

A Genome-Wide Association Study into the Aetiology of Congenital Solitary Functioning Kidney
 , , , , , and
, , , , , and
Abstract
:1. Introduction
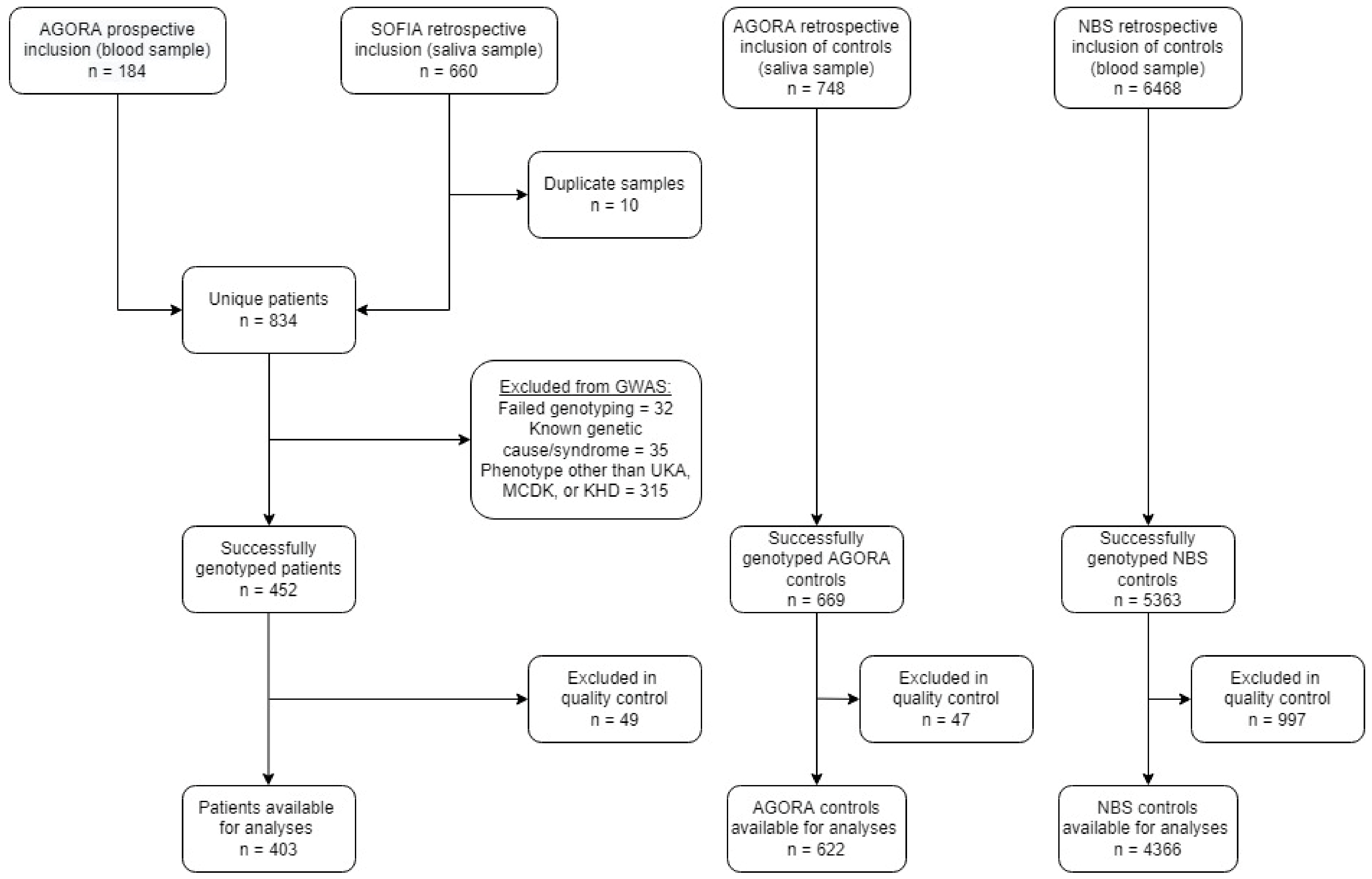
2. Patients and Methods
2.1. Patients
2.2. Genotyping
2.3. Quality Control
2.4. Imputation
2.5. Statistical Analyses
3. Results
3.1. Participants
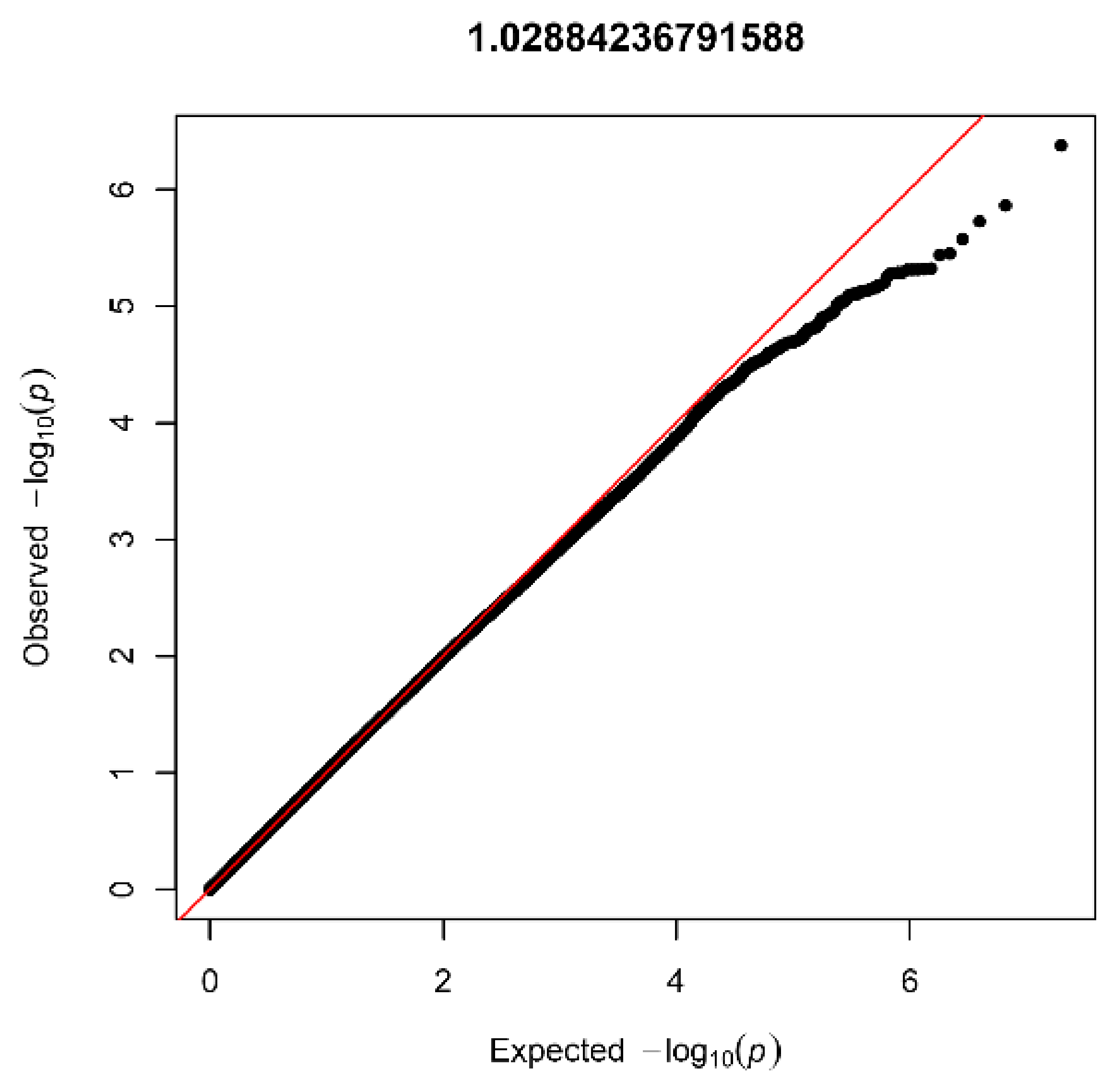
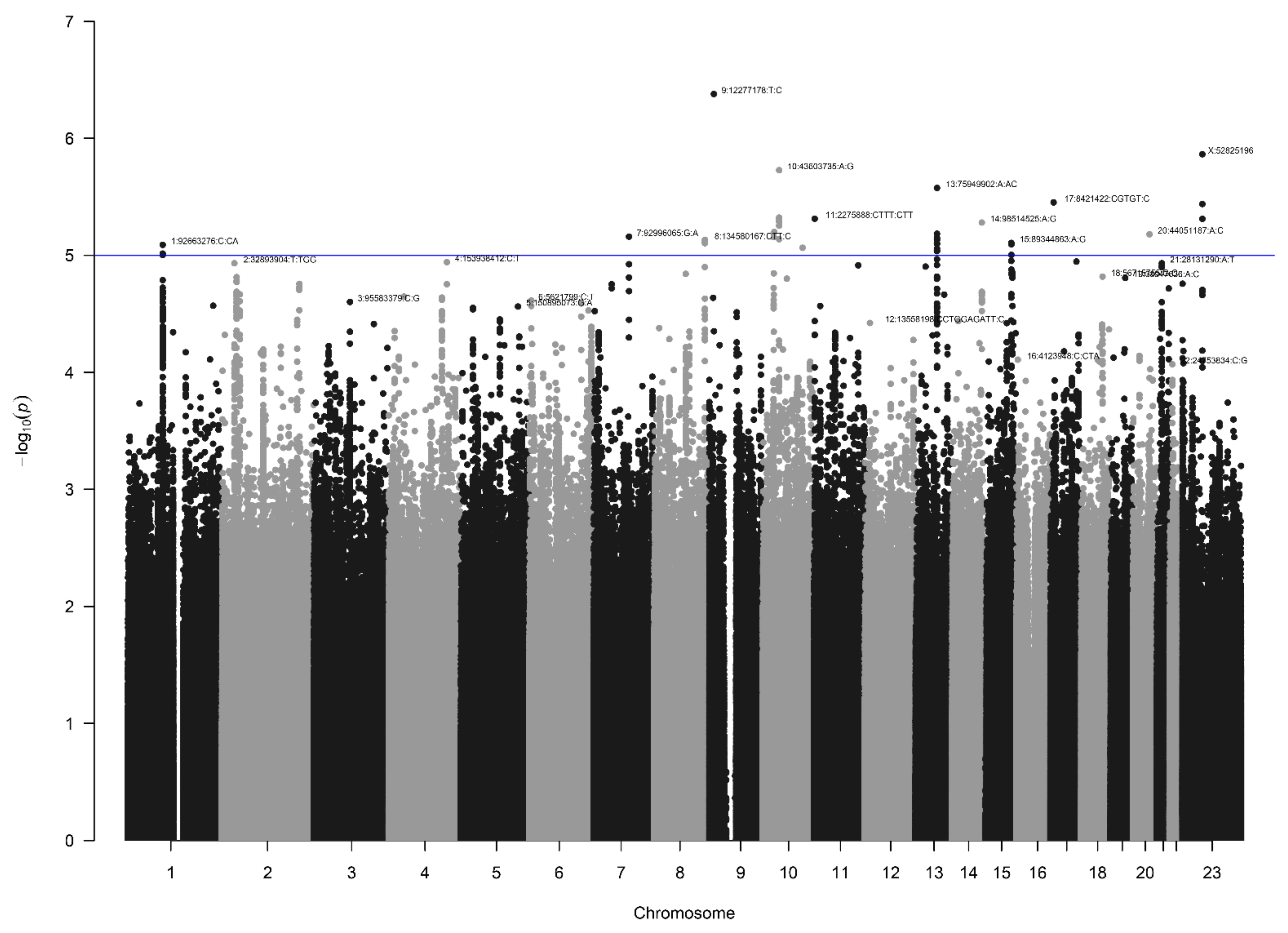
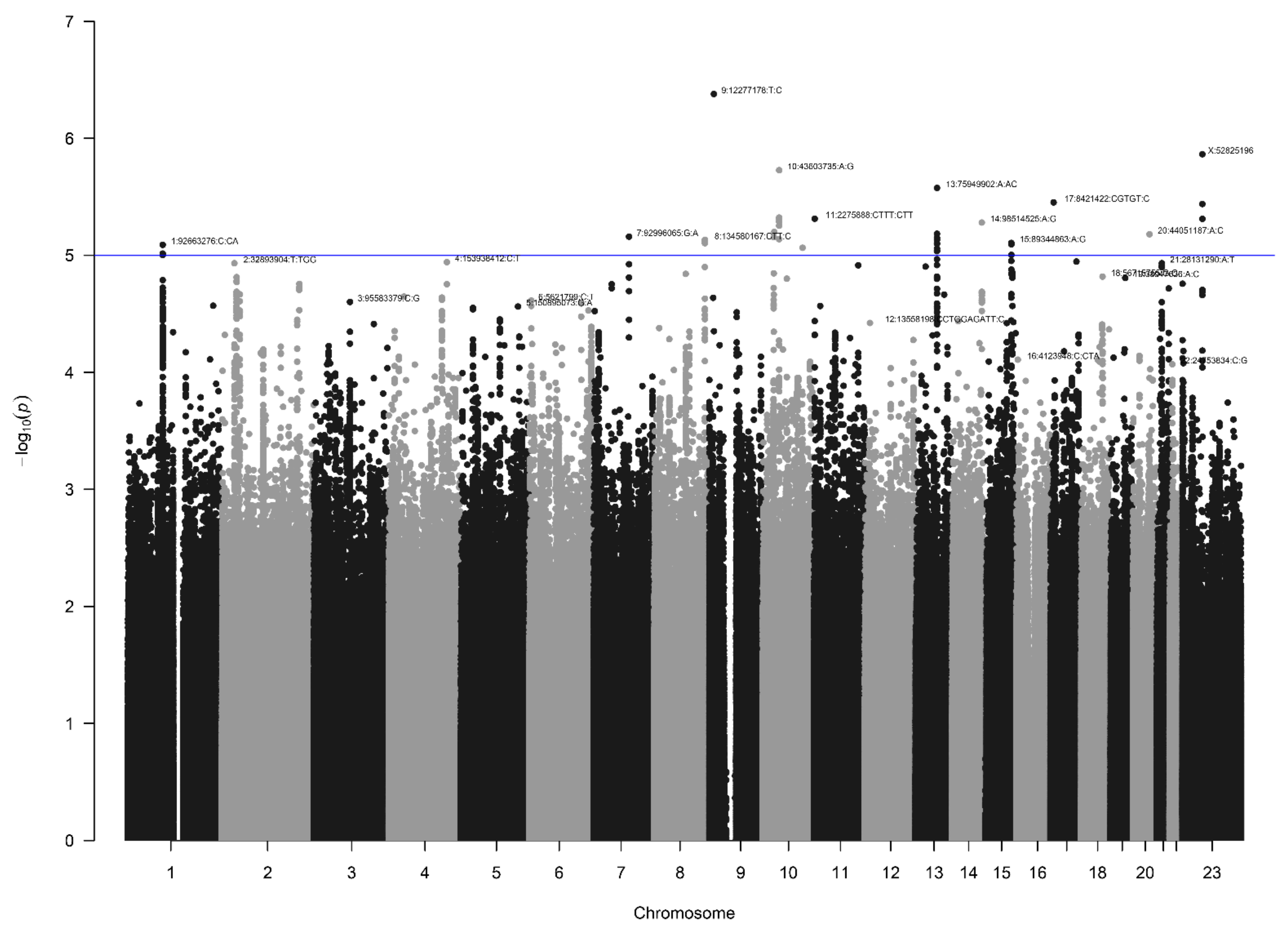
3.2. AGORA Dataset Results
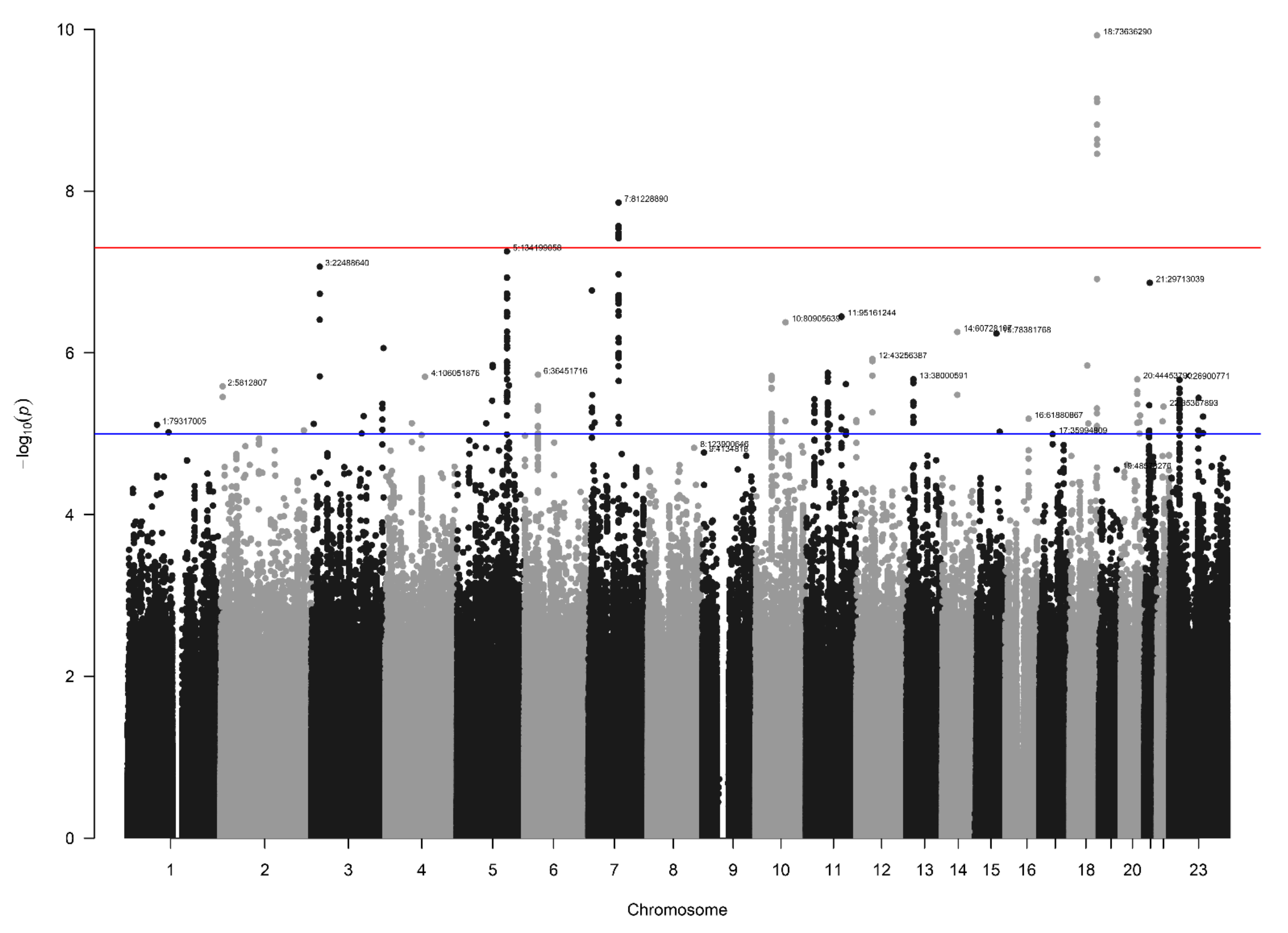
3.3. NBS Dataset Results
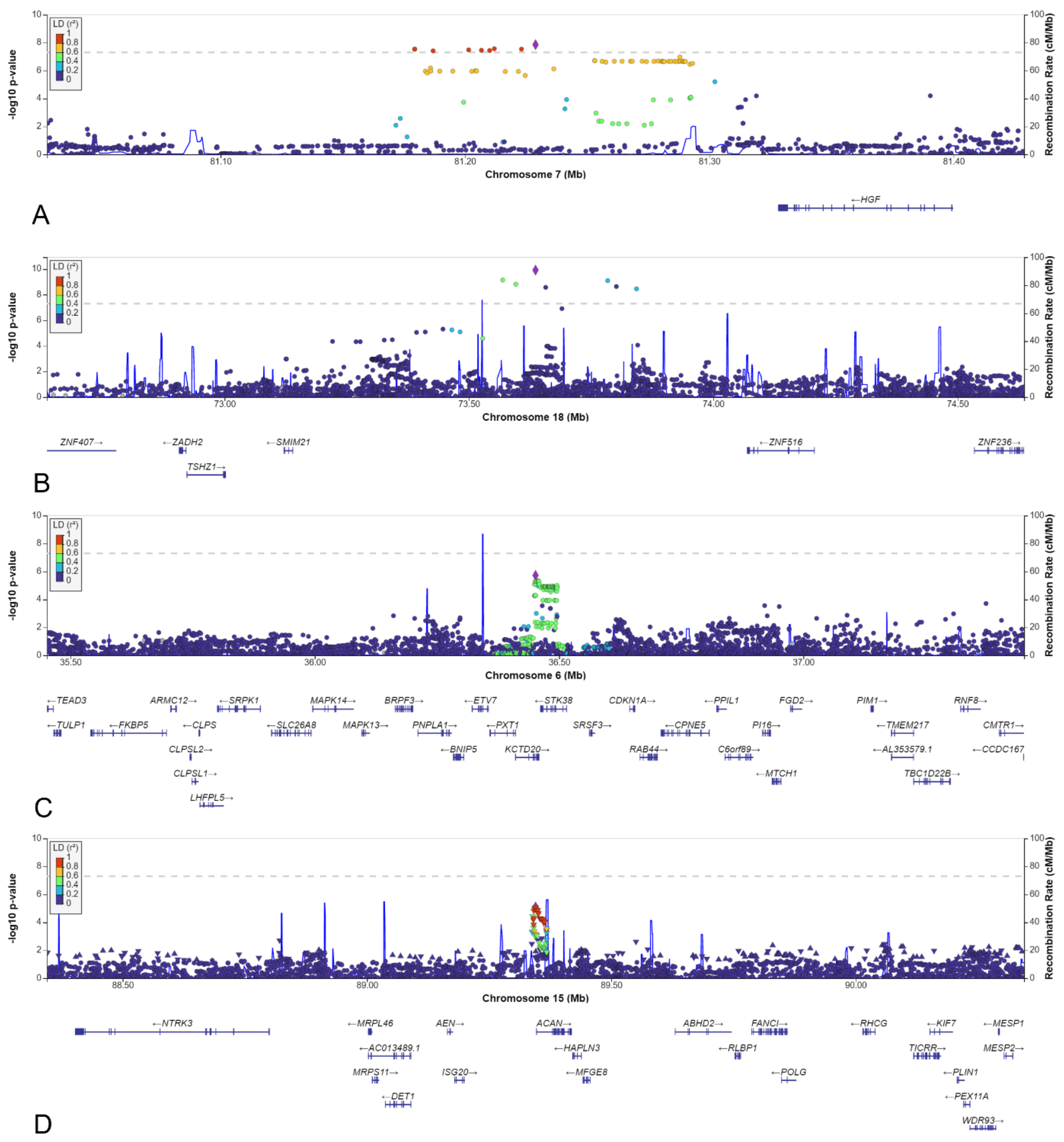
3.4. Loci with a Genome-Wide Statistically Significant p-Value
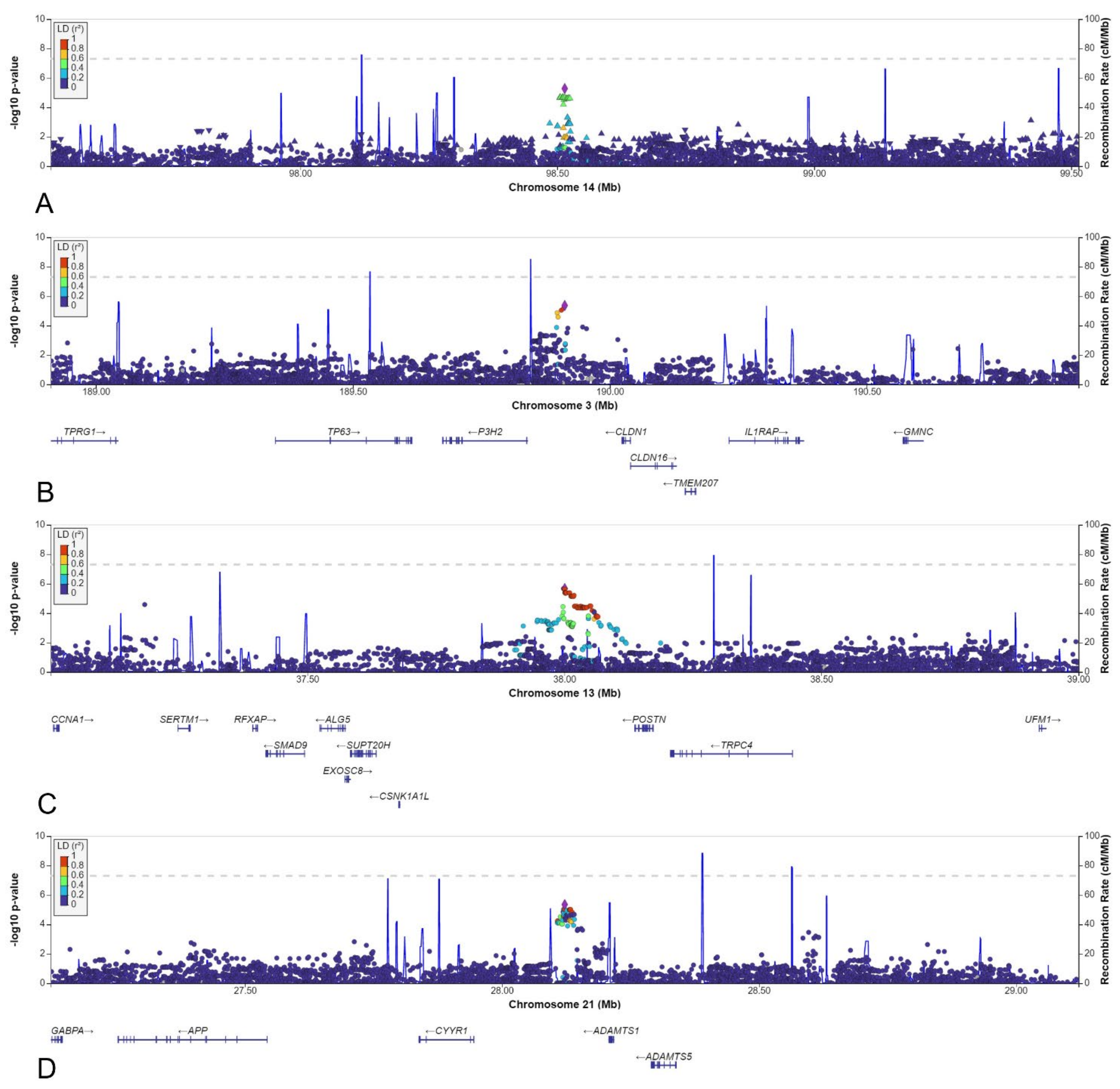
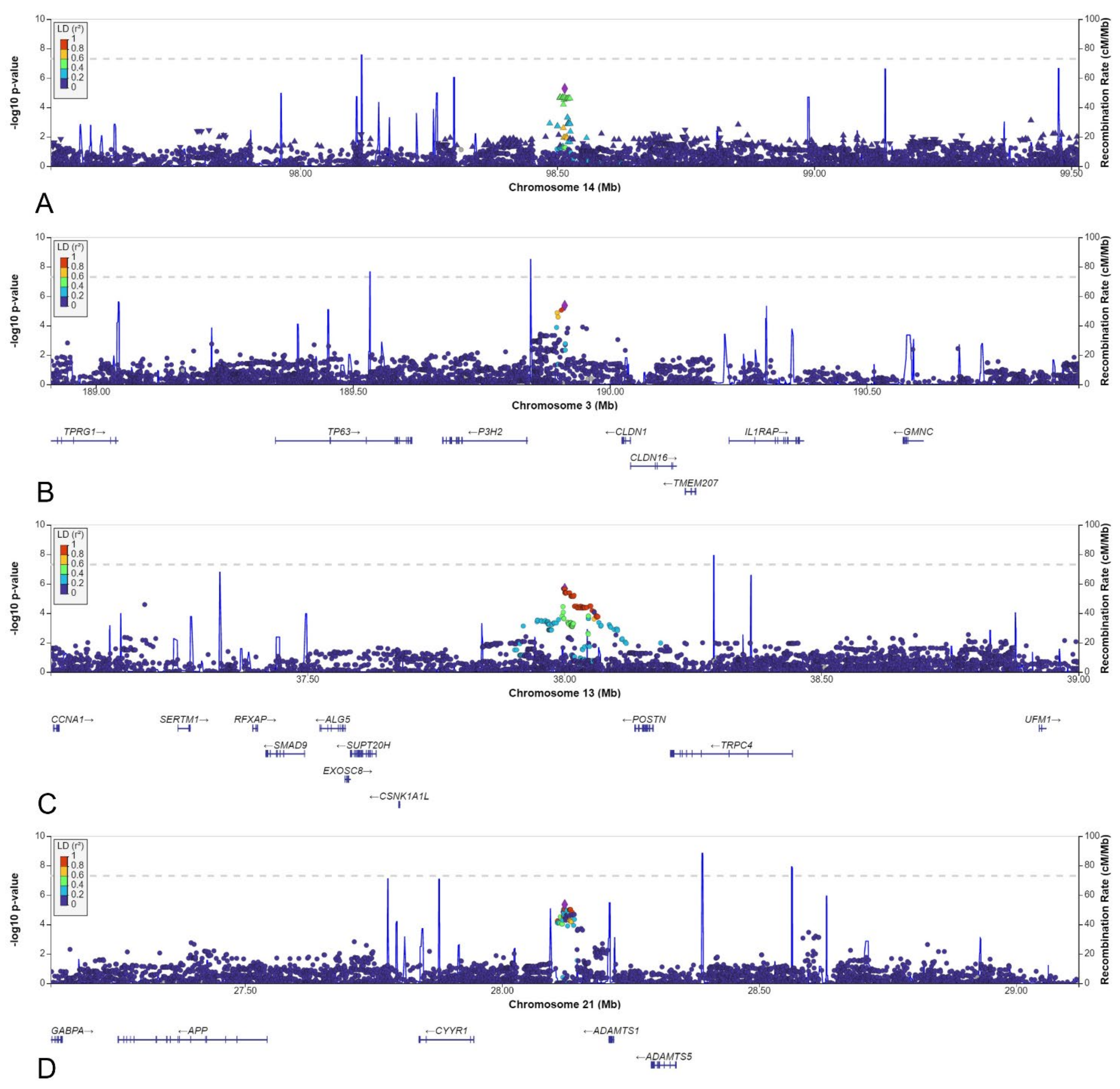
3.5. Other Selected Loci
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Groen in ’t Woud, S.R.; Westland, R.; Renkema, K.Y.; Steffens, M.G.; Gracchi, V.; Lilien, M.R.; van Wijk, J.A.E.; Feitz, W.F.J.; Schreuder, M.F.; van der Zanden, L.F.M.; for the SOFIA Study Group. Risk factors for kidney injury in children with solitary functioning kidney. Kidney Int. 2022, in press. [Google Scholar] [CrossRef] [PubMed]
- Sanna-Cherchi, S.; Ravani, P.; Corbani, V.; Parodi, S.; Haupt, R.; Piaggio, G.; Innocenti, M.L.; Somenzi, D.; Trivelli, A.; Caridi, G.; et al. Renal outcome in patients with congenital anomalies of the kidney and urinary tract. Research Support, Non-U.S. Gov’t. Kidney Int. 2009, 76, 528–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, D.R., Jr.; Woo, J.G.; Sinaiko, A.R.; Daniels, S.R.; Ikonen, J.; Juonala, M.; Kartiosuo, N.; Lehtimaki, T.; Magnussen, C.G.; Viikari, J.S.A.; et al. Childhood Cardiovascular Risk Factors and Adult Cardiovascular Events. N. Engl. J. Med. 2022, 386, 1877–1888. [Google Scholar] [CrossRef] [PubMed]
- Knoers, N. The term CAKUT has outlived its usefulness: The case for the defense. Pediatr. Nephrol. 2022, 37, 2793–2798. [Google Scholar] [CrossRef]
- Nicolaou, N.; Renkema, K.Y.; Bongers, E.M.H.F.; Giles, R.H.; Knoers, N.V.A.M. Genetic, environmental, and epigenetic factors involved in CAKUT. Nat. Rev. Nephrol. 2015, 11, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Dart, A.; Ruth, C.; Sellers, E.; Au, W.; Dean, H. Maternal diabetes mellitus and congenital anomalies of the kidney and urinary tract (CAKUT) in the child. Am. J. Kidney Dis. 2015, 65, 684–691. [Google Scholar] [CrossRef] [PubMed]
- Groen in ’t Woud, S.; Renkema, K.; Schreuder, M.; Wijers, C.; van der Zanden, L.F.M.; Knoers, N.V.A.M.; Feitz, W.F.J.; Bongers, E.; Roeleveld, N.; van Rooij, I.A.L.M. Maternal risk factors involved in specific congenital anomalies of the kidney and urinary tract: A case-control study. Birth Defects Res. Part A Clin. Mol. Teratol. 2016, 106, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Macumber, I.; Schwartz, S.; Leca, N. Maternal obesity is associated with congenital anomalies of the kidney and urinary tract in offspring. Pediatr. Nephrol. 2017, 32, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Groen in ’t Woud, S.R.; van Rooij, A.L.M.; Feitz, W.F.J.; Schreuder, M.F.; van der Zanden, L.F.M. Environmental risk factors for congenital solitary functioning kidney—A case-control study. 2022; submitted for publication. [Google Scholar]
- Sanna-Cherchi, S.; Westland, R.; Ghiggeri, G.M.; Gharavi, A.G. Genetic basis of human congenital anomalies of the kidney and urinary tract. J. Clin. Investig. 2018, 128, 4–15. [Google Scholar] [CrossRef]
- Seltzsam, S.; Wang, C.; Zheng, B.; Mann, N.; Connaughton, D.M.; Wu, C.W.; Schneider, S.; Schierbaum, L.; Kause, F.; Kolvenbach, C.M.; et al. Reverse phenotyping facilitates disease allele calling in exome sequencing of patients with CAKUT. Genet. Med. 2022, 24, 307–318. [Google Scholar] [CrossRef]
- Westland, R.; Schreuder, M.F.; van Goudoever, J.B.; Sanna-Cherchi, S.; van Wijk, J.A.E. Clinical implications of the solitary functioning kidney. Clin. J. Am. Soc. Nephrol. 2014, 9, 978–986. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Li, J.; Li, Y.; Li, H.; Yang, B.; Fan, H.; Wang, J.; Gu, Y.; Yu, H.; Bai, M.; et al. Genetic diagnosis of common fetal renal abnormalities detected on prenatal ultrasound. Prenat. Diagn. 2022, 42, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Lei, T.Y.; Fu, F.; Li, R.; Yu, Q.X.; Du, K.; Zhang, W.W.; Deng, Q.; Li, L.S.; Wang, D.; Yang, X.; et al. Whole-exome sequencing in the evaluation of fetal congenital anomalies of the kidney and urinary tract detected by ultrasonography. Prenat. Diagn. 2020, 40, 1290–1299. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Xu, Q.; Xie, J.; Ma, J.; Qiao, P.; Zhang, W.; Yu, H.; Wang, W.; Qian, Y.; Zhang, Q.; et al. Identification of 8 Novel Mutations in Nephrogenesis-Related Genes in Chinese Han Patients with Unilateral Renal Agenesis. Am. J. Nephrol. 2017, 46, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Sanna-Cherchi, S.; Kiryluk, K.; Burgess, K.E.; Bodria, M.; Sampson, M.G.; Hadley, D.; Nees, S.N.; Verbitsky, M.; Perry, B.J.; Sterken, R.; et al. Copy-Number Disorders Are a Common Cause of Congenital Kidney Malformations. Am. J. Hum. Genet. 2012, 91, 987–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbitsky, M.; Westland, R.; Perez, A.; Kiryluk, K.; Liu, Q.X.; Krithivasan, P.; Mitrotti, A.; Fasel, D.A.; Batourina, E.; Sampson, M.G.; et al. The copy number variation landscape of congenital anomalies of the kidney and urinary tract. Nat. Genet. 2019, 51, 117–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westland, R.; Verbitsky, M.; Vukojevic, K.; Perry, B.J.; Fasel, D.A.; Zwijnenburg, P.J.G.; Bokenkamp, A.; Gille, J.J.P.; Saraga-Babic, M.; Ghiggeri, G.M.; et al. Copy number variation analysis identifies novel CAKUT candidate genes in children with a solitary functioning kidney. Kidney Int. 2015, 88, 1402–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [Green Version]
- Verbitsky, M.; Krithivasan, P.; Batourina, E.; Khan, A.; Graham, S.E.; Marasa, M.; Kim, H.; Lim, T.Y.; Weng, P.L.; Sanchez-Rodriguez, E.; et al. Copy Number Variant Analysis and Genome-wide Association Study Identify Loci with Large Effect for Vesicoureteral Reflux. J. Am. Soc. Nephrol. 2021, 2, 805–820. [Google Scholar] [CrossRef]
- Darlow, J.M.; Dobson, M.G.; Darlay, R.; Molony, C.M.; Hunziker, M.; Green, A.J.; Cordell, H.J.; Puri, P.; Barton, D.E. A new genome scan for primary nonsyndromic vesicoureteric reflux emphasizes high genetic heterogeneity and shows linkage and association with various genes already implicated in urinary tract development. Mol. Genet. Genom. Med. 2014, 2, 7–29. [Google Scholar] [CrossRef]
- Van der Zanden, L.F.M.; Borisov, O.; van Rooij, I.A.L.M.; Quaedackers, J.S.L.T.; Steffens, M.G.; Schierbaum, L.; Schneider, S.; Waffenschmidt, L.; Kiemeney, L.A.L.M.; de Wall, L.L.L.; et al. Genome-wide association study in patients with posterior urethral valves. Front. Pediatr. 2022, 10, 988374. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.M.Y.; Sadeghi-Alavijeh, O.; Lopes, F.M.; Hilger, A.C.; Stanescu, H.C.; Voinescu, C.D.; Beaman, G.M.; Newman, W.G.; Zaniew, M.; Weber, S.; et al. Diverse ancestry whole-genome sequencing association study identifies TBX5 and PTK7 as susceptibility genes for posterior urethral valves. eLife 2022, 11, E74777. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, I.A.; van der Zanden, L.F.; Bongers, E.M.; Renkema, K.Y.; Wijers, C.H.; Thonissen, M.; Dokter, E.M.; Marcelis, C.L.; de Blaauw, I.; Wijnen, M.H.; et al. AGORA, a data- and biobank for birth defects and childhood cancer. Birth Defects Res. A Clin. Mol. Teratol. 2016, 106, 675–684. [Google Scholar] [CrossRef]
- Galesloot, T.E.; Vermeulen, S.H.; Swinkels, D.W.; de Vegt, F.; Franke, B.; den Heijer, M.; de Graaf, J.; Verbeek, A.L.M.; Kiemeney, L. Cohort Profile: The Nijmegen Biomedical Study (NBS). Int. J. Epidemiol. 2017, 46, 1099–1100j. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Manichaikul, A.; Mychaleckyj, J.C.; Rich, S.S.; Daly, K.; Sale, M.; Chen, W.M. Robust relationship inference in genome-wide association studies. Bioinformatics 2010, 26, 2867–2873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Forer, L.; Schonherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef] [Green Version]
- The 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loh, P.R.; Danecek, P.; Palamara, P.F.; Fuchsberger, C.; Reshef, Y.A.; Finucane, H.K.; Schoenherr, S.; Forer, L.; McCarthy, S.; Abecasis, G.R.; et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 2016, 48, 1443–1448. [Google Scholar] [CrossRef] [Green Version]
- Privé, F. Ancestry inference and grouping from principal component analysis of genetic data. bioRxiv 2020, 109, 12–23. [Google Scholar]
- Watanabe, K.; Taskesen, E.; van Bochoven, A.; Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 2017, 8, 1826. [Google Scholar] [CrossRef] [PubMed]
- Boomsma, D.I.; Wijmenga, C.; Slagboom, E.P.; Swertz, M.A.; Karssen, L.C.; Abdellaoui, A.; Ye, K.; Guryev, V.; Vermaat, M.; van Dijk, F.; et al. The Genome of the Netherlands: Design, and project goals. Eur. J. Hum. Genet. 2014, 22, 221–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruim, R.J.; Welch, R.P.; Sanna, S.; Teslovich, T.M.; Chines, P.S.; Gliedt, T.P.; Boehnke, M.; Abecasis, G.R.; Willer, C.J. LocusZoom: Regional visualization of genome-wide association scan results. Bioinformatics 2010, 26, 2336–2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Porter, J.; Zhao, C.; Zhu, H.; Wang, N.; Sun, Z.; Mo, Y.Y.; Wang, Z. TADKB: Family classification and a knowledge base of topologically associating domains. BMC Genom. 2019, 20, 217. [Google Scholar] [CrossRef] [Green Version]
- Boyle, A.P.; Hong, E.L.; Hariharan, M.; Cheng, Y.; Schaub, M.A.; Kasowski, M.; Karczewski, K.J.; Park, J.; Hitz, B.C.; Weng, S.; et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012, 22, 1790–1797. [Google Scholar] [CrossRef] [Green Version]
- Cardoso-Moreira, M.; Halbert, J.; Valloton, D.; Velten, B.; Chen, C.; Shao, Y.; Liechti, A.; Ascencao, K.; Rummel, C.; Ovchinnikova, S.; et al. Gene expression across mammalian organ development. Nature 2019, 571, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Van der Zanden, L.F.; van Rooij, I.A.; Feitz, W.F.; Knight, J.; Donders, A.R.; Renkema, K.Y.; Bongers, E.M.; Vermeulen, S.H.; Kiemeney, L.A.; Veltman, J.A.; et al. Common variants in DGKK are strongly associated with risk of hypospadias. Nat. Genet. 2011, 43, 48–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoenwolf, G.C.; Bleyl, S.B.; Brauer, P.R.; Francis-West, P.H. Larsen’s Human Embryology, 5th ed.; Churchill Livingstone: London, UK, 2015. [Google Scholar]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, O.; Guo, M.H.; Dunbar, N.; Popovic, J.; Flynn, D.; Jacobsen, C.; Lui, J.C.; Hirschhorn, J.N.; Baron, J.; Dauber, A. Short stature, accelerated bone maturation, and early growth cessation due to heterozygous aggrecan mutations. J. Clin. Endocrinol. Metab. 2014, 99, E1510–E1518. [Google Scholar] [CrossRef] [PubMed]
- Santos, O.F.; Barros, E.J.; Yang, X.M.; Matsumoto, K.; Nakamura, T.; Park, M.; Nigam, S.K. Involvement of hepatocyte growth factor in kidney development. Dev. Biol. 1994, 163, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Woolf, A.S.; Kolatsi-Joannou, M.; Hardman, P.; Andermarcher, E.; Moorby, C.; Fine, L.G.; Jat, P.S.; Noble, M.D.; Gherardi, E. Roles of hepatocyte growth factor/scatter factor and the met receptor in the early development of the metanephros. J. Cell Biol. 1995, 128, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Uehara, Y.; Minowa, O.; Mori, C.; Shiota, K.; Kuno, J.; Noda, T.; Kitamura, N. Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature 1995, 373, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Ishibe, S.; Karihaloo, A.; Ma, H.; Zhang, J.; Marlier, A.; Mitobe, M.; Togawa, A.; Schmitt, R.; Czyczk, J.; Kashgarian, M.; et al. Met and the epidermal growth factor receptor act cooperatively to regulate final nephron number and maintain collecting duct morphology. Development 2009, 136, 337–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulte, L.; Schulz, A.; Unland, J.; Schulz, H.; Hubner, N.; Schmidt-Ott, K.M.; Kreutz, R. MWF rats with spontaneous albuminuria inherit a reduced efficiency of nephron induction during early nephrogenesis in comparison to SHR rats. J. Hypertens. 2012, 30, 2031–2038. [Google Scholar] [CrossRef] [PubMed]
- Skoblov, M.; Marakhonov, A.; Marakasova, E.; Guskova, A.; Chandhoke, V.; Birerdinc, A.; Baranova, A. Protein partners of KCTD proteins provide insights about their functional roles in cell differentiation and vertebrate development. BioEssays 2013, 35, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Nawa, M.; Matsuoka, M. KCTD20, a relative of BTBD10, is a positive regulator of Akt. BMC Biochem. 2013, 14, 27. [Google Scholar] [CrossRef] [Green Version]
- Awazu, M.; Hida, M. Maternal nutrient restriction inhibits ureteric bud branching but does not affect the duration of nephrogenesis in rats. Pediatr. Res. 2015, 77, 633–639. [Google Scholar] [CrossRef] [Green Version]
- De Barros Sene, L.; Lamana, G.L.; Schwambach Vieira, A.; Scarano, W.R.; Gontijo, J.A.R.; Boer, P.A. Gestational Low Protein Diet Modulation on miRNA Transcriptome and Its Target During Fetal and Breastfeeding Nephrogenesis. Front. Physiol. 2021, 12, 648056. [Google Scholar] [CrossRef]
- Chen, J.K.; Chen, J.; Neilson, E.G.; Harris, R.C. Role of mammalian target of rapamycin signaling in compensatory renal hypertrophy. J. Am. Soc. Nephrol. 2005, 16, 1384–1391. [Google Scholar] [CrossRef] [Green Version]
- Apostolou, A.; Poreau, B.; Delrieu, L.; Thevenon, J.; Jouk, P.S.; Lallemand, G.; Emadali, A.; Sartelet, H. High Activation of the AKT Pathway in Human Multicystic Renal Dysplasia. Pathobiology 2020, 87, 302–310. [Google Scholar] [CrossRef]
- Martin, A.P.; Aushev, V.N.; Zalcman, G.; Camonis, G.H. The STK38-XPO1 axis, a new actor in physiology and cancer. Cell Mol. Life Sci. 2021, 78, 1943–1955. [Google Scholar] [CrossRef] [PubMed]
- Reginensi, A.; Enderle, L.; Gregorieff, A.; Johnson, R.L.; Wrana, J.L.; McNeill, H. A critical role for NF2 and the Hippo pathway in branching morphogenesis. Nat. Commun. 2016, 7, 12309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reginensi, A.; Scott, R.P.; Gregorieff, A.; Bagherie-Lachidan, M.; Chung, C.; Lim, D.S.; Pawson, T.; Wrana, J.; McNeill, H. Yap- and Cdc42-dependent nephrogenesis and morphogenesis during mouse kidney development. PLoS Genet. 2013, 9, e1003380. [Google Scholar] [CrossRef] [Green Version]
- Drake, K.A.; Chaney, C.; Patel, M.; Das, A.; Bittencourt, J.; Cohn, M.; Carroll, T.J. Transcription Factors YAP/TAZ and SRF Cooperate To Specify Renal Myofibroblasts in the Developing Mouse Kidney. J. Am. Soc. Nephrol. 2022, 33, 1694–1707. [Google Scholar] [CrossRef] [PubMed]
- Holloway, E.M.; Wu, J.H.; Czerwinski, M.; Sweet, C.W.; Wu, A.; Tsai, Y.H.; Huang, S.; Stoddard, A.E.; Capeling, M.M.; Glass, I.; et al. Differentiation of Human Intestinal Organoids with Endogenous Vascular Endothelial Cells. Dev. Cell. 2020, 54, 516–528.e7. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chr | Position | rsID | Discovery Dataset | MAF Patients | MAF Reference | AGORA Dataset | NBS Dataset | n SNVs | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MAF Controls | OR | p-Value | MAF Controls | OR | p-Value | |||||||
| Genome-wide significant SNVs | ||||||||||||
| 7 | 81228890 | rs140804918 | NBS | 0.048 | 0.031 | 0.029 | 1.91 | 8.4 × 10−3 | 0.020 | 3.10 | 1.4 × 10−8 | 51 |
| 18 | 73636290 | rs184382636 | NBS | 0.017 | 0.002 | 0.003 | 5.93 | 2.4 × 10−3 | 0.002 | 12.9 | 1.2 × 10−10 | 42 |
| SNVs selected based on MAF in discovery and reference populations | ||||||||||||
| 3 | 189912222 | rs10433490 | NBS | 0.092 | 0.169 | 0.153 | 0.59 | 4.0 × 10−4 | 0.150 | 0.53 | 4.3 × 10−6 | 6 |
| 6 | 36451716 | rs148413365 | NBS | 0.106 | 0.188 * | 0.156 | 0.94 | 7.0 × 10−1 | 0.166 | 0.51 | 1.9 × 10−6 | 84 |
| 13 | 38000591 | rs9547854 | NBS | 0.087 | 0.047 | 0.052 | 1.83 | 1.6 × 10−3 | 0.051 | 1.97 | 2.1 × 10−6 | 66 |
| 14 | 98514525 | rs148251525 | AGORA | 0.127 | 0.048 | 0.075 | 2.08 | 5.2 × 10−6 | 0.106 | 1.22 | 8.2 × 10−2 | 5 |
| 15 | 89344863 | rs111283115 | AGORA | 0.333 | 0.449 | 0.499 | 0.65 | 7.8 × 10−6 | 0.387 | 0.71 | 3.5 × 10−4 | 24 |
| 21 | 28122146 | rs2830456 | NBS | 0.281 | 0.370 | 0.410 | 0.64 | 2.5 × 10−5 | 0.350 | 0.63 | 4.5 × 10−6 | 46 |
| Other suggestively significant SNVs | ||||||||||||
| 1 | 79317005 | rs140198115 | NBS | 0.025 | 0.009 | 0.011 | 2.37 | 1.9 × 10−2 | 0.010 | 3.79 | 7.8 × 10−6 | 17 |
| 1 | 92663276 | rs11406716 | AGORA | 0.319 | 0.283 * | 0.246 | 1.64 | 8.1 × 10−6 | 0.277 | 1.27 | 8.4 × 10−3 | 76 |
| 3 | 22488640 | rs182382581 | NBS | 0.025 | 0.012 | 0.009 | 4.10 | 1.5 × 10−3 | 0.008 | 5.04 | 8.6 × 10−8 | 7 |
| 3 | 6328767 | rs7629003 | NBS | 0.009 | 0.003 | 0.003 | 4.68 | 4.9 × 10−2 | 0.002 | 14.48 | 7.6 × 10−6 | 20 |
| 4 | 106051876 | rs190397903 | NBS | 0.020 | 0.007 | 0.013 | 1.97 | 8.5 × 10−2 | 0.007 | 5.23 | 2.0 × 10−6 | 9 |
| 4 | 70836149 | rs146356251 | NBS | 0.018 | 0.008 | 0.008 | 2.79 | 2.1 × 10−2 | 0.006 | 4.84 | 7.4 × 10−6 | 7 |
| 5 | 78674386 | rs138487999 | NBS | 0.034 | 0.019 | 0.016 | 2.33 | 5.5 × 10−3 | 0.014 | 2.84 | 7.4 × 10−6 | 125 |
| 5 | 139646076 | rs145922969 | NBS | 0.030 | 0.025 | 0.013 | 3.15 | 1.6 × 10−3 | 0.015 | 3.20 | 2.5 × 10−6 | 8 |
| 5 | 96015902 | rs181945740 | NBS | 0.018 | 0.001 | 0.008 | 4.20 | 7.0 × 10−3 | 0.005 | 5.90 | 1.4 × 10−6 | 7 |
| 5 | 134199058 | rs73282857 | NBS | 0.019 | 0.006 | 0.005 | 4.68 | 9.4 × 10−3 | 0.006 | 7.00 | 5.6 × 10−8 | 150 |
| 7 | 10014656 | rs190937828 | NBS | 0.090 | 0.063 | 0.066 | 1.76 | 1.3 × 10−3 | 0.054 | 2.30 | 1.7 × 10−6 | 14 |
| 8 | 134595232 | rs10112722 | AGORA | 0.437 | 0.366 | 0.347 | 1.55 | 7.5 × 10−6 | 0.370 | 1.34 | 2.3 × 10−4 | 13 |
| 10 | 44117049 | rs60001082 | NBS | 0.085 | 0.069 * | 0.065 | 1.46 | 4.3 × 10−2 | 0.054 | 2.00 | 7.3 × 10−6 | 17 |
| 11 | 2275888 | rs1325019515 | AGORA | 0.380 | n/a | 0.305 | 1.61 | 4.9 × 10−6 | 0.312 | 1.40 | 1.1 × 10−4 | 19 |
| 11 | 58767505 | rs11229723 | NBS | 0.022 | 0.006 | 0.011 | 2.23 | 3.4 × 10−2 | 0.007 | 4.41 | 1.8 × 10−6 | 64 |
| 11 | 95161244 | rs192152837 | NBS | 0.018 | 0.004 | 0.004 | 10.2 | 2.4 × 10−4 | 0.005 | 6.35 | 3.6 × 10−7 | 8 |
| 12 | 43256387 | rs181072352 | NBS | 0.012 | 0.005 | 0.008 | 1.44 | 4.8 × 10−1 | 0.002 | 10.40 | 1.2 × 10−6 | 5 |
| 12 | 385846 | rs554290784 | NBS | 0.015 | 0.005 | 0.004 | 4.23 | 1.1 × 10−2 | 0.003 | 5.59 | 6.8 × 10−6 | 5 |
| 13 | 75949902 | rs144419778 | AGORA | 0.267 | 0.212 * | 0.19 | 1.71 | 2.7 × 10−6 | 0.205 | 1.47 | 1.9 × 10−5 | 24 |
| 14 | 60728107 | rs146557102 | NBS | 0.041 | n/a | 0.018 | 2.75 | 4.8 × 10−4 | 0.017 | 2.96 | 5.5 × 10−7 | 8 |
| 18 | 50196707 | rs77493404 | NBS | 0.022 | 0.009 | 0.014 | 2.25 | 1.2 × 10−2 | 0.008 | 4.23 | 7.5 × 10−6 | 22 |
| 20 | 44453790 | rs201841698 | NBS | 0.033 | 0.029 | 0.020 | 2.42 | 6.1 × 10−3 | 0.013 | 3.40 | 2.1 × 10−6 | 7 |
| 20 | 51885312 | rs6126804 | NBS | 0.030 | 0.008 | 0.016 | 2.58 | 7.3 × 10−3 | 0.013 | 3.30 | 6.0 × 10−6 | 8 |
| 22 | 32945612 | rs73172143 | NBS | 0.095 | 0.078 | 0.068 | 1.47 | 2.7 × 10−2 | 0.059 | 1.88 | 7.0 × 10−6 | 6 |
| Chr | rsID | Dataset | Candidate Affected Gene | Distance from SNV to TSS | Regulome Score [36] | Variant Effect Predictor [40] | Gene Function * |
|---|---|---|---|---|---|---|---|
| Genome-wide significant SNVs | |||||||
| 7 | rs140804918 | NBS | HGF | 171 kbp | 0.247 | Intergenic | Regulates cell growth, motility and morphogenesis in various cell types and tissues |
| 18 | rs184382636 | NBS | SMIM21 | 497 kbp | 0.071 | Intergenic | Integral membrane component |
| SNVs selected based on MAF in discovery and reference populations | |||||||
| 3 | rs10433490 | NBS | P3H2 | 72 kbp | 0.154 | Intergenic | Posttranslational modifier of collagen IV |
| 6 | rs148413365 | NBS | KCTD20 | 41 kbp | 0.163 | Intronic | Member of AKT-mTOR-p70 S6k signalling cascade |
| STK38 | 64 kbp | YAP/TAZ inhibitor in hippo pathway | |||||
| 13 | rs9547854 | NBS | POSTN | 172 kbp | 0.112 | Intergenic | Induces cell attachment and spreading and plays a role in cell adhesion |
| 14 | rs148251525 | AGORA | BCL11B | 1224 kbp | 0.000 | Intergenic | T cell transcription factor |
| 15 | rs111283115 | AGORA | ACAN | 1.8 kbp | 0.145 | Upstream gene variant | Major component of extracellular matrix of cartilaginous tissues |
| 21 | rs2830456 | NBS | ADAMTS1 | 96 kbp | 0.184 | Intergenic | Has antiangiogenic activity, involved in inflammation and cancer cachexia |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Groen in ’t Woud, S.; Maj, C.; Renkema, K.Y.; Westland, R.; Galesloot, T.; van Rooij, I.A.L.M.; Vermeulen, S.H.; Feitz, W.F.J.; Roeleveld, N.; Schreuder, M.F.; et al. A Genome-Wide Association Study into the Aetiology of Congenital Solitary Functioning Kidney. Biomedicines 2022, 10, 3023. https://doi.org/10.3390/biomedicines10123023
Groen in ’t Woud S, Maj C, Renkema KY, Westland R, Galesloot T, van Rooij IALM, Vermeulen SH, Feitz WFJ, Roeleveld N, Schreuder MF, et al. A Genome-Wide Association Study into the Aetiology of Congenital Solitary Functioning Kidney. Biomedicines. 2022; 10(12):3023. https://doi.org/10.3390/biomedicines10123023
Chicago/Turabian StyleGroen in ’t Woud, Sander, Carlo Maj, Kirsten Y. Renkema, Rik Westland, Tessel Galesloot, Iris A. L. M. van Rooij, Sita H. Vermeulen, Wout F. J. Feitz, Nel Roeleveld, Michiel F. Schreuder, and et al. 2022. "A Genome-Wide Association Study into the Aetiology of Congenital Solitary Functioning Kidney" Biomedicines 10, no. 12: 3023. https://doi.org/10.3390/biomedicines10123023