed in embryos injected with endonucleases [217,218]. This provides the same expediency and versatility as morpholinos, but without the drawbacks of toxicity and transient phenotypes [217,218]. Therefore when phenotypes are present, knockout models should be used as the primary model for studying genetic causes of human disorders.
6. The Integrated Model: Utilizing Emerging Technology to Integrate Anatomy, Physiology and Behavior
These initial ASD-ID developmental models provide windows into the anatomical changes, mechanisms and critical periods that relate to prenatal neuropathology. However, zebrafish neuropsychiatric models can reach their full potential by using an integrative methodological approach. Future studies can bridge the gap between anatomy, physiology and behavior by combining systems level developmental changes (e.g., decreased proliferation), with specificity to neuronal subclasses (e.g., decreased dopaminergic neurons) and behavior (e.g., decreased dopaminergic drive affects motivated behavior). Ideally the next generation of models will utilize innovative methods including, brain atlases providing molecular marker libraries to interrogate anatomical and physiological causes of behavioral phenotypes, and small molecule screens for drug discovery.
6.1. Characteristics of Behaviors during Zebrafish Embryonic and Larval Development
Zebrafish are a powerful prenatal model because precocious embryonic and larval behavior provide an early indicator for disruptions of neural circuits. Prenatal disruptions of neural circuits may go undetected in humans, because ASD-ID diagnostic behaviors emerge slowly during post-natal development. Therefore, a delay between circuit disruption and the emergence of diagnostic behavior might result in prenatal etiological knowledge gaps. These gaps can easily be addressed by studying emerging behavior in zebrafish gene models of ASD-ID. During embryogenesis zebrafish tails begin to bend spontaneously (18 hpf), due to large calcium depolarizations flowing from head to tail [23,219]. Early synchronous behavior then transitions into fully functional swimming by 28 hpf, including touch evoked evasive movements [23]. Importantly, these behaviors utilize conserved midbrain, hindbrain and spinal cord circuits that drive ambulatory movement in humans [220,221]. Zebrafish begin to wire and integrate sensory-motor systems during larval development needed to avoid predators and capture prey [24]. These sensory provoked behaviors have extensive background literature, including functional studies detailing the relationship of sensory input to motor output [22,111,222,223]. This attribute could provide the most compelling translational research from zebrafish ASD-ID models, as sensory based deficits are common in ASD and were recently added to the diagnostic criteria [224,225]. Therefore, we argue that emergent behavioral phenotypes can be used in zebrafish ASD-ID models as a means to assay human prenatal developmental milestones.
Zebrafish also develop social and memory-based behaviors during larval development [24,226]. Social behaviors are important for modeling ASD-ID and several groups have established larval assays, that include shoaling and kin recognition [107,227,228]. However, these behaviors emerge at later stages (>9 DPF) and, although likely conserved with mammals, the neural circuitry remains largely unknown [229]. Studies in adult zebrafish have established assays and provided insight into oxytocinergic and dopaminergic circuits involved in social interactions [230]. Additionally, a novel social interaction paradigm was recently developed that identified forebrain cholinergic neurons essential for social orientating [231]. These emerging behaviors also evoke similar comparative questions discussed in Section 3, such as, what are the neurodevelopmental events and circuits that correspond to these complex behaviors? The zebrafish community will need to address these knowledge gaps in order to interpret circuit-level changes in larval social behavior.
6.2. Brain Atlases and High Resolution Morphometry
Research focusing on structural and functional mapping of the brain has recently made impressive progress. Subtle anatomical changes are common in ASD-ID and will require analyzing microstructure of brain circuits in zebrafish ASD-ID gene models [33,232,233]. Zebrafish may be well suited for studying these subtle anatomical changes due to their smaller brains. Several zebrafish groups have established community brain atlases and libraries using brain registration methods [19,234,235,236,237]. Confocal stacks of larval brains are orientated to a reference brain in a specified x, y, z axis using non-linear registration [238]. These registration methods provide sub-cellular accuracy and ensure that brain regions and neuronal populations overlap properly. Reference brains are then used to map libraries of molecular markers, to delineate neuronal subpopulations and circuits within the brain. Therefore, users can register confocal stacks of neuronal activity (e.g., genetically encoded calcium indicator, GECI) or transgenic backgrounds (e.g., vesicular glutamate transporter 2, vGluT2a:DsRed transgenic line [239]), to determine discrete sources of physiological and anatomical phenotypes. Furthermore, some of these analytical methods provide fine structural detail that could be missed analyzing gross anatomical regions by hand. For example, many studies looking at macro- and microcephaly measure changes to the entire brain or conspicuous regions (e.g., between the optic tecta [160]). However, innovations in mapping allow users to analyze specific somas or neuropil in fine detail. A new registration and brain segmentation method was recently used to provide a neuroanatomical map based on transgene expression profiles [235]. This method was able to determine significant differences in microstructure of larval brain regions exposed to VPA [235]. VPA exposed larvae displayed several anatomical deficits, including a decrease in excitatory components of the statoacoustic ganglion. Consistent with this structural alteration, VPA-exposed larvae showed decreased responsiveness following an acoustic startle stimulus. These resources have overcome challenges in analyzing circuits, allowing the community to link anatomical and physiological changes to behavioral phenotypes.
6.3. Whole-Brain Visual Outputs of Neuronal Activity
Larval zebrafish have a small brain (~100,000 neuron at 6 dpf) in comparison to adult humans (80,000,000,000), allowing for brain-wide neuronal activity monitoring using a neuronal activity indicator [22]. This includes immediate early genes (IEGs) and genetically encoded calcium indicators (GECIs). Although both are readily available for any lab to use, the former (IEGs) are more accessible to be captured on a typical confocal microscope and analyzed using a community brain atlas.
IEGs are genes and proteins whose expression increases exponentially following an action potential [240]. IEGs, such as cFOS, are commonly used as a proxy for neuronal firing. However, cFOS expression peaks around an hour following an action potential, making it impossible to use for transient stimulus provoked behavior [241]. In contrast, ERK protein is phosphorylated (pERK) within seconds of neuronal firing and phosphorylation peaks within five minutes [242]. pERK expression can provide information on activity changes following a temporally short stimulus, such as a light flash or vibration. Recently, pERK expression was coupled with a brain mapping technique to provide regional discrimination of changes in neuronal activity [234]. This MAP-Mapping technique takes normalized pERK expression in two groups and computes the difference in expression between comparable anatomical regions [234]. For example, the GABA potentiator pentylenetetrazol (PTZ) causes uninhibited excitatory activity throughout the brain that can be visualized in MAP-Map showing an increase in pERK throughout the brain in comparison to pERK expression with no PTZ. This method provides an unbiased way to survey activity changes in the entire brain during a disorder relevant behavior.
Calcium indicators become more fluorescent upon binding calcium and are used to study nervous system activity. Calcium indicator dyes have been utilized by zebrafish researchers for decades, because dyes can be injected into blastomeres and backfilled in the spinal cord to label neurons [243,244,245]. This system has advantages over Genetically Encoded Calcium Indicators (GECI), because mutant backgrounds do not have to be outcrossed to GECIs. However, GECI methods have been refined and are now sensitive enough to record whole-brain activity in awake behaving fish [22,246]. Labs have provided a wealth of information on zebrafish brain activity underlying specific behaviors during early larval behavior (6–7 dpf), including response to visual and auditory stimuli [17,20,22,25]. Typically, zebrafish need to be paralyzed or restrained in low melt agarose for recording, however two labs have recently built robotic light-sheet microscopes that can follow freely swimming zebrafish larvae in a dish, while recording GCaMP whole-brain imaging [247,248]. Future studies will be able to follow zebrafish ASD-ID models during complex sensory or social behavior, allowing researchers to define, in real-time, the neural circuit deficits underlying ASD-ID behavioral phenotypes. These methods will be revolutionary in determining whether conserved brain regions are utilized during larval social behavior and memory to model core ASD-ID deficits.
Finally, a new approach called Multi-MAP has combined fixed and live imaging techniques to assess modulations of brain activity by arousal pathways [249]. This approach uses GCaMP6s transgenic fish to record neuronal activity in live fish, followed by fixation and immunostaining to identify changes in different neuromodulatory neuronal sub-classes. A looming stimulus was used to measure reaction time and determine alertness [249]. Fast and slow alertness was found to be correlated with the firing of specific subtypes of neuromodulatory neurons in the locus coeruleus, tegmentum and hypothalamus [249]. Importantly, alertness correlated brain regions showed similar activity patterns in mice, suggesting that the relationship between alertness circuits and behavior are conserved [249]. Furthermore, these conserved circuits and behaviors are important for ASD-ID research because arousal pathways have been implicated in ASD-ID [191,250,251]. This method could be strengthened by functionally testing circuits using lines from a Gal4 or Cre-Lox library [19,252]. The multi-MAP approach leverages the power of live whole-brain activity monitoring and molecularly defined circuits to discover cellular elements underlying behavior.
6.4. Small Molecule Screening and Drug Discovery
Zebrafish larvae are small and take up small molecules through the water column, macking zebrafish attractive for medium-throughput drug screening. Small molecules can be dissolved into water or embryo media, and automated behavioral rigs can test 96 fish simultaneously [253]. Additionally, outbred zebrafish lines provide a heterogeneous animal model that reflects the genetic diversity seen in humans [254]. A potential drug discovery should therefore be attractive for preclinical trials when a phenotype-drug relationship is clearly reproducible. Seminal work in small molecule screens have discovered novel compounds that affect sleep [125,255], the photomotor response [256], seizures [193] and behavioral effects of neuropsychological drugs [257]. A drug screen recently correlated changes in behavior between known neuropsychiatric drugs and novel molecules [257]. By using the drug haloperidol, this method validated the therapeutic efficacy of structurally similar compounds [257]. Identifying alternative molecules to treat neuropsychiatric disorders is important, because many neuropsychiatric drugs target multiple neuromodulatory systems that results in unwanted side-effects. Using this method, zebrafish ASD-ID gene models could be screened for small molecules that recapitulate the therapeutic effect of a marketed drug, while lessoning the effect of unwanted side effects. This strategy could be important for currently prescribed ASD medications, such as risperidone and lithium, that cause severe side effects [258,259]. Finally, whole-brain activity methods could be combined with drug screens to determine relationships between drug influenced circuits and improvements in behavioral deficits.
7. Conclusions
Past proof-of-concept studies have established zebrafish as a powerful model for studying prenatal neuropathologies underlying ASD-ID. Prenatal risk factors for developing ASD-ID have been well-documented for over a half century, yet there is still little known about the earliest developmental origins of these disorders. In addition, geneticists are beginning to identify prenatally expressed genes that constitute risk factors for ASD-ID. The current body of zebrafish ASD-ID literature has provided a groundwork for studying broad mechanisms underlying ASD-ID neuropathology, such as brain morphogenesis and circuit development. However, the majority of these studies involved gene knockdowns and future genetic mutants will be needed to determine the link between ASD-ID gene mutations and developmental phenotypes. These mutant studies will also need to integrate the current technological leaps, linking anatomical phenotypes to physiological mechanisms underlying ASD-ID relevant behavior.
Funding
University of Miami’s Department of Biology Teaching Assistantship.
Acknowledgments
I would like to thank Julia Dallman for feedback and guidance during the writing and editing processes.
Conflicts of Interest
The author declares no conflict of interest.
References
- Srivastava, A.K.; Schwartz, C.E. Intellectual disability and autism spectrum disorders: Causal genes and molecular mechanisms. Neurosci. Biobehav. Rev. 2014, 46, 161–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Association, A.P. Intellectual Disability. In Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; DSM: Heerlen, The Netherlands, 2013. [Google Scholar] [Green Version]
- Fombonne, E. Epidemiological surveys of autism and other pervasive developmental disorders: An update. J. Autism. Dev. Disord. 2003, 33, 365–382. [Google Scholar] [CrossRef] [PubMed]
- Matson, J.L.; Shoemaker, M. Intellectual disability and its relationship to autism spectrum disorders. Res. Dev. Disabil. 2009, 30, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Kou, Y.; Betancur, C.; Xu, H.; Buxbaum, J.D.; Ma’ayan, A. Network- and attribute-based classifiers can prioritize genes and pathways for autism spectrum disorders and intellectual disability. Am. J. Med. Genet. C Semin. Med. Genet. 2012, 160C, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Baio, J.; Wiggins, L.; Christensen, D.L.; Maenner, M.J.; Daniels, J.; Warren, Z.; Kurzius-Spencer, M.; Zahorodny, W.; Rosenberg, C.R.; White, T.; et al. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2014. MMWR Surveillance Summer 2018, 67, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.M.; Geschwind, D.H. Autism genetics: Searching for specificity and convergence. Genome Biol. 2012, 13, 247. [Google Scholar] [CrossRef] [PubMed]
- Peca, J.; Feng, G. Cellular and synaptic network defects in autism. Curr. Opin. Neurobiol. 2012, 22, 866–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.A.; Penagarikano, O.; Belgard, T.G.; Swarup, V.; Geschwind, D.H. The emerging picture of autism spectrum disorder: Genetics and pathology. Annu. Rev. Pathol. 2015, 10, 111–144. [Google Scholar] [CrossRef] [PubMed]
- Ehninger, D.; Sano, Y.; de Vries, P.J.; Dies, K.; Franz, D.; Geschwind, D.H.; Kaur, M.; Lee, Y.S.; Li, W.; Lowe, J.K.; et al. Gestational immune activation and Tsc2 haploinsufficiency cooperate to disrupt fetal survival and may perturb social behavior in adult mice. Mol. Psychiatry 2012, 17, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Belinson, H.; Nakatani, J.; Babineau, B.A.; Birnbaum, R.Y.; Ellegood, J.; Bershteyn, M.; McEvilly, R.J.; Long, J.M.; Willert, K.; Klein, O.D.; et al. Prenatal beta-catenin/Brn2/Tbr2 transcriptional cascade regulates adult social and stereotypic behaviors. Mol. Psychiatry 2016, 21, 1417–1433. [Google Scholar] [CrossRef] [PubMed]
- Kozol, R.A.; Abrams, A.J.; James, D.M.; Buglo, E.; Yan, Q.; Dallman, J.E. Function Over Form: Modeling Groups of Inherited Neurological Conditions in Zebrafish. Front. Mol. Neurosci. 2016, 9, 55. [Google Scholar] [CrossRef] [PubMed]
- Sakai, C.; Ijaz, S.; Hoffman, E.J. Zebrafish Models of Neurodevelopmental Disorders: Past, Present, and Future. Front. Mol. Neurosci. 2018, 11, 294. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, C.B.; Ballard, W.W.; Kimmel, S.R.; Ullmann, B.; Schilling, T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995, 203, 253–310. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, C.B.; Hatta, K.; Metcalfe, W.K. Early axonal contacts during development of an identified dendrite in the brain of the zebrafish. Neuron 1990, 4, 535–545. [Google Scholar] [CrossRef]
- Higashijima, S.; Masino, M.A.; Mandel, G.; Fetcho, J.R. Imaging neuronal activity during zebrafish behavior with a genetically encoded calcium indicator. J. Neurophysiol. 2003, 90, 3986–3997. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, M.B.; Orger, M.B.; Robson, D.N.; Li, J.M.; Keller, P.J. Whole-brain functional imaging at cellular resolution using light-sheet microscopy. Nat. Methods 2013, 10, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Satou, C.; Kimura, Y.; Hirata, H.; Suster, M.L.; Kawakami, K.; Higashijima, S. Transgenic tools to characterize neuronal properties of discrete populations of zebrafish neurons. Development 2013, 140, 3927–3931. [Google Scholar] [CrossRef] [PubMed]
- Marquart, G.D.; Tabor, K.M.; Brown, M.; Strykowski, J.L.; Varshney, G.K.; LaFave, M.C.; Mueller, T.; Burgess, S.M.; Higashijima, S.; Burgess, H.A. A 3D Searchable Database of Transgenic Zebrafish Gal4 and Cre Lines for Functional Neuroanatomy Studies. Front. Neural Circuits 2015, 9, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akerboom, J.; Chen, T.W.; Wardill, T.J.; Tian, L.; Marvin, J.S.; Mutlu, S.; Calderon, N.C.; Esposti, F.; Borghuis, B.G.; Sun, X.R.; et al. Optimization of a GCaMP calcium indicator for neural activity imaging. J. Neurosci. 2012, 32, 13819–13840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fosque, B.F.; Sun, Y.; Dana, H.; Yang, C.T.; Ohyama, T.; Tadross, M.R.; Patel, R.; Zlatic, M.; Kim, D.S.; Ahrens, M.B.; et al. Neural circuits. Labeling of active neural circuits in vivo with designed calcium integrators. Science 2015, 347, 755–760. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, M.B.; Li, J.M.; Orger, M.B.; Robson, D.N.; Schier, A.F.; Engert, F.; Portugues, R. Brain-wide neuronal dynamics during motor adaptation in zebrafish. Nature 2012, 485, 471–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brustein, E.; Saint-Amant, L.; Buss, R.R.; Chong, M.; McDearmid, J.R.; Drapeau, P. Steps during the development of the zebrafish locomotor network. J. Physiol. Paris 2003, 97, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Fero, K.Y.T.; Burgess, H.A. The Behavioral Repertoire of Larval Zebrafish. In Zebrafish Models in Neurobehavioral Research; Humana Press: New York, NY, USA, 2011; pp. 249–291. [Google Scholar]
- Warp, E.; Agarwal, G.; Wyart, C.; Friedmann, D.; Oldfield, C.S.; Conner, A.; Del Bene, F.; Arrenberg, A.B.; Baier, H.; Isacoff, E.Y. Emergence of patterned activity in the developing zebrafish spinal cord. Curr. Biol. 2012, 22, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Orger, M.B.; de Polavieja, G.G. Zebrafish Behavior: Opportunities and Challenges. Annu. Rev. Neurosci. 2017, 40, 125–147. [Google Scholar] [CrossRef] [PubMed]
- Arndt, T.L.; Stodgell, C.J.; Rodier, P.M. The teratology of autism. Int. J. Dev. Neurosci. 2005, 23, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.T.; Stromland, K.; Ventura, L.; Johansson, M.; Bandim, J.M.; Gillberg, C. Autism associated with conditions characterized by developmental errors in early embryogenesis: A mini review. Int. J. Dev. Neurosci. 2005, 23, 201–219. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, R.; Jaffe, A.E.; Hyde, T.M.; Kleinman, J.E.; Weinberger, D.R. Prenatal expression patterns of genes associated with neuropsychiatric disorders. Am. J. Psychiatry 2014, 171, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, M.V.; Moon, H.M.; Su, J.; Palmer, T.D.; Courchesne, E.; Pramparo, T. Maternal immune activation dysregulation of the fetal brain transcriptome and relevance to the pathophysiology of autism spectrum disorder. Mol. Psychiatry 2018, 23, 1001–1013. [Google Scholar] [CrossRef] [PubMed]
- Willsey, A.J.; Sanders, S.J.; Li, M.; Dong, S.; Tebbenkamp, A.T.; Muhle, R.A.; Reilly, S.K.; Lin, L.; Fertuzinhos, S.; Miller, J.A.; et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 2013, 155, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.L.; Croen, L.A.; Yoshida, C.K.; Heuer, L.; Hansen, R.; Zerbo, O.; DeLorenze, G.N.; Kharrazi, M.; Yolken, R.; Ashwood, P.; et al. Autism with intellectual disability is associated with increased levels of maternal cytokines and chemokines during gestation. Mol. Psychiatry 2017, 22, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Bauman, M.L. Microscopic neuroanatomic abnormalities in autism. Pediatrics 1991, 87, 791–796. [Google Scholar] [PubMed]
- Bauman, M.L.; Kemper, T.L. Neuroanatomic observations of the brain in autism: A review and future directions. Int. J. Dev. Neurosci. 2005, 23, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, S.H.; Aldinger, K.A.; Ashwood, P.; Bauman, M.L.; Blaha, C.D.; Blatt, G.J.; Chauhan, A.; Chauhan, V.; Dager, S.R.; Dickson, P.E.; et al. Consensus paper: Pathological role of the cerebellum in autism. Cerebellum 2012, 11, 777–807. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, J.; Kuchna, I.; Nowicki, K.; Imaki, H.; Wegiel, J.; Marchi, E.; Ma, S.Y.; Chauhan, A.; Chauhan, V.; Bobrowicz, T.W.; et al. The neuropathology of autism: Defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. 2010, 119, 755–770. [Google Scholar] [CrossRef] [PubMed]
- Chess, S.; Fernandez, P. Neurologic damage and behavior disorder in rubella children. Am. Ann. Deaf 1980, 125, 998–1001. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.J.; Turnpenny, P.; Quinn, A.; Glover, S.; Lloyd, D.J.; Montgomery, T.; Dean, J.C. A clinical study of 57 children with fetal anticonvulsant syndromes. J. Med. Genet. 2000, 37, 489–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stromland, K.; Nordin, V.; Miller, M.; Akerstrom, B.; Gillberg, C. Autism in thalidomide embryopathy: A population study. Dev. Med. Child. Neurol. 1994, 36, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, C.; Fahnestock, M. The valproic acid-induced rodent model of autism. Exp. Neurol. 2018, 299, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Seung, H.; Kwon, K.J.; Ko, M.J.; Lee, E.J.; Oh, H.A.; Choi, C.S.; Kim, K.C.; Gonzales, E.L.; You, J.S.; et al. Subchronic treatment of donepezil rescues impaired social, hyperactive, and stereotypic behavior in valproic acid-induced animal model of autism. PLoS ONE 2014, 9, e104927. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a primary target of thalidomide teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lei, L.; Tian, L.; Hou, F.; Roper, C.; Ge, X.; Zhao, Y.; Chen, Y.; Dong, Q.; Tanguay, R.L.; et al. Developmental and behavioral alterations in zebrafish embryonically exposed to valproic acid (VPA): An aquatic model for autism. Neurotoxicol. Teratol. 2018, 66, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Baronio, D.; Puttonen, H.A.J.; Sundvik, M.; Semenova, S.; Lehtonen, E.; Panula, P. Embryonic exposure to valproic acid affects the histaminergic system and the social behaviour of adult zebrafish (Danio rerio). Br. J. Pharmacol. 2018, 175, 797–809. [Google Scholar] [CrossRef] [PubMed]
- Krakowiak, P.; Goines, P.E.; Tancredi, D.J.; Ashwood, P.; Hansen, R.L.; Hertz-Picciotto, I.; Van de Water, J. Neonatal Cytokine Profiles Associated With Autism Spectrum Disorder. Biol. Psychiatry 2017, 81, 442–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Vaart, M.; Svoboda, O.; Weijts, B.G.; Espin-Palazon, R.; Sapp, V.; Pietri, T.; Bagnat, M.; Muotri, A.R.; Traver, D. Mecp2 regulates tnfa during zebrafish embryonic development and acute inflammation. Dis. Model Mech. 2017, 10, 1439–1451. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Folstein, S.E.; Piven, J. Etiology of autism: Genetic influences. Pediatrics 1991, 87, 767–773. [Google Scholar] [PubMed]
- Tassone, F.; De Rubeis, S.; Carosi, C.; La Fata, G.; Serpa, G.; Raske, C.; Willemsen, R.; Hagerman, P.J.; Bagni, C. Differential usage of transcriptional start sites and polyadenylation sites in FMR1 premutation alleles. Nucleic Acids Res. 2011, 39, 6172–6185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, P.; Schoenfeld, B.P.; Bell, A.J.; Choi, C.H.; Bradley, M.P.; Hinchey, P.; Kollaros, M.; Park, J.H.; McBride, S.M.; Dockendorff, T.C. Short- and long-term memory are modulated by multiple isoforms of the fragile X mental retardation protein. J. Neurosci. 2010, 30, 6782–6792. [Google Scholar] [CrossRef] [PubMed]
- Boland, M.J.; Nazor, K.L.; Tran, H.T.; Szucs, A.; Lynch, C.L.; Paredes, R.; Tassone, F.; Sanna, P.P.; Hagerman, R.J.; Loring, J.F. Molecular analyses of neurogenic defects in a human pluripotent stem cell model of fragile X syndrome. Brain 2017, 140, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-H.; Pepkowitz, S.H.; Kerfoot, C.; de Rosa, M.J.; Poukens, V.; Wienecke, R.; de Clue, J.E.; Vinters, H.V. Tuberous sclerosis in a 20-week gestation fetus: Immunohistochemical study. Acta Neuropathol. 1997, 94, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Li, P.; Chiriboga, L.; Mizuguchi, M.; Yee, H.; Miller, D.C.; Greco, M.A. Tuberous sclerosis in a 19-week fetus: Immunohistochemical and molecular study of hamartin and tuberin. Pediatr. Dev. Pathol. 2002, 5, 448–464. [Google Scholar] [CrossRef] [PubMed]
- Bordarier, C.; Lellouch-Tubiana, A.; Robain, O. Cardiac rhabdomyoma and tuberous sclerosis in three fetuses: A neuropathological study. Brain Dev. 1994, 16, 467–471. [Google Scholar] [CrossRef]
- Durak, O.; Gao, F.; Kaeser-Woo, Y.J.; Rueda, R.; Martorell, A.J.; Nott, A.; Liu, C.Y.; Watson, L.A.; Tsai, L.-H. Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nat. Neurosci. 2016, 19, 1477–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durak, O.; de Anda, F.C.; Singh, K.K.; Leussis, M.P.; Petryshen, T.L.; Sklar, P.; Tsai, L.-H. Ankyrin-G regulates neurogenesis and Wnt signaling by altering the subcellular localization of beta-catenin. Mol. Psychiatry 2015, 20, 388–397. [Google Scholar] [CrossRef] [PubMed]
- O’Roak, B.J.; Vives, L.; Fu, W.; Egertson, J.D.; Stanaway, I.B.; Phelps, I.G.; Carvill, G.; Kumar, A.; Lee, C.; Ankenman, K.; et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 2012, 338, 1619–1622. [Google Scholar] [CrossRef] [PubMed]
- O’Roak, B.J.; Vives, L.; Girirajan, S.; Karakoc, E.; Krumm, N.; Coe, B.P.; Levy, R.; Ko, A.; Lee, C.; Smith, J.D.; et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012, 485, 246–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werling, D.M.; Brand, H.; An, J.Y.; Stone, M.R.; Zhu, L.; Glessner, J.T.; Collins, R.L.; Dong, S.; Layer, R.M.; Markenscoff-Papadimitriou, E.; et al. An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nat. Genet. 2018, 50, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Geschwind, D.H.; State, M.W. Gene hunting in autism spectrum disorder: On the path to precision medicine. Lancet Neurol. 2015, 14, 1109–1120. [Google Scholar] [CrossRef]
- De Rubeis, S.; Buxbaum, J.D. Genetics and genomics of autism spectrum disorder: Embracing complexity. Hum. Mol. Genet. 2015, 24, R24–R31. [Google Scholar] [CrossRef] [PubMed]
- Ionita-Laza, I.; Capanu, M.; De Rubeis, S.; McCallum, K.; Buxbaum, J.D. Identification of rare causal variants in sequence-based studies: Methods and applications to VPS13B, a gene involved in Cohen syndrome and autism. PLoS Genet. 2014, 10, e1004729. [Google Scholar] [CrossRef] [PubMed]
- Parikshak, N.N.; Luo, R.; Zhang, A.; Won, H.; Lowe, J.K.; Chandran, V.; Horvath, S.; Geschwind, D.H. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 2013, 155, 1008–1021. [Google Scholar] [CrossRef] [PubMed]
- Cristino, A.S.; Williams, S.M.; Hawi, Z.; An, J.Y.; Bellgrove, M.A.; Schwartz, C.E.; Costa Lda, F.; Claudianos, C. Neurodevelopmental and neuropsychiatric disorders represent an interconnected molecular system. Mol. Psychiatry 2014, 19, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Hormozdiari, F.; Penn, O.; Borenstein, E.; Eichler, E.E. The discovery of integrated gene networks for autism and related disorders. Genome Res. 2015, 25, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Voineagu, I.; Wang, X.; Johnston, P.; Lowe, J.K.; Tian, Y.; Horvath, S.; Mill, J.; Cantor, R.M.; Blencowe, B.J.; Geschwind, D.H. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011, 474, 380–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vorstman, J.A.S.; Parr, J.R.; Moreno-De-Luca, D.; Anney, R.J.L.; Nurnberger, J.I., Jr.; Hallmayer, J.F. Autism genetics: Opportunities and challenges for clinical translation. Nat. Rev. Genet. 2017, 18, 362–376. [Google Scholar] [CrossRef] [PubMed]
- Niemi, M.E.K.; Martin, H.C.; Rice, D.L.; Gallone, G.; Gordon, S.; Kelemen, M.; McAloney, K.; McRae, J.; Radford, E.J.; Yu, S.; et al. Common genetic variants contribute to risk of rare severe neurodevelopmental disorders. Nature 2018, 562, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.B.; Wade, M.J. What are maternal effects (and what are they not)? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 1107–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilmatre, A.; Sharp, A.J. Parent of origin effects. Clin. Genet. 2012, 81, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Connolly, S.; Anney, R.; Gallagher, L.; Heron, E.A. A genome-wide investigation into parent-of-origin effects in autism spectrum disorder identifies previously associated genes including SHANK3. Eur. J. Hum. Genet. 2017, 25, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Greenblatt, E.J.; Spradling, A.C. Fragile X mental retardation 1 gene enhances the translation of large autism-related proteins. Science 2018, 361, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Kindler, S.; Kreienkamp, H.J. Dendritic mRNA targeting and translation. Adv. Exp. Med. Biol. 2012, 970, 285–305. [Google Scholar] [PubMed]
- Rice, D.; Barone, S., Jr. Critical periods of vulnerability for the developing nervous system: Evidence from humans and animal models. Environ. Health Perspect. 2000, 108 (Suppl. 3), 511–533. [Google Scholar] [PubMed]
- Clancy, B.; Darlington, R.B.; Finlay, B.L. Translating developmental time across mammalian species. Neuroscience 2001, 105, 7–17. [Google Scholar] [CrossRef] [Green Version]
- Vaillancourt, C.; Lafond, J. Human embryogenesis: Overview. Methods Mol. Biol. 2009, 550, 3–7. [Google Scholar] [PubMed]
- Workman, A.D.; Charvet, C.J.; Clancy, B.; Darlington, R.B.; Finlay, B.L. Modeling transformations of neurodevelopmental sequences across mammalian species. J. Neurosci. 2013, 33, 7368–7383. [Google Scholar] [CrossRef] [PubMed]
- Moens, C.B.; Prince, V.E. Constructing the hindbrain: Insights from the zebrafish. Dev. Dyn. 2002, 224, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, M.; Hibi, M. Development and evolution of cerebellar neural circuits. Dev. Growth Differ. 2012, 54, 373–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wurst, W.; Bally-Cuif, L. Neural plate patterning: Upstream and downstream of the isthmic organizer. Nat. Rev. Neurosci. 2001, 2, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.W.; Ross, L.S.; Parrett, T.; Easter, S.S., Jr. The development of a simple scaffold of axon tracts in the brain of the embryonic zebrafish, Brachydanio rerio. Development 1990, 108, 121–145. [Google Scholar] [PubMed]
- Ross, L.S.; Parrett, T.; Easter, S.S., Jr. Axonogenesis and morphogenesis in the embryonic zebrafish brain. J. Neurosci. 1992, 12, 467–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golden, G.S. A review of the neuroembryology of monoamine systems. Brain Res. Bull. 1982, 9, 553–558. [Google Scholar] [CrossRef]
- Bonnin, A.; Levitt, P. Fetal, maternal, and placental sources of serotonin and new implications for developmental programming of the brain. Neuroscience 2011, 197, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, D.L.; Fetcho, J.R. Relationship of tyrosine hydroxylase and serotonin immunoreactivity to sensorimotor circuitry in larval zebrafish. J. Comp. Neurol. 2004, 480, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Wilson, S.W.; Cooke, S.; Chitnis, A.B.; Driever, W.; Rosenthal, A. Mutations in the zebrafish unmask shared regulatory pathways controlling the development of catecholaminergic neurons. Dev. Biol. 1999, 208, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Holzschuh, J.; Ryu, S.; Aberger, F.; Driever, W. Dopamine transporter expression distinguishes dopaminergic neurons from other catecholaminergic neurons in the developing zebrafish embryo. Mech. Dev. 2001, 101, 237–243. [Google Scholar] [CrossRef]
- Bae, Y.K.; Kani, S.; Shimizu, T.; Tanabe, K.; Nojima, H.; Kimura, Y.; Higashijima, S.; Hibi, M. Anatomy of zebrafish cerebellum and screen for mutations affecting its development. Dev. Biol. 2009, 330, 406–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chitnis, A.B.; Kuwada, J.Y. Axonogenesis in the brain of zebrafish embryos. J. Neurosci. 1990, 10, 1892–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, T. What is the Thalamus in Zebrafish? Front. Neurosci. 2012, 6, 64. [Google Scholar] [CrossRef] [PubMed]
- Laessing, U.; Stuermer, C.A. Spatiotemporal pattern of retinal ganglion cell differentiation revealed by the expression of neurolin in embryonic zebrafish. J. Neurobiol. 1996, 29, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Sakata, Y.; Olson, J.K.; Michel, W.C. Assessment of neuronal maturation and acquisition of functional competence in the developing zebrafish olfactory system. Methods Cell Sci. 2003, 25, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Mueller, T.; Vernier, P.; Wullimann, M.F. A phylotypic stage in vertebrate brain development: GABA cell patterns in zebrafish compared with mouse. J. Comp. Neurol. 2006, 494, 620–634. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, C.B.; Patterson, J.; Kimmel, R.O. The development and behavioral characteristics of the startle response in the zebra fish. Dev. Psychobiol. 1974, 7, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Lumsden, A.; Krumlauf, R. Patterning the vertebrate neuraxis. Science 1996, 274, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Strahle, U.; Scholpp, S. Neurogenesis in zebrafish—From embryo to adult. Neural Dev. 2013, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Strahle, U.; Blader, P. Early neurogenesis in the zebrafish embryo. FASEB J. 1994, 8, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Folgueira, M.; Bayley, P.; Navratilova, P.; Becker, T.S.; Wilson, S.W.; Clarke, J.D. Morphogenesis underlying the development of the everted teleost telencephalon. Neural Dev. 2012, 7, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, T.; Wullimann, M.F. An evolutionary interpretation of teleostean forebrain anatomy. Brain Behav. Evol. 2009, 74, 30–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieuwenhuys, R. The forebrain of actinopterygians revisited. Brain Behav. Evol. 2009, 73, 229–252. [Google Scholar] [CrossRef] [PubMed]
- Ganz, J.; Kroehne, V.; Freudenreich, D.; Machate, A.; Geffarth, M.; Braasch, I.; Kaslin, J.; Brand, M. Subdivisions of the adult zebrafish pallium based on molecular marker analysis. F1000Res 2014, 3, 308. [Google Scholar] [CrossRef] [PubMed]
- Portavella, M.; Torres, B.; Salas, C.; Papini, M.R. Lesions of the medial pallium, but not of the lateral pallium, disrupt spaced-trial avoidance learning in goldfish (Carassius auratus). Neurosci. Lett. 2004, 362, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Lal, P.; Tanabe, H.; Suster, M.L.; Ailani, D.; Kotani, Y.; Muto, A.; Itoh, M.; Iwasaki, M.; Wada, H.; Yaksi, E.; et al. Identification of a neuronal population in the telencephalon essential for fear conditioning in zebrafish. BMC Biol. 2018, 16, 45. [Google Scholar] [CrossRef] [PubMed]
- Broglio, C.; Gomez, A.; Duran, E.; Ocana, F.M.; Jimenez-Moya, F.; Rodriguez, F.; Salas, C. Hallmarks of a common forebrain vertebrate plan: Specialized pallial areas for spatial, temporal and emotional memory in actinopterygian fish. Brain Res. Bull. 2005, 66, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Broglio, C.; Rodriguez, F.; Gomez, A.; Arias, J.L.; Salas, C. Selective involvement of the goldfish lateral pallium in spatial memory. Behav. Brain Res. 2010, 210, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.M.; Ullmann, J.F.; Norton, W.H.; Parker, M.O.; Brennan, C.H.; Gerlai, R.; Kalueff, A.V. Molecular psychiatry of zebrafish. Mol. Psychiatry 2015, 20, 2–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norton, W.H. Toward developmental models of psychiatric disorders in zebrafish. Front. Neural Circuits 2013, 7, 79. [Google Scholar] [CrossRef] [PubMed]
- Wullimann, M.F.; Mueller, T.; Distel, M.; Babaryka, A.; Grothe, B.; Koster, R.W. The long adventurous journey of rhombic lip cells in jawed vertebrates: A comparative developmental analysis. Front. Neuroanat 2011, 5, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, M.; Yamaguchi, S.; Sakakibara, Y.; Hayashi, T.; Matsuda, K.; Hara, Y.; Tanegashima, C.; Shimizu, T.; Kuraku, S.; Hibi, M. Gene expression profiling of granule cells and Purkinje cells in the zebrafish cerebellum. J. Comp. Neurol. 2017, 525, 1558–1585. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Namikawa, K.; Babaryka, A.; Koster, R.W. Functional regionalization of the teleost cerebellum analyzed in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, 11846–11851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sillitoe, R.V.; Joyner, A.L. Morphology, molecular codes, and circuitry produce the three-dimensional complexity of the cerebellum. Annu. Rev. Cell Dev. Biol. 2007, 23, 549–577. [Google Scholar] [CrossRef] [PubMed]
- Kani, S.; Bae, Y.K.; Shimizu, T.; Tanabe, K.; Satou, C.; Parsons, M.J.; Scott, E.; Higashijima, S.; Hibi, M. Proneural gene-linked neurogenesis in zebrafish cerebellum. Dev. Biol. 2010, 343, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hibi, M.; Matsuda, K.; Takeuchi, M.; Shimizu, T.; Murakami, Y. Evolutionary mechanisms that generate morphology and neural-circuit diversity of the cerebellum. Dev. Growth Differ. 2017, 59, 228–243. [Google Scholar] [CrossRef] [PubMed]
- Sylvester, S.J.G.; Lee, M.M.; Ramirez, A.D.; Lim, S.; Goldman, M.S.; Aksay, E.R.F. Population-scale organization of cerebellar granule neuron signaling during a visuomotor behavior. Sci. Rep. 2017, 7, 16240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Severi, K.E.; Bohm, U.L.; Wyart, C. Investigation of hindbrain activity during active locomotion reveals inhibitory neurons involved in sensorimotor processing. Sci. Rep. 2018, 8, 13615. [Google Scholar] [CrossRef] [PubMed]
- Kheradmand, A.; Zee, D.S. Cerebellum and ocular motor control. Front. Neurol. 2011, 2, 53. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, L.M.; Cook, E.H.; Sweeney, J.A.; Mosconi, M.W. Saccadic eye movement abnormalities in autism spectrum disorder indicate dysfunctions in cerebellum and brainstem. Mol. Autism 2014, 5, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, J.Y.; Ulrich, B.; Issa, F.A.; Wan, J.; Papazian, D.M. Rapid development of Purkinje cell excitability, functional cerebellar circuit, and afferent sensory input to cerebellum in zebrafish. Front. Neural Circuits 2014, 8, 147. [Google Scholar] [CrossRef] [PubMed]
- McKay, B.E.; Turner, R.W. Physiological and morphological development of the rat cerebellar Purkinje cell. J. Physiol. 2005, 567, 829–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tropepe, V.; Sive, H.L. Can zebrafish be used as a model to study the neurodevelopmental causes of autism? Genes Brain Behav. 2003, 2, 268–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Rienzo, G.; Bishop, J.A.; Mao, Y.; Pan, L.; Ma, T.P.; Moens, C.B.; Tsai, L.H.; Sive, H. Disc1 regulates both beta-catenin-mediated and noncanonical Wnt signaling during vertebrate embryogenesis. FASEB J. 2011, 25, 4184–4197. [Google Scholar] [CrossRef] [PubMed]
- Kalueff, A.V.; Stewart, A.M.; Gerlai, R. Zebrafish as an emerging model for studying complex brain disorders. Trends Pharmacol. Sci. 2014, 35, 63–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ijaz, S.; Hoffman, E.J. Zebrafish: A Translational Model System for Studying Neuropsychiatric Disorders. J. Am. Acad. Child. Adolesc. Psychiatry 2016, 55, 746–748. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.J.; Turner, K.J.; Fernandez, J.M.; Cifuentes, D.; Ghosh, M.; Ijaz, S.; Jain, R.A.; Kubo, F.; Bill, B.R.; Baier, H.; et al. Estrogens Suppress a Behavioral Phenotype in Zebrafish Mutants of the Autism Risk Gene, CNTNAP2. Neuron 2016, 89, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Kozol, R.A.; Cukier, H.N.; Zou, B.; Mayo, V.; De Rubeis, S.; Cai, G.; Griswold, A.J.; Whitehead, P.L.; Haines, J.L.; Gilbert, J.R.; et al. Two knockdown models of the autism genes SYNGAP1 and SHANK3 in zebrafish produce similar behavioral phenotypes associated with embryonic disruptions of brain morphogenesis. Hum. Mol. Genet. 2015, 24, 4006–4023. [Google Scholar] [CrossRef] [PubMed]
- Blaker-Lee, A.; Gupta, S.; McCammon, J.M.; De Rienzo, G.; Sive, H. Zebrafish homologs of genes within 16p11.2, a genomic region associated with brain disorders, are active during brain development, and include two deletion dosage sensor genes. Dis. Model Mech. 2012, 5, 834–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsen, G.E.; Choi, L.Y.; Prince, V.E.; Ho, R.K. The autism susceptibility gene met regulates zebrafish cerebellar development and facial motor neuron migration. Dev. Biol. 2009, 335, 78–92. [Google Scholar] [CrossRef] [PubMed]
- Psychiatric, G.C.C.C.; Cichon, S.; Craddock, N.; Daly, M.; Faraone, S.V.; Gejman, P.V.; Kelsoe, J.; Lehner, T.; Levinson, D.F.; Moran, A.; et al. Genomewide association studies: History, rationale, and prospects for psychiatric disorders. Am. J. Psychiatry 2009, 166, 540–556. [Google Scholar]
- Volkmar, F.R.; McPartland, J.C. From Kanner to DSM-5: Autism as an evolving diagnostic concept. Annu. Rev. Clin. Psychol. 2014, 10, 193–212. [Google Scholar] [CrossRef] [PubMed]
- DiBella, L.M.; Park, A.; Sun, Z. Zebrafish Tsc1 reveals functional interactions between the cilium and the TOR pathway. Hum. Mol. Genet. 2009, 18, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Tucker, B.; Richards, R.I.; Lardelli, M. Contribution of mGluR and Fmr1 functional pathways to neurite morphogenesis, craniofacial development and fragile X syndrome. Hum. Mol. Genet. 2006, 15, 3446–3458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, J.; Champagne, N.; Lafreniere, R.G.; Xiong, L.; Spiegelman, D.; Brustein, E.; Lapointe, M.; Peng, H.; Cote, M.; Noreau, A.; et al. De novo mutations in the gene encoding the synaptic scaffolding protein SHANK3 in patients ascertained for schizophrenia. Proc. Natl. Acad. Sci. USA 2010, 107, 7863–7868. [Google Scholar] [CrossRef] [PubMed]
- Golzio, C.; Willer, J.; Talkowski, M.E.; Oh, E.C.; Taniguchi, Y.; Jacquemont, S.; Reymond, A.; Sun, M.; Sawa, A.; Gusella, J.F.; et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature 2012, 485, 363–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisen, J.S.; Smith, J.C. Controlling morpholino experiments: Don’t stop making antisense. Development 2008, 135, 1735–1743. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Kontarakis, Z.; Gerri, C.; Nolte, H.; Holper, S.; Kruger, M.; Stainier, D.Y. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 2015, 524, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Y.; Spitzer, N.C. Phenotypic checkpoints regulate neuronal development. Trends Neurosci. 2010, 33, 485–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lubs, H.A. A marker X chromosome. Am. J. Hum. Genet. 1969, 21, 231–244. [Google Scholar] [PubMed]
- Lejeune, J.; Maunoury, C.; Prieur, M.; Van den Akker, J. A jumping translocation (5p;15q), (8q;15q), and (12q;15q) (author’s transl). Ann. Genet. 1979, 22, 210–213. [Google Scholar] [PubMed]
- Archidiacono, N.; Rett, A.; Rocchi, M.; Rolando, S.; Lugaresi, E.; Romeo, G. Rett syndrome and fragile site in Xp22. Lancet 1985, 2, 1242–1243. [Google Scholar] [CrossRef]
- Fryer, A.E.; Chalmers, A.; Connor, J.M.; Fraser, I.; Povey, S.; Yates, A.D.; Yates, J.R.; Osborne, J.P. Evidence that the gene for tuberous sclerosis is on chromosome 9. Lancet 1987, 1, 659–661. [Google Scholar] [CrossRef]
- Kandt, R.S.; Haines, J.L.; Smith, M.; Northrup, H.; Gardner, R.J.; Short, M.P.; Dumars, K.; Roach, E.S.; Steingold, S.; Wall, S.; et al. Linkage of an important gene locus for tuberous sclerosis to a chromosome 16 marker for polycystic kidney disease. Nat. Genet. 1992, 2, 37–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, J.B.; Konecki, D.S.; Munstermann, E.; Bolton, P.; Poustka, A.; Poustka, F.; Gurling, H.M. Point mutation analysis of the FMR-1 gene in autism. Mol. Psychiatry 1996, 1, 227–231. [Google Scholar] [PubMed]
- van Slegtenhorst, M.; de Hoogt, R.; Hermans, C.; Nellist, M.; Janssen, B.; Verhoef, S.; Lindhout, D.; van den Ouweland, A.; Halley, D.; Young, J.; et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 1997, 277, 805–808. [Google Scholar] [CrossRef] [PubMed]
- van Slegtenhorst, M.; Nellist, M.; Nagelkerken, B.; Cheadle, J.; Snell, R.; van den Ouweland, A.; Reuser, A.; Sampson, J.; Halley, D.; van der Sluijs, P. Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum. Mol. Genet. 1998, 7, 1053–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persico, A.M.; D’Agruma, L.; Maiorano, N.; Totaro, A.; Militerni, R.; Bravaccio, C.; Wassink, T.H.; Schneider, C.; Melmed, R.; Trillo, S.; et al. Reelin gene alleles and haplotypes as a factor predisposing to autistic disorder. Mol. Psychiatry 2001, 6, 150–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, L.A.; Escayg, A.; Kearney, J.A.; Trudeau, M.; MacDonald, B.T.; Mori, M.; Reichert, J.; Buxbaum, J.D.; Meisler, M.H. Sodium channels SCN1A, SCN2A and SCN3A in familial autism. Mol. Psychiatry 2003, 8, 186–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, M.G.; Dasouki, M.J.; Zhou, X.P.; Talebizadeh, Z.; Brown, M.; Takahashi, T.N.; Miles, J.H.; Wang, C.H.; Stratton, R.; Pilarski, R.; et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J. Med. Genet. 2005, 42, 318–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonaglia, M.C.; Giorda, R.; Mani, E.; Aceti, G.; Anderlid, B.M.; Baroncini, A.; Pramparo, T.; Zuffardi, O. Identification of a recurrent breakpoint within the SHANK3 gene in the 22q13.3 deletion syndrome. J. Med. Genet. 2006, 43, 822–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrijenhoek, T.; Buizer-Voskamp, J.E.; van der Stelt, I.; Strengman, E.; Genetic, R.; Outcome in Psychosis, C.; Sabatti, C.; Geurts van Kessel, A.; Brunner, H.G.; Ophoff, R.A.; et al. Recurrent CNVs disrupt three candidate genes in schizophrenia patients. Am. J. Hum. Genet. 2008, 83, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Nord, A.S.; Roeb, W.; Dickel, D.E.; Walsh, T.; Kusenda, M.; O’Connor, K.L.; Malhotra, D.; McCarthy, S.E.; Stray, S.M.; Taylor, S.M.; et al. Reduced transcript expression of genes affected by inherited and de novo CNVs in autism. Eur. J. Hum. Genet. 2011, 19, 727–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Roak, B.J.; Deriziotis, P.; Lee, C.; Vives, L.; Schwartz, J.J.; Girirajan, S.; Karakoc, E.; Mackenzie, A.P.; Ng, S.B.; Baker, C.; et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 2011, 43, 585–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iossifov, I.; Ronemus, M.; Levy, D.; Wang, Z.; Hakker, I.; Rosenbaum, J.; Yamrom, B.; Lee, Y.H.; Narzisi, G.; Leotta, A.; et al. De novo gene disruptions in children on the autistic spectrum. Neuron 2012, 74, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Neale, B.M.; Kou, Y.; Liu, L.; Ma’ayan, A.; Samocha, K.E.; Sabo, A.; Lin, C.F.; Stevens, C.; Wang, L.S.; Makarov, V.; et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012, 485, 242–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chahrour, M.H.; Yu, T.W.; Lim, E.T.; Ataman, B.; Coulter, M.E.; Hill, R.S.; Stevens, C.R.; Schubert, C.R.; Collaboration, A.A.S.; Greenberg, M.E.; et al. Whole-exome sequencing and homozygosity analysis implicate depolarization-regulated neuronal genes in autism. PLoS Genet. 2012, 8, e1002635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinwiddie, D.L.; Soden, S.E.; Saunders, C.J.; Miller, N.A.; Farrow, E.G.; Smith, L.D.; Kingsmore, S.F. De novo frameshift mutation in ASXL3 in a patient with global developmental delay, microcephaly, and craniofacial anomalies. BMC Med. Genom. 2013, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Beunders, G.; Voorhoeve, E.; Golzio, C.; Pardo, L.M.; Rosenfeld, J.A.; Talkowski, M.E.; Simonic, I.; Lionel, A.C.; Vergult, S.; Pyatt, R.E.; et al. Exonic deletions in AUTS2 cause a syndromic form of intellectual disability and suggest a critical role for the C terminus. Am. J. Hum. Genet. 2013, 92, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Suls, A.; Jaehn, J.A.; Kecskes, A.; Weber, Y.; Weckhuysen, S.; Craiu, D.C.; Siekierska, A.; Djemie, T.; Afrikanova, T.; Gormley, P.; et al. De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am. J. Hum. Genet. 2013, 93, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Bernier, R.; Golzio, C.; Xiong, B.; Stessman, H.A.; Coe, B.P.; Penn, O.; Witherspoon, K.; Gerdts, J.; Baker, C.; Vulto-van Silfhout, A.T.; et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell 2014, 158, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Hofmeister, W.; Nilsson, D.; Topa, A.; Anderlid, B.M.; Darki, F.; Matsson, H.; Tapia Paez, I.; Klingberg, T.; Samuelsson, L.; Wirta, V.; et al. CTNND2-a candidate gene for reading problems and mild intellectual disability. J. Med. Genet. 2015, 52, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.X.; Li, C.Y.; Hu, C.C.; Wang, Y.; Lin, J.; Jiang, Y.H.; Li, Q.; Xu, X. CRISPR/Cas9-induced shank3b mutant zebrafish display autism-like behaviors. Mol. Autism 2018, 9, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warga, R.M.; Kimmel, C.B. Cell movements during epiboly and gastrulation in zebrafish. Development 1990, 108, 569–580. [Google Scholar] [PubMed]
- Ho, R.K.; Kimmel, C.B. Commitment of cell fate in the early zebrafish embryo. Science 1993, 261, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Feldman, B.; Gates, M.A.; Egan, E.S.; Dougan, S.T.; Rennebeck, G.; Sirotkin, H.I.; Schier, A.F.; Talbot, W.S. Zebrafish organizer development and germ-layer formation require nodal-related signals. Nature 1998, 395, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Bachiller, D.; Klingensmith, J.; Kemp, C.; Belo, J.A.; Anderson, R.M.; May, S.R.; McMahon, J.A.; McMahon, A.P.; Harland, R.M.; Rossant, J.; et al. The organizer factors Chordin and Noggin are required for mouse forebrain development. Nature 2000, 403, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Houart, C.; Westerfield, M.; Wilson, S.W. A small population of anterior cells patterns the forebrain during zebrafish gastrulation. Nature 1998, 391, 788–792. [Google Scholar] [CrossRef] [PubMed]
- Lun, K.; Brand, M. A series of no isthmus (noi) alleles of the zebrafish pax2.1 gene reveals multiple signaling events in development of the midbrain-hindbrain boundary. Development 1998, 125, 3049–3062. [Google Scholar] [PubMed]
- Grinblat, Y.; Gamse, J.; Patel, M.; Sive, H. Determination of the zebrafish forebrain: Induction and patterning. Development 1998, 125, 4403–4416. [Google Scholar] [PubMed]
- Brand, M.; Heisenberg, C.P.; Jiang, Y.J.; Beuchle, D.; Lun, K.; Furutani-Seiki, M.; Granato, M.; Haffter, P.; Hammerschmidt, M.; Kane, D.A.; et al. Mutations in zebrafish genes affecting the formation of the boundary between midbrain and hindbrain. Development 1996, 123, 179–190. [Google Scholar] [PubMed]
- Piccolo, S.; Sasai, Y.; Lu, B.; De Robertis, E.M. Dorsoventral patterning in Xenopus: Inhibition of ventral signals by direct binding of chordin to BMP-4. Cell 1996, 86, 589–598. [Google Scholar] [CrossRef]
- Turner, T.N.; Sharma, K.; Oh, E.C.; Liu, Y.P.; Collins, R.L.; Sosa, M.X.; Auer, D.R.; Brand, H.; Sanders, S.J.; Moreno-De-Luca, D.; et al. Loss of delta-catenin function in severe autism. Nature 2015, 520, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Sokol, S.Y. Graded amounts of Xenopus dishevelled specify discrete anteroposterior cell fates in prospective ectoderm. Mech. Dev. 1997, 61, 113–125. [Google Scholar] [CrossRef]
- Backman, M.; Machon, O.; Mygland, L.; van den Bout, C.J.; Zhong, W.; Taketo, M.M.; Krauss, S. Effects of canonical Wnt signaling on dorso-ventral specification of the mouse telencephalon. Dev. Biol. 2005, 279, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Sugathan, A.; Biagioli, M.; Golzio, C.; Erdin, S.; Blumenthal, I.; Manavalan, P.; Ragavendran, A.; Brand, H.; Lucente, D.; Miles, J.; et al. CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc. Natl. Acad. Sci. USA 2014, 111, E4468–4477. [Google Scholar] [CrossRef] [PubMed]
- Oksenberg, N.; Stevison, L.; Wall, J.D.; Ahituv, N. Function and regulation of AUTS2, a gene implicated in autism and human evolution. PLoS Genet. 2013, 9, e1003221. [Google Scholar] [CrossRef] [PubMed]
- Kathuria, A.; Nowosiad, P.; Jagasia, R.; Aigner, S.; Taylor, R.D.; Andreae, L.C.; Gatford, N.J.F.; Lucchesi, W.; Srivastava, D.P.; Price, J. Stem cell-derived neurons from autistic individuals with SHANK3 mutation show morphogenetic abnormalities during early development. Mol. Psychiatry 2018, 23, 735–746. [Google Scholar] [CrossRef] [PubMed]
- McCammon, J.M.; Blaker-Lee, A.; Chen, X.; Sive, H. The 16p11.2 homologs fam57ba and doc2a generate certain brain and body phenotypes. Hum. Mol. Genet. 2017, 26, 3699–3712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escamilla, C.O.; Filonova, I.; Walker, A.K.; Xuan, Z.X.; Holehonnur, R.; Espinosa, F.; Liu, S.; Thyme, S.B.; Lopez-Garcia, I.A.; Mendoza, D.B.; et al. Kctd13 deletion reduces synaptic transmission via increased RhoA. Nature 2017, 551, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Kuwada, J.Y.; Bernhardt, R.R. Axonal outgrowth by identified neurons in the spinal cord of zebrafish embryos. Exp. Neurol. 1990, 109, 29–34. [Google Scholar] [CrossRef]
- Metcalfe, W.K.; Myers, P.Z.; Trevarrow, B.; Bass, M.B.; Kimmel, C.B. Primary neurons that express the L2/HNK-1 carbohydrate during early development in the zebrafish. Development 1990, 110, 491–504. [Google Scholar] [PubMed]
- Borodinsky, L.N.; Root, C.M.; Cronin, J.A.; Sann, S.B.; Gu, H.L.; Spitzer, N.C. Activity-dependent homeostatic specification of transmitter expression in embryonic neurons. Nature 2004, 429, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, N.C. Electrical activity in early neuronal development. Nature 2006, 444, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Gatto, C.L.; Broadie, K. Genetic controls balancing excitatory and inhibitory synaptogenesis in neurodevelopmental disorder models. Front. Synaptic Neurosci. 2010, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.B.; Valakh, V. Excitatory/Inhibitory Balance and Circuit Homeostasis in Autism Spectrum Disorders. Neuron 2015, 87, 684–698. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.F.; Yang, L.; Li, S.; Wang, Q.; Yuan, X.B.; Gao, X.; Bao, L.; Zhang, X. Fibroblast growth factor 13 is a microtubule-stabilizing protein regulating neuronal polarization and migration. Cell 2012, 149, 1549–1564. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; De Rienzo, G.; Drane, L.; Mao, Y.; Flood, Z.; Madison, J.; Ferreira, M.; Bergen, S.; King, C.; Sklar, P.; et al. Common DISC1 polymorphisms disrupt Wnt/GSK3beta signaling and brain development. Neuron 2011, 72, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Reijnders, M.R.F.; Ansor, N.M.; Kousi, M.; Yue, W.W.; Tan, P.L.; Clarkson, K.; Clayton-Smith, J.; Corning, K.; Jones, J.R.; Lam, W.W.K.; et al. RAC1 Missense Mutations in Developmental Disorders with Diverse Phenotypes. Am. J. Hum. Genet. 2017, 101, 466–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peca, J.; Feliciano, C.; Ting, J.T.; Wang, W.; Wells, M.F.; Venkatraman, T.N.; Lascola, C.D.; Fu, Z.; Feng, G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 2011, 472, 437–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clement, J.P.; Aceti, M.; Creson, T.K.; Ozkan, E.D.; Shi, Y.; Reish, N.J.; Almonte, A.G.; Miller, B.H.; Wiltgen, B.J.; Miller, C.A.; et al. Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell 2012, 151, 709–723. [Google Scholar] [CrossRef] [PubMed]
- Golden, C.E.; Buxbaum, J.D.; De Rubeis, S. Disrupted circuits in mouse models of autism spectrum disorder and intellectual disability. Curr. Opin. Neurobiol. 2018, 48, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Dutton, S.B.; Makinson, C.D.; Papale, L.A.; Shankar, A.; Balakrishnan, B.; Nakazawa, K.; Escayg, A. Preferential inactivation of Scn1a in parvalbumin interneurons increases seizure susceptibility. Neurobiol. Dis. 2013, 49, 211–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baraban, S.C.; Dinday, M.T.; Hortopan, G.A. Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet syndrome treatment. Nat. Commun. 2013, 4, 2410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herget, U.; Gutierrez-Triana, J.A.; Salazar Thula, O.; Knerr, B.; Ryu, S. Single-Cell Reconstruction of Oxytocinergic Neurons Reveals Separate Hypophysiotropic and Encephalotropic Subtypes in Larval Zebrafish. eNeuro 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Herget, U.; Wolf, A.; Wullimann, M.F.; Ryu, S. Molecular neuroanatomy and chemoarchitecture of the neurosecretory preoptic-hypothalamic area in zebrafish larvae. J. Comp. Neurol. 2014, 522, 1542–1564. [Google Scholar] [CrossRef] [PubMed]
- Filippi, A.; Mueller, T.; Driever, W. vglut2 and gad expression reveal distinct patterns of dual GABAergic versus glutamatergic cotransmitter phenotypes of dopaminergic and noradrenergic neurons in the zebrafish brain. J. Comp. Neurol. 2014, 522, 2019–2037. [Google Scholar] [CrossRef] [PubMed]
- Haehnel-Taguchi, M.; Fernandes, A.M.; Bohler, M.; Schmitt, I.; Tittel, L.; Driever, W. Projections of the Diencephalospinal Dopaminergic System to Peripheral Sense Organs in Larval Zebrafish (Danio rerio). Front. Neuroanat. 2018, 12, 20. [Google Scholar] [CrossRef] [PubMed]
- Braasch, I.; Gehrke, A.R.; Smith, J.J.; Kawasaki, K.; Manousaki, T.; Pasquier, J.; Amores, A.; Desvignes, T.; Batzel, P.; Catchen, J.; et al. The spotted gar genome illuminates vertebrate evolution and facilitates human-teleost comparisons. Nat. Genet. 2016, 48, 427–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stainier, D.Y.; Kontarakis, Z.; Rossi, A. Making sense of anti-sense data. Dev. Cell 2015, 32, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postlethwait, J.; Amores, A.; Cresko, W.; Singer, A.; Yan, Y.L. Subfunction partitioning, the teleost radiation and the annotation of the human genome. Trends Genet. 2004, 20, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Catchen, J.M.; Braasch, I.; Postlethwait, J.H. Conserved synteny and the zebrafish genome. Methods Cell. Biol. 2011, 104, 259–285. [Google Scholar] [PubMed]
- Pasquier, J.; Braasch, I.; Batzel, P.; Cabau, C.; Montfort, J.; Nguyen, T.; Jouanno, E.; Berthelot, C.; Klopp, C.; Journot, L.; et al. Evolution of gene expression after whole-genome duplication: New insights from the spotted gar genome. J. Exp. Zool. B Mol. Dev. Evol. 2017, 328, 709–721. [Google Scholar] [CrossRef] [PubMed]
- McWhorter, M.L.; Monani, U.R.; Burghes, A.H.; Beattie, C.E. Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J. Cell Biol. 2003, 162, 919–931. [Google Scholar] [CrossRef] [PubMed]
- Shepard, J.L.; Amatruda, J.F.; Stern, H.M.; Subramanian, A.; Finkelstein, D.; Ziai, J.; Finley, K.R.; Pfaff, K.L.; Hersey, C.; Zhou, Y.; et al. A zebrafish bmyb mutation causes genome instability and increased cancer susceptibility. Proc. Natl. Acad. Sci. USA 2005, 102, 13194–13199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, J.Y.; Einhorn, Z.; Mercurio, S.; Lee, S.; Lau, B.; Mione, M.; Wilson, S.W.; Guo, S. Neurogenin1 is a determinant of zebrafish basal forebrain dopaminergic neurons and is regulated by the conserved zinc finger protein Tof/Fezl. Proc. Natl. Acad. Sci. USA 2006, 103, 5143–5148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omran, H.; Kobayashi, D.; Olbrich, H.; Tsukahara, T.; Loges, N.T.; Hagiwara, H.; Zhang, Q.; Leblond, G.; O’Toole, E.; Hara, C.; et al. Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature 2008, 456, 611–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, M.; Gloy, J.; Ganner, A.; Bullerkotte, A.; Bashkurov, M.; Kronig, C.; Schermer, B.; Benzing, T.; Cabello, O.A.; Jenny, A.; et al. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat. Genet. 2005, 37, 537–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kok, F.O.; Shin, M.; Ni, C.W.; Gupta, A.; Grosse, A.S.; van Impel, A.; Kirchmaier, B.C.; Peterson-Maduro, J.; Kourkoulis, G.; Male, I.; et al. Reverse genetic screening reveals poor correlation between morpholino-induced and mutant phenotypes in zebrafish. Dev. Cell 2015, 32, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Robu, M.E.; Larson, J.D.; Nasevicius, A.; Beiraghi, S.; Brenner, C.; Farber, S.A.; Ekker, S.C. p53 activation by knockdown technologies. PLoS Genet. 2007, 3, e78. [Google Scholar] [CrossRef] [PubMed]
- Nasevicius, A.; Ekker, S.C. Effective targeted gene ‘knockdown’ in zebrafish. Nat. Genet. 2000, 26, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Karlen, S.; Rebagliati, M. A morpholino phenocopy of the cyclops mutation. Genesis 2001, 30, 126–128. [Google Scholar] [CrossRef] [PubMed]
- Stainier, D.Y.R.; Raz, E.; Lawson, N.D.; Ekker, S.C.; Burdine, R.D.; Eisen, J.S.; Ingham, P.W.; Schulte-Merker, S.; Yelon, D.; Weinstein, B.M.; et al. Guidelines for morpholino use in zebrafish. PLoS Genet. 2017, 13, e1007000. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhao, L.; Page-McCaw, P.S.; Chen, W. Zebrafish Genome Engineering Using the CRISPR-Cas9 System. Trends Genet. 2016, 32, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Simone, B.W.; Martinez-Galvez, G.; WareJoncas, Z.; Ekker, S.C. Fishing for understanding: Unlocking the zebrafish gene editor’s toolbox. Methods 2018, 150, 3–10. [Google Scholar] [CrossRef] [PubMed]
- El-Brolosy, M.A.; Stainier, D.Y.R. Genetic compensation: A phenomenon in search of mechanisms. PLoS Genet. 2017, 13, e1006780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankaran, S.S.; Dahlem, T.J.; Bisgrove, B.W.; Yost, H.J.; Tristani-Firouzi, M. CRISPR/Cas9-Directed Gene Editing for the Generation of Loss-of-Function Mutants in High-Throughput Zebrafish F0 Screens. Curr. Protoc. Mol. Biol. 2017, 119, 31–39. [Google Scholar] [PubMed]
- Shah, A.N.; Moens, C.B.; Miller, A.C. Targeted candidate gene screens using CRISPR/Cas9 technology. Methods Cell Biol. 2016, 135, 89–106. [Google Scholar] [PubMed]
- Saint-Amant, L.; Drapeau, P. Time course of the development of motor behaviors in the zebrafish embryo. J. Neurobiol. 1998, 37, 622–632. [Google Scholar] [CrossRef] [Green Version]
- Grillner, S. The motor infrastructure: From ion channels to neuronal networks. Nat. Rev. Neurosci. 2003, 4, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Goulding, M.D. Circuits controlling vertebrate locomotion: Moving in a new direction. Nat. Rev. Neurosci. 2009, 10, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Burgess, H.A.; Granato, M. Sensorimotor gating in larval zebrafish. J. Neurosci. 2007, 27, 4984–4994. [Google Scholar] [CrossRef] [PubMed]
- Portugues, R.; Feierstein, C.E.; Engert, F.; Orger, M.B. Whole-brain activity maps reveal stereotyped, distributed networks for visuomotor behavior. Neuron 2014, 81, 1328–1343. [Google Scholar] [CrossRef] [PubMed]
- Tavassoli, T.; Bellesheim, K.; Siper, P.M.; Wang, A.T.; Halpern, D.; Gorenstein, M.; Grodberg, D.; Kolevzon, A.; Buxbaum, J.D. Measuring Sensory Reactivity in Autism Spectrum Disorder: Application and Simplification of a Clinician-Administered Sensory Observation Scale. J. Autism. Dev. Disord. 2016, 46, 287–293. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association; American Psychiatric Association. DSM-5 Task Force. Diagnostic and Statistical Manual of Mental Disorders: DSM-5, 5th ed. American Psychiatric Association: Washington, DC, USA, 2013. [Google Scholar]
- Engeszer, R.E.; Barbiano, L.A.; Ryan, M.J.; Parichy, D.M. Timing and plasticity of shoaling behaviour in the zebrafish, Danio rerio. Anim. Behav. 2007, 74, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Dreosti, E.; Lopes, G.; Kampff, A.R.; Wilson, S.W. Development of social behavior in young zebrafish. Front. Neural Circuits 2015, 9, 39. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, G.; Hodgins-Davis, A.; Avolio, C.; Schunter, C. Kin recognition in zebrafish: A 24-hour window for olfactory imprinting. Proc. Biol. Sci. 2008, 275, 2165–2170. [Google Scholar] [CrossRef] [PubMed]
- Wircer, E.; Blechman, J.; Borodovsky, N.; Tsoory, M.; Nunes, A.R.; Oliveira, R.F.; Levkowitz, G. Homeodomain protein Otp affects developmental neuropeptide switching in oxytocin neurons associated with a long-term effect on social behavior. Elife 2017, 6, e22170. [Google Scholar] [CrossRef] [PubMed]
- Engeszer, R.E.; Ryan, M.J.; Parichy, D.M. Learned social preference in zebrafish. Curr. Biol. 2004, 14, 881–884. [Google Scholar] [CrossRef] [PubMed]
- Stednitz, S.J.; McDermott, E.M.; Ncube, D.; Tallafuss, A.; Eisen, J.S.; Washbourne, P. Forebrain Control of Behaviorally Driven Social Orienting in Zebrafish. Curr. Biol. 2018, 28, 2445–2451. [Google Scholar] [CrossRef] [PubMed]
- Myers, L.; Anderlid, B.M.; Nordgren, A.; Willfors, C.; Kuja-Halkola, R.; Tammimies, K.; Bolte, S. Minor physical anomalies in neurodevelopmental disorders: A twin study. Child. Adolesc. Psychiatry Ment. Health 2017, 11, 57. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, R.; DeVito, T.J.; Vidal, C.N.; Sui, Y.; Hayashi, K.M.; Drost, D.J.; Williamson, P.C.; Rajakumar, N.; Toga, A.W.; Thompson, P.M. Detection and mapping of hippocampal abnormalities in autism. Psychiatry Res. 2006, 148, 11–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randlett, O.; Wee, C.L.; Naumann, E.A.; Nnaemeka, O.; Schoppik, D.; Fitzgerald, J.E.; Portugues, R.; Lacoste, A.M.; Riegler, C.; Engert, F.; et al. Whole-brain activity mapping onto a zebrafish brain atlas. Nat. Methods 2015, 12, 1039–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, T.; Marquart, G.D.; Horstick, E.J.; Tabor, K.M.; Pajevic, S.; Burgess, H.A. Morphometric analysis and neuroanatomical mapping of the zebrafish brain. Methods 2018. [Google Scholar] [CrossRef] [PubMed]
- Marquart, G.D.; Tabor, K.M.; Horstick, E.J.; Brown, M.; Geoca, A.K.; Polys, N.F.; Nogare, D.D.; Burgess, H.A. High-precision registration between zebrafish brain atlases using symmetric diffeomorphic normalization. Gigascience 2017, 6, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forster, D.; Arnold-Ammer, I.; Laurell, E.; Barker, A.J.; Fernandes, A.M.; Finger-Baier, K.; Filosa, A.; Helmbrecht, T.O.; Kolsch, Y.; Kuhn, E.; et al. Genetic targeting and anatomical registration of neuronal populations in the zebrafish brain with a new set of BAC transgenic tools. Sci. Rep. 2017, 7, 5230. [Google Scholar] [CrossRef] [PubMed]
- Jefferis, G.S.; Potter, C.J.; Chan, A.M.; Marin, E.C.; Rohlfing, T.; Maurer, C.R., Jr.; Luo, L. Comprehensive maps of Drosophila higher olfactory centers: Spatially segregated fruit and pheromone representation. Cell 2007, 128, 1187–1203. [Google Scholar] [CrossRef] [PubMed]
- Satou, C.; Kimura, Y.; Higashijima, S. Generation of multiple classes of V0 neurons in zebrafish spinal cord: Progenitor heterogeneity and temporal control of neuronal diversity. J. Neurosci. 2012, 32, 1771–1783. [Google Scholar] [CrossRef] [PubMed]
- Guzowski, J.F.; Timlin, J.A.; Roysam, B.; McNaughton, B.L.; Worley, P.F.; Barnes, C.A. Mapping behaviorally relevant neural circuits with immediate-early gene expression. Curr. Opin. Neurobiol. 2005, 15, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Barros, V.N.; Mundim, M.; Galindo, L.T.; Bittencourt, S.; Porcionatto, M.; Mello, L.E. The pattern of c-Fos expression and its refractory period in the brain of rats and monkeys. Front. Cell Neurosci. 2015, 9, 72. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Dudek, H.; Miranti, C.K.; Greenberg, M.E. Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J. Neurosci. 1996, 16, 5425–5436. [Google Scholar] [CrossRef] [PubMed]
- Cox, K.J.; Fetcho, J.R. Labeling blastomeres with a calcium indicator: A non-invasive method of visualizing neuronal activity in zebrafish. J. Neurosci. Methods 1996, 68, 185–191. [Google Scholar] [CrossRef]
- Takahashi, M.; Narushima, M.; Oda, Y. In vivo imaging of functional inhibitory networks on the mauthner cell of larval zebrafish. J. Neurosci. 2002, 22, 3929–3938. [Google Scholar] [CrossRef] [PubMed]
- Sumbre, G.; Muto, A.; Baier, H.; Poo, M.M. Entrained rhythmic activities of neuronal ensembles as perceptual memory of time interval. Nature 2008, 456, 102–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyawaki, A.; Llopis, J.; Heim, R.; McCaffery, J.M.; Adams, J.A.; Ikura, M.; Tsien, R.Y. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 1997, 388, 882–887. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Kim, J.; Marques, J.C.; Grama, A.; Hildebrand, D.G.C.; Gu, W.; Li, J.M.; Robson, D.N. Pan-neuronal calcium imaging with cellular resolution in freely swimming zebrafish. Nat. Methods 2017, 14, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Wang, Z.; Chai, Y.; Hang, W.; Shang, C.; Yang, W.; Bai, L.; Du, J.; Wang, K.; Wen, Q. Rapid whole brain imaging of neural activity in freely behaving larval zebrafish (Danio rerio). Elife 2017, 6, e28158. [Google Scholar] [CrossRef] [PubMed]
- Lovett-Barron, M.; Andalman, A.S.; Allen, W.E.; Vesuna, S.; Kauvar, I.; Burns, V.M.; Deisseroth, K. Ancestral Circuits for the Coordinated Modulation of Brain State. Cell 2017, 171, 1411–1423. [Google Scholar] [CrossRef] [PubMed]
- Jawinski, P.; Kirsten, H.; Sander, C.; Spada, J.; Ulke, C.; Huang, J.; Burkhardt, R.; Scholz, M.; Hensch, T.; Hegerl, U. Human brain arousal in the resting state: A genome-wide association study. Mol. Psychiatry 2018. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.; Mollinedo-Gajate, I.; Penagarikano, O. Neural Circuits for Social Cognition: Implications for Autism. Neuroscience 2018, 370, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K.; Asakawa, K.; Hibi, M.; Itoh, M.; Muto, A.; Wada, H. Gal4 Driver Transgenic Zebrafish: Powerful Tools to Study Developmental Biology, Organogenesis, and Neuroscience. Adv. Genet. 2016, 95, 65–87. [Google Scholar] [PubMed]
- Wiley, D.S.; Redfield, S.E.; Zon, L.I. Chemical screening in zebrafish for novel biological and therapeutic discovery. Methods Cell Biol. 2017, 138, 651–679. [Google Scholar] [PubMed] [Green Version]
- Svenson, K.L.; Gatti, D.M.; Valdar, W.; Welsh, C.E.; Cheng, R.; Chesler, E.J.; Palmer, A.A.; McMillan, L.; Churchill, G.A. High-resolution genetic mapping using the Mouse Diversity outbred population. Genetics 2012, 190, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Rihel, J.; Prober, D.A.; Arvanites, A.; Lam, K.; Zimmerman, S.; Jang, S.; Haggarty, S.J.; Kokel, D.; Rubin, L.L.; Peterson, R.T.; et al. Zebrafish behavioral profiling links drugs to biological targets and rest/wake regulation. Science 2010, 327, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Kokel, D.; Peterson, R.T. Using the zebrafish photomotor response for psychotropic drug screening. Methods Cell. Biol. 2011, 105, 517–524. [Google Scholar] [PubMed]
- Bruni, G.; Rennekamp, A.J.; Velenich, A.; McCarroll, M.; Gendelev, L.; Fertsch, E.; Taylor, J.; Lakhani, P.; Lensen, D.; Evron, T.; et al. Zebrafish behavioral profiling identifies multitarget antipsychotic-like compounds. Nat. Chem. Biol. 2016, 12, 559–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, M.; Beresford, C.A.; Bunker, M.; Verdi, M.; Vishnevetsky, D.; Karlsson, C.; Teer, O.; Stedman, A.; Smith, K.A. Preliminary investigation of lithium for mood disorder symptoms in children and adolescents with autism spectrum disorder. J. Child Adolesc. Psychopharmacol. 2014, 24, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.E.; Gregas, M.; Jeste, S.S. Risperidone use in autism spectrum disorders: A retrospective review of a clinic-referred patient population. J. Child Neurol. 2011, 26, 428–432. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A timeline of ASD-ID genetic discoveries and ASD-ID modeling in zebrafish. Names represent the characterization of a gene associated with ASD-ID or the first report of a genetic variant associated with ASD-ID (bottom). ASD-ID genes where chosen from the Simons Autism Research Initiative (tier 1 genes) and common ASD-ID syndromes. Zebrafish genetic models denote the first reported embryonic or larval model and the method of genetic manipulation, KD = morpholino knock-down, KO = mutant allele. Histograms describe publications per year for PubMed keywords ASD, ID, Genes, Genetics, Loci, Chromosome (top zebrafish, bottom human). Non-linear intervals of years were averaged. Human timeline references: “Marker X” [138]; 15q Prader Wili/Angelman [139]; Xp22 RTT [140]; TSC Ch. 9 [141], Ch. 16 [142]; FMR1 [143]; TSC1/2 [144,145]; MeCP2 [47]; RELN [146]; SCN2A [147]; PTEN [148]; SHANK3 [149]; MYT1L [150]; ARID1B [151]; GRIN2B [152]; CHD8, ADNP, DYRK1A, TBR1 [57]; KMT5B, KATNAL2, SETD5, POGZ [153,154]; TRIP12 [155]; ASH1L [31]; ASXL3 [156]; DSCAM, NAA15, KMT2A [157]. Zebrafish timeline references: zebrafish-ASD perspective [121]; fmr1 KD [132], shank3 KD [133], 16p11.2 KD [127], kctd13 KD [134], chd7 KD, auts2 KD [158], chd2 KD [159], mecp2 KO, chd8 KD/KO [160], ctnnd2 KD [161], syngap1b KD [126], mecp2 KD, cntnap2ab KO [125], dyrk1a KO, shank3b KO [162], chd7 KO.
Figure 1.
A timeline of ASD-ID genetic discoveries and ASD-ID modeling in zebrafish. Names represent the characterization of a gene associated with ASD-ID or the first report of a genetic variant associated with ASD-ID (bottom). ASD-ID genes where chosen from the Simons Autism Research Initiative (tier 1 genes) and common ASD-ID syndromes. Zebrafish genetic models denote the first reported embryonic or larval model and the method of genetic manipulation, KD = morpholino knock-down, KO = mutant allele. Histograms describe publications per year for PubMed keywords ASD, ID, Genes, Genetics, Loci, Chromosome (top zebrafish, bottom human). Non-linear intervals of years were averaged. Human timeline references: “Marker X” [138]; 15q Prader Wili/Angelman [139]; Xp22 RTT [140]; TSC Ch. 9 [141], Ch. 16 [142]; FMR1 [143]; TSC1/2 [144,145]; MeCP2 [47]; RELN [146]; SCN2A [147]; PTEN [148]; SHANK3 [149]; MYT1L [150]; ARID1B [151]; GRIN2B [152]; CHD8, ADNP, DYRK1A, TBR1 [57]; KMT5B, KATNAL2, SETD5, POGZ [153,154]; TRIP12 [155]; ASH1L [31]; ASXL3 [156]; DSCAM, NAA15, KMT2A [157]. Zebrafish timeline references: zebrafish-ASD perspective [121]; fmr1 KD [132], shank3 KD [133], 16p11.2 KD [127], kctd13 KD [134], chd7 KD, auts2 KD [158], chd2 KD [159], mecp2 KO, chd8 KD/KO [160], ctnnd2 KD [161], syngap1b KD [126], mecp2 KD, cntnap2ab KO [125], dyrk1a KO, shank3b KO [162], chd7 KO.


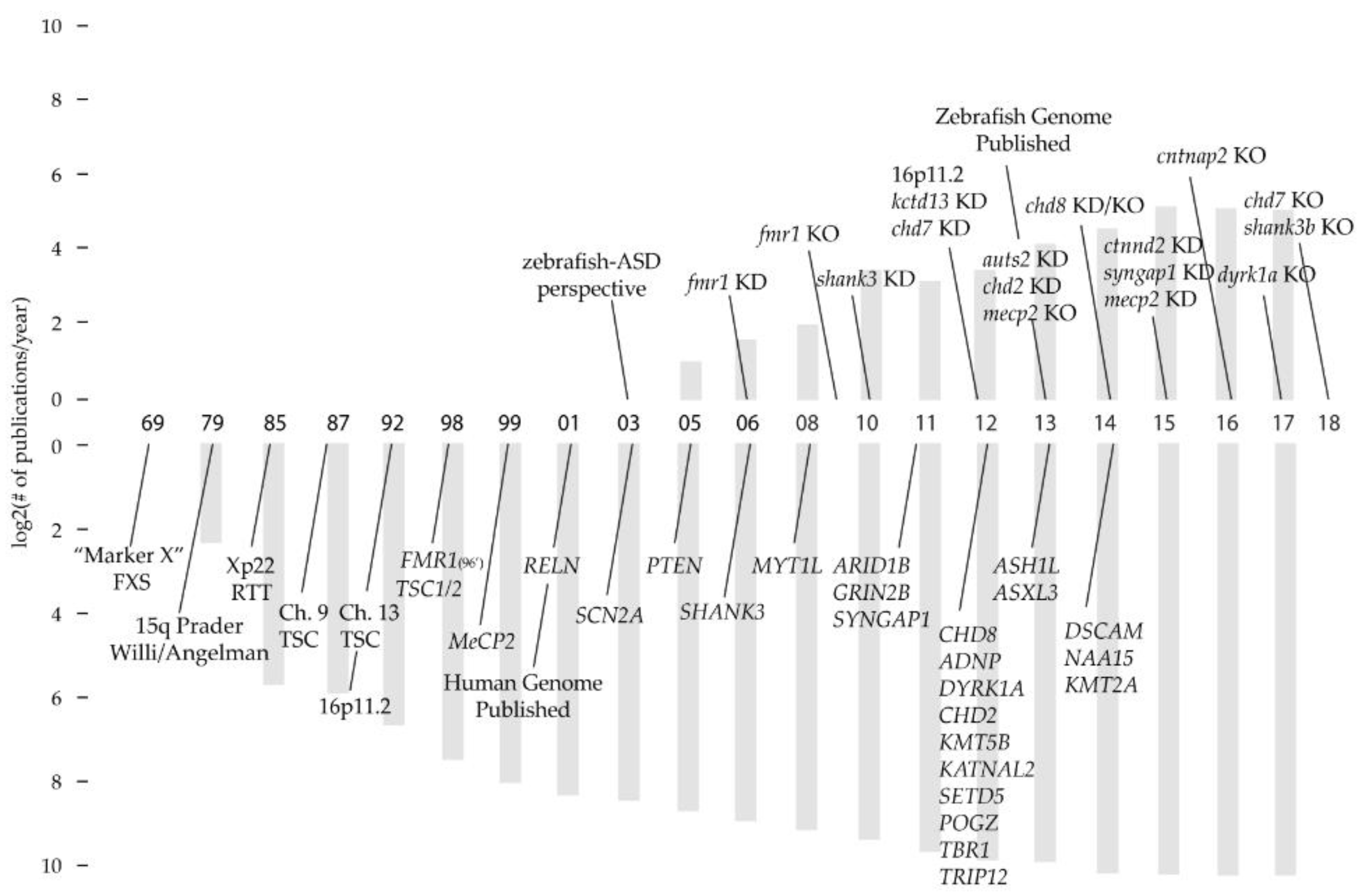{kind=link}
Table 1.
A timeline of neurogenic events in mammals and zebrafish. Time periods represent approximate dates related to morphological delineation, onset of neurogenesis and onset of behavior. Time periods are approximated as days post-fertilization (dpf).
Table 1.
A timeline of neurogenic events in mammals and zebrafish. Time periods represent approximate dates related to morphological delineation, onset of neurogenesis and onset of behavior. Time periods are approximated as days post-fertilization (dpf).
| Structure or Region | Estimated Onset of Development (dpf) | ||
|---|---|---|---|
| Human [75,78] | Rat [75,78] | Zebrafish | |
| Neural tube | 21 | 10 | 0.4–0.8 [14] |
| Telencephalon, Diencephalon, Mesencephalon, Rhombencephalon | 28–33 | 11 | 0.7–0.75 [14] |
| Cerebellar primordia | 28 | 12 | 0.8 [14] |
| Optic tectum | 28 | 12 | 1 [82] |
| Hypothalamus, pineal (epiphysis) | 28 | 12 | 0.75, 1 [83] |
| Raphe complex | 28 [84] | 10.5 [85] | 3 [86] |
| Locus coeruleus | 28 | 11 | 1 [87,88] |
| Inferior olive | 30 | 11 | 3 [89] |
| Medial longitudinal fasciculus | 30 | 11 | 1.2 [90] |
| Thalamus | 36 | 14 | 2 [91] |
| Cell type | |||
| Retinal ganglion cells | 33 | 12 | 1.2 [92] |
| Mitral cells | 33 | 12 | 1 [93] |
| GABAergic neurons | 52 | 15 | 1.2 [94] |
| Behavior | |||
| Walking (zebrafish, swimming) | 455 | 48 | 1.13 [23] |
| Acoustic startle response | 265 | 29 | 4 [95] |
Table 2.
Critical periods in the development of the cerebellum. Time periods represent approximate dates related to onset of neurogenesis, function and formation of structure. Time periods are approximated as days post-fertilization (dpf).
Table 2.
Critical periods in the development of the cerebellum. Time periods represent approximate dates related to onset of neurogenesis, function and formation of structure. Time periods are approximated as days post-fertilization (dpf).
| Developmental Event | Estimated Onset of Development (dpf) | |
|---|---|---|
| Rat [112] | Zebrafish [109,113,119] | |
| Cerebellar primordia (En1/2, Wnt1, Pax2, Gbx2) | 12.5 | 0.8 |
| Progenitor pools specified (Atoh1a/Ptf1a) | 10.5 | 1–2 |
| GC/PC migration-differentiation | 12.5–15 | 3 |
| Purkinje cells functionally mature | 29.5 [120] | 4 |
| Trilaminar structure formed | 35–42 | 5 |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kozol, R.A. Prenatal Neuropathologies in Autism Spectrum Disorder and Intellectual Disability: The Gestation of a Comprehensive Zebrafish Model. J. Dev. Biol. 2018, 6, 29. https://doi.org/10.3390/jdb6040029
AMA Style
Kozol RA. Prenatal Neuropathologies in Autism Spectrum Disorder and Intellectual Disability: The Gestation of a Comprehensive Zebrafish Model. Journal of Developmental Biology. 2018; 6(4):29. https://doi.org/10.3390/jdb6040029
Chicago/Turabian StyleKozol, Robert A. 2018. "Prenatal Neuropathologies in Autism Spectrum Disorder and Intellectual Disability: The Gestation of a Comprehensive Zebrafish Model" Journal of Developmental Biology 6, no. 4: 29. https://doi.org/10.3390/jdb6040029
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.