
Proteomic Discovery and Validation of Novel Fluid Biomarkers for Improved Patient Selection and Prediction of Clinical Outcomes in Alzheimer’s Disease Patient Cohorts


Abstract
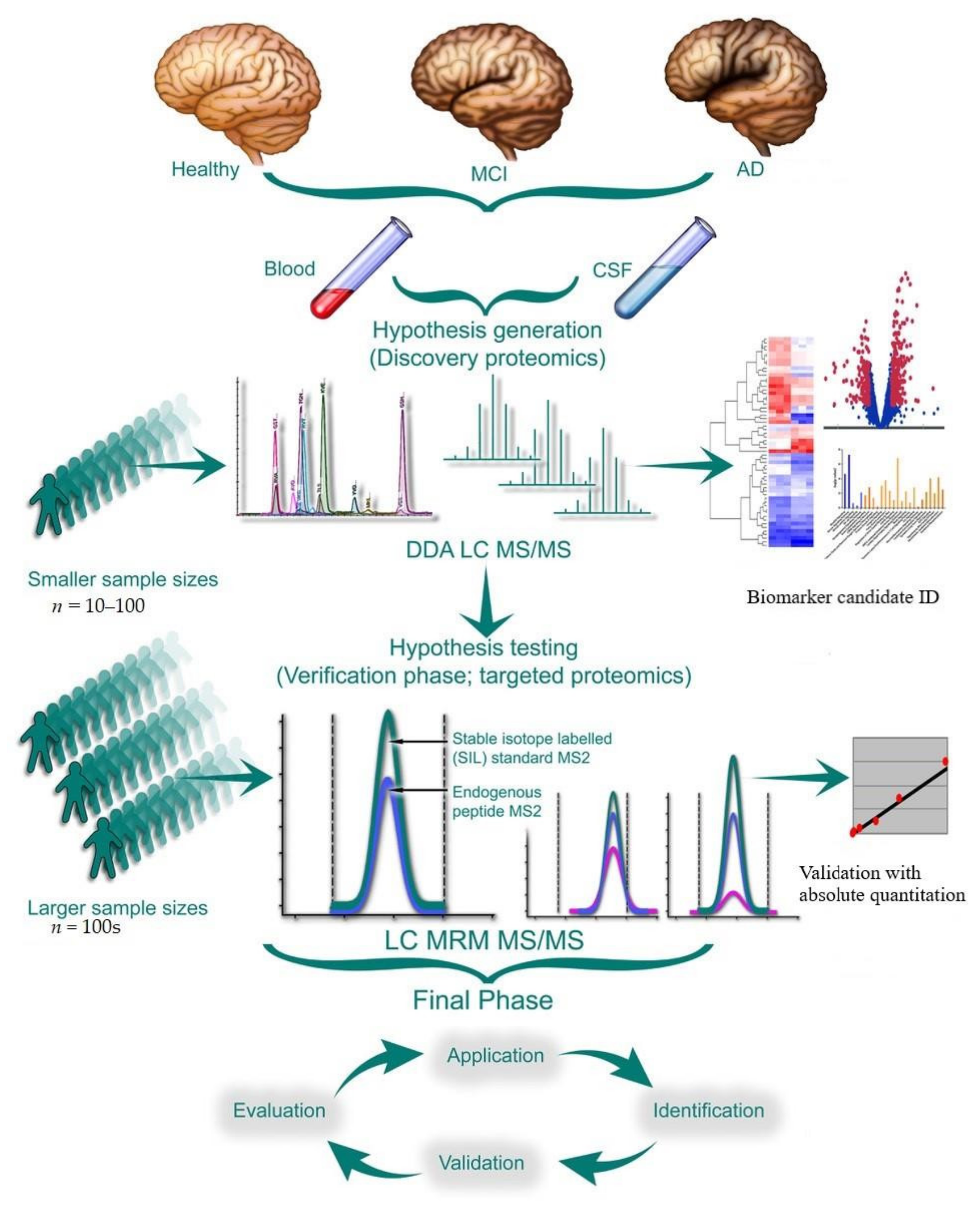
:1. Introduction
2. Clinically Validated Fluid Biomarkers for AD
2.1. AD Epidemiology
2.2. AD Pathology and Diagnosis
2.3. Clinical Utility of CSF Aβ42, Total Tau, and Phospho-Tau
2.4. The Unmet Medical Need
3. Mass Spectrometry-Based Discovery Proteomics
3.1. Bottom-Up Shotgun Proteomics (DDA vs. DIA)
3.2. Quantitative Proteomics for Differential Feature Identification
3.2.1. Labeling Strategies for Quantitative Proteomic Comparisons
3.2.2. Label-Free Feature Extraction and Quantitation
3.3. Data Processing and Bioinformatics
3.4. Analytical Instrument Considerations
3.5. Emerging Technologies
4. Biofluid Sample Preparation
4.1. Considerations for Sample Integrity
4.2. CSF vs. Blood
4.3. Immunodepletion
5. Experimental Design
5.1. Analytical Validation, Optimization, and Quality Control
5.2. Biomarker Validation
5.2.1. Validation of Discovery Proteomic Data Using Targeted Quantification

{kind=link}
| Discovery Proteomics Studies | |||
|---|---|---|---|
| Sample | LC MS Technique | Summary | Ref. |
| Plasma AD (n = 17) MCI (n = 12) Control (n = 11) | IP-MS coupled to MALDI-TOF | Immuno-Affinity purification (IP) MS method developed to measure Aβs; Aβ1–40 and Aβ1–42) and Aβ approximate peptides. APP/Aβ (−3–40)/Aβ1–42 ratio was increased in amyloid PET-positive AD patients and was proposed as biomarker to surrogate cerebral amyloid deposition. | [164] |
| CSF AD (n = 100) MCI (n = 40) Control (n = 80) | Label free LC MS | Anti-neurogranin antibodies were developed and used to show a marked increased level of neurogranin in AD dementia as well as MCI. | [165] |
| CSF AD patients (n = 14) Control (n = 14) | IP-PRM-MS | Significantly higher levels of CSF lysosomal protein LAMP2 were reported in AD patients when compared to controls | [166] |
| CSF Familial AD mutation carries (PSEN1 and APP, n = 14) Non-carriers (n = 5) | Label free LC MS | Comparative analysis identified 56 significantly differentially-expressed proteins between groups. Fourteen of these aligned with the previous findings. Novel proteins reported include calsyntenin-3, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor, CD99 antigen, di-N-acetyl-chitobiase, and secreted phosphoprotein-1. Protein expression changes in symptomatic and asymptomatic mutation carriers overlapped with those seen in late-onset AD. | [167] |
| CSF AD patients (n = 8) Controls (n = 8) | TMT labeling coupled to LC MS | The integrated proteomic and endopeptidomic approach simultaneously analyzed the abundances of 437 endogenous peptides and 374 proteins. The proteins that differed between groups include mesothelin, Ig alpha-1 chain C region, neurexin-1-beta, N-acetyllactosaminide beta-1,3-N-acetylglucosaminyltransferase, neurosecretory protein VGF, isoform 3 of neurotrimin, metalloproteinase inhibitor 2, and UPF0454 protein C12orf49. | [168] |
| CSF and cultured cells AD patients (n = 3) Control (n = 3) | CSF and cells combined in the same TMT multiplexed workflow | The optimized TMTcalibrator workflow allowed identification of lowly abundant peptides. Of the 77 proteins identified, 41 that are regulated in AD hadn’t been previously reported. | [169] |
| CSF AD patients (n = 20) Control (n = 20) | Endopeptidomic approach with stepwise protein and peptide precipitation followed by MALDI TOF MS and nLC MS | Peptides from VGF nerve growth factor-inducible precursor and α-2-HS-glycoprotein were downregulated in AD and a peptide from complement C4 factor and an O-glycosylated peptide from α-2-HS glycoprotein were found to be elevated | [170] |
| CSF Healthy volunteers (n = 50) | Endopeptidomic approach with TMT labeling coupled to LC MS | Changes in CSF peptidome were measured longitudinally following administration of a γ-secretase inhibitor. Many peptides showed dose-dependent changes in expression, including one derived from APP and one from amyloid precursor-like protein-1, which are known γ-secretase substrates. | [171] |
| CSF Pooled aliquots (n = 14) | Label free LC MS | Quantitative label-free proteomic technique coupled to multi-affinity fractionation was used to assess technical variability as well as inter-subject variation. The technique was also evaluated for its ability to distinguish samples based on the dried biomarker criteria | [172] |
| CSF Dementia patients (n = 159) Controls (n = 17) | CE MS to identify differential peptide pattern for early differential diagnosis of various dementias | Using CSF measurements of A β 42, tau, and phospho-tau, the AD pattern was diagnosed with a sensitivity of 87% and a specificity of 83%. Potential synaptic biomarkers identified: Apo-J, chromogranin A, phospholemman, synaptic protein-like proSAAS and neuronal secretory protein VGF | [173] |
| CSF AD patients (n = 4) Controls (n = 22) | Label free LC MS | Aβ42 to Aβ40 ratio was estimated in PSEN1 mutant AD using surrogate amyloid precursor-like protein-1-derived Aβ-like peptide (APL1β), including APL1β28. Relatively high ratio of CSF Aβ42 surrogate in PSEN1 mutant AD without an increase of Aβ42 secretion in the brain. | [174] |
| CSF (n = 2) | Label free LC MS for extracellular vesicles (EV) characterization | Exosomal markers identified were alixand syntenin-1, heat shock proteins and tetraspanins and several brain -derived proteins. Known biomarkers of neurodegeneration were also identified in the EV fractions., e.g., amyloid precursor protein, the prion protein, and DJ-1 | [175] |
| CSF Postmortem CSF (n = 4) Antemortem CSF (n = 4) | TMT 6-plex coupled to LC MS | Discovery analyses found 78 identified proteins to be significantly upregulated in post-mortem CSF samples when compared to antemortem. Previously identified brain damage biomarkers were identified like glial fibrillary acidic protein (GFAP), protein S100B, and protein DJ-1 (PARK7) | [176] |
| Plasma Non-demented controls (ND, n = 36) Non demented subjects with AD family history (ND-FH, n = 44) AD (n = 40) | Label free LC MS | Aβ-binding proteins circulating in the plasma were isolated and identified by LC MS. Many apolipoproteins were identified, i.e., apoA-I, apoB-100, apoC-III, and apoE. ApoA-I was reduced in AD and was proposed as an AD biomarker. ApoC-III was reduced in both ND-FH and AD and was proposed as a predictive marker for AD | [177] |
| Plasma Cohort 1 AD (n = 24) MCI (n = 261) Control (n = 411) Cohort 2 MCI (n = 180) Control (n = 153) | iTRAQ coupled to LC MS | AD-relevant biological pathways enriched in MCI included complement system, the coagulation cascade, lipid metabolism, and metal and vitamin D and E transport. Significant downregulation of potential markers fibronectin and C1 inhibitor was seen in the MCI cohorts. | [178] |
| Plasma AD (n = 15) MCI (n = 15) Control (n = 15) Validation cohort AD (n = 60) Control (n = 35) | Isobaric labeling coupled to LC MS | Plasma levels of gelsolin were found to be decreased in AD subjects when compared to controls. This finding was validated via western blotting in the bigger validation cohort. However, additional validation from three different regions of the brain failed to replicate this finding. | [179] |
| Plasma AD (n = 15) Control (n = 15) | iTRAQ coupled to LC MS | Differential expression of zinc-alpha-2-glycoprotein (AZGP1), fibulin-1 (FBLN1), platelet basic protein (PPBP), thrombospondin-1 (THBS1), S100 calcium-binding protein A8 (S100A8), and S100 calcium-binding protein A9 (S100A9) seen in the AD patients when compared to controls. | [180] |
| Plasma Stable MCI (n = 58) Progressive MCI (n = 34) Control (n = 23) AD (n = 31) | Label free LC MS | Both inflammation mediating proteins and pro-inflammatory IgG Fc glycoforms were significantly upregulated in AD subjects. | [181] |
| CSF Delirium (n = 17) AD (n = 17) Control (n = 8) | iTRAQ coupled to LC MS | Discovery analyses of patients with delirium, a risk factor for development of dementia and patients with mild AD identified several interesting protein families, including apolipoproteins, secretogranins, chromogranins, clotting factors, serine protease inhibitors, and acute-phase response elements. | [182] |
5.2.2. Higher Throughput Quantitative Assays for Use in Validation Study Cohorts
5.3. Multisite Variability Assessment: Quantitative Proteomic Data Reporting, Sharing, and the Need for Standardization
6. Case Study—Longitudinal Proteomic Changes in CSF from ADNI: Towards Better Defining the Trajectory of Early Alzheimer’s Disease
7. The Promise of Fluid Biomarkers of CNS-Related Diseases
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Mattsson, N.; Carrillo, M.C.; Dean, A.; Devous, M.D., Sr.; Nikolcheva, T.; Pesini, P.; Salter, H.; Potter, W.Z.; Sperling, R.S.; Bateman, R.; et al. Revolutionizing Alzheimer’s disease and clinical trials through biomarkers. Alzheimers Dement. 2015, 1, 412–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewczuk, P.; Riederer, P.; O’Bryant, S.E.; Verbeek, M.M.; Dubois, B.; Visser, P.J.; Jellinger, K.A.; Engelborghs, S.; Ramirez, A.; Parnetti, L.; et al. Cerebrospinal fluid and blood biomarkers for neurodegenerative dementias: An update of the Consen-sus of the Task Force on Biological Markers in Psychiatry of the World Federation of Societies of Biological Psychiatry. World J. Biol. Psychiatry 2018, 19, 244–328. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Disease Facts and Figures. Available online: https://www.alz.org/alzheimers-dementia/facts-figures (accessed on 10 June 2022).
- Glenner, G.G.; Wong, C.W.; Quaranta, V.; Eanes, E.D. The amyloid deposits in Alzheimer’s disease: Their nature and pathogenesis. Appl. Pathol. 1984, 2, 357–369. [Google Scholar] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Selkoe, D.J. Biochemistry of amyloid beta-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006262. [Google Scholar] [CrossRef]
- Kosik, K.S.; Joachim, C.L.; Selkoe, D.J. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1986, 83, 4044–4048. [Google Scholar] [CrossRef] [Green Version]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskele-tal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, K.; del Alonso, A.C.; Gondal, J.A.; Gong, C.-X.; Haque, N.; Khatoon, S.; Sengupta, A.; Wang, J.-Z.; Grundke-Iqbal, I. Mechanism of neurofibrillary degeneration and pharmacologic therapeutic approach. J. Neural. Transm. Suppl. 2000, 59, 213–222. [Google Scholar] [PubMed]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef]
- Reitz, C.; Brayne, C.; Mayeux, R. Epidemiology of Alzheimer disease. Nat. Rev. Neurol. 2011, 7, 137–152. [Google Scholar] [CrossRef]
- Dubois, B.; Feldman, H.H.; Jacova, C.; DeKosky, S.T.; Barberger-Gateau, P.; Cummings, J.L.; Delacourte, A.; Galasko, D.; Gauthier, S.; Jicha, G.A.; et al. Research Criteria for the Diagnosis of Alzheimer’s Disease: Revising the NINCDS–ADRDA Criteria. Lancet Neurol. 2007, 6, 734–746. [Google Scholar] [CrossRef]
- Petersen, R.C. Mild cognitive impairment as a diagnostic entity. J. Intern. Med. 2004, 256, 183–194. [Google Scholar] [CrossRef]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. J. Alzheimers Assoc. 2011, 7, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storandt, M.; Grant, E.A.; Miller, J.P.; Morris, J.C. Longitudinal course and neuropathologic outcomes in original vs revised MCI and in pre-MCI. Neurology 2006, 67, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Duara, R.; Loewenstein, D.A.; Potter, E.; Barker, W.; Raj, A.; Schoenberg, M.; Wu, Y.; Banko, J.; Potter, H.; Greig, M.T.; et al. Pre-MCI and MCI: Neuropsychological, Clinical, and Imaging Features and Progression Rates. Am. J. Geriatr. Psychiatry 2011, 19, 951–960. [Google Scholar] [CrossRef] [Green Version]
- Migliaccio, R.; Agosta, F.; Possin, K.L.; Canu, E.; Filippi, M.; Rabinovici, G.D.; Rosen, H.J.; Miller, B.L.; Gorno-Tempini, M.L. Mapping the Progression of Atrophy in Early- and Late-Onset Alzheimer’s Disease. J. Alzheimers Dis. 2015, 46, 351–364. [Google Scholar] [CrossRef]
- Koss, E.; Edland, S.; Fillenbaum, G.; Mohs, R.; Clark, C.; Galasko, D.; Morris, J.C. Clinical and neuropsychological differences between patients with earlier and later onset of Alzheimer’s disease: A CERAD analysis, Part XII. Neurology 1996, 46, 136–141. [Google Scholar] [CrossRef]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Hampel, H.; Molinuevo, J.L.; Blennow, K.; DeKosky, S.T.; Gauthier, S.; Selkoe, D.; Bateman, R.; et al. Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. Lancet Neurol. 2014, 13, 614–629. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Feldman, H.H.; Frisoni, G.B.; Hampel, H.; Jagust, W.J.; Johnson, K.A.; Knopman, D.S.; et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016, 87, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Motter, R.; Vigo-Pelfrey, C.; Kholodenko, D.; Barbour, R.; Johnson-Wood, K.; Galasko, D.; Chang, L.; Miller, B.; Clark, C.; Green, R. Reduction of beta-amyloid peptide42 in the cerebrospinal fluid of patients with Alzheimer’s disease. Ann. Neurol. 1995, 38, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Strozyk, D.; Blennow, K.; White, L.R.; Launer, L.J. CSF Abeta 42 levels correlate with amyloid-neuropathology in a population-based autopsy study. Neurology 2003, 60, 652–656. [Google Scholar] [CrossRef]
- Blennow, K.; Wallin, A.; Agren, H.; Spenger, C.; Siegfried, J.; Vanmechelen, E. Tau protein in cerebrospinal fluid: A biochemical marker for axonal degeneration in Alzheimer disease? Mol. Chem. Neuropathol. 1995, 26, 231–245. [Google Scholar] [CrossRef]
- Shaw, L.M.; Vanderstichele, H.; Knapik-Czajka, M.; Clark, C.M.; Aisen, P.S.; Petersen, R.C.; Blennow, K.; Soares, H.; Simon, A.; Lewczuk, P.; et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann. Neurol. 2009, 65, 403–413. [Google Scholar] [CrossRef] [Green Version]
- Hansson, O.; Zetterberg, H.; Buchhave, P.; Londos, E.; Blennow, K.; Minthon, L. Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: A follow-up study. Lancet Neurol. 2006, 5, 228–234. [Google Scholar] [CrossRef] [Green Version]
- Barthélemy, N.R.; Li, Y.; Joseph-Mathurin, N.; Gordon, B.A.; Hassenstab, J.; Benzinger, T.L.S.; Buckles, V.; Fagan, A.M.; Per-rin, R.J.; Goate, A.M.; et al. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of domi-nantly inherited Alzheimer’s disease. Nat. Med. 2020, 26, 398–407. [Google Scholar] [CrossRef]
- Barthélemy, N.R.; Li, Y.; Wang, G.; Fagan, A.M.; Morris, J.C.; Benzinger, T.L.S.; Goate, A.; Hassenstab, J.; Xiong, C.; Sato, C.; et al. P1-023: Mass Spectrometry–Based Measurement of Longitudinal Csf Tau Identifies Different Phosphorylated Sites That Track Distinct Stages of Presymptomatic Dominantly Inherited Ad. Alzheimers Dement. 2018, 14, P273–P274. [Google Scholar] [CrossRef]
- Barthélemy, N.R.; Horie, K.; Sato, C.; Bateman, R.J. Blood plasma phosphorylated-tau isoforms track CNS change in Alzheimer’s disease. J. Exp. Med. 2020, 217, e20200861. [Google Scholar] [CrossRef] [PubMed]
- Mattsson-Carlgren, N.; Janelidze, S.; Palmqvist, S.; Cullen, N.; Svenningsson, A.L.; Strandberg, O.; Mengel, D.; Walsh, D.M.; Stomrud, E.; Dage, J.L.; et al. Longitudinal plasma p-tau217 is increased in early stages of Alzheimer’s disease. Brain 2020, 143, 3234–3241. [Google Scholar] [CrossRef]
- Wennström, M.; Janelidze, S.; Nilsson, K.P.R.; Serrano, G.E.; Beach, T.G.; Dage, J.L.; Hansson, O.; Bank, T.N.B. Cellular localization of p-tau217 in brain and its association with p-tau217 plasma levels. Acta Neuropathol. Commun. 2022, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Frank, R.; Broich, K.; Teipel, S.J.; Katz, R.G.; Hardy, J.; Herholz, K.; Bokde, A.L.; Jessen, F.; Hoessler, Y.C.; et al. Biomarkers for Alzheimer’s disease: Academic, industry and regulatory perspectives. Nat. Rev. Drug Discov. 2010, 9, 560–574. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration (2018) Early Alzheimer’s Disease: Developing Drugs for Treatment; Draft Guidance for Industry. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/alzheimers-disease-developing-drugs-treatment-guidance-industy (accessed on 10 November 2021).
- European Medicines Agency, Committee for Medicinal Products for Human Use. Guideline on the Clinical Investigation of Medicines for the Treatment of Alzheimer’s Disease. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Scientifc_guideline/2018/02/WC500244609.pdf (accessed on 10 November 2021).
- Bertram, L.; Hampel, H. The role of genetics for biomarker development in neurodegeneration. Prog. Neurobiol. 2011, 95, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Zetzsche, T.; Rujescu, D.; Hardy, J.; Hampel, H. Advances and perspectives from genetic research: Development of biological markers in Alzheimer’s disease. Expert Rev. Mol. Diagn. 2010, 10, 667–690. [Google Scholar] [CrossRef] [PubMed]
- Teipel, S.J.; Grothe, M.; Lista, S.; Toschi, N.; Garaci, F.G.; Hampel, H. Relevance of Magnetic Resonance Imaging for Early Detection and Diagnosis of Alzheimer Disease. Med. Clin. N. Am. 2013, 97, 399–424. [Google Scholar] [CrossRef] [PubMed]
- Ewers, M.; Sperling, R.A.; Klunk, W.E.; Weiner, M.W.; Hampel, H. Neuroimaging markers for the prediction and early diagnosis of Alzheimer’s disease dementia. Trends Neurosci. 2011, 34, 430–442. [Google Scholar] [CrossRef] [Green Version]
- Blennow, K.; Dubois, B.; Fagan, A.M.; Lewczuk, P.; de Leon, M.J.; Hampel, H. Clinical utility of cerebrospinal fluid biomarkers in the diagnosis of early Alzheimer’s disease. Alzheimers Dement. 2014, 11, 58–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, H.M.; Carrillo, M.C.; Grodstein, F.; Henriksen, K.; Jeromin, A.; Lovestone, S.; Mielke, M.M.; O’Bryant, S.; Sarasa, M.; Sjogren, M.; et al. Developing novel blood-based biomarkers for Alzheimer’s disease. Alzheimers Dement. 2013, 10, 109–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teipel, S.J.; Sabri, O.; Grothe, M.; Barthel, H.; Prvulovic, D.; Buerger, K.; Bokde, A.L.; Ewers, M.; Hoffmann, W.; Hampel, H. Perspectives for Multimodal Neurochemical and Imaging Biomarkers in Alzheimer’s Disease. J. Alzheimers Dis. 2012, 33, S329–S347. [Google Scholar] [CrossRef] [Green Version]
- Beach, T.G.; Monsell, S.E.; Phillips, L.E.; Kukull, W. Accuracy of the Clinical Diagnosis of Alzheimer Disease at National Institute on Aging Alzheimer Disease Centers, 2005–2010. J. Neuropathol. Exp. Neurol. 2012, 71, 266–273. [Google Scholar] [CrossRef]
- Archer, M.C.; Hall, P.H.; Morgan, J.C. Accuracy of Clinical Diagnosis of Alzheimer’s Disease in Alzheimer’s Disease Centers (Adcs). Alzheimers Dement. J. Alzheimers Assoc. 2017, 13, P800–P801. [Google Scholar] [CrossRef]
- Martorana, A.; di Lorenzo, F.; Belli, L.; Sancesario, G.; Toniolo, S.; Sallustio, F.; Sancesario, G.M.; Koch, G. Cerebrospinal Fluid Aβ42 Levels: When Physiological Become Pathological State. CNS Neurosci. Ther. 2015, 21, 921–925. [Google Scholar] [CrossRef]
- Franklin, E.E.; Perrin, R.J.; Vincent, B.; Baxter, M.; Morris, J.C.; Cairns, N.J. Brain collection, standardized neuropathologic assessment, and comorbidity in Alzheimer’s Disease Neuroimaging Initiative 2 participants. Alzheimers Dement. 2015, 11, 815–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hjalmarsson, C.; Bjerke, M.; Andersson, B.; Blennow, K.; Zetterberg, H.; Åberg, N.D.; Olsson, B.; Eckerström, C.; Bokemark, L.; Wallin, A. Neuronal and Glia-Related Biomarkers in Cerebrospinal Fluid of Patients with Acute Ischemic Stroke. J. Central Nerv. Syst. Dis. 2014, 6, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Lattanzio, F.; Abu-Rumeileh, S.; Franceschini, A.; Kai, H.; Amore, G.; Poggiolini, I.; Rossi, M.; Baiardi, S.; McGuire, L.; Ladogana, A.; et al. Prion-specific and surrogate CSF biomarkers in Creutzfeldt-Jakob disease: Diagnostic accuracy in rela-tion to molecular subtypes and analysis of neuropathological correlates of p-tau and Aβ42 levels. Acta Neuropathol. 2017, 133, 559–578. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.R.; Miller, R.A.; Spellman, D.S. Mass Spectrometry-Based Biomarkers in Drug Development. Adv. Exp. Med. Biol. 2019, 1140, 435–449. [Google Scholar] [PubMed]
- Hosp, F.; Mann, M. A Primer on Concepts and Applications of Proteomics in Neuroscience. Neuron 2017, 96, 558–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartwell, L.H.; Hopfield, J.J.; Leibler, S.; Murray, A.W. From molecular to modular cell biology. Nature 1999, 402, C47–C52. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, M.R.; Sanchez, J.-C.; Gooley, A.A.; Appel, R.D.; Humphery-Smith, I.; Hochstrasser, D.F.; Williams, K.L. Progress with Proteome Projects: Why all Proteins Expressed by a Genome Should be Identified and How to Do It. Biotechnol. Genet. Eng. Rev. 1996, 13, 19–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aebersold, R.; Mann, M. Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. [Google Scholar] [CrossRef]
- Cravatt, B.F.; Simon, G.M.; Iii, J.R.Y. The biological impact of mass-spectrometry-based proteomics. Nature 2007, 450, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Kulak, N.A.; Nagaraj, N.; Cox, J. The Coming Age of Complete, Accurate, and Ubiquitous Proteomes. Mol. Cell 2013, 49, 583–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, J.R.; Ruse, C.I.; Nakorchevsky, A. Proteomics by Mass Spectrometry: Approaches, Advances, and Applications. Annu. Rev. Biomed. Eng. 2009, 11, 49–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, M.; Jensen, O.N. Proteomic analysis of post-translational modifications. Nat. Biotechnol. 2003, 21, 255–261. [Google Scholar] [CrossRef]
- Doll, S.; Burlingame, A.L. Mass Spectrometry-Based Detection and Assignment of Protein Posttranslational Modifications. ACS Chem. Biol. 2014, 10, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Gingras, A.-C.; Gstaiger, M.; Raught, B.; Aebersold, R. Analysis of protein complexes using mass spectrometry. Nat. Rev. Mol. Cell Biol. 2007, 8, 645–654. [Google Scholar] [CrossRef]
- Zhang, Y.; Fonslow, B.R.; Shan, B.; Baek, M.-C.; Yates, J.R., 3rd. Protein Analysis by Shotgun/Bottom-up Proteomics. Chem. Rev. 2013, 113, 2343–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelstrup, C.D.; Bekker-Jensen, D.B.; Arrey, T.N.; Hogrebe, A.; Harder, A.; Olsen, J.V. Performance Evaluation of the Q Exactive HF-X for Shotgun Proteomics. J. Proteome Res. 2018, 17, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Michalski, A.; Damoc, E.; Hauschild, J.P.; Lange, O.; Wieghaus, A.; Makarov, A.; Nagaraj, N.; Cox, J.; Mann, M.; Horning, S. Mass spectrometry-based proteomics using Q Exactive, a high-performance benchtop quadrupole Or-bitrap mass spectrometer. Mol. Cell. Proteom. 2011, 10, M111.011015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, J.R., 3rd. The revolution and evolution of shotgun proteomics for large-scale proteome analysis. J. Am. Chem. Soc. 2013, 135, 1629–1640. [Google Scholar] [CrossRef] [Green Version]
- Fenn, J.B.; Mann, M.; Meng, C.K.; Wong, S.F.; Whitehouse, C.M. Electrospray Ionization for Mass Spectrometry of Large Biomolecules. Science 1989, 246, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Fenn, J.B. Electrospray ion source. Another variation on the free-jet theme. J. Phys. Chem. 1984, 88, 4451–4459. [Google Scholar] [CrossRef]
- Tanaka, K.; Waki, H.; Ido, Y.; Akita, S.; Yoshida, Y.; Yoshida, T.; Matsuo, T. Protein and polymer analyses up to m/z 100,000 by laser ionization time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 1988, 2, 151–153. [Google Scholar] [CrossRef]
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347. [Google Scholar] [CrossRef] [PubMed]
- Tolley, L.; Jorgenson, J.W.; Moseley, M.A. Very High Pressure Gradient LC/MS/MS. Anal. Chem. 2001, 73, 2985–2991. [Google Scholar] [CrossRef] [PubMed]
- Hunt, D.F.; Henderson, R.A.; Shabanowitz, J.; Sakaguchi, K.; Michel, H.; Sevilir, N.; Cox, A.L.; Appella, E.; Engelhard, V.H. Characterization of Peptides Bound to the Class I MHC Molecule HLA-A2.1 by Mass Spectrometry. Science 1992, 255, 1261–1263. [Google Scholar] [CrossRef] [Green Version]
- Olsen, J.; Macek, B.; Lange, O.; Makarov, A.; Horning, S.; Mann, M. Higher-energy C-trap dissociation for peptide modification analysis. Nat. Methods 2007, 4, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Steen, H.; Mann, M. The abc’s (and xyz’s) of peptide sequencing. Nat. Rev. Mol. Cell Biol. 2004, 5, 699–711. [Google Scholar] [CrossRef]
- Eliuk, S.; Makarov, A. Evolution of Orbitrap Mass Spectrometry Instrumentation. Annu. Rev. Anal. Chem. 2015, 8, 61–80. [Google Scholar] [CrossRef] [PubMed]
- Andrews, G.L.; Simons, B.L.; Young, J.B.; Hawkridge, A.M.; Muddiman, D.C. Performance Characteristics of a New Hybrid Quadrupole Time-of-Flight Tandem Mass Spectrometer (TripleTOF 5600). Anal. Chem. 2011, 83, 5442–5446. [Google Scholar] [CrossRef] [Green Version]
- Beck, S.; Michalski, A.; Raether, O.; Lubeck, M.; Kaspar, S.; Goedecke, N.; Baessmann, C.; Hornburg, D.; Meier, F.; Paron, I.; et al. The Impact II, a Very High-Resolution Quadrupole Time-of-Flight Instrument (QTOF) for Deep Shotgun Proteomics. Mol. Cell. Proteom. 2015, 14, 2014–2029. [Google Scholar] [CrossRef] [Green Version]
- Harper, J.; Bennett, E.J. Proteome complexity and the forces that drive proteome imbalance. Nature 2016, 537, 328–338. [Google Scholar] [CrossRef] [Green Version]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Schwenk, J.M.; Omenn, G.S.; Sun, Z.; Campbell, D.S.; Baker, M.S.; Overall, C.M.; Aebersold, R.; Moritz, R.L.; Deutsch, E.W. The Human Plasma Proteome Draft of 2017: Building on the Human Plasma PeptideAtlas from Mass Spectrometry and Complementary Assays. J. Proteome Res. 2017, 16, 4299–4310. [Google Scholar] [CrossRef] [Green Version]
- Michalski, A.; Cox, J.; Mann, M. More than 100,000 Detectable Peptide Species Elute in Single Shotgun Proteomics Runs but the Majority is Inaccessible to Data-Dependent Lc−Ms/Ms. J. Proteome Res. 2011, 10, 1785–1793. [Google Scholar] [CrossRef]
- Gillet, L.C.; Leitner, A.; Aebersold, R. Mass Spectrometry Applied to Bottom-Up Proteomics: Entering the High-Throughput Era for Hypothesis Testing. Annu. Rev. Anal. Chem. 2016, 9, 449–472. [Google Scholar] [CrossRef]
- Ong, S.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expres-sion proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Ong, S.-E.; Foster, L.J.; Mann, M. Mass spectrometric-based approaches in quantitative proteomics. Methods 2003, 29, 124–130. [Google Scholar] [CrossRef]
- Thompson, A.; Schafer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Johnstone, R.; Mohammed, A.K.; Hamon, C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mix-tures by Ms/Ms. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef]
- Gerber, S.A.; Rush, J.; Stemman, O.; Kirschner, M.W.; Gygi, S.P. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc. Natl. Acad. Sci. USA 2003, 100, 6940–6945. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, J.D.; Paulo, J.A.; O’Brien, J.J.; Gygi, S.P. Proteome-Wide Evaluation of Two Common Protein Quantification Methods. J. Proteome Res. 2018, 17, 1934–1942. [Google Scholar] [CrossRef]
- Tabb, D.L.; Wang, X.; Carr, S.A.; Clauser, K.R.; Mertins, P.; Chambers, M.C.; Holman, J.D.; Wang, J.; Zhang, B.; Zimmerman, L.J.; et al. Reproducibility of Differential Proteomic Technologies in CPTAC Fractionated Xenografts. J. Proteome Res. 2015, 15, 691–706. [Google Scholar] [CrossRef]
- Nahnsen, S.; Bielow, C.; Reinert, K.; Kohlbacher, O. Tools for Label-free Peptide Quantification. Mol. Cell. Proteom. 2013, 12, 549–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and pro-teome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Claassen, M.; Reiter, L.; Hengartner, M.; Buhmann, J.M.; Aebersold, R. Generic Comparison of Protein Inference Engines. Mol. Cell. Proteom. 2012, 11, O110.007088. [Google Scholar] [CrossRef] [Green Version]
- Nesvizhskii, A.I.; Aebersold, R. Interpretation of shotgun proteomic data: The protein inference problem. Mol. Cell. Proteom. 2005, 4, 1419–1440. [Google Scholar] [CrossRef] [Green Version]
- Ting, Y.; Egertson, J.D.; Payne, S.; Kim, S.; MacLean, B.; Käll, L.; Aebersold, R.; Smith, R.D.; Noble, W.S.; MacCoss, M.J. Peptide-Centric Proteome Analysis: An Alternative Strategy for the Analysis of Tandem Mass Spectrometry Data. Mol. Cell. Proteom. 2015, 14, 2301–2307. [Google Scholar] [CrossRef] [Green Version]
- Wilm, M.; Mann, M. Analytical Properties of the Nanoelectrospray Ion Source. Anal. Chem. 1996, 68, 1–8. [Google Scholar] [CrossRef]
- Zhou, F.; Lu, Y.; Ficarro, S.B.; Webber, J.T.; Marto, J.A. Nanoflow Low Pressure High Peak Capacity Single Dimension LC-MS/MS Platform for High-Throughput, In-Depth Analysis of Mammalian Proteomes. Anal. Chem. 2012, 84, 5133–5139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacNair, J.E.; Lewis, K.C.; Jorgenson, J.W. Ultrahigh-Pressure Reversed-Phase Liquid Chromatography in Packed Capillary Columns. Anal. Chem. 1997, 69, 983–989. [Google Scholar] [CrossRef]
- Motoyama, A.; Venable, J.D.; Ruse, C.I.; Yates, J.R., 3rd. Automated ultra-high-pressure multidimensional protein identification technology (UHP-MudPIT) for improved peptide identification of proteomic samples. Anal. Chem. 2006, 78, 5109–5118. [Google Scholar] [CrossRef]
- Chen, C.J.; Chen, W.Y.; Tseng, M.C.; Chen, Y.R. Tunnel frit: A nonmetallic in-capillary frit for nanoflow ultra high-performance liquid chromatog-raphy-mass spectrometry applications. Anal. Chem. 2012, 84, 297–303. [Google Scholar] [CrossRef]
- Iwasaki, M.; Sugiyama, N.; Tanaka, N.; Ishihama, Y. Human proteome analysis by using reversed phase monolithic silica capillary columns with enhanced sensitivity. J. Chromatogr. A 2011, 1228, 292–297. [Google Scholar] [CrossRef]
- Shi, T.; Fillmore, T.L.; Gao, Y.; Zhao, R.; He, J.; Schepmoes, A.A.; Nicora, C.D.; Wu, C.; Chambers, J.L.; Moore, R.J.; et al. Long-Gradient Separations Coupled with Selected Reaction Monitoring for Highly Sensitive, Large Scale Targeted Protein Quantification in a Single Analysis. Anal. Chem. 2013, 85, 9196–9203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, M.W.; Keshishian, H.; Mani, D.R.; Gillette, M.A.; Carr, S.A. Simplified and Efficient Quantification of Low-abundance Proteins at Very High Multiplex via Targeted Mass Spectrometry. Mol. Cell. Proteom. 2014, 13, 1137–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Kuromitsu, J.; Oda, Y. Evaluation of Comprehensive Multidimensional Separations Using Reversed-Phase, Reversed-Phase Liquid Chromatography/Mass Spectrometry for Shotgun Proteomics. J. Proteome Res. 2008, 7, 1007–1011. [Google Scholar] [CrossRef]
- Gilar, M.; Olivova, P.; Daly, A.E.; Gebler, J.C. Orthogonality of Separation in Two-Dimensional Liquid Chromatography. Anal. Chem. 2005, 77, 6426–6434. [Google Scholar] [CrossRef]
- Essader, A.S.; Cargile, B.J.; Bundy, J.L.; Stephenson, J.L., Jr. A comparison of immobilized pH gradient isoelectric focusing and strong-cation-exchange chromatog-raphy as a first dimension in shotgun proteomics. Proteomics 2005, 5, 24–34. [Google Scholar] [CrossRef]
- Dai, J.; Shieh, C.H.; Sheng, Q.-H.; Zhou, A.H.; Zeng, R. Proteomic Analysis with Integrated Multiple Dimensional Liquid Chromatography/Mass Spectrometry Based on Elution of Ion Exchange Column Using pH Steps. Anal. Chem. 2005, 77, 5793–5799. [Google Scholar] [CrossRef]
- Zhou, H.; Dai, J.; Sheng, Q.-H.; Li, R.-X.; Shieh, C.-H.; Guttman, A.; Zeng, R. A fully automated 2-D LC-MS method utilizing online continuous pH and RP gradients for global proteome analysis. Electrophoresis 2007, 28, 4311–4319. [Google Scholar] [CrossRef] [PubMed]
- Gilar, M.; Olivova, P.; Daly, A.E.; Gebler, J. Two-dimensional separation of peptides using RP-RP-HPLC system with different pH in first and second separation dimensions. J. Sep. Sci. 2005, 28, 1694–1703. [Google Scholar] [CrossRef]
- Boersema, P.J.; Mohammed, S.; Heck, A.J.R. Hydrophilic interaction liquid chromatography (HILIC) in proteomics. Anal. Bioanal. Chem. 2008, 391, 151–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boersema, P.J.; Divecha, N.; Heck, A.J.R.; Mohammed, S. Evaluation and Optimization of ZIC-HILIC-RP as an Alternative MudPIT Strategy. J. Proteome Res. 2007, 6, 937–946. [Google Scholar] [CrossRef] [Green Version]
- Hao, P.; Guo, T.; Li, X.; Adav, S.S.; Yang, J.; Wei, M.; Sze, S.K. Novel application of electrostatic repulsion-hydrophilic interaction chromatography (ERLIC) in shotgun pro-teomics: Comprehensive profiling of rat kidney proteome. J. Proteome Res. 2010, 9, 3520–3526. [Google Scholar] [CrossRef] [PubMed]
- Hao, P.; Guo, T.; Sze, S.K. Simultaneous analysis of proteome, phospho- and glycoproteome of rat kidney tissue with elec-trostatic repulsion hydrophilic interaction chromatography. PLoS ONE 2011, 6, e16884. [Google Scholar] [CrossRef] [PubMed]
- Pfammatter, S.; Bonneil, E.; McManus, F.P.; Prasad, S.; Bailey, D.J.; Belford, M.; Dunyach, J.J.; Thibault, P. A Novel Differential Ion Mobility Device Expands the Depth of Proteome Coverage and the Sensitivi-ty of Multiplex Proteomic Measurements. Mol. Cell. Proteom. 2018, 17, 2051–2067. [Google Scholar] [CrossRef] [Green Version]
- Meier, F.; Beck, S.; Grassl, N.; Lubeck, M.; Park, M.A.; Raether, O.; Mann, M. Parallel Accumulation–Serial Fragmentation (Pasef): Multiplying Sequencing Speed and Sensitivity by Synchronized Scans in a Trapped Ion Mobility Device. J. Proteome Res. 2015, 14, 5378–5387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, F.; Brunner, A.D.; Koch, S.; Koch, H.; Lubeck, M.; Krause, M.; Goedecke, N.; Decker, J.; Kosinski, T.; Park, M.A.; et al. Online Parallel Accumulation–Serial Fragmentation (Pasef) with a Novel Trapped Ion Mobility Mass Spec-trometer. Mol. Cell. Proteom. 2018, 17, 2534. [Google Scholar] [CrossRef] [Green Version]
- Meier, F.; Geyer, P.E.; Winter, S.V.; Cox, J.; Mann, M. BoxCar acquisition method enables single-shot proteomics at a depth of 10,000 proteins in 100 minutes. Nat. Methods 2018, 15, 440–448. [Google Scholar] [CrossRef]
- Stolz, A.; Jooß, K.; Höcker, O.; Römer, J.; Schlecht, J.; Neusüß, C. Recent advances in capillary electrophoresis-mass spectrometry: Instrumentation, methodology and applications. Electrophoresis 2018, 40, 79–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, F.P.; Yates, J.R., III. Recent trends of capillary electrophoresis-mass spectrometry in proteomics research. Mass Spectrom. Rev. 2019, 38, 445–460. [Google Scholar] [CrossRef] [PubMed]
- Jansson, E.T. Strategies for analysis of isomeric peptides. J. Sep. Sci. 2017, 41, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Haselberg, R.; De Jong, G.J.; Somsen, G.W. CE-MS for the analysis of intact proteins 2010–2012. Electrophoresis 2012, 34, 99–112. [Google Scholar] [CrossRef]
- Pontillo, C.; Filip, S.; Borràs, D.M.; Mullen, W.; Vlahou, A.; Mischak, H. CE-MS-based proteomics in biomarker discovery and clinical application. Proteom. Clin. Appl. 2015, 9, 322–334. [Google Scholar] [CrossRef] [PubMed]
- Faserl, K.; Sarg, B.; Gruber, P.; Lindner, H.H. Investigating capillary electrophoresis-mass spectrometry for the analysis of common post-translational modifications. Electrophoresis 2018, 39, 1208–1215. [Google Scholar] [CrossRef]
- Váradi, C.; Mittermayr, S.; Millán-Martín, S.; Bones, J. Quantitative twoplex glycan analysis using 12C6 and 13C6 stable isotope 2-aminobenzoic acid labelling and capillary electrophoresis mass spectrometry. Anal. Bioanal. Chem. 2016, 408, 8691–8700. [Google Scholar] [CrossRef]
- Del Campo, M.; Mollenhauer, B.; Bertolotto, A.; Engelborghs, S.; Hampel, H.; Simonsen, A.H.; Kapaki, E.; Kruse, N.; le Bas-tard, N.; Lehmann, S.; et al. Recommendations to standardize preanalytical confounding factors in Alzheimer’s and Parkinson’s disease cerebrospinal fluid biomarkers: An update. Biomark. Med. 2012, 6, 419–430. [Google Scholar] [CrossRef]
- Leitão, M.J.; Baldeiras, I.; Eherukka, S.-K.; Epikkarainen, M.; Eleinonen, V.; Simonsen, A.H.; Eperret-Liaudet, A.; Efourier, A.; Quadrio, I.; Veiga, P.M.; et al. Chasing the Effects of Pre-Analytical Confounders—A Multicenter Study on CSF-AD Biomarkers. Front. Neurol. 2015, 6, 153. [Google Scholar] [CrossRef] [PubMed]
- Fourier, A.; Portelius, E.; Zetterberg, H.; Blennow, K.; Quadrio, I.; Perret-Liaudet, A. Pre-analytical and analytical factors influencing Alzheimer’s disease cerebrospinal fluid biomarker varia-bility. Clin. Chim. Acta 2015, 449, 9–15. [Google Scholar] [CrossRef]
- Le Bastard, N.; De Deyn, P.P.; Engelborghs, S. Importance and impact of preanalytical variables on Alzheimer disease bi-omarker concentrations in cerebrospinal fluid. Clin. Chem. 2015, 61, 734–743. [Google Scholar] [CrossRef] [Green Version]
- Comstock, G.W.; Burke, A.E.; Norkus, E.P.; Gordon, G.B.; Hoffman, S.C.; Helzlsouer, K.J. Effects of repeated freeze-thaw cycles on concentrations of cholesterol, micronutrients, and hormones in human plasma and serum. Am. J. Epidemiol. 2008, 168, 827–830. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.J.; Wen, G.; Morris, J.C.; Holtzman, D.M. Fluctuations of CSF amyloid-β levels: Implications for a diagnostic and therapeutic biomarker. Neurology 2007, 68, 666–669. [Google Scholar] [CrossRef]
- Vanderstichele, H.M.; Janelidze, S.; Demeyer, L.; Coart, E.; Stoops, E.; Herbst, V.; Mauroo, K.; Brix, B.; Hansson, O. Optimized Standard Operating Procedures for the Analysis of Cerebrospinal Fluid Abeta42 and the Ratios of Abeta Isoforms Using Low Protein Binding Tubes. J. Alzheimers Dis. 2016, 53, 1121–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewczuk, P.; Beck, G.; Esselmann, H.; Bruckmoser, R.; Zimmermann, R.; Fiszer, M.; Bibl, M.; Maler, J.M.; Kornhuber, J.; Wiltfang, J. Effect of Sample Collection Tubes on Cerebrospinal Fluid Concentrations of Tau Proteins and Amyloid β Peptides. Clin. Chem. 2006, 52, 332–334. [Google Scholar] [CrossRef] [PubMed]
- Perret-Liaudet, A.; Pelpel, M.; Tholance, Y.; Dumont, B.; Vanderstichele, H.; Zorzi, W.; ElMoualij, B.; Schraen, S.; Moreaud, O.; Gabelle, A.; et al. Risk of Alzheimer’s Disease Biological Misdiagnosis Linked to Cerebrospinal Collection Tubes. J. Alzheimers Dis. 2012, 31, 13–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, J.-S.; Gelfanova, V.; Knierman, M.D.; Witzmann, F.A.; Wang, M.; Hale, J.E. The impact of blood contamination on the proteome of cerebrospinal fluid. Proteomics 2005, 5, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Bjerke, M.; Portelius, E.; Minthon, L.; Wallin, A.; Anckarsäter, H.; Anckarsäter, R.; Andreasen, N.; Zetterberg, H.; Andreasson, U.; Blennow, K. Confounding Factors Influencing Amyloid Beta Concentration in Cerebrospinal Fluid. Int. J. Alzheimers Dis. 2010, 2010, 986310. [Google Scholar] [CrossRef] [Green Version]
- Schoonenboom, N.S.; Mulder, C.; Vanderstichele, H.; van Elk, E.J.; Kok, A.; van Kamp, G.J.; Scheltens, P.; Blankenstein, M.A. Effects of processing and storage conditions on amyloid beta (1-42) and tau concentrations in cerebrospinal fluid: Implications for use in clinical practice. Clin. Chem. 2005, 51, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Vanderstichele, H.; Bibl, M.; Engelborghs, S.; Le Bastard, N.; Lewczuk, P.; Molinuevo, J.L.; Parnetti, L.; Perret-Liaudet, A.; Shaw, L.M.; Teunissen, C.; et al. Standardization of preanalytical aspects of cerebrospinal fluid biomarker testing for Alzheimer’s disease diagnosis: A consensus paper from the Alzheimer’s Biomarkers Standardization Initiative. Alzheimers Dement. 2011, 8, 65–73. [Google Scholar] [CrossRef]
- Zimmermann, R.; Lelental, N.; Ganslandt, O.; Maler, J.M.; Kornhuber, J.; Lewczuk, P. Preanalytical sample handling and sample stability testing for the neurochemical dementia diag-nostics. J. Alzheimers Dis. 2011, 25, 739–745. [Google Scholar] [CrossRef]
- West-Nielsen, M.; Høgdall, E.V.; Marchiori, E.; Høgdall, C.K.; Schou, C.; Heegaard, N.H.H. Sample Handling for Mass Spectrometric Proteomic Investigations of Human Sera. Anal. Chem. 2005, 77, 5114–5123. [Google Scholar] [CrossRef]
- Hokfelt, T.; Broberger, C.; Xu, Z.Q.; Sergeyev, V.; Ubink, R.; Diez, M. Neuropeptides—An overview. Neuropharmacology 2000, 39, 1337–1356. [Google Scholar] [CrossRef]
- Reiber, H. Dynamics of brain-derived proteins in cerebrospinal fluid. Clin. Chim. Acta 2001, 310, 173–186. [Google Scholar] [CrossRef] [Green Version]
- Engelborghs, S.; Niemantsverdriet, E.; Struyfs, H.; Blennow, K.; Brouns, R.; Comabella, M.; Dujmovic, I.; Van Der Flier, W.; Frölich, L.; Galimberti, D.; et al. Consensus guidelines for lumbar puncture in patients with neurological diseases. Alzheimers Dement. 2017, 8, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.; Koziarz, A.; Badhiwala, J.H.; Alhazzani, W.; Jaeschke, R.; Sharma, S.; Banfield, L.; Shoamanesh, A.; Singh, S.; Nassiri, F.; et al. Atraumatic versus conventional lumbar puncture needles: A systematic review and meta-analysis. Lancet 2018, 391, 1197–1204. [Google Scholar] [CrossRef]
- Peskind, E.; Nordberg, A.; Darreh-Shori, T.; Soininen, H. Safety of Lumbar Puncture Procedures in Patients with Alzheimers Disease. Curr. Alzheimer Res. 2009, 6, 290–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zetterberg, H.; Tullhög, K.; Hansson, O.; Minthon, L.; Londos, E.; Blennow, K. Low Incidence of Post-Lumbar Puncture Headache in 1,089 Consecutive Memory Clinic Patients. Eur. Neurol. 2010, 63, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Desiderio, D.M. Proteomics analysis of human cerebrospinal fluid. J. Chromatogr. B 2005, 815, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.L.; Anderson, N.G. The human plasma proteome: History, character, and diagnostic prospects. Mol. Cell. Proteomics 2002, 1, 845–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henrik, Z. Applying Fluid Biomarkers to Alzheimer’s Disease. Am. J. Physiol.-Cell Physiol. 2017, 1, C3–C10. [Google Scholar]
- Schutzer, S.E.; Liu, T.; Natelson, B.H.; Angel, T.E.; Schepmoes, A.A.; Purvine, S.; Hixson, K.K.; Lipton, M.S.; Camp, D.G.; Coyle, P.K.; et al. Establishing the Proteome of Normal Human Cerebrospinal Fluid. PLoS ONE 2010, 5, e10980. [Google Scholar] [CrossRef] [Green Version]
- Gillette, M.A.; Mani, D.R.; Carr, S.A. Place of Pattern in Proteomic Biomarker Discovery. J. Proteome Res. 2005, 4, 1143–1154. [Google Scholar] [CrossRef]
- Boschetti, E.; Lomas, L.; Citterio, A.; Righetti, P.G. Romancing the “hidden proteome”, Anno Domini two zero zero seven. J. Chromatogr. A 2007, 1153, 277–290. [Google Scholar] [CrossRef]
- Kroksveen, A.; Opsahl, J.; Aye, T.; Ulvik, R.; Berven, F. Proteomics of human cerebrospinal fluid: Discovery and verification of biomarker candidates in neurodegenerative diseases using quantitative proteomics. J. Proteom. 2011, 74, 371–388. [Google Scholar] [CrossRef]
- Thambisetty, M.; Lovestone, S. Blood-based biomarkers of Alzheimer’s disease: Challenging but feasible. Biomarkers Med. 2010, 4, 65–79. [Google Scholar] [CrossRef]
- O’Bryant, S.E.; Gupta, V.; Henriksen, K.; Edwards, M.; Jeromin, A.; Lista, S.; Bazenet, C.; Soares, H.; Lovestone, S.; Hampel, H.; et al. Guidelines for the Standardization of Preanalytic Variables for Blood-Based Biomarker Studies in Alzheimer’s Disease Research. Alzheimers Dement. 2015, 1, 549–560. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Duan, J.; Liu, T.; Smith, R.D.; Qian, W.-J. Contributions of immunoaffinity chromatography to deep proteome profiling of human biofluids. J. Chromatogr. B 2016, 1021, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Pieper, R.; Su, Q.; Gatlin, C.L.; Huang, S.-T.; Anderson, N.L.; Steiner, S. Multi-component immunoaffinity subtraction chromatography: An innovative step towards a comprehensive survey of the human plasma proteome. Proteomics 2003, 3, 422–432. [Google Scholar] [CrossRef]
- Liu, T.; Qian, W.J.; Mottaz, H.M.; Gritsenko, M.A.; Norbeck, A.D.; Moore, R.J.; Purvine, S.O.; Ii, D.G.C.; Smith, R.D. Evaluation of Multiprotein Immunoaffnity Subtraction for Plasma Proteomics and Candidate Biomaker Discovery Using Mass Spectrometry. Mol. Cell. Proteom. 2006, 1, 2167–2174. [Google Scholar] [CrossRef] [Green Version]
- Boschetti, E.; Righetti, P.G. The ProteoMiner in the proteomic arena: A non-depleting tool for discovering low-abundance species. J. Proteom. 2008, 71, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Righetti, P.G.; Candiano, G.; Citterio, A.; Boschetti, E. Combinatorial Peptide Ligand Libraries as a “Trojan Horse” in Deep Discovery Proteomics. Anal. Chem. 2014, 87, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Jankovska, E.; Svitek, M.; Holada, K.; Petrak, J. Affinity depletion versus relative protein enrichment: A side-by-side comparison of two major strategies for increasing human cerebrospinal fluid proteome coverage. Clin. Proteom. 2019, 16, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forshed, J. Experimental Design in Clinical ‘Omics Biomarker Discovery. J. Proteome Res. 2017, 16, 3954–3960. [Google Scholar] [CrossRef]
- Prasad, B.; Achour, B.; Artursson, P.; Hop, C.E.; Lai, Y.; Smith, P.C.; Barber, J.; Wisniewski, J.R.; Spellman, D.; Uchida, Y.; et al. Toward a Consensus on Applying Quantitative Liquid Chromatography-Tandem Mass Spectrometry Proteomics in Translational Pharmacology Research: A White Paper. Clin. Pharmacol. Ther. 2019, 106, 525–543. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, P.A.; Clauser, K.; Kilpatrick, L.E.; Tchekhovskoi, D.V.; Neta, P.; Blonder, N.; Billheimer, D.D.; Blackman, R.K.; Bunk, D.M.; Cardasis, H.L.; et al. Performance Metrics for Liquid Chromatography-Tandem Mass Spectrometry Systems in Proteomics Analyses. Mol. Cell. Proteom. 2010, 9, 225–241. [Google Scholar] [CrossRef] [Green Version]
- Bereman, M.S.; Beri, J.; Sharma, V.; Nathe, C.; Eckels, J.; MacLean, B.; MacCoss, M.J. An Automated Pipeline to Monitor System Performance in Liquid Chromatography–Tandem Mass Spectrometry Proteomic Experiments. J. Proteome Res. 2016, 15, 4763–4769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, S.A.; Abbatiello, S.E.; Ackermann, B.L.; Borchers, C.; Domon, B.; Deutsch, E.W.; Grant, R.P.; Hoofnagle, A.N.; Hüttenhain, R.; Koomen, J.M.; et al. Targeted Peptide Measurements in Biology and Medicine: Best Practices for Mass Spectrometry-based Assay Development Using a Fit-for-Purpose Approach. Mol. Cell. Proteom. 2014, 13, 907–917. [Google Scholar] [CrossRef] [Green Version]
- Abbatiello, S.; Ackermann, B.L.; Borchers, C.; Bradshaw, R.A.; Carr, S.A.; Chalkley, R.; Choi, M.; Deutsch, E.; Domon, B.; Hoofnagle, A.N.; et al. New Guidelines for Publication of Manuscripts Describing Development and Application of Targeted Mass Spectrometry Measurements of Peptides and Proteins. Mol. Cell. Proteom. 2017, 16, 327–328. [Google Scholar] [CrossRef] [Green Version]
- Pedrero-Prieto, C.M.; García-Carpintero, S.; Frontiñán-Rubio, J.; Llanos-González, E.; García, C.A.; Alcaín, F.J.; Lindberg, I.; Durán-Prado, M.; Peinado, J.R.; Rabanal-Ruiz, Y. A Comprehensive Systematic Review of Csf Proteins and Peptides That Define Alzheimer’s Disease. Clin. Proteom. 2020, 1, 21. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, N.; Nakamura, A.; Washimi, Y.; Kato, T.; Sakurai, T.; Arahata, Y.; Bundo, M.; Takeda, A.; Niida, S.; Ito, K.; et al. Novel plasma biomarker surrogating cerebral amyloid deposition. Proc. Jpn. Acad. Ser. B 2014, 90, 353–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvartsberg, H.; Duits, F.H.; Ingelsson, M.; Andreasen, N.; Ohrfelt, A.; Andersson, K.; Brinkmalm, G.; Lannfelt, L.; Minthon, L.; Hansson, O.; et al. Cerebrospinal Fluid Levels of the Synaptic Protein Neurogranin Correlates with Cognitive Decline in Prodromal Alzheimer’s Disease. Alzheimers Dement 2015, 1, 1180–1190. [Google Scholar] [CrossRef] [PubMed]
- Sjödin, S.; Öhrfelt, A.; Brinkmalm, G.; Zetterberg, H.; Blennow, K.; Brinkmalm, A. Targeting LAMP2 in human cerebrospinal fluid with a combination of immunopurification and high resolution parallel reaction monitoring mass spectrometry. Clin. Proteom. 2016, 13, 4. [Google Scholar] [CrossRef] [Green Version]
- Ringman, J.M.; Schulman, H.; Becker, C.; Jones, T.; Bai, Y.; Immermann, F.; Cole, G.; Sokolow, S.; Gylys, K.; Geschwind, D.H.; et al. Proteomic Changes in Cerebrospinal Fluid of Presymptomatic and Affected Persons Carrying Familial Alzheimer Disease Mutations. Arch. Neurol. 2012, 1, 96–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hölttä, M.; Minthon, L.; Hansson, O.; Holmén-Larsson, J.; Pike, I.; Ward, M.; Kuhn, K.; Rüetschi, U.; Zetterberg, H.; Blennow, K.; et al. An Integrated Workflow for Multiplex CSF Proteomics and Peptidomics—Identification of Candidate Cerebrospinal Fluid Biomarkers of Alzheimer’s Disease. J. Proteome Res. 2014, 14, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Russell, C.L.; Heslegrave, A.; Mitra, V.; Zetterberg, H.; Pocock, J.M.; Ward, M.A.; Pike, I. Combined Tissue and Fluid Proteomics with Tandem Mass Tags to Identify Low-Abundance Protein Biomarkers of Disease in Peripheral Body Fluid: An Alzheimer’s Disease Case Study. Rapid Commun. Mass Spectrom. 2017, 1, 153–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijte, D.; McDonnell, L.A.; Balog, C.I.; Bossers, K.; Deelder, A.M.; Swaab, D.F.; Verhaagen, J.; Mayboroda, O.A. A novel peptidomics approach to detect markers of Alzheimer’s disease in cerebrospinal fluid. Methods 2012, 56, 500–507. [Google Scholar] [CrossRef]
- Holtta, M.; Dean, R.A.; Siemers, E.; Mawuenyega, K.G.; Sigurdson, W.; May, P.C.; Holtzman, D.M.; Portelius, E.; Zetterberg, H.; Bateman, R.J.; et al. A Single Dose of the Gamma-Secretase Inhibitor Semagacestat Alters the Cerebrospinal Fluid Peptidome in Humans. Alzheimers Res. Ther. 2016, 1, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrin, R.J.; Payton, J.E.; Malone, J.P.; Gilmore, P.; Davis, A.E.; Xiong, C.; Fagan, A.M.; Townsend, R.R.; Holtzman, D.M. Quantitative Label-Free Proteomics for Discovery of Biomarkers in Cerebrospinal Fluid: Assessment of Technical and Inter-Individual Variation. PLoS ONE 2013, 1, e64314. [Google Scholar]
- Jahn, H.; Wittke, S.; Zurbig, P.; Raedler, T.J.; Arlt, S.; Kellmann, M.; Mullen, W.; Eichenlaub, M.; Mischak, H.; Wiedemann, K. Peptide Fingerprinting of Alzheimer’s Disease in Cerebrospinal Fluid: Identification and Prospective Evaluation of New Synaptic Biomarkers. PLoS ONE 2011, 1, e26540. [Google Scholar] [CrossRef] [PubMed]
- Tagami, S.; Okochi, M.; Yanagida, K.; Kodama, T.; Arai, T.; Kuwano, R.; Ikeuchi, T.; Takeda, M. Relative Ratio and Level of Amyloid-Beta 42 Surrogate in Cerebrospinal Fluid of Familial Alzheimer Disease Patients with Presenilin 1 Mutations. Neurodegener. Dis. 2014, 13, 166–170. [Google Scholar] [CrossRef]
- Chiasserini, D.; van Weering, J.R.; Piersma, S.R.; Pham, T.V.; Malekzadeh, A.; Teunissen, C.E.; de Wit, H.; Jiménez, C.R. Proteomic analysis of cerebrospinal fluid extracellular vesicles: A comprehensive dataset. J. Proteom. 2014, 106, 191–204. [Google Scholar] [CrossRef]
- Dayon, L.; Hainard, A.; Licker, V.; Turck, N.; Kuhn, K.; Hochstrasser, D.F.; Burkhard, A.P.R.; Sanchez, J.-C. Relative Quantification of Proteins in Human Cerebrospinal Fluids by MS/MS Using 6-Plex Isobaric Tags. Anal. Chem. 2008, 80, 2921–2931. [Google Scholar] [CrossRef] [PubMed]
- Shih, Y.H.; Tsai, K.J.; Lee, C.W.; Shiesh, S.C.; Chen, W.T.; Pai, M.C.; Kuo, Y.M. Apolipoprotein C-Iii Is an Amyloid-Beta-Binding Protein and an Early Marker for Alzheimer’s Disease. J. Alzheimers Dis. 2014, 1, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Muenchhoff, J.; Poljak, A.; Song, F.; Raftery, M.; Brodaty, H.; Duncan, M.; McEvoy, M.; Attia, J.; Schofield, P.W.; Sachdev, P.S. Plasma Protein Profiling of Mild Cognitive Impairment and Alzheimer’s Disease across Two Independent Cohorts. J. Alzheimers Dis. 2015, 1, 1355–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guntert, A.; Campbell, J.; Saleem, M.; O’Brien, D.P.; Thompson, A.J.; Byers, H.L.; Ward, M.A.; Lovestone, S. Plasma Gelsolin Is Decreased and Correlates with Rate of Decline in Alzheimer’s Disease. J. Alzheimers Dis. 2010, 1, 585–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, L.; Liao, L.; Chen, C.; Guo, Y.; Song, D.; Wang, Y.; Chen, Y.; Zhang, K.; Ying, M.; Li, S.; et al. Proteomics Analysis of Blood Serums from Alzheimer’s Disease Patients Using iTRAQ Labeling Technology. J. Alzheimers Dis. 2017, 56, 361–378. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, S.L.; Yang, H.; Lyutvinskiy, Y.; Rutishauser, D.; Herukka, S.K.; Soininen, H.; Zubarev, R.A. Blood Plasma Igg Fc Glycans Are Significantly Altered in Alzheimer’s Disease and Progressive Mild Cognitive Impairment. J. Alzheimers Dis. 2014, 1, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Poljak, A.; Hill, M.N.; Hall, R.J.; MacLullich, A.; Raftery, M.J.; Tai, J.; Yan, S.; Caplan, G.A. Quantitative proteomics of delirium cerebrospinal fluid. Transl. Psychiatry 2014, 4, e477. [Google Scholar] [CrossRef]
- Kockmann, T.; Trachsel, C.; Panse, C.; Wahlander, A.; Selevsek, N.; Grossmann, J.; Wolski, W.E.; Schlapbach, R. Targeted proteomics coming of age—SRM, PRM and DIA performance evaluated from a core facility perspective. Proteomics 2016, 16, 2183–2192. [Google Scholar] [CrossRef] [PubMed]
- Picotti, P.; Aebersold, R. Selected Reaction Monitoring-Based Proteomics: Workflows, Potential, Pitfalls and Future Directions. Nat. Methods 2012, 1, 555–566. [Google Scholar] [CrossRef]
- Liebler, D.C.; Zimmerman, L.J. Targeted Quantitation of Proteins by Mass Spectrometry. Biochemistry 2013, 52, 3797–3806. [Google Scholar] [CrossRef]
- Gallien, S.; Duriez, E.; Crone, C.; Kellmann, M.; Moehring, T.; Domon, B. Targeted Proteomic Quantification on Quadrupole-Orbitrap Mass Spectrometer. Mol. Cell. Proteom. 2012, 11, 1709–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronsein, G.E.; Pamir, N.; von Haller, P.D.; Kim, D.S.; Oda, M.N.; Jarvik, G.P.; Vaisar, T.; Heinecke, J.W. Parallel reaction monitoring (PRM) and selected reaction monitoring (SRM) exhibit comparable linearity, dynamic range and precision for targeted quantitative HDL proteomics. J. Proteom. 2014, 113, 388–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogeberg, M.; Almdahl, I.S.; Wettergreen, M.; Nilsson, L.N.; Fladby, T. Isobaric Quantification of Cerebrospinal Fluid Amyloid-Beta Peptides in Alzheimer’s Disease: C-Terminal Truncation Relates to Early Measures of Neurodegeneration. J. Proteome Res. 2015, 1, 4834–4843. [Google Scholar] [CrossRef]
- Rezeli, M.; Zetterberg, H.; Blennow, K.; Brinkmalm, A.; Laurell, T.; Hansson, O.; Marko-Varga, G. Quantification of total apolipoprotein E and its specific isoforms in cerebrospinal fluid and blood in Alzheimer’s disease and other neurodegenerative diseases. EuPA Open Proteom. 2015, 8, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Simon, R.; Girod, M.; Fonbonne, C.; Salvador, A.; Clément, Y.; Lantéri, P.; Amouyel, P.; Lambert, J.C.; Lemoine, J. Total Apoe and Apoe4 Isoform Assays in an Alzheimer’s Disease Case-Control Study by Targeted Mass Spectrometry (N=669): A Pilot Assay for Methionine-Containing Proteotypic Peptides. Mol. Cell. Proteom. MCP 2012, 1, 1389–1403. [Google Scholar] [CrossRef] [Green Version]
- Lundström, S.L.; Zhang, B.; Rutishauser, D.; Aarsland, D.; Zubarev, R.A. SpotLight Proteomics: Uncovering the hidden blood proteome improves diagnostic power of proteomics. Sci. Rep. 2017, 7, srep41929. [Google Scholar] [CrossRef] [Green Version]
- Portelius, E.; Dean, R.A.; Gustavsson, M.K.; Andreasson, U.; Zetterberg, H.; Siemers, E.; Blennow, K. A Novel Abeta Isoform Pattern in Csf Reflects Gamma-Secretase Inhibition in Alzheimer Disease. Alzheimers Res. Ther. 2010, 1, 7. [Google Scholar] [CrossRef] [Green Version]
- Portelius, E.; Dean, R.A.; Andreasson, U.; Mattsson, N.; Westerlund, A.; Olsson, M.; DeMattos, R.B.; Racke, M.M.; Zetterberg, H.; May, P.C.; et al. β-site amyloid precursor protein-cleaving enzyme 1(BACE1) inhibitor treatment induces Aβ5-X peptides through alternative amyloid precursor protein cleavage. Alzheimers Res. Ther. 2014, 6, 75. [Google Scholar] [CrossRef] [Green Version]
- Portelius, E.; Tran, A.J.; Andreasson, U.; Persson, R.; Brinkmalm, G.; Zetterberg, H.; Blennow, K.; Westman-Brinkmalm, A. Characterization of Amyloid Beta Peptides in Cerebrospinal Fluid by an Automated Immunoprecipitation Procedure Followed by Mass Spectrometry. J. Proteome Res. 2007, 1, 4433–4439. [Google Scholar] [CrossRef] [PubMed]
- Brinkmalm, A.; Brinkmalm, G.; Honer, W.G.; Frölich, L.; Hausner, L.; Minthon, L.; Hansson, O.; Wallin, A.; Zetterberg, H.; Blennow, K.; et al. Snap-25 Is a Promising Novel Cerebrospinal Fluid Biomarker for Synapse Degeneration in Alzheimer’s Disease. Mol. Neurodegener. 2014, 1, 53. [Google Scholar] [CrossRef] [Green Version]
- Öhrfelt, A.; Brinkmalm, A.; Dumurgier, J.; Brinkmalm, G.; Hansson, O.; Zetterberg, H.; Bouaziz-Amar, E.; Hugon, J.; Paquet, C.; Blennow, K. The pre-synaptic vesicle protein synaptotagmin is a novel biomarker for Alzheimer’s disease. Alzheimers Res. Ther. 2016, 8, 41. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Morillo, E.; Hansson, O.; Atagi, Y.; Bu, G.; Minthon, L.; Diamandis, E.P.; Nielsen, H.M. Total apolipoprotein E levels and specific isoform composition in cerebrospinal fluid and plasma from Alzheimer’s disease patients and controls. Acta Neuropathol. 2014, 127, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Heslegrave, A.; Heywood, W.; Paterson, R.W.; Magdalinou, N.; Svensson, J.; Johansson, P.; Öhrfelt, A.; Blennow, K.; Hardy, J.; Schott, J.M.; et al. Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ternent, T.; Csordas, A.; Qi, D.; Gómez-Baena, G.; Beynon, R.J.; Jones, A.R.; Hermjakob, H.; Vizcaíno, J.A. How to submit MS proteomics data to ProteomeXchange via the PRIDE database. Proteomics 2014, 14, 2233–2241. [Google Scholar] [CrossRef] [PubMed]
- Jarnuczak, A.F.; Ternent, T.; Vizcaíno, J.A. Quantitative Proteomics Data in the Public Domain: Challenges and Opportunities. Methods Mol. Biol. 2019, 1977, 217–235. [Google Scholar] [PubMed]
- Sharma, V.; Eckels, J.; Schilling, B.; Ludwig, C.; Jaffe, J.D.; MacCoss, M.J.; MacLean, B. Panorama Public: A Public Repository for Quantitative Data Sets Processed in Skyline. Mol. Cell. Proteom. 2018, 17, 1239–1244. [Google Scholar] [CrossRef] [Green Version]
- Ellis, M.J.; Gillette, M.; Carr, S.A.; Paulovich, A.G.; Smith, R.D.; Rodland, K.K.; Townsend, R.R.; Kinsinger, C.; Mesri, M.; Rodriguez, H.; et al. Connecting Genomic Alterations to Cancer Biology with Proteomics: The NCI Clinical Proteomic Tumor Analysis Consortium. Cancer Discov. 2013, 3, 1108–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudnick, P.A.; Markey, S.P.; Roth, J.; Mirokhin, Y.; Yan, X.; Tchekhovskoi, D.V.; Edwards, N.J.; Thangudu, R.R.; Ketchum, K.A.; Kinsinger, C.R.; et al. A Description of the Clinical Proteomic Tumor Analysis Consortium (CPTAC) Common Data Analysis Pipeline. J. Proteome Res. 2016, 15, 1023–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overview of Assay Characterization for the CPTAC Assay Portal. Available online: https://proteomics.cancer.gov/sites/default/files/assay-characterization-guidance-document.pdf (accessed on 30 November 2021).
- Mueller, S.G.; Weiner, M.W.; Thal, L.J.; Petersen, R.C.; Jack, C.; Jagust, W.; Trojanowski, J.Q.; Toga, A.W.; Beckett, L. The Alzheimer’s Disease Neuroimaging Initiative. Neuroimaging Clin. N. Am. 2005, 15, 869–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiner, M.W.; Aisen, P.S.; Jack, C.R., Jr.; Jagust, W.J.; Trojanowski, J.Q.; Shaw, L.; Saykin, A.J.; Morris, J.C.; Cairns, N.; Beckett, L.A.; et al. The Alzheimer’s Disease Neuroimaging Initiative: Progress Report and Future Plans. Alzheimers Dement. J. Alzheimer’s Assoc. 2010, 1, 202–211.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiner, M.W.; Veitch, D.P.; Aisen, P.S.; Beckett, L.A.; Cairns, N.J.; Cedarbaum, J.; Donohue, M.C.; Green, R.C.; Harvey, D.; Jack, C.R., Jr.; et al. Impact of the Alzheimer’s Disease Neuroimaging Initiative, 2004 to 2014. Alzheimers Dement. J. Alzheimer’s Assoc. 2015, 1, 865–884. [Google Scholar] [CrossRef] [Green Version]
- Weiner, M.W.; Veitch, D.P.; Aisen, P.S.; Beckett, L.A.; Cairns, N.J.; Green, R.C.; Harvey, D.; Jack, C.R., Jr.; Jagust, W.; Morris, J.C.; et al. The Alzheimer’s Disease Neuroimaging Initiative 3: Continued Innovation for Clinical Trial Improvement. Alzheimers Dement. J. Alzheimer’s Assoc. 2017, 1, 561–571. [Google Scholar] [CrossRef] [Green Version]
- Toga, A.W.; Crawford, K.L. The Alzheimer’s Disease Neuroimaging Initiative Informatics Core: A Decade in Review. Alzheimers Dement. J. Alzheimer’s Assoc. 2015, 1, 832–839. [Google Scholar] [CrossRef] [Green Version]
- Thompson, P.M.; Stein, J.L.; Medland, S.E.; Hibar, D.P.; Vasquez, A.A.; Renteria, M.E.; Toro, R.; Jahanshad, N.; Schumann, G.; Franke, B.; et al. The Enigma Consortium: Large-Scale Collaborative Analyses of Neuroimaging and Genetic Data. Brain Imaging Behav. 2014, 1, 153–182. [Google Scholar] [CrossRef] [Green Version]
- Global CEO Initiative on Alzheimer’s Disease Big Data Challenge for Alzheimer’s Disease Launches in Global Effort to Use Innovative Open Science Techniques to Improve Diagnosis and Treatment. Available online: https://sagebionetworks.org/in-the-news/big-data-challenge-for-alzheimers-disease-launches-in-global-effort-to-use-innovative-open-science-techniques-to-improve-diagnosis-and-treatment/ (accessed on 30 November 2021).
- Spellman, D.S.; Wildsmith, K.R.; Honigberg, L.A.; Tuefferd, M.; Baker, D.; Raghavan, N.; Nairn, A.C.; Croteau, P.; Schirm, M.; Allard, R.; et al. Development and Evaluation of a Multiplexed Mass Spectrometry Based Assay for Measuring Candidate Peptide Biomarkers in Alzheimer’s Disease Neuroimaging Initiative (Adni) Csf. Proteomics. Clin. Appl. 2015, 9, 715–731. [Google Scholar] [CrossRef]
- Libiger, O.; Shaw, L.M.; Watson, M.H.; Nairn, A.C.; Umaña, K.L.; Biarnes, M.C.; Canet-Avilés, R.M.; Jack, C.R., Jr.; Breton, Y.A.; Cortes, L.; et al. Longitudinal Csf Proteomics Identifies Nptx2 as a Prognostic Biomarker of Alzheimer’s Disease. Alzheimers Dement. 2021, 1, 1976–1987. [Google Scholar] [CrossRef]

| Discovery Proteomics Studies | |||
|---|---|---|---|
| Sample | LC MS Technique | Summary | Ref. |
| CSF AD dementia (n = 8), MCI (n = 11), controls (n = 19) | TMT coupled to IP-MS | Robust assay developed for parallel relative quantification of 27 Aβ peptides in CSF. Although no statistical difference was seen between diseased and control groups. | [188] |
| CSF and blood AD patients (n = 39) Control patients (n = 38) | SRM: Absolute quant with heavy isotope standards | ApoE proteoforms quantified using stable isotope dilution. Total ApoE in CSF or blood doesn’t distinguish AD from non-AD subjects. ApoE e4 carriers have lower blood ApoE irrespective of clinical diagnosis. | [189] |
| Plasma Case-control (n = 669) | SRM-MS | Total ApoE and ApoE e4 proteoform quantified. ApoE e4 specific peptide contained a single methionine, which was chemically oxidized after tryptic digestion, completeness of oxidation was thoroughly evaluated. Chemical oxidation allowed unbiased monitoring of ApoE e4 unique proteotypic peptide. Neither total ApoE and ApoE e4 levels nor ApoE/APOE e4 ratio consistent with AD diagnosis | [190] |
| Serum DLB patients (n = 47) AD patients (n = 97) | SpotLight Melon Gel kit enriches polyclonal IgGs. | De-novo sequencing identifies peptides from variable regions of IgGs and uncovers “hidden proteome”. SpotLight peptide quantification generated a predictive model with 95% accuracy to distinguish AD and dementia with Lewy bodies | [191] |
| CSF (Sample size not listed) | IP-MS with heavy isotope internal standards coupled to MALDI-TOF. Confirmation carried out on LIT-FT ICR | Affinity purification MS method optimized for Aβ using Aβ specific crosslinked antibodies. Two novel Ab peptides identified: Aβ2-17 and Aβ3-17 (probable cleavage products of neprilysin and ECE) The developed assay facilitated target engagement clinical studies | [192,193,194] |
| CSF (three separate cohorts) Cohort 1: AD subjects (n = 9), prodromal AD (n = 7), non-demented controls (n = 9) Cohort 2: AD (n = 10), non-demented controls (n = 6) Cohort 3: AD (n = 17), non-demented controls (n = 17) Brain tissue Autopsy confirmed AD patients (n = 15) Age-matched controls (n = 15) | IP-SRM-MS with heavy isotope internal standards | Affinity purification MS method developed to measure levels of the presynaptic protein synaptosomal-associated protein 25 (SNAP-25) in CSF. SNAP-25 levels were significantly higher in prodormal AD and AD when compared to controls. CSF SNAP-25 differentiated AD from controls and was proposed as novel biomarker for synapse degeneration. | [195] |
| CSF (2 cohorts) Cohort 1: CSF AD dementia (n = 15), MCI (n = 5), controls (n = 17) Cohort 2: CSF AD (n = 24), MCI (n = 18), controls (n = 36) | IP-PRM-MS with heavy isotope internal standards | Affinity purification MS method developed to measure levels of the presynaptic vesicle protein synaptotagmin-1 in CSF Synaptotagmin-1 levels were significantly higher in MCI AD and AD dementia when compared to controls. CSF synaptotagmin-1 was proposed as a biomarker of synaptic dysfunction and degeneration in AD | [196] |
| CSF and plasma AD patients (n = 43) Control (n = 43) | SRM MS with heavy isotope internal standards | Previously developed ApoE quantification assay was used to measure ApoE proteoforms ApoE2, ApoE3 and ApoE4. No distinction was found between AD patients aid controls. | [197] |
| CSF AD patients (n = 37) Control (n = 22) Validation cohort AD patients (n = 24) Control (n = 16) | SRM with heavy isotope internal standards | Significantly higher concentration of soluble triggering receptor expressed on myeloid cells 2 (sTREM2) was found in AD patients when compared to controls. This finding was replicated in the validation sample set. sTREM2 was found to correlate with markers of neurodegeneration and glial activation. | [198] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Awasthi, S.; Spellman, D.S.; Hatcher, N.G. Proteomic Discovery and Validation of Novel Fluid Biomarkers for Improved Patient Selection and Prediction of Clinical Outcomes in Alzheimer’s Disease Patient Cohorts. Proteomes 2022, 10, 26. https://doi.org/10.3390/proteomes10030026
Awasthi S, Spellman DS, Hatcher NG. Proteomic Discovery and Validation of Novel Fluid Biomarkers for Improved Patient Selection and Prediction of Clinical Outcomes in Alzheimer’s Disease Patient Cohorts. Proteomes. 2022; 10(3):26. https://doi.org/10.3390/proteomes10030026
Chicago/Turabian StyleAwasthi, Shivangi, Daniel S. Spellman, and Nathan G. Hatcher. 2022. "Proteomic Discovery and Validation of Novel Fluid Biomarkers for Improved Patient Selection and Prediction of Clinical Outcomes in Alzheimer’s Disease Patient Cohorts" Proteomes 10, no. 3: 26. https://doi.org/10.3390/proteomes10030026