

TCR-Engineered Lymphocytes Targeting NY-ESO-1: In Vitro Assessment of Cytotoxicity against Tumors
 , , , , , , and
, , , , , , and
Abstract
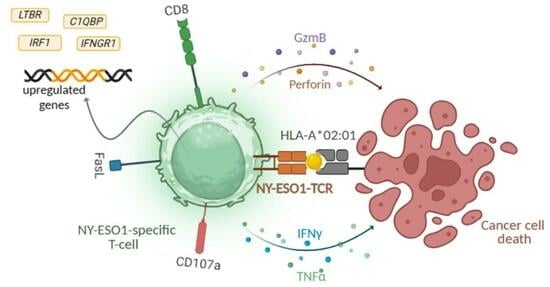
:
1. Introduction
2. Materials and Methods
2.1. Study Population and Interventions
2.2. Isolation of Peripheral Blood Mononuclear Cells
2.3. Retroviral Vector
2.4. Triggering T-Cell Proliferation
2.5. Transduction of Anti-CD3 Primed PBMCs via Retroviral Vectors
2.6. Evaluation of the Efficiency of Transduction
2.7. Cell Lines
2.8. Phenotyping for Markers of Activation and Cytotoxicity by Flow Cytometry
2.9. HSNE Dimensionality Reduction and Clustering
2.10. Magnetic Separation of Transduced T Cells after Co-Culturing with Tumor Cells
2.11. Total RNA Extraction
2.12. Gene Expression Profiling by Nanostring
2.13. Differential Gene Expression Testing
2.14. LDH Cytotoxicity Assay
2.15. Quantification of Cytokine Production
3. Results
3.1. Anti-NY-ESO-1-Construct-Transduced T Cells Exhibit Cytotoxicity Indicators
3.2. Transcriptional Profiling Unveils Immunological Signatures within Transduced T Cells
3.3. Anti-NY-ESO-1-Construct-Transduced T Cells Induce Antigen-Specific Cytotoxicity
3.4. Anti-NY-ESO-1 Construct-Transduced T Cells Release Cytotoxicity-Related Cytokines upon Encountering Tumor Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Manfredi, F.; Cianciotti, B.C.; Potenza, A.; Tassi, E.; Noviello, M.; Biondi, A.; Ciceri, F.; Bonini, C.; Ruggiero, E. TCR Redirected T Cells for Cancer Treatment: Achievements, Hurdles, and Goals. Front. Immunol. 2020, 11, 1689. [Google Scholar] [CrossRef] [PubMed]
- Want, M.Y.; Bashir, Z.; Najar, R.A. T Cell Based Immunotherapy for Cancer: Approaches and Strategies. Vaccines 2023, 11, 835. [Google Scholar] [CrossRef] [PubMed]
- Cappell, K.M.; Kochenderfer, J.N. Long-Term Outcomes Following CAR T Cell Therapy: What We Know so Far. Nat. Rev. Clin. Oncol. 2023, 20, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Jiang, G. The Journey of CAR-T Therapy in Hematological Malignancies. Mol. Cancer 2022, 21, 194. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Lu, L.; Ma, W. Efficacy, Safety, and Challenges of CAR T-Cells in the Treatment of Solid Tumors. Cancers 2022, 14, 5983. [Google Scholar] [CrossRef]
- Hiltensperger, M.; Krackhardt, A.M. Current and Future Concepts for the Generation and Application of Genetically Engineered CAR-T and TCR-T Cells. Front. Immunol. 2023, 14, 1121030. [Google Scholar] [CrossRef]
- Edes, I.; Clauss, J.; Stahn, R.; Japp, A.S.; Lorenz, F.K.M. TCR and CAR Engineering of Primary Human T Cells BT—Gene Therapy of Cancer: Methods and Protocols; Walther, W., Ed.; Springer: New York, NY, USA, 2022; pp. 85–93. ISBN 978-1-0716-2441-8. [Google Scholar]
- Jungbluth, A.A.; Chen, Y.T.; Stockert, E.; Busam, K.J.; Kolb, D.; Iversen, K.; Coplan, K.; Williamson, B.; Altorki, N.; Old, L.J. Immunohistochemical Analysis of NY-ESO-1 Antigen Expression in Normal and Malignant Human Tissues. Int. J. Cancer 2001, 92, 856–860. [Google Scholar] [CrossRef]
- Theurillat, J.-P.; Ingold, F.; Frei, C.; Zippelius, A.; Varga, Z.; Seifert, B.; Chen, Y.-T.; Jäger, D.; Knuth, A.; Moch, H. NY-ESO-1 Protein Expression in Primary Breast Carcinoma and Metastases: Correlation with CD8+ T-Cell and CD79a+ Plasmacytic/B-Cell Infiltration. Int. J. Cancer 2007, 120, 2411–2417. [Google Scholar] [CrossRef]
- Aung, P.P.; Liu, Y.-C.; Ballester, L.Y.; Robbins, P.F.; Rosenberg, S.A.; Lee, C.-C.R. Expression of New York Esophageal Squamous Cell Carcinoma-1 in Primary and Metastatic Melanoma. Hum. Pathol. 2014, 45, 259–267. [Google Scholar] [CrossRef]
- Prasad, M.L.; Jungbluth, A.A.; Patel, S.G.; Iversen, K.; Hoshaw-Woodard, S.; Busam, K.J. Expression and Significance of Cancer Testis Antigens in Primary Mucosal Melanoma of the Head and Neck. Head Neck 2004, 26, 1053–1057. [Google Scholar] [CrossRef]
- Grigoriadis, A.; Caballero, O.L.; Hoek, K.S.; da Silva, L.; Chen, Y.-T.; Shin, S.J.; Jungbluth, A.A.; Miller, L.D.; Clouston, D.; Cebon, J.; et al. CT-X Antigen Expression in Human Breast Cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 13493–13498. [Google Scholar] [CrossRef] [PubMed]
- Satie, A.-P.; Rajpert-De Meyts, E.; Spagnoli, G.C.; Henno, S.; Olivo, L.; Jacobsen, G.K.; Rioux-Leclercq, N.; Jégou, B.; Samson, M. The Cancer-Testis Gene, NY-ESO-1, Is Expressed in Normal Fetal and Adult Testes and in Spermatocytic Seminomas and Testicular Carcinoma in Situ. Lab. Investig. 2002, 82, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Melsen, J.E.; van Ostaijen-Ten Dam, M.M.; Lankester, A.C.; Schilham, M.W.; van den Akker, E.B. A Comprehensive Workflow for Applying Single-Cell Clustering and Pseudotime Analysis to Flow Cytometry Data. J. Immunol. 2020, 205, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Höllt, T.; Pezzotti, N.; van Unen, V.; Koning, F.; Eisemann, E.; Lelieveldt, B.; Vilanova, A. Cytosplore: Interactive Immune Cell Phenotyping for Large Single-Cell Datasets. Comput. Graph. Forum 2016, 35, 171–180. [Google Scholar] [CrossRef]
- Fang, Z.; Liu, X.; Peltz, G. GSEApy: A Comprehensive Package for Performing Gene Set Enrichment Analysis in Python. Bioinformatics 2023, 39, btac757. [Google Scholar] [CrossRef]
- Raza, A.; Merhi, M.; Inchakalody, V.P.; Krishnankutty, R.; Relecom, A.; Uddin, S.; Dermime, S. Unleashing the Immune Response to NY-ESO-1 Cancer Testis Antigen as a Potential Target for Cancer Immunotherapy. J. Transl. Med. 2020, 18, 140. [Google Scholar] [CrossRef]
- Peng, J.; Leng, X. NY-ESO-1 and cancer immunotherapy. Zhongguo Yi Xue Ke Xue Yuan Xue Bao 2008, 30, 371–377. [Google Scholar]
- Thomas, R.; Al-Khadairi, G.; Roelands, J.; Hendrickx, W.; Dermime, S.; Bedognetti, D.; Decock, J. NY-ESO-1 Based Immunotherapy of Cancer: Current Perspectives. Front. Immunol. 2018, 9, 947. [Google Scholar] [CrossRef]
- Ishihara, M.; Kitano, S.; Kageyama, S.; Miyahara, Y.; Yamamoto, N.; Kato, H.; Mishima, H.; Hattori, H.; Funakoshi, T.; Kojima, T.; et al. NY-ESO-1-Specific Redirected T Cells with Endogenous TCR Knockdown Mediate Tumor Response and Cytokine Release Syndrome. J. Immunother. Cancer 2022, 10, e003811. [Google Scholar] [CrossRef]
- Ishihara, M.; Nishida, Y.; Kitano, S.; Kawai, A.; Muraoka, D.; Momose, F.; Harada, N.; Miyahara, Y.; Seo, N.; Hattori, H.; et al. A Phase 1 Trial of NY-ESO-1-Specific TCR-Engineered T-Cell Therapy Combined with a Lymph Node-Targeting Nanoparticulate Peptide Vaccine for the Treatment of Advanced Soft Tissue Sarcoma. Int. J. Cancer 2023, 152, 2554–2566. [Google Scholar] [CrossRef]
- Olivo Pimentel, V.; Yaromina, A.; Marcus, D.; Dubois, L.J.; Lambin, P. A Novel Co-Culture Assay to Assess Anti-Tumor CD8+ T Cell Cytotoxicity via Luminescence and Multicolor Flow Cytometry. J. Immunol. Methods 2020, 487, 112899. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo-Herrero, S.; Sordo-Bahamonde, C.; Gonzalez, S.; López-Soto, A. CD107a Degranulation Assay to Evaluate Immune Cell Antitumor Activity. Methods Mol. Biol. 2019, 1884, 119–130. [Google Scholar] [CrossRef]
- Nagata, S. Apoptosis by Death Factor. Cell 1997, 88, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Fernández, M.; de la Fuente, H.; Martín, P.; Cibrián, D.; Sánchez-Madrid, F. Unraveling CD69 Signaling Pathways, Ligands and Laterally Associated Molecules. EXCLI J. 2023, 22, 334–351. [Google Scholar] [PubMed]
- Xie, Y.; Akpinarli, A.; Maris, C.; Hipkiss, E.L.; Lane, M.; Kwon, E.-K.M.; Muranski, P.; Restifo, N.P.; Antony, P.A. Naive Tumor-Specific CD4(+) T Cells Differentiated in Vivo Eradicate Established Melanoma. J. Exp. Med. 2010, 207, 651–667. [Google Scholar] [CrossRef]
- Quezada, S.A.; Simpson, T.R.; Peggs, K.S.; Merghoub, T.; Vider, J.; Fan, X.; Blasberg, R.; Yagita, H.; Muranski, P.; Antony, P.A.; et al. Tumor-Reactive CD4(+) T Cells Develop Cytotoxic Activity and Eradicate Large Established Melanoma after Transfer into Lymphopenic Hosts. J. Exp. Med. 2010, 207, 637–650. [Google Scholar] [CrossRef]
- Lee, B.O.; Haynes, L.; Eaton, S.M.; Swain, S.L.; Randall, T.D. The Biological Outcome of CD40 Signaling Is Dependent on the Duration of CD40 Ligand Expression: Reciprocal Regulation by Interleukin (IL)-4 and IL-12. J. Exp. Med. 2002, 196, 693–704. [Google Scholar] [CrossRef]
- Iqbal, M.M.; Serralha, M.; Kaur, P.; Martino, D. Mapping the Landscape of Chromatin Dynamics during Naïve CD4+ T-Cell Activation. Sci. Rep. 2021, 11, 14101. [Google Scholar] [CrossRef]
- Bhat, M.Y.; Solanki, H.S.; Advani, J.; Khan, A.A.; Keshava Prasad, T.S.; Gowda, H.; Thiyagarajan, S.; Chatterjee, A. Comprehensive Network Map of Interferon Gamma Signaling. J. Cell Commun. Signal. 2018, 12, 745–751. [Google Scholar] [CrossRef]
- Legut, M.; Gajic, Z.; Guarino, M.; Daniloski, Z.; Rahman, J.A.; Xue, X.; Lu, C.; Lu, L.; Mimitou, E.P.; Hao, S.; et al. A Genome-Scale Screen for Synthetic Drivers of T Cell Proliferation. Nature 2022, 603, 728–735. [Google Scholar] [CrossRef]
- Jiang, J.; Zhang, Y.; Krainer, A.R.; Xu, R.M. Crystal Structure of Human P32, a Doughnut-Shaped Acidic Mitochondrial Matrix Protein. Proc. Natl. Acad. Sci. USA 1999, 96, 3572–3577. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Liu, K.; Fang, H.; Zhang, Q.; Gao, X.; Liu, F.; Zhou, S.; Wang, X.; Niu, Y.; Hong, Y.; et al. Mitochondrial C1qbp Promotes Differentiation of Effector CD8(+) T Cells via Metabolic-Epigenetic Reprogramming. Sci. Adv. 2021, 7, eabk0490. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Zagouras, P.; Fischer, J.E.; Lu, J.; Li, B.; Flavell, R.A. Kinetic Analysis of Genomewide Gene Expression Reveals Molecule Circuitries That Control T Cell Activation and Th1/2 Differentiation. Proc. Natl. Acad. Sci. USA 2004, 101, 3023–3028. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, M.; Martín-Serrano, J.; Oberlin, E.; Pedraza, M.A.; Serrano, A.; Santiago, B.; Caruz, A.; Loetscher, P.; Baggiolini, M.; Arenzana-Seisdedos, F.; et al. Activation of Blood T Lymphocytes Down-Regulates CXCR4 Expression and Interferes with Propagation of X4 HIV Strains. Eur. J. Immunol. 1998, 28, 3192–3204. [Google Scholar] [CrossRef]
- Rheinländer, A.; Schraven, B.; Bommhardt, U. CD45 in Human Physiology and Clinical Medicine. Immunol. Lett. 2018, 196, 22–32. [Google Scholar] [CrossRef]
- Larbi, A.; Fulop, T. From “Truly Naïve” to “Exhausted Senescent” T Cells: When Markers Predict Functionality. Cytom. Part A J. Int. Soc. Anal. Cytol. 2014, 85, 25–35. [Google Scholar] [CrossRef]
- Saxena, A.; Dagur, P.K.; Biancotto, A. Multiparametric Flow Cytometry Analysis of Naïve, Memory, and Effector T Cells. Methods Mol. Biol. 2019, 2032, 129–140. [Google Scholar] [CrossRef]
- Yang, S.; Liu, F.; Wang, Q.J.; Rosenberg, S.A.; Morgan, R.A. The Shedding of CD62L (L-Selectin) Regulates the Acquisition of Lytic Activity in Human Tumor Reactive T Lymphocytes. PLoS ONE 2011, 6, e22560. [Google Scholar] [CrossRef]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.-J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of Peripheral CD4+CD25- Naive T Cells to CD4+CD25+ Regulatory T Cells by TGF-Beta Induction of Transcription Factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of Regulatory T Cell Development by the Transcription Factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef]
- Sad, S.; Mosmann, T.R. Interleukin (IL) 4, in the Absence of Antigen Stimulation, Induces an Anergy-like State in Differentiated CD8+ TC1 Cells: Loss of IL-2 Synthesis and Autonomous Proliferation but Retention of Cytotoxicity and Synthesis of Other Cytokines. J. Exp. Med. 1995, 182, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Levings, M.K.; Bacchetta, R.; Schulz, U.; Roncarolo, M.G. The Role of IL-10 and TGF-Beta in the Differentiation and Effector Function of T Regulatory Cells. Int. Arch. Allergy Immunol. 2002, 129, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Laufer, J.M.; Legler, D.F. Beyond Migration-Chemokines in Lymphocyte Priming, Differentiation, and Modulating Effector Functions. J. Leukoc. Biol. 2018, 104, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Holgado, M.P.; Sananez, I.; Raiden, S.; Geffner, J.R.; Arruvito, L. CD32 Ligation Promotes the Activation of CD4(+) T Cells. Front. Immunol. 2018, 9, 2814. [Google Scholar] [CrossRef]
- Krishnan, S.; Warke, V.G.; Nambiar, M.P.; Tsokos, G.C.; Farber, D.L. The FcR Gamma Subunit and Syk Kinase Replace the CD3 Zeta-Chain and ZAP-70 Kinase in the TCR Signaling Complex of Human Effector CD4 T Cells. J. Immunol. 2003, 170, 4189–4195. [Google Scholar] [CrossRef]
- Penna, D.; Müller, S.; Martinon, F.; Demotz, S.; Iwashima, M.; Valitutti, S. Degradation of ZAP-70 Following Antigenic Stimulation in Human T Lymphocytes: Role of Calpain Proteolytic Pathway. J. Immunol. 1999, 163, 50–56. [Google Scholar] [CrossRef]
- Yang, Y.; Kohler, M.E.; Chien, C.D.; Sauter, C.T.; Jacoby, E.; Yan, C.; Hu, Y.; Wanhainen, K.; Qin, H.; Fry, T.J. TCR Engagement Negatively Affects CD8 but Not CD4 CAR T Cell Expansion and Leukemic Clearance. Sci. Transl. Med. 2017, 9, eaag1209. [Google Scholar] [CrossRef]
- Kum, W.W.; Cameron, S.B.; Hung, R.W.; Kalyan, S.; Chow, A.W. Temporal Sequence and Kinetics of Proinflammatory and Anti-Inflammatory Cytokine Secretion Induced by Toxic Shock Syndrome Toxin 1 in Human Peripheral Blood Mononuclear Cells. Infect. Immun. 2001, 69, 7544–7549. [Google Scholar] [CrossRef]
- Morvan, P.Y.; Picot, C.; Dejour, R.; Genetet, B.; Genetet, N. Distinct Pattern of IL-2 and IFN-Gamma Gene Expression in CD4 and CD8 T Cells: Cytofluorometric Analysis at a Single Cell Level Using Non-Radioactive Probes. Cell. Mol. Biol. 1995, 41, 945–957. [Google Scholar]
- Abdalla, A.O.; Kiaii, S.; Hansson, L.; Rossmann, E.D.; Jeddi-Tehrani, M.; Shokri, F.; Osterborg, A.; Mellstedt, H.; Rabbani, H. Kinetics of Cytokine Gene Expression in Human CD4+ and CD8+ T-Lymphocyte Subsets Using Quantitative Real-Time PCR. Scand. J. Immunol. 2003, 58, 601–606. [Google Scholar] [CrossRef]
- Golstein, P.; Griffiths, G.M. An Early History of T Cell-Mediated Cytotoxicity. Nat. Rev. Immunol. 2018, 18, 527–535. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Donor ID | Age (Years) | Gender | Previous Treatments | Race |
|---|---|---|---|---|
| D001 | 33 | Male | No | Caucasian |
| D002 | 24 | Male | No | Caucasian |
| D003 | 28 | Male | No | Caucasian |
| D004 | 31 | Male | No | Caucasian |
| D005 | 24 | Male | No | Caucasian |
| D006 | 24 | Male | No | Caucasian |
| Term | Overlap | q-Value | Combined Score | Genes |
|---|---|---|---|---|
| Cytokine-mediated signaling pathway | 37/621 | 0.0 | 2443.644 | CIITA, CSF1, CXCL1, IL1RAP, CXCL2, ICAM1, SOCS3, PSMB7, PSMB5, IRAK2, CCL2, NCAM1, GBP1, HLA-DQA1, HLA-DPA1, IL13RA1, CCL20, IFNGR1, TNFRSF9, PRKCD, LIF, FN1, NFKBIA, BST2, IL1A, CXCL10, CXCL11, IL6, BCL6, IRF1, IL1B, HLA-DPB1, HLA-DRA, LTBR, IL6ST, HLA-DRB3, HLA-DRB1 |
| Cellular response to interferon-gamma | 16/121 | 0.0 | 2272.529 | CIITA, CCL20, IFNGR1, PRKCD, ICAM1, IRF1, HLA-DPB1, HLA-DRA, CCL2, NCAM1, HLA-DRB3, GBP1, HLA-DQA1, HLA-DRB1, TLR2, HLA-DPA1 |
| Cellular response to cytokine stimulus | 23/482 | 0.0 | 772.551 | CSF1, CCL20, IFNGR1, LIF, FN1, CXCL1, IL1RAP, CXCL2, ICAM1, IL1A, CXCL10, SOCS3, IL6, BCL6, IRAK2, IRF1, IL1B, CCL2, IL6ST, GBP1, TLR2, HLA-DPA1, IL13RA1 |
| Interferon-gamma-mediated signaling pathway | 13/68 | 0.0 | 3113.543 | CIITA, IFNGR1, PRKCD, ICAM1, IRF1, HLA-DPB1, HLA-DRA, NCAM1, HLA-DRB3, GBP1, HLA-DQA1, HLA-DRB1, HLA-DPA1 |
| Positive regulation of T cell activation | 13/75 | 0.0 | 2671.611 | CD274, TFRC, CD81, PDCD1LG2, THY1, IL6, HLA-DMB, IL1B, HLA-DPB1, CCL2, IL6ST, HLA-DPA1, CD276 |
| Regulation of T cell proliferation | 13/76 | 0.0 | 2617.25 | CD274, CEBPB, TFRC, PDCD1LG2, IL6, HLA-DMB, IL1B, HLA-DPB1, IL6ST, HLA-DRB1, HLA-DPA1, IDO1, CD276 |
| Positive regulation of lymphocyte proliferation | 12/75 | 0.0 | 2148.229 | CD274, BST1, IL6, HLA-DMB, TFRC, CD81, IL1B, HLA-DPB1, PDCD1LG2, IL6ST, HLA-DPA1, CD276 |
| Positive regulation of T cell proliferation | 10/66 | 0.0 | 1604.452 | CD274, IL6, HLA-DMB, TFRC, IL1B, HLA-DPB1, PDCD1LG2, IL6ST, HLA-DPA1, CD276 |
| Regulation of immune effector process | 9/53 | 0.0 | 1700.874 | C3, C1S, CD81, C1R, C1QBP, CFI, HLA-DRA, CD59, HLA-DRB1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsalloum, A.; Alrhmoun, S.; Shevchenko, J.; Fisher, M.; Philippova, J.; Perik-Zavodskii, R.; Perik-Zavodskaia, O.; Lopatnikova, J.; Kurilin, V.; Volynets, M.; et al. TCR-Engineered Lymphocytes Targeting NY-ESO-1: In Vitro Assessment of Cytotoxicity against Tumors. Biomedicines 2023, 11, 2805. https://doi.org/10.3390/biomedicines11102805
Alsalloum A, Alrhmoun S, Shevchenko J, Fisher M, Philippova J, Perik-Zavodskii R, Perik-Zavodskaia O, Lopatnikova J, Kurilin V, Volynets M, et al. TCR-Engineered Lymphocytes Targeting NY-ESO-1: In Vitro Assessment of Cytotoxicity against Tumors. Biomedicines. 2023; 11(10):2805. https://doi.org/10.3390/biomedicines11102805
Chicago/Turabian StyleAlsalloum, Alaa, Saleh Alrhmoun, Julia Shevchenko, Marina Fisher, Julia Philippova, Roman Perik-Zavodskii, Olga Perik-Zavodskaia, Julia Lopatnikova, Vasily Kurilin, Marina Volynets, and et al. 2023. "TCR-Engineered Lymphocytes Targeting NY-ESO-1: In Vitro Assessment of Cytotoxicity against Tumors" Biomedicines 11, no. 10: 2805. https://doi.org/10.3390/biomedicines11102805