
Population Variation and Phylogeography of Cherry Blossom (Prunus conradinae) in China
 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ecological Niche Model
2.2. Plant Materials
2.3. DNA Extraction, Polymerase Chain Reaction (PCR) Amplification, Sequencing, and Sequence Alignment
2.4. Data Analysis
2.5. Molecular Dating and Demographic Analyses
3. Results
3.1. Ecological Niche Model
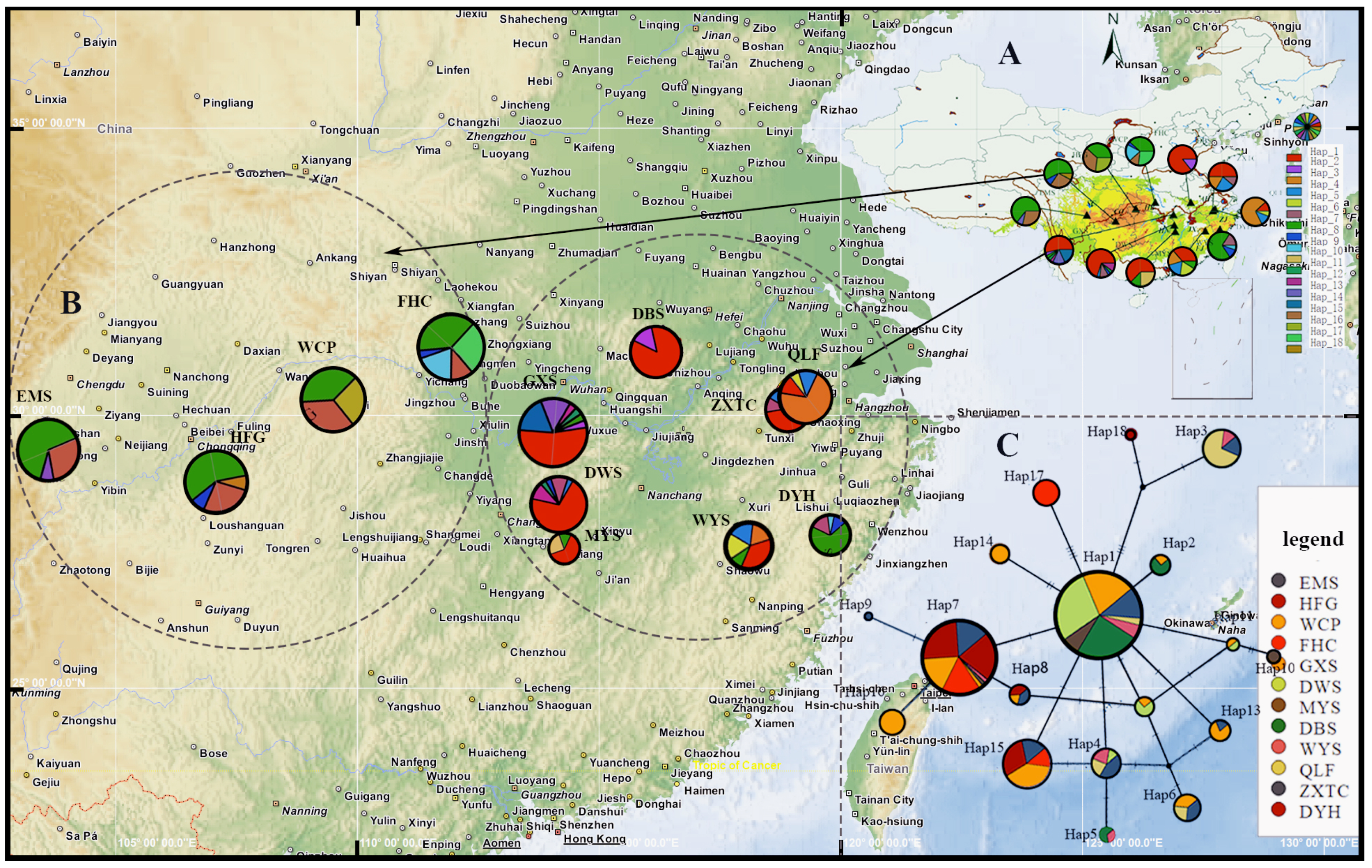
3.2. Sequence Variation, Haplotype Frequency, and Distribution
3.3. Haplotype Network
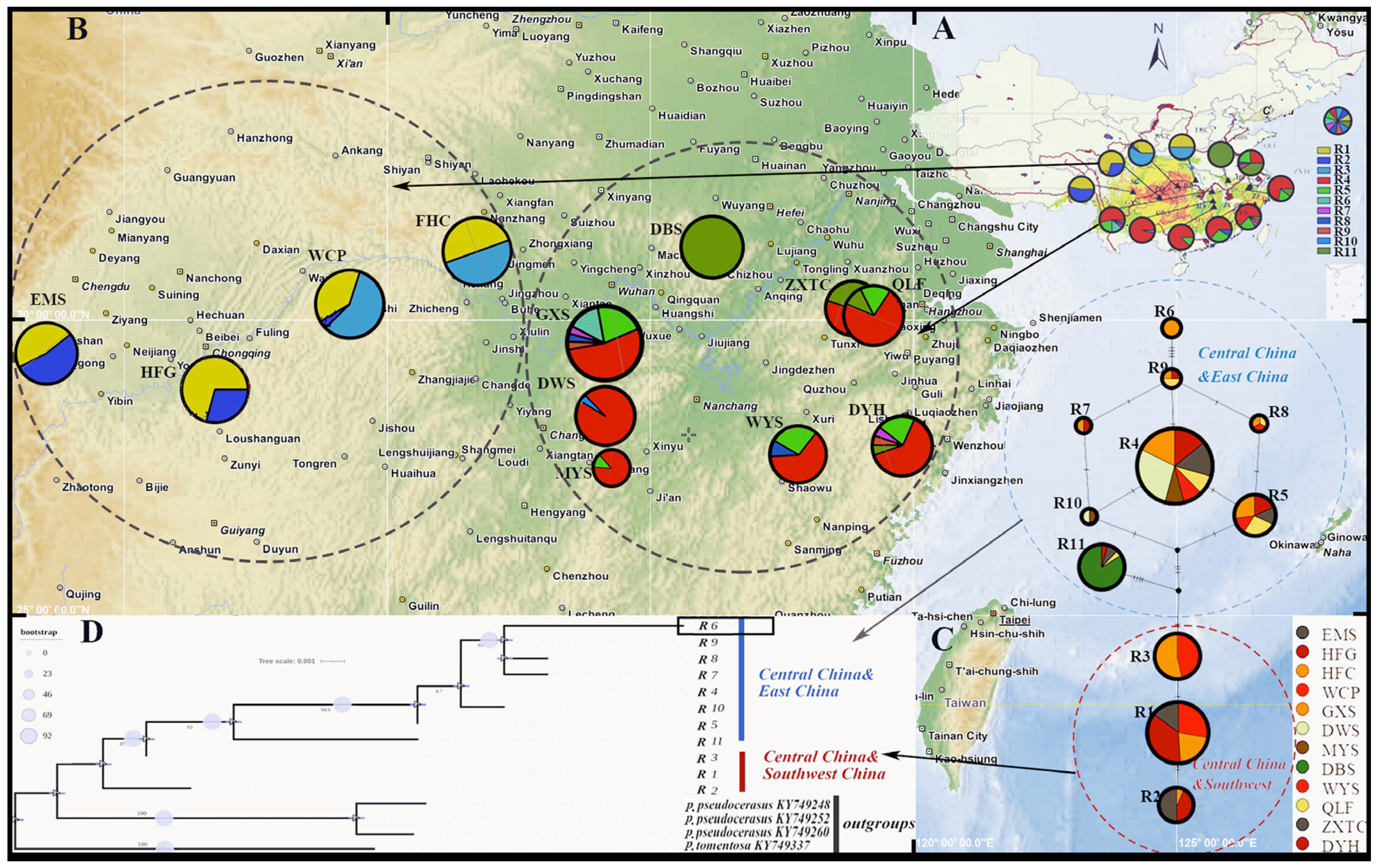
3.4. Genetic Diversity and Population Genetic Structure
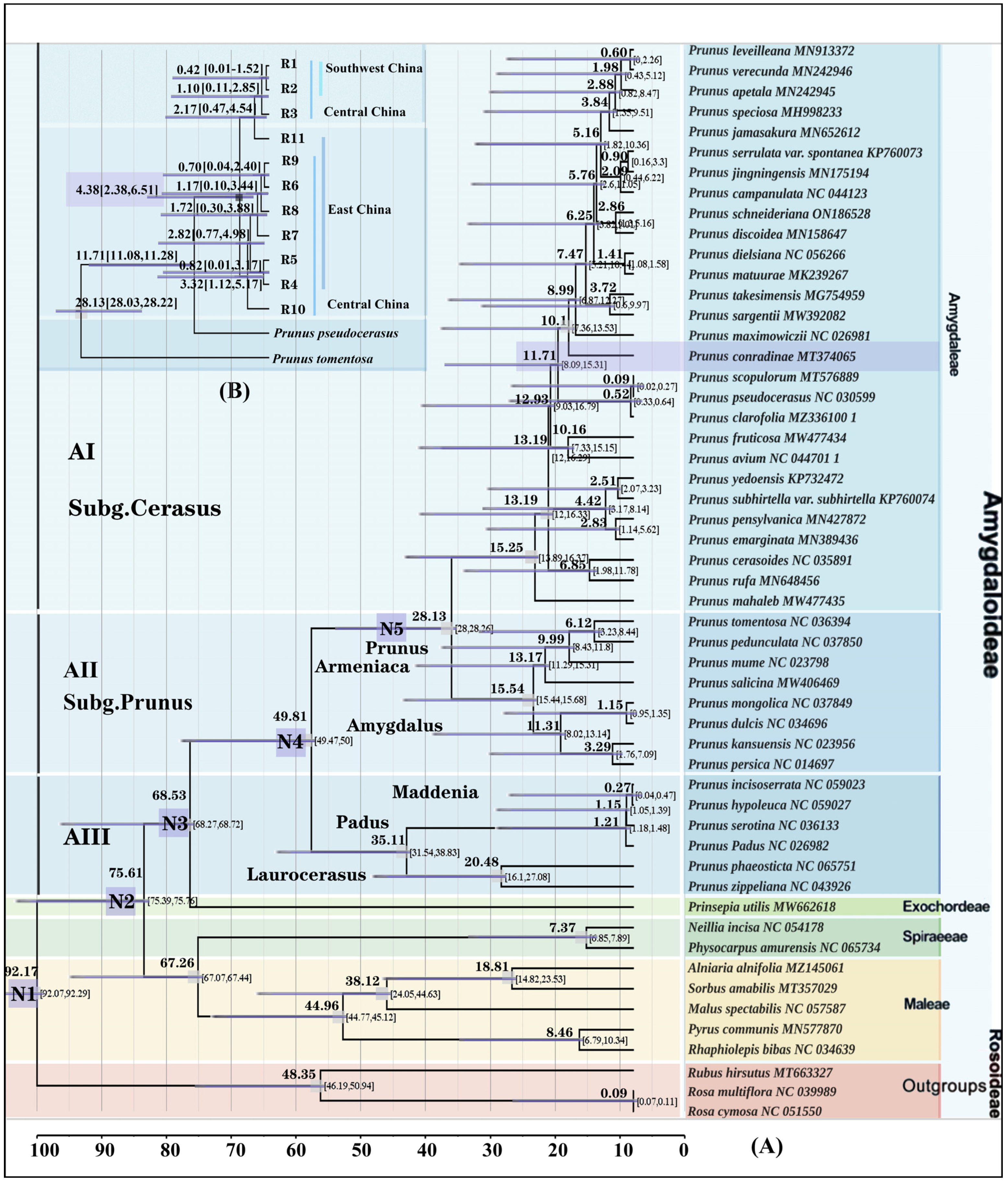
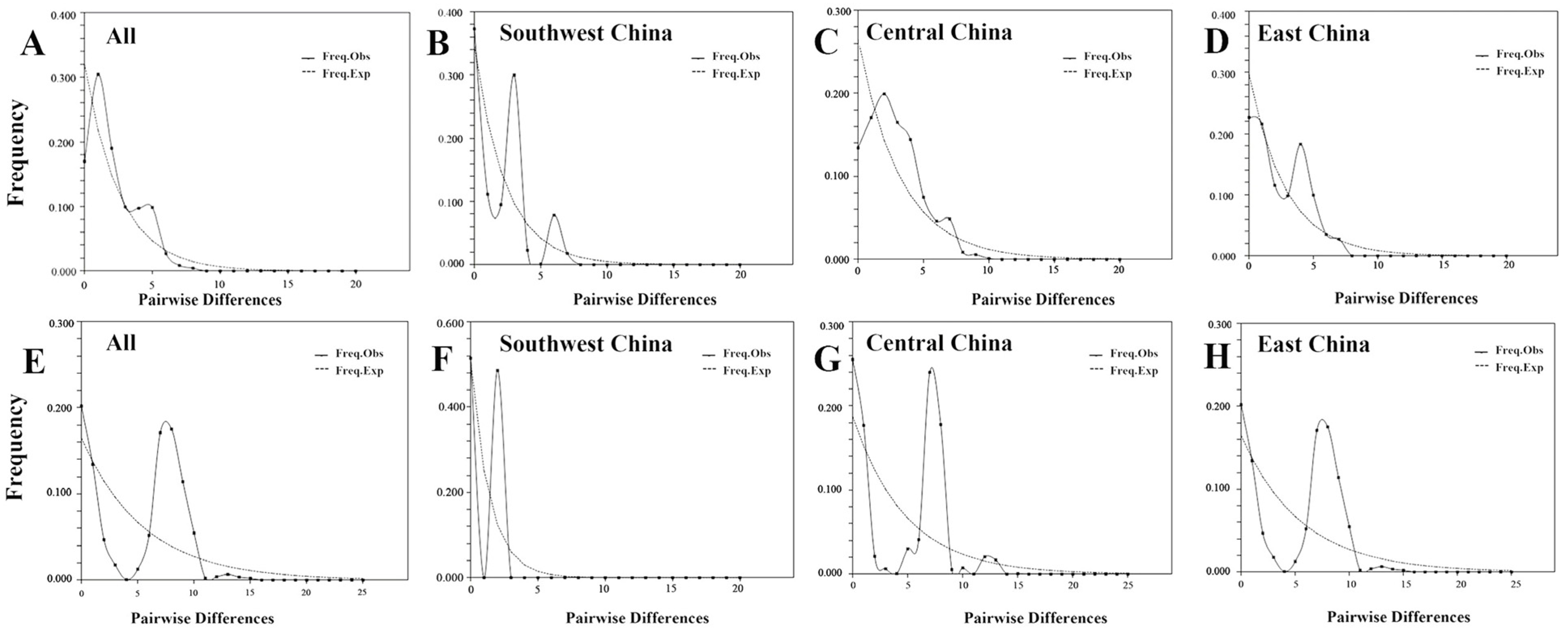
3.5. Molecular Dating and Demographic Analyses
3.6. Population Variation and Taxonomic Status
4. Discussion
4.1. Genetic Diversity and Population Genetic Structure
4.2. Geographical Structure of Pedigree
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yü, D.J.; Lu, L.T.; Ku, T.C.; Li, C.L.; Chen, S.X. Flora of China; Science Press: Beijing, China, 1986; Volume 38. [Google Scholar]
- Shulaev, V.; Korban, S.S.; Sosinski, B.; Abbott, A.G.; Aldwinckle, H.S.; Folta, K.M. Multiple models for Rosaceae genomics. Plant Physiol. 2008, 147, 985–1003. [Google Scholar] [CrossRef]
- Phipps, J.B. Flora of North America North of Mexico; Oxford University Press: Oxford, UK, 2014; Volume 9. [Google Scholar]
- Wang, R.X. An Illustrated Monograph of Cherry Cultivars in China; Science Press: Beijing, China, 2014; Volume 12, pp. 24–28. [Google Scholar]
- Chen, N.L. Evaluation of Ornamental Traits of Cerasus Species and Study on Suitable Areas of Chinese Wild Species in Zhejiang; Zhejiang Science and Technology University: Hangzhou, China, 2015. [Google Scholar]
- Bai, F.W.; Yu, L.; Li, H.J.; Nei, L.D.; Yan, W.J.; Wu, Z.S.; Li, C.J.; Xiao, D.J. Investigation of Cerasus resources on Dawei Mountain. Hunan For. Sci. Technol. 2019, 46, 85–90. [Google Scholar]
- Chen, T.; Li, L.; Zhang, J.; Huang, Z.L.; Zhang, H.W.; Liu, Y. Investigation, collection and preliminary evaluation of genetic resources of Chinese cherry [Cerasus pseudocerasus (Lindl.) G. Don]. J. Fruit Sci. 2016, 33, 917–933. [Google Scholar] [CrossRef]
- Potter, D.; Eriksson, T.; Evans, R.C.; Oh, S.; Smedmark, J.E.; Morgan, D.R. Phylogeny and classification of Rosaceae. Plant Syst. Evol. 2007, 266, 5–43. [Google Scholar] [CrossRef]
- Chen, T.; Hu, G.P.; Wang, Y.; Chen, Q.; Zhang, J.; Wang, L. Survey, collection and conservation of wild Cerasus Mill. germplasm resources in China. J. Plant Genet. Resour. 2020, 21, 532–541. [Google Scholar] [CrossRef]
- Zhang, S.D.; Jin, J.J.; Chen, S.Y.; Chase, M.W.; Soltis, D.E.; Li, H.T. Diversification of Rosaceae since the Late Cretaceous based on plastid phylogenomics. New Phytol. 2017, 214, 1355–1367. [Google Scholar] [CrossRef]
- Aranzana, M.J.; Decroocq, V.; Dirlewanger, E.; Eduardo, I.; Gao, Z.S.; Gasic, K. Prunus genetics and applications after de novo genome sequencing: Achievements and prospects. Hortic. Res. 2019, 6, 58. [Google Scholar] [CrossRef]
- Chen, F.; Song, Y.F.; Li, X.J.; Chen, J.H.; Mo, L.; Zhang, X.T. Genome sequences of horticultural plants: Past, present, and future. Hortic. Res. 2019, 6, 112. [Google Scholar] [CrossRef]
- Yu, L.; Li, H.J.; Bai, F.W.; Nei, D.L.; Zhang, X.W.; Wu, Z.S.; Han, L.X. Investigation and Utilization Evaluation of Wild Cerasus Resources in Daxiong Mountain of Hunan Province. For. Environ. Sci. 2019, 35, 38–43. [Google Scholar]
- Nybom, H.; Weising, K.; Rotter, B. DNA fingerprinting in botany: Past, present, future. Investig. Genet. 2014, 5, 1. [Google Scholar] [CrossRef]
- Avise, J.C.; Arnold, J.; Ball, R.M.; Bermingham, E.; Lamb, T.; Neigel, J.E.; Reeb, C.A.; Saunders, N.C. Intraspecific phylogeography: The mitochondrial DNA bridge between population genetics and systematics. Annu. Rev. Ecol. Syst. 1987, 18, 489–522. [Google Scholar] [CrossRef]
- Avise, J.C. Phylogeography: The History and Formation of Species; Harvard University Press: Cambridge, MA, USA, 2000. [Google Scholar]
- Vander Wall, S.B.; Beck, M.J. A comparison of frugivory and scatter-hoardin seed- dispersal syndromes. Bot. Rev. 2012, 78, 10–31. [Google Scholar] [CrossRef]
- Perdereau, A.C.; Kelleher, C.T.; Douglas, G.C.; Hodkinson, T.R. High levels of gene flow and genetic diversity in Irish populations of Salix caprea L. inferred from chloroplast and nuclear SSR markers. BMC Plant Biol. 2014, 14, 202. [Google Scholar] [CrossRef]
- Chan, L.M.; Brown, J.L.; Yoder, A.D. Integrating statistical genetic and geospatial methods brings new power to phylogeography. Mol. Phylogenet. Evol. 2011, 59, 523–537. [Google Scholar] [CrossRef]
- Phillips, S.; Dudík, M.; Schapire, R. A maximum entropy approach to species distribution modeling. In Proceedings of the Twenty-First International Conference on Machine Learning, Banff, AB, Canada, 4–8 July 2004; ACM: New York, NY, USA, 2004. [Google Scholar]
- Turkoglu, Z.; Bilgener, S.; Ercisli, S.; Bakir, M.; Koc, A.; Akbulut, M.; Gercekcioglu, R.; Gunes, M.; Esitken, A. Simple sequence repeat-based assessment of genetic relationships among Prunus rootstocks. Genet. Mol. Res. 2010, 9, 2156–2165. [Google Scholar] [CrossRef]
- Bai, N.W.; Zhang, Y.D. Current status and future directions in plant phylogeography. Chin. Bull. Life Sci. 2014, 26, 125–137. [Google Scholar] [CrossRef]
- Yi, G.X.; Yu, X.; Chen, J.; Zhang, M.; Liu, S.W.; Zhu, H.; Li, M.; Duan, Y.F.; Chen, L.; Wu, L.; et al. The genome of Chinese flowering cherry (Cerasus serrulata) provides new insights into Cerasus species. Hortic. Res. 2020, 7, 14. [Google Scholar] [CrossRef]
- Qiu, J.; Zhu, H.; Chen, X. Modeling the suitable areas and ecological characteristics of Sorbus alnifolia using DIVA-GIS software. J. Beijing For. Univ. 2018, 40, 25–32. [Google Scholar]
- Zhu, H.; You, L.X.; Li, F.Y. Modeling the Geographical Distribution Pattern and Climatic Limited Factors of Cerasus schneideriana. J. Trop. Subtrop. Bot. 2017, 25, 315–322. [Google Scholar] [CrossRef]
- Garah, K.; Bentouati, A. Using the MaxEnt model for assessing the impact of climate change on the Aurasian Aleppo pine distribution in Algeria. Afr. J. Ecol. 2019, 57, 500–511. [Google Scholar] [CrossRef]
- Heckenhauer, J.; Barfuss, M.; Samuel, R. Universal multiplexable matK primers for DNA barcoding of angiosperms. Appl. Plant Sci. 2016, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Liu, T.; Wang, X.Y.; Li, B.B.; Liang, C.; Cai, Y. Characterization of the complete chloroplast genome of the Chinese cherry Prunus pseudocerasus, (Rosaceae). Conserv. Genet. Resour. 2018, 10, 85–88. [Google Scholar] [CrossRef]
- Yi, G.X. Study on Population Variation and Phylogeography of Cerasus Serrulata; Nanjing Forestry University: Nanjing, China, 2018. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Excoffier, L.; Lischer, H.E. Arlequin suite version 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Clement, M.; Podada, D.; Crandall, K.A. TCS: A computer program to estimate gene genealogies. Mol. Ecol. 2000, 9, 1657–1659. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Harpending, H.C. Signature of ancient population growth in a low- resolution mitochondrial DNA mismatch distribution. Hum. Biol. 1994, 66, 591–600. [Google Scholar]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef]
- Nogués-Bravo, D. Predicting the past distribution of species climatic niches. Glob. Ecol. Biogeogr. 2009, 18, 521–531. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, Y.; Chen, T.; Chen, Q.; Wang, L.; Liu, Z.S.; Wang, H.; Xie, R.; He, W.; Li, M.; et al. Evolution of Rosaceae Plastomes Highlights Unique Cerasus Diversification and Independent Origins of Fruiting Cherry. Front. Plant Sci. 2021, 12, 736053. [Google Scholar] [CrossRef] [PubMed]
- Wehr, W.C.; Hopkins, D.Q. The Eocene orchards and gardens of Republic, Washington. Wash. Geol. 1994, 22, 27–34. [Google Scholar]
- Xiang, Y.Z.; Huang, C.H.; Hu, Y.; Wen, J.; Li, S.S.; Yi, T.S.; Chen, H.; Xiang, J.; Ma, H. Evolution of Rosaceae fruit types based on nuclear phylogeny in the context of geological times and genome duplication. Mol. Biol. Evol. 2017, 34, 262–281. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Smith, T.; Liu, C.J.; Awasthi, N.; Yang, J.; Wang, Y.F.; Cheng, S.L. Endocarps of Prunus (Rosaceae: Prunoideae) from the early Eocene of Wutu, Shandong Province, China. Taxon 2011, 60, 555–564. [Google Scholar] [CrossRef]
- Chin, S.W.; Shaw, J.; Haberle, R.; Wen, J.; Potter, D. Diversification of almonds, peaches, plums and cherries-molecular systematics and biogeographic history of Prunus (Rosaceae). Mol. Phylogenet. Evol. 2014, 76, 34–48. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree 2009, 1.3.1. Available online: http://tree.bio.ed.ac.uk/software/figtree (accessed on 20 February 2023).
- Verde, I.; The International Peach Genome Initiative; Abbott, A.G.; Scalabrin, S.; Jung, S.; Shu, S.; Marroni, F.; Zhebentyayeva, T.; Dettori, M.T.; Grimwood, J.; et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat. Genet. 2013, 45, 487–494. [Google Scholar] [CrossRef]
- Hodel, R.; Zimmer, E.; Wen, J. A phylogenomic approach resolves the backbone of Prunus (Rosaceae) and identifies signals of hybridization and allopolyploidy. Mol. Phylogenet. Evol. 2021, 160, 107118. [Google Scholar] [CrossRef]
- Petit, R.J.; Duminil, J.; Fineschi, S.; Hampe, A.; Salvini, D.; Vendramin, G.G. Invited review: Comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Mol. Ecol. 2005, 14, 689–701. [Google Scholar] [CrossRef]
- Varvio, S.L.; Chakraborty, R.; Nei, M. Genetic variation in subdivided populations and conservation genetics. Heredity 1986, 57, 189–198. [Google Scholar] [CrossRef]
- Qiu, Y.X.; Guan, B.C.; Fu, C.X.; Comes, H.P. Did glacials and/or interglacials promote allopatric incipient speciation in East Asian temperate plants? Phylogeographic and coalescent analyses on refugial isolation and divergence in Dysosma versipellis. Mol. Phylogenet. Evol. 2009, 51, 281–293. [Google Scholar] [CrossRef]
- Zhang, Q.; Chiang, T.; George, M.; Liu, J.Q.; Abbott, R.J. Phylogeography of the Qinghai-Tibetan Plateau endemic Juniperus przewalskii (Cupressaceae) inferred from chloroplast DNA sequence variation. Mol. Ecol. 2010, 14, 3513–3524. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, G.M. Post-glacial re-colonization of European biota. Biol. J. Linn. Soc. 1999, 68, 87–112. [Google Scholar] [CrossRef]
- Wang, Y.H.; Jiang, W.M.; Hans, P.C.; Hu, F.S.; Qiu, Y.-X.; Fu, C.-X. Molecular phylogeography and ecological niche modelling of a widespread herbaceous climber, Tetrastigma hemsleyanum (Vitaceae): Insights into Plio-Pleistocene range dynamics of evergreen forest in subtropical China. New Phytol. 2015, 206, 852–867. [Google Scholar] [CrossRef]
- Qiu, X.Y.; Lu, X.Q.; Zhang, H.Y.; Cao, N.Y. Phylogeography of East Asia’s Tertiary relict plants: Current progress and future prospects. Biodivers. Sci. 2017, 25, 136–146. [Google Scholar] [CrossRef]
- Arenas, M.; Ray, N.; Currat, M.; Excoffier, L. Consequences of range contractions and range shifts on molecular diversity. Mol. Biol. Evol. 2012, 29, 207–218. [Google Scholar] [CrossRef]
- Mona, S.; Ray, N.; Arenas, M.; Excoffier, L. Genetic consequences of habitat fragmentation during a range expansion. Heredity 2014, 112, 291–299. [Google Scholar] [CrossRef]
- Alvarado-Serrano, D.F.; Knowles, L.L. Ecological niche models in phylogeographic studies: Applications, advances and precautions. Mol. Ecol. Resour. 2014, 14, 233–248. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxa | Code | Country | Location | GPS | Altitude/m | Sample Size |
|---|---|---|---|---|---|---|
| P. conradinae | EMS | China | Mount Emei, Leshan City, Sichuan Province | 103.4680 E 29.5770 N | 680 | 14 |
| HFG | China | Heifeng Valley, Heishan Town, Chongqing City | 106.9800 E 28.8690 N | 580 | 26 | |
| DWS | China | Dawei Mountain, Changsha City, Hunan Province | 114.1640 E 28.3620 N | 692 | 30 | |
| MYS | China | Mingyue Mountain, Yichun City, Jiangxi Provin | 114.2970 E 27.5870 N | 1100 | 8 | |
| DBS | China | Dabie Mountain, Lu ‘an City, Anhui Province | 116.2010 E 31.1300 N | 890 | 21 | |
| WYS | China | Wuyi Mountain, Nanping City, Fujian Province | 117.9630 E 27.6680 N | 980 | 11 | |
| QLF | China | Qingliangfeng Peak, Lin ‘an City, Zhejiang Province | 118.9140 E 30.1150 N | 1050 | 17 | |
| ZXTC | China | Tianchi Lake, West Zhejiang, Lin ‘an City, Zhejiang Province | 119.1270 E 30.3000 N | 1200 | 18 | |
| DYH | China | Dayang Lake in Lishui City, Zhejiang Province | 119.7480 E 27.8740 N | 1230 | 19 | |
| P. conradinae var. ruburm | FHC | China | Phoenix Pool, Yichang City, Hubei Province | 111.8470 E 31.1440 N | 780 | 26 |
| GXS | China | Gexian Mountain, Xianning City, Hubei Province | 114.0710 E 29.6380 N | 680 | 28 | |
| P. conradinae var. pubescens | WCP | China | Wangcheng slope, Enshi Autonomous Prefecture, Hubei Province | 109.4520 E 30.3480 N | 850 | 26 |
| Node | Mean Values/Standard Deviation Used at Calibration Points | References |
|---|---|---|
| N1# Rosaceae Crown | 90.18/0.05 | Zhang et al., 2017 [38] |
| N2# (Tribe Maleae + Tribe Spiraeeae) + | 75.62/0.05 | Zhang et al., 2017; Zhang et al., 2021 [10,41] |
| Tribe Amygdaleae | ||
| N3# Tribe Amygdaleae | 68.58/0.01 | Wehr and Hopkins, 1994; Xiang et al., 2017; Zhang et al., 2021 [41,42,43] |
| N4# Node Prunus | 55.0/0.09 | Li et al., 2011; Chin et al., 2014 [44,45] |
| N5# Node Sub.g Cerasus | 28.21/0.05 | Zhang et al., 2021 [41] |
| Haplotypes | Number of Individuals | Frequencies in Individuals(%) | Number of Populations | Frequencies in Populations(%) | Geographical Distributions |
|---|---|---|---|---|---|
| Hap1 | 74 | 30.33% | 7 | 58.33% | GXS DWS MYS DBS WYS QLF ZXTC |
| Hap2 | 4 | 1.64% | 1 | 8.33% | GXS |
| Hap3 | 17 | 6.97% | 3 | 25.00% | WYS QLF ZXTC |
| Hap4 | 9 | 3.69% | 4 | 33.33% | DWS WYS QLF ZXTC |
| Hap5 | 3 | 1.23% | 2 | 16.67% | WYS QLF |
| Hap6 | 8 | 3.28% | 3 | 25.00% | DWS ZXTC DYH |
| Hap7 | 60 | 24.59% | 8 | 66.67% | EMS HFG WCP FHC GXS MYS WYS DYH |
| Hap8 | 6 | 2.46% | 4 | 33.33% | HFG WCP FHC GXS |
| Hap9 | 6 | 2.46% | 2 | 16.67% | FHC DYH |
| Hap10 | 2 | 0.82% | 1 | 8.33% | MYS |
| Hap11 | 2 | 0.82% | 2 | 16.67% | GXS DWS |
| Hap12 | 4 | 1.64% | 2 | 16.67% | GXS DWS |
| Hap13 | 5 | 2.05% | 1 | 8.33% | GXS |
| Hap14 | 5 | 2.05% | 1 | 8.33% | GXS |
| Hap15 | 23 | 9.43% | 4 | 33.33% | EMS HFG WCP FHC |
| Hap16 | 7 | 2.87% | 1 | 8.33% | WCP |
| Hap17 | 7 | 2.87% | 1 | 8.33% | FHC |
| Hap18 | 2 | 0.82% | 1 | 8.33% | HFG |
| Ribotypes | Number of individuals | Frequencies in individuals(%) | Number of populations | Frequencies in populations(%) | Geographical Distributions |
| Rib 1 | 47 | 19.75% | 4 | 33.33% | EMS HFG WCP FHC |
| Rib 2 | 16 | 6.72% | 3 | 25.00% | EMS HFG WCP |
| Rib 3 | 28 | 11.76% | 2 | 16.67% | WCP FHC |
| Rib 4 | 85 | 35.71% | 7 | 58.33% | GXS DWS MYS WYS QLF ZXTC DYH |
| Rib 5 | 22 | 9.24% | 6 | 50.00% | GXS MYS WYS QLF ZXTC DYH |
| Rib 6 | 4 | 1.68% | 1 | 8.33% | GXS |
| Rib 7 | 2 | 0.84% | 2 | 16.67% | GXS DYH |
| Rib 8 | 3 | 1.26% | 3 | 25.00% | GXS WYS QLF |
| Rib 9 | 4 | 1.68% | 3 | 25.00% | GXS WYS DYH |
| Rib10 | 2 | 0.84% | 1 | 8.33% | MYS |
| Rib11 | 25 | 10.50% | 4 | 33.33% | DBS QLF ZXTC DYH |
| Population Code | Pop. Size | Hd | Pi × 10−3 | Haplotypes/Ribotypes (no. of Individuals) | |
|---|---|---|---|---|---|
| Genetic diversity parameters of sampled populations and their chloroplast genes | |||||
| 1 | EMS | 1 4 | 0.538 | 0.680 | H7(9)H13(1)H15(4) |
| 2 | HFG | 26 | 0.683 | 0.960 | H7(15)H8(2)H15(7)H18(2) |
| 3 | WCP | 26 | 0.686 | 0.810 | H7(10)H15(9)H16(7) |
| 4 | FHC | 26 | 0.785 | 1.780 | H7(10)H8(1)H9(5)H15(3)H17(7) |
| 5 | GXS | 28 | 0.680 | 0.540 | H1(15)H2(1)H7(1)H11(1)H12(1)H13(4)H14(5) |
| 6 | DWS | 30 | 0.687 | 0.950 | H1(21)H4(1)H6(3)H8(1)H11(1)H12(3) |
| 7 | MYS | 8 | 0.607 | 0.690 | H1(5)H7(1)H10(2) |
| 8 | DBS | 21 | 0.257 | 0.230 | H1(18)H2(3) |
| 9 | WYS | 11 | 0.836 | 1.113 | H1(4)H3(2)H4(2)H5(2)H7(1) |
| 10 | QLF | 17 | 0.500 | 1.070 | H1(2)H3(12)H4(2)H5(1) |
| 11 | ZXTC | 18 | 0.699 | 1.010 | H1(9)H3(3)H4(4)H6(2) |
| 12 | DYH | 19 | 0.526 | 0.700 | H6(3)H7(13)H8(2)H9(1) |
| Southwest China | 40 | 0.627 | 1.050 | H7(24)H8(2)H13(1)H15(11)H18(2) | |
| Central China | 110 | 0.866 | 1.240 | H1(41)H2(1)H7(21)H8(2)H9(5)H11(2)H12(4) H13(4)H14(5)H15(12)H16(7)H17(7) | |
| East China | 94 | 0.774 | 1.150 | H1(36)H2(1)H4(1)H6(3)H7(21)H8(2)H9(5)H10(2) | |
| Mean | 0.623 | 0.878 | |||
| All | 244 | 0.830 | 1.04 | ||
| Genetic diversity parameters of sampled populations and their nuclear genes (ITS) | |||||
| 1 | EMS | 15 | 0.533 | 0.186 | R1(7), R2(8) |
| 2 | HFG | 24 | 0.431 | 0.150 | R1(17), R2(7) |
| 3 | WCP | 26 | 0.538 | 0.115 | R1(10), R2(1), R3(15) |
| 4 | FHC | 26 | 0.520 | 0.091 | R1(13), R3(13) |
| 5 | GXS | 28 | 0.667 | 0.325 | R4(15), R5(6), R6(4), R7(1), R8(1), R9(1) |
| 6 | DWS | 25 | 0.080 | 0.014 | R4(24), R10(1) |
| 7 | MYS | 8 | 0.250 | 0.044 | R4(7), R5(1) |
| 8 | DBS | 21 | 0.000 | 0.000 | R11(21) |
| 9 | WYS | 11 | 0.564 | 0.120 | R4(7), R5(3), R8(1) |
| 10 | QLF | 17 | 0.728 | 0.311 | R4(7), R5(6), R8(1), R9(2), R11(1) |
| 11 | ZXTC | 18 | 0.464 | 0.368 | R4(13), R5(3), R11(2) |
| 12 | DYH | 19 | 0.579 | 0.269 | R4(12), R5(4), R7(1), R9(1), R11(1) |
| Southwest China | 39 | 0.486 | 0.169 | R1(24), R2(15) | |
| Central China | 105 | 0.745 | 0.756 | R1(23), R2(1), R3(28), R4(39), R5(6), R6(4), R7(1), R8(1), R9(1), R10(1) | |
| East China | 94 | 0.666 | 0.669 | R4(46), R5(17), R7(1), R8(2), R9(3), R11(25) | |
| All | 238 | 0.798 | 0.886 | R1(24), R2(15) | |
| Source of Variation | d.f. | Sum of Squares | Variance Components | Percentage of Variation | Fixation Indices | GST/NST | ||
|---|---|---|---|---|---|---|---|---|
| chloroplast DNA fragments | ||||||||
| All groups | ||||||||
| Among populations | 11 | 1560.531 | 6.69964 | 48.89 | FST = 0.48886 | 0.28176/0.29843 (p < 0.05) | ||
| Within populations | 232 | 1625.161 | 7.00501 | 51.11 | ||||
| Southwest China | ||||||||
| Among populations | 1 | 0.402 | 0.1090 | 11.01 | FST = 0.08126 | 0.072/0.081 (p < 0.05) | ||
| Within populations | 38 | 36.651 | 0.96451 | 88.99 | ||||
| Central China | ||||||||
| Among populations | 3 | 27.065 | 0.29098 | 22.04 | FST = 0.22038 | 0.18051/0.22810 (p < 0.05) | ||
| Within populations | 106 | 109.116 | 1.02940 | 77.96 | ||||
| East China | ||||||||
| Among populations | 5 | 1173.349 | 14.16449 | 46.05 | FST = 0.46046 | 0.30473/0.33970 (p < 0.05) | ||
| Within populations | 88 | 1460.534 | 16.59698 | 53.95 | ||||
| Southwest & Central & East | ||||||||
| Among regions | 2 | 359.203 | 0.42291 | 3.06 | FSC = 0.47782 | |||
| Within regions | 9 | 1201.328 | 6.41002 | 46.32 | FST =0.49378 | |||
| Within populations | 232 | 1625.161 | 7.00501 | 50.62 | FCT =0.03056 | |||
| nuclear DNA fragments | ||||||||
| All groups | ||||||||
| Among populations | 11 | 499.251 | 2.28341 | 82.51 | FST = 0.74918 | 0.37339/0.74924 | ||
| Within populations | 226 | 109.380 | 0.48398 | 17.49 | (p < 0.05) | |||
| Southwest China | ||||||||
| Among populations | 1 | 1.213 | 0.03707 | 6.55 | FST = 0.06157 | 0.03221/0.06157 | ||
| Within populations | 37 | 19.556 | 0.52853 | 93.45 | (p < 0.05) | |||
| Central China | ||||||||
| Among populations | 3 | 255.125 | 3.23420 | 94.38 | FST = 0.85663 | 0.38164/0.85743 | ||
| Within populations | 101 | 19.451 | 0.19258 | 5.62 | (p < 0.05) | |||
| East China | ||||||||
| Among populations | 5 | 126.356 | 1.60432 | 73.73 | FST = 0.73734 | 0.29616/0.55987 | ||
| Within populations | 88 | 50.293 | 0.57151 | 26.27 | (p < 0.05) | |||
| Southwest & Central & East | ||||||||
| Among regions | 2 | 197.775 | 0.46342 | 16.51 | FSC = 0.67382 | |||
| Within regions | 12 | 577.426 | 1.57873 | 56.25 | FST = 0.72768 | |||
| Within populations | 461 | 352.317 | 0.76424 | 27.23 | FCT = 0.16513 | |||
| Groups | Tajima’s D (p-Value) | Fu’s Fs (p-Value) | Demographic Expansion | Spatial Expansion | ||
|---|---|---|---|---|---|---|
| (SSD) (p-Value) | Raggedness Index (p-Value) | (SSD) (p-Value) | Raggedness Index (p-Value) | |||
| Based on the detection results of chloroplast DNA fragments | ||||||
| 12 populations 244 individuals | 0.09900 | 0.10000 | 0.38800 | 0.44200 | 0.26900 | 0.43600 |
| Southwest China | 0.26400 | 0.64200 | 0.14900 | 0.10000 | 0.19300 | 0.27900 |
| Central China | 0.24400 | 0.08000 | 0.12400 | 0.39100 | 0.25400 | 0.43400 |
| East China | 0.40800 | 0.50900 | 0.68500 | 0.84200 | 0.55700 | 0.87500 |
| Based on the detection results of nuclear DNA fragments | ||||||
| 12 populations 238 individuals | 0.35291 | N.A. | 0.17772 | 0.03300 | 0.08500 | 0.12093 |
| Southwest China | 1.00000 | 0.95300 | 0.32324 | 0.00000 | 0.12400 | 0.32879 |
| Central China | 0.98600 | 0.79100 | 0.12764 | 0.02900 | 0.21900 | 0.17021 |
| East China | 0.62164 | N.A. | 0.28450 | 0.11800 | 0.56100 | 0.39557 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, J.; Yi, X.; Wang, X.; Li, M.; Chen, X.; Gao, S.; Fu, W.; Qian, S.; Zeng, X.; Yun, Y. Population Variation and Phylogeography of Cherry Blossom (Prunus conradinae) in China. Plants 2024, 13, 974. https://doi.org/10.3390/plants13070974
Dong J, Yi X, Wang X, Li M, Chen X, Gao S, Fu W, Qian S, Zeng X, Yun Y. Population Variation and Phylogeography of Cherry Blossom (Prunus conradinae) in China. Plants. 2024; 13(7):974. https://doi.org/10.3390/plants13070974
Chicago/Turabian StyleDong, Jingjing, Xiangui Yi, Xianrong Wang, Meng Li, Xiangzhen Chen, Shucheng Gao, Wenyi Fu, Siyu Qian, Xinglin Zeng, and Yingke Yun. 2024. "Population Variation and Phylogeography of Cherry Blossom (Prunus conradinae) in China" Plants 13, no. 7: 974. https://doi.org/10.3390/plants13070974