

Development of a Comprehensive Gene Signature Linking Hypoxia, Glycolysis, Lactylation, and Metabolomic Insights in Gastric Cancer through the Integration of Bulk and Single-Cell RNA-Seq Data


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
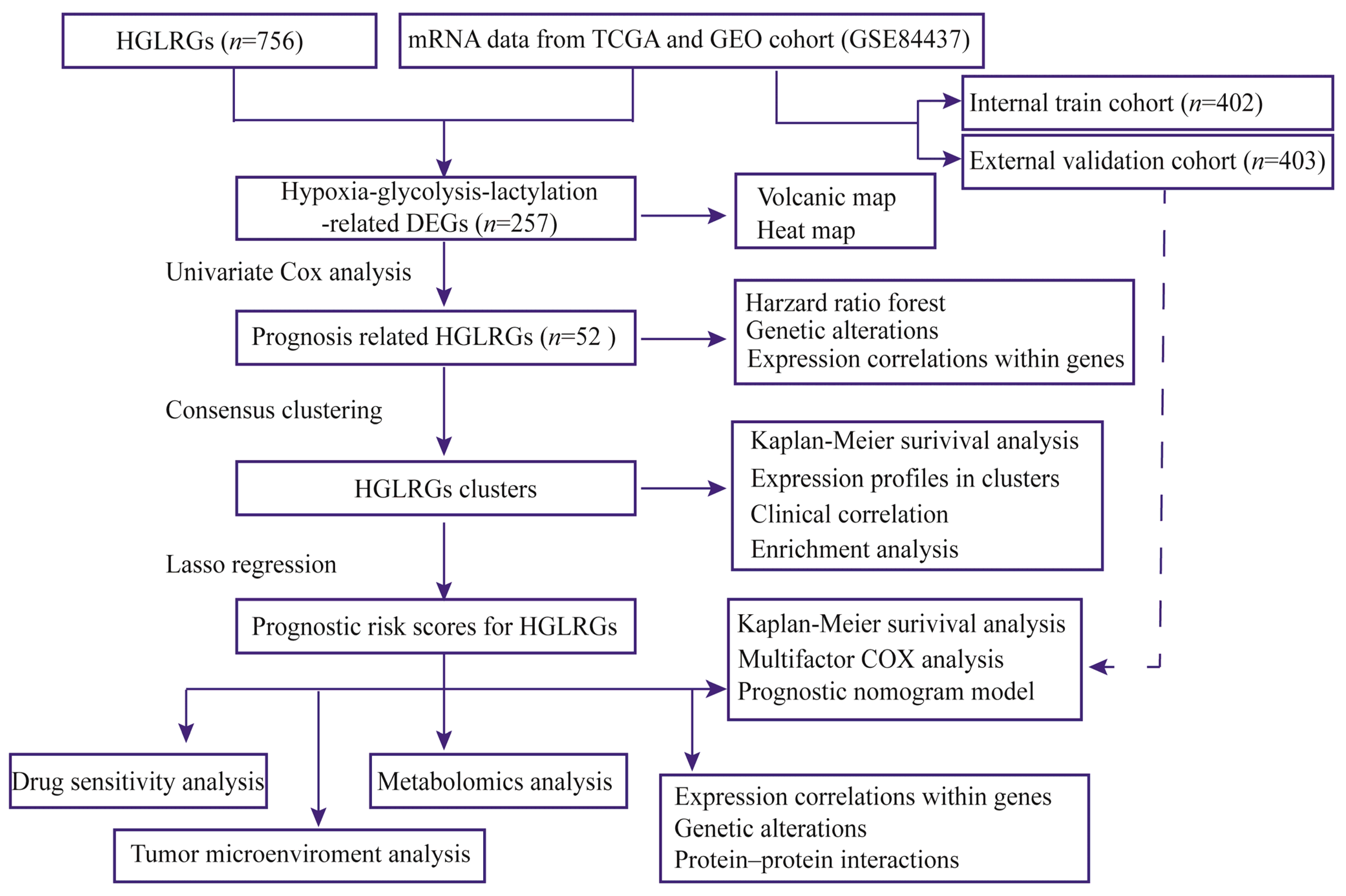
2. Materials and Methods
2.1. Acquisition of Stomach Carcinoma Data
2.2. Identification and Characteristics of HGLRG Molecular Subtypes
2.3. Development and Validation of a Prognostic Signature Based on HGLRGs
2.4. Relationship between HGLRG Risk Score and Drug Sensitivity
2.5. Relationship between HGLRG Risk Score and the Tumor Immune Microenvironment
2.6. Single-Cell Transcriptome Analysis
2.7. Statistical Analysis
3. Results
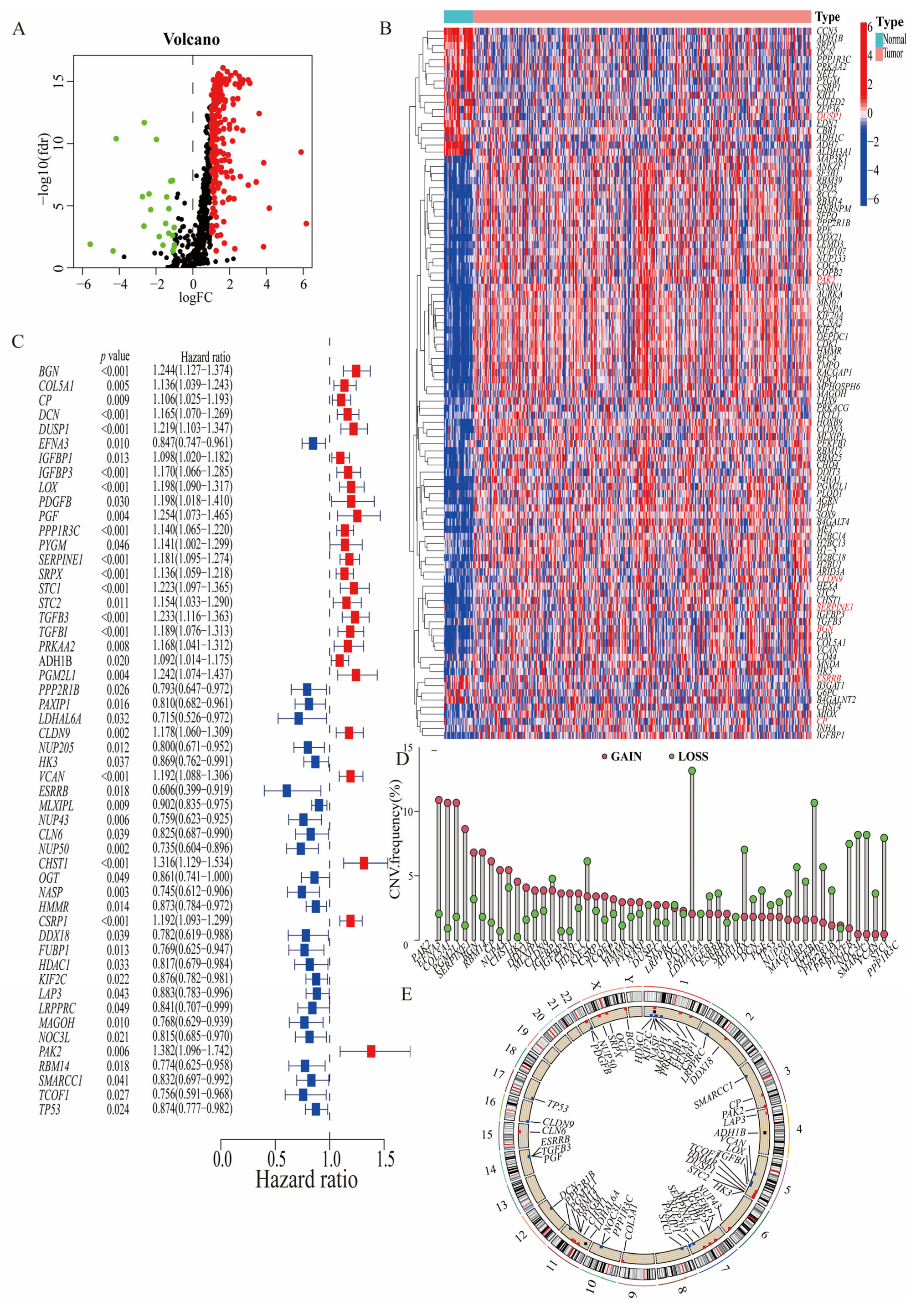
3.1. Identification of Prognosis-Related Hypoxia–Glycolysis–Lactylation-Related Genes
3.2. Consistency Clustering and Pathway Enrichment Analysis among 52 Prognostically Relevant HGLRGs
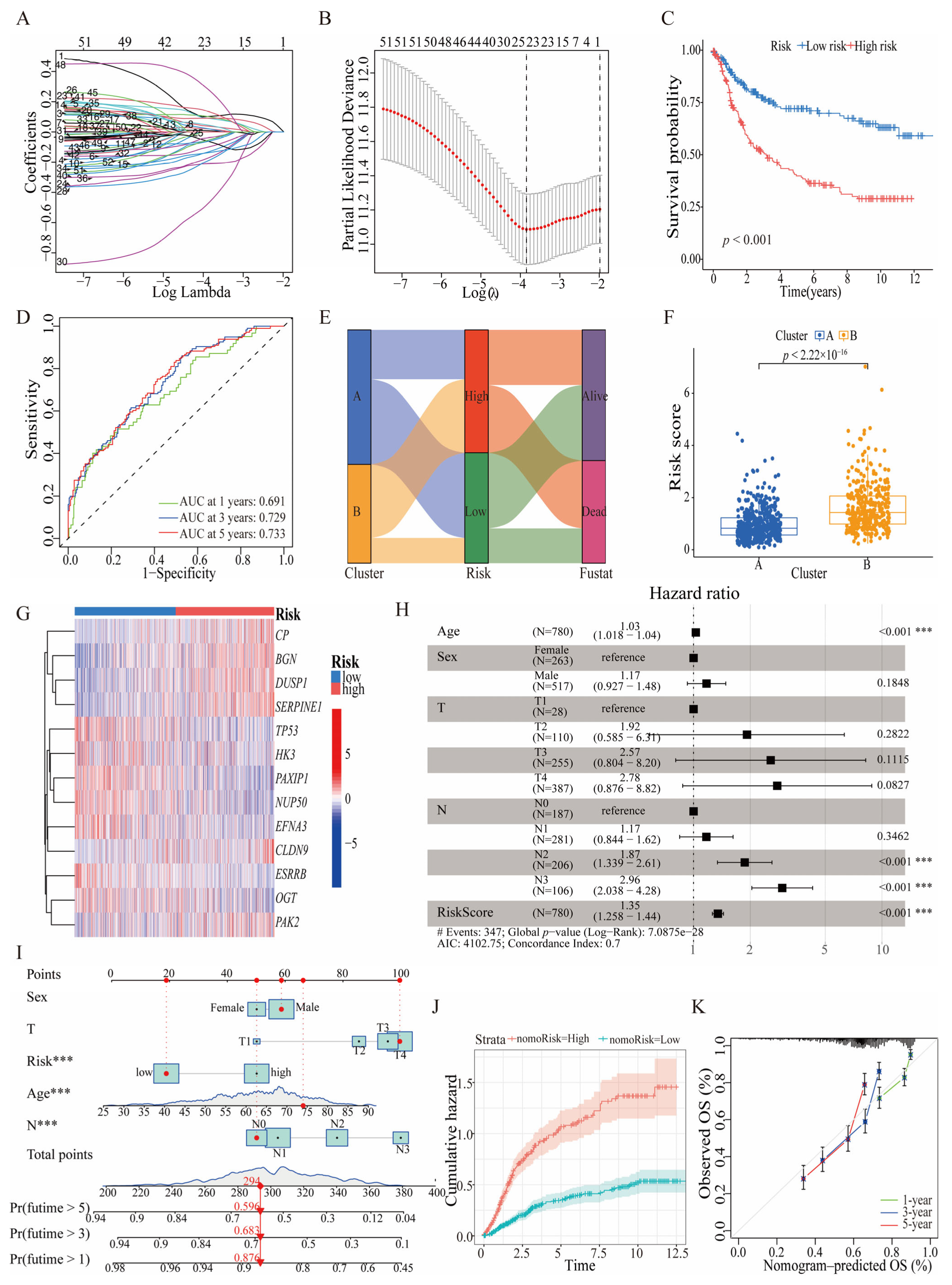
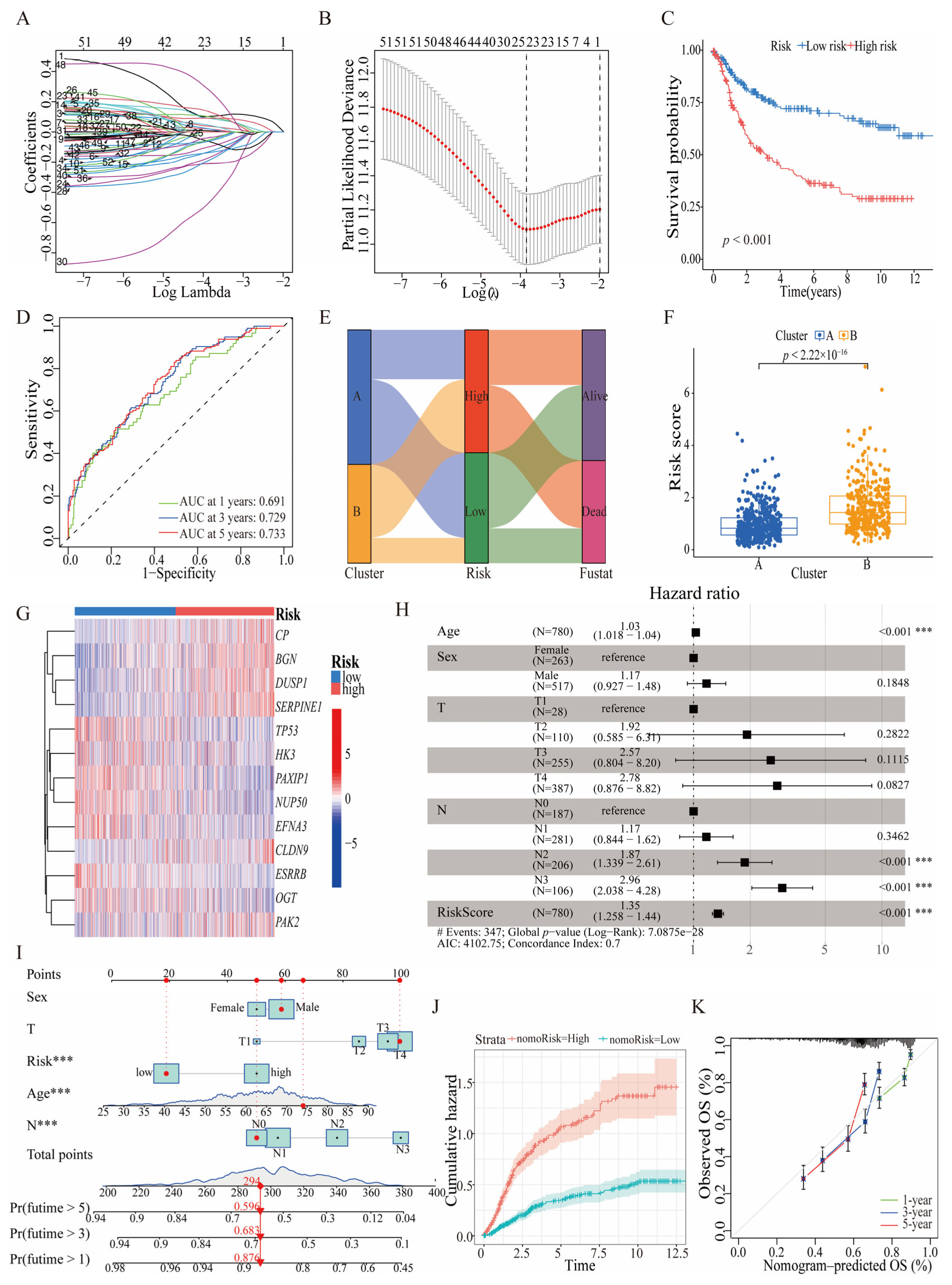
3.3. Prognostic Significance and Predictive Efficacy of HGLRG-Based Models in GC Survival
3.4. HGLRG Signature Correlates with Tumor Metabolism
3.5. HGLRG Signature Predicts Drug Sensitivity in GC
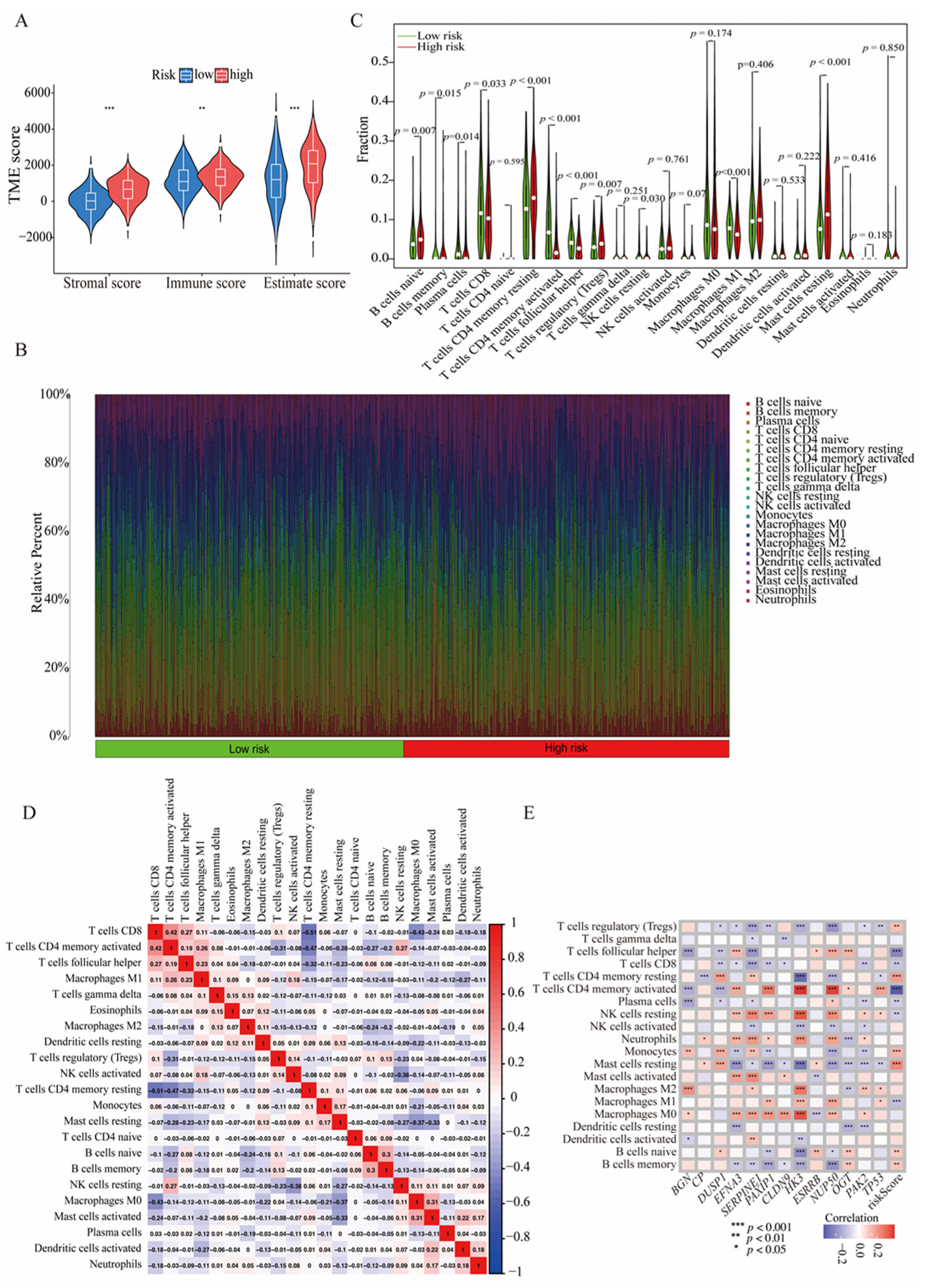
3.6. The HGLRG Signature Shapes the Tumor Microenvironment and Predicts Immunotherapy Response in GC
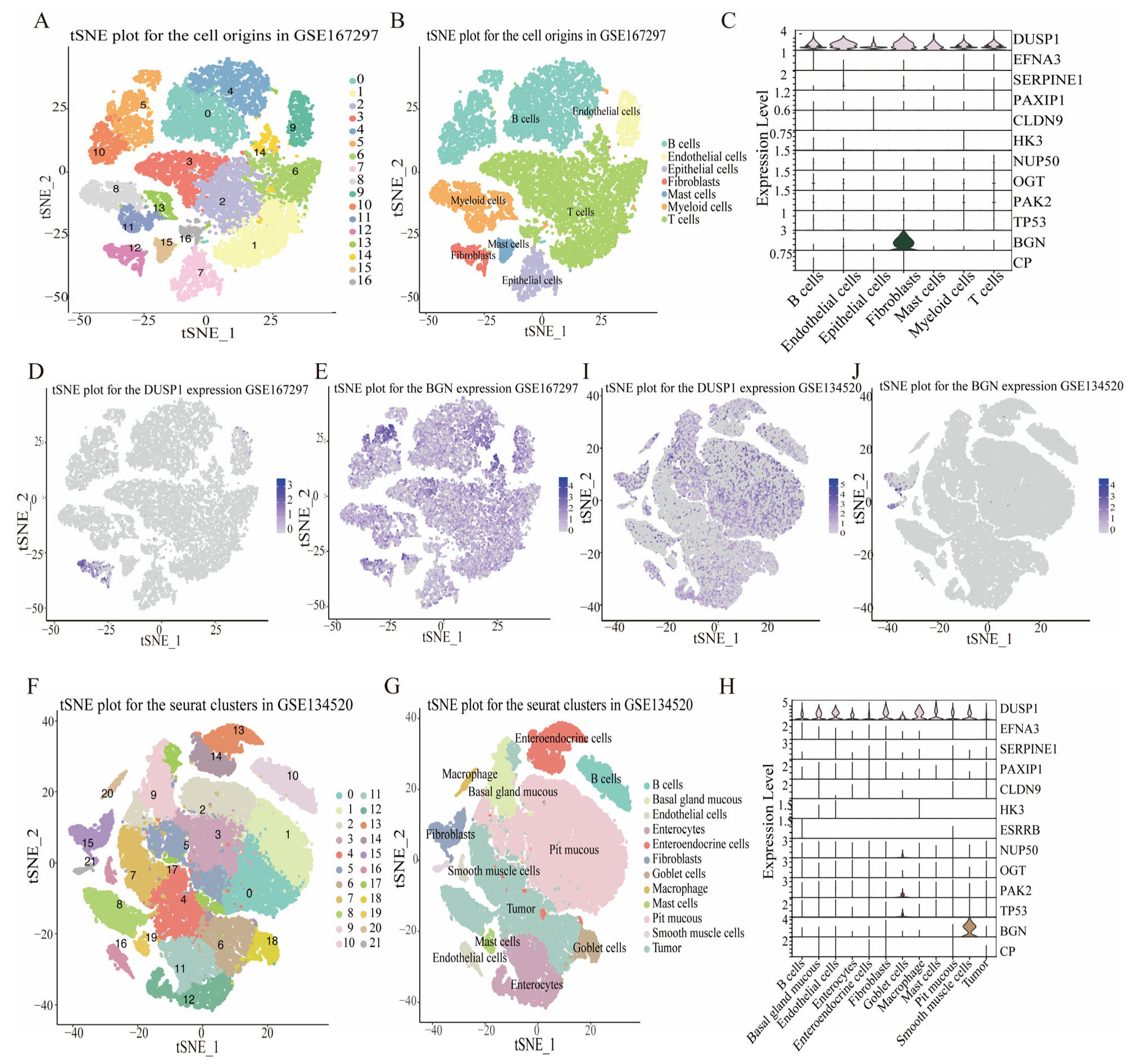
3.7. HGLRG Signature in GC via Single-Cell Transcriptomics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Cancer Today. Available online: http://gco.iarc.fr/today/home (accessed on 15 May 2023).
- Thrift, A.P.; Wenker, T.N.; El-Serag, H.B. Global Burden of Gastric Cancer: Epidemiological Trends, Risk Factors, Screening and Prevention. Nat. Rev. Clin. Oncol. 2023, 20, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhu, Y.; Lu, N.-H. The Management of Helicobacter Pylori Infection and Prevention and Control of Gastric Cancer in China. Front. Cell Infect. Microbiol. 2022, 12, 1049279. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Badgwell, B.D. Current Treatment and Recent Progress in Gastric Cancer. CA Cancer J. Clin. 2021, 71, 264–279. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.-P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair–Deficient Cancer: Results from the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Cai, Y.; Wang, Z.; Guo, S.; Lin, C.; Yao, H.; Yang, Q.; Wang, Y.; Yu, X.; He, X.; Sun, W.; et al. Detection, Mechanisms, and Therapeutic Implications of Oncometabolites. Trends Endocrinol. Metab. 2023. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Bai, X.; Zhang, M.; Wu, S.; Xiao, J.; Zeng, X.; Li, Y.; Zhang, Z. Energy Metabolism: A New Target for Gastric Cancer Treatment. Clin. Transl. Oncol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Zhang, Y.; Peng, Q.; Zheng, J.; Yang, Y.; Zhang, X.; Ma, A.; Qin, Y.; Qin, Z.; Zheng, X. The Function and Mechanism of Lactate and Lactylation in Tumor Metabolism and Microenvironment. Genes. Dis. 2023, 10, 2029–2037. [Google Scholar] [CrossRef]
- Wang, T.; Ye, Z.; Li, Z.; Jing, D.-S.; Fan, G.-X.; Liu, M.-Q.; Zhuo, Q.-F.; Ji, S.-R.; Yu, X.-J.; Xu, X.-W.; et al. Lactate-Induced Protein Lactylation: A Bridge between Epigenetics and Metabolic Reprogramming in Cancer. Cell Prolif. 2023, 56, e13478. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic Regulation of Gene Expression by Histone Lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Cen, K.; Song, Y.; Zhang, X.; Liou, Y.-C.; Liu, P.; Huang, J.; Ruan, J.; He, J.; Ye, W.; et al. NUSAP1-LDHA-Glycolysis-Lactate Feedforward Loop Promotes Warburg Effect and Metastasis in Pancreatic Ductal Adenocarcinoma. Cancer Lett. 2023, 567, 216285. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Yin, J.; Shan, L.; Yi, X.; Zhang, W.; Ding, Y. Identification of Lysine-Lactylated Substrates in Gastric Cancer Cells. iScience 2022, 25, 104630. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zou, X.; Yang, S.; Zhang, A.; Li, N.; Ma, Z. Identification of Lactylation Related Model to Predict Prognostic, Tumor Infiltrating Immunocytes and Response of Immunotherapy in Gastric Cancer. Front. Immunol. 2023, 14, 1149989. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Huang, H.; Li, M.; Liang, X.; Tan, Y.; Chen, Y. Lactylation-Related Gene Signature Effectively Predicts Prognosis and Treatment Responsiveness in Hepatocellular Carcinoma. Pharmaceuticals 2023, 16, 644. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Program (TCGA)—NCI. Available online: https://www.cancer.gov/ccg/research/genome-sequencing/tcga (accessed on 13 June 2023).
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI Gene Expression and Hybridization Array Data Repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- The Human Protein Atlas. Available online: http://www.proteinatlas.org/ (accessed on 6 April 2023).
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular Signatures Database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA Prediction Server: Biological Network Integration for Gene Prioritization and Predicting Gene Function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef]
- DepMap: The Cancer Dependency Map Project at Broad Institute. Available online: https://depmap.org/portal/ (accessed on 3 July 2023).
- Ye, L.; Jiang, Y.; Zhang, M. Crosstalk between Glucose Metabolism, Lactate Production and Immune Response Modulation. Cytokine Growth Factor. Rev. 2022, 68, 81–92. [Google Scholar] [CrossRef]
- Huo, J.; Wu, L.; Zang, Y. Eight-Gene Prognostic Signature Associated with Hypoxia and Ferroptosis for Gastric Cancer with General Applicability. Epigenomics 2021, 13, 875–890. [Google Scholar] [CrossRef]
- Yu, S.; Hu, C.; Cai, L.; Du, X.; Lin, F.; Yu, Q.; Liu, L.; Zhang, C.; Liu, X.; Li, W.; et al. Seven-Gene Signature Based on Glycolysis Is Closely Related to the Prognosis and Tumor Immune Infiltration of Patients with Gastric Cancer. Front. Oncol. 2020, 10, 1778. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.-P.; Zhang, C.-D.; Yusupu, M.; Zhang, C.; Dai, D.-Q. Screening and Validation of the Hypoxia-Related Signature of Evaluating Tumor Immune Microenvironment and Predicting Prognosis in Gastric Cancer. Front. Immunol. 2021, 12, 705511. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zou, J.; Wang, Z.; Li, G.; Lei, Y. Identifying Novel Cell Glycolysis-Related Gene Signature Predictive of Overall Survival in Gastric Cancer. BioMed Res. Int. 2021, 2021, 9656947. [Google Scholar] [CrossRef]
- Ghojazadeh, M.; Somi, M.H.; Naseri, A.; Salehi-Pourmehr, H.; Hassannezhad, S.; Hajikamanaj Olia, A.; Kafshdouz, L.; Nikniaz, Z. Systematic Review and Meta-Analysis of TP53, HER2/ERBB2, KRAS, APC, and PIK3CA Genes Expression Pattern in Gastric Cancer. Middle East. J. Dig. Dis. 2022, 14, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Fenoglio-Preiser, C.M.; Wang, J.; Stemmermann, G.N.; Noffsinger, A. TP53 and Gastric Carcinoma: A Review. Hum. Mutat. 2003, 21, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Yu, W.; Su, H.; Pang, X. Genomic Landscape of Gastric Cancer: Molecular Classification and Potential Targets. Sci. China Life Sci. 2017, 60, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Cai, H.; Huang, H.; Long, Z.; Shi, Y.; Wang, Y. The Prognostic Significance of Apoptosis-Related Biological Markers in Chinese Gastric Cancer Patients. PLoS ONE 2011, 6, e29670. [Google Scholar] [CrossRef] [PubMed]
- Callen, E.; Faryabi, R.B.; Luckey, M.; Hao, B.; Daniel, J.A.; Yang, W.; Sun, H.-W.; Dressler, G.; Peng, W.; Chi, H.; et al. The DNA Damage- and Transcription-Associated Protein Paxip1 Controls Thymocyte Development and Emigration. Immunity 2012, 37, 971–985. [Google Scholar] [CrossRef]
- De Gregoriis, G.; Ramos, J.A.; Fernandes, P.V.; Vignal, G.M.; Brianese, R.C.; Carraro, D.M.; Monteiro, A.N.; Struchiner, C.J.; Suarez-Kurtz, G.; Vianna-Jorge, R.; et al. DNA Repair Genes PAXIP1 and TP53BP1 Expression Is Associated with Breast Cancer Prognosis. Cancer Biol. Ther. 2017, 18, 439–449. [Google Scholar] [CrossRef]
- Lawan, A.; Shi, H.; Gatzke, F.; Bennett, A.M. Diversity and Specificity of the Mitogen-Activated Protein Kinase Phosphatase-1 Functions. Cell Mol. Life Sci. 2013, 70, 223–237. [Google Scholar] [CrossRef]
- Jang, T.J.; Kim, U.J. O-GlcNAcylation Is Associated with the Development and Progression of Gastric Carcinoma. Pathol. Res. Pract. 2016, 212, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Xu, Z.; Sun, J.; Liu, B.; Ruan, Y.; Gu, J.; Song, S. O-GlcNAcylation Enhances Reticulon 2 Protein Stability and Its Promotive Effects on Gastric Cancer Progression. Cell Signal. 2023, 108, 110718. [Google Scholar] [CrossRef] [PubMed]
- Zavala-Zendejas, V.E.; Torres-Martinez, A.C.; Salas-Morales, B.; Fortoul, T.I.; Montaño, L.F.; Rendon-Huerta, E.P. Claudin-6, 7, or 9 Overexpression in the Human Gastric Adenocarcinoma Cell Line AGS Increases Its Invasiveness, Migration, and Proliferation Rate. Cancer Investig. 2011, 29, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rendón-Huerta, E.; Teresa, F.; Teresa, G.M.; Xochitl, G.-S.; Georgina, A.-F.; Veronica, Z.-Z.; Montaño, L.F. Distribution and Expression Pattern of Claudins 6, 7, and 9 in Diffuse- and Intestinal-Type Gastric Adenocarcinomas. J. Gastrointest. Cancer 2010, 41, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Olkowicz, M.; Czyzynska-Cichon, I.; Szupryczynska, N.; Kostogrys, R.B.; Kochan, Z.; Debski, J.; Dadlez, M.; Chlopicki, S.; Smolenski, R.T. Multi-Omic Signatures of Atherogenic Dyslipidaemia: Pre-Clinical Target Identification and Validation in Humans. J. Transl. Med. 2021, 19, 6. [Google Scholar] [CrossRef] [PubMed]
- Wight, T.N. A Role for Proteoglycans in Vascular Disease. Matrix Biol. 2018, 71–72, 396–420. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.C.; Tang, T.; Wilson, P.G.; Yoder, M.H.; Tannock, L.R. Increased Atherosclerosis in Mice with Increased Vascular Biglycan Content. Atherosclerosis 2014, 235, 71–75. [Google Scholar] [CrossRef]
- Yoon, B.K.; Kim, H.; Oh, T.G.; Oh, S.K.; Jo, S.; Kim, M.; Chun, K.-H.; Hwang, N.; Lee, S.; Jin, S.; et al. PHGDH Preserves One-Carbon Cycle to Confer Metabolic Plasticity in Chemoresistant Gastric Cancer during Nutrient Stress. Proc. Natl. Acad. Sci. USA 2023, 120, e2217826120. [Google Scholar] [CrossRef]
- Liu, Y.; Li, C.; Lu, Y.; Liu, C.; Yang, W. Tumor Microenvironment-Mediated Immune Tolerance in Development and Treatment of Gastric Cancer. Front. Immunol. 2022, 13, 1016817. [Google Scholar] [CrossRef]
- Wang, R.; Song, S.; Qin, J.; Yoshimura, K.; Peng, F.; Chu, Y.; Li, Y.; Fan, Y.; Jin, J.; Dang, M.; et al. Evolution of Immune and Stromal Cell States and Ecotypes during Gastric Adenocarcinoma Progression. Cancer Cell 2023, 14, 1407–1426.e9. [Google Scholar] [CrossRef]
- Pinto, F.; Santos-Ferreira, L.; Pinto, M.T.; Gomes, C.; Reis, C.A. The Extracellular Small Leucine-Rich Proteoglycan Biglycan Is a Key Player in Gastric Cancer Aggressiveness. Cancers 2021, 13, 1330. [Google Scholar] [CrossRef]
- Brina, D.; Ponzoni, A.; Troiani, M.; Calì, B.; Pasquini, E.; Attanasio, G.; Mosole, S.; Mirenda, M.; D’Ambrosio, M.; Colucci, M.; et al. The Akt/mTOR and MNK/eIF4E Pathways Rewire the Prostate Cancer Translatome to Secrete HGF, SPP1 and BGN and Recruit Suppressive Myeloid Cells. Nat. Cancer 2023, 4, 1102–1121. [Google Scholar] [CrossRef]
- Zheng, S.; Liang, J.-Y.; Tang, Y.; Xie, J.; Zou, Y.; Yang, A.; Shao, N.; Kuang, X.; Ji, F.; Liu, X.; et al. Dissecting the Role of Cancer-Associated Fibroblast-Derived Biglycan as a Potential Therapeutic Target in Immunotherapy Resistance: A Tumor Bulk and Single-Cell Transcriptomic Study. Clin. Transl. Med. 2023, 13, e1189. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Li, Y.; Chen, Y. Development of a Comprehensive Gene Signature Linking Hypoxia, Glycolysis, Lactylation, and Metabolomic Insights in Gastric Cancer through the Integration of Bulk and Single-Cell RNA-Seq Data. Biomedicines 2023, 11, 2948. https://doi.org/10.3390/biomedicines11112948
Zhang X, Li Y, Chen Y. Development of a Comprehensive Gene Signature Linking Hypoxia, Glycolysis, Lactylation, and Metabolomic Insights in Gastric Cancer through the Integration of Bulk and Single-Cell RNA-Seq Data. Biomedicines. 2023; 11(11):2948. https://doi.org/10.3390/biomedicines11112948
Chicago/Turabian StyleZhang, Xiangqian, Yun Li, and Yongheng Chen. 2023. "Development of a Comprehensive Gene Signature Linking Hypoxia, Glycolysis, Lactylation, and Metabolomic Insights in Gastric Cancer through the Integration of Bulk and Single-Cell RNA-Seq Data" Biomedicines 11, no. 11: 2948. https://doi.org/10.3390/biomedicines11112948