
Ca2+-Mediated Signaling Pathways: A Promising Target for the Successful Generation of Mature and Functional Stem Cell-Derived Pancreatic Beta Cells In Vitro
{kind=link}
{kind=link}
Abstract
:1. Introduction
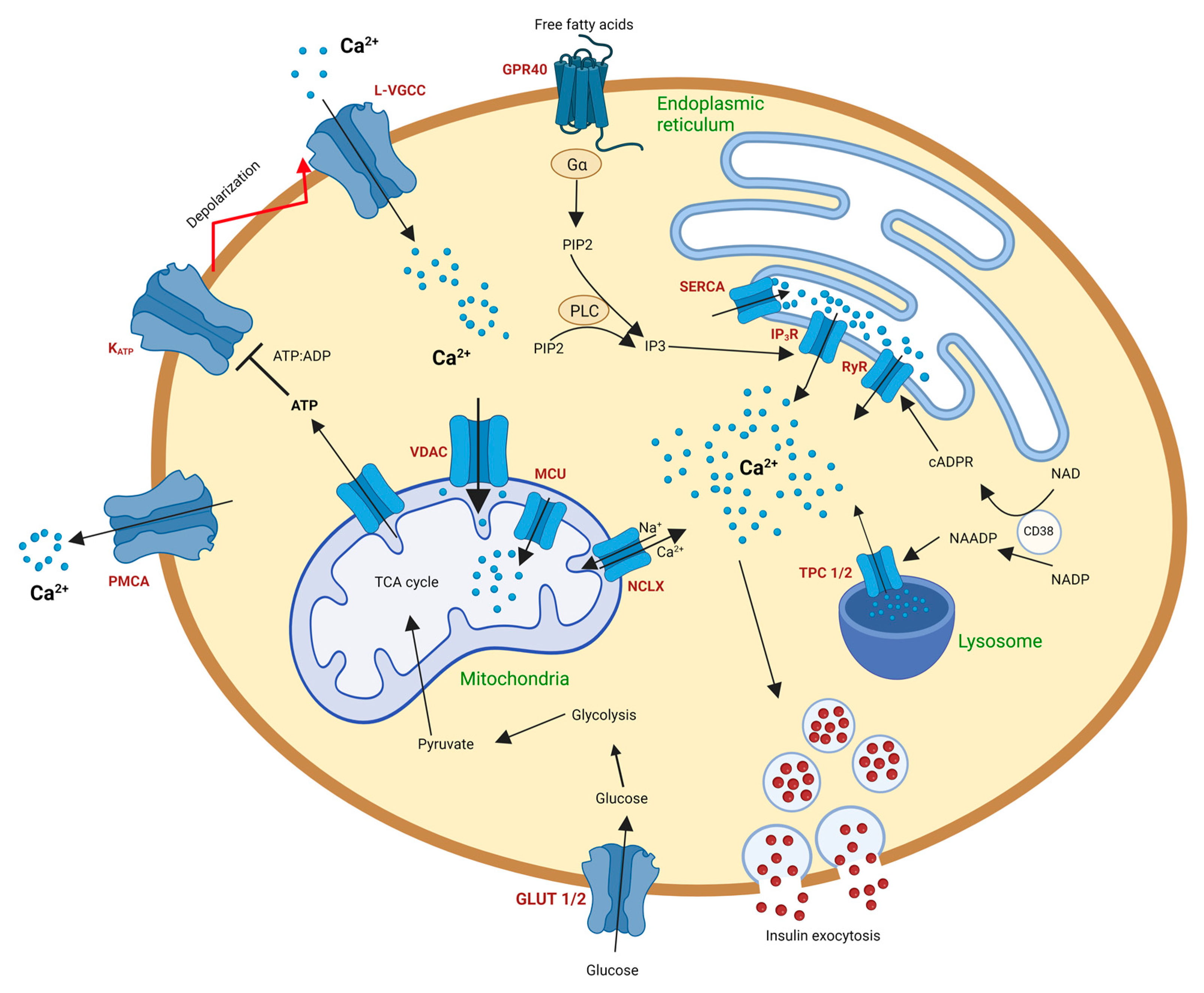
2. Calcium Signaling in Pancreatic Beta Cells (Basics)
2.1. Calcium Signaling in Beta Cell Endoplasmic Reticulum
2.2. Calcium Signaling in the Beta Cell Lysosomes
2.3. Calcium Signaling in the Beta Cell Mitochondria
3. Role of Ca2+ in Beta Cell Proliferation
4. Role of Ca2+ in Beta Cell Survival
5. Calcium Signaling in Diabetes
6. Calcium Signaling in Stem Cell-Derived Beta Cells
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Atkinson, M.A.; Bluestone, J.A.; Eisenbarth, G.S.; Hebrok, M.; Herold, K.C.; Accili, D.; Pietropaolo, M.; Arvan, P.R.; Von Herrath, M.; Markel, D.S.; et al. How does type 1 diabetes develop? The notion of homicide or beta-cell suicide revisited. Diabetes 2011, 60, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Eizirik, D.L.; Pasquali, L.; Cnop, M. Pancreatic beta-cells in type 1 and type 2 diabetes mellitus: Different pathways to failure. Nat. Rev. Endocrinol. 2020, 16, 349–362. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2014, 37 (Suppl. 1), S81–S90. [Google Scholar] [CrossRef] [PubMed]
- Maahs, D.M.; West, N.A.; Lawrence, J.M.; Mayer-Davis, E.J. Epidemiology of type 1 diabetes. Endocrinol. Metab. Clin. N. Am. 2010, 39, 481–497. [Google Scholar] [CrossRef]
- Craig, M.E.; Hattersley, A.; Donaghue, K.C. Definition, epidemiology and classification of diabetes in children and adolescents. Pediatr. Diabetes 2009, 10 (Suppl. 12), 3–12. [Google Scholar] [CrossRef]
- Dabelea, D.; Mayer-Davis, E.J.; Saydah, S.; Imperatore, G.; Linder, B.; Divers, J.; Bell, R.; Badaru, A.; Talton, J.W.; Crume, T.; et al. Prevalence of type 1 and type 2 diabetes among children and adolescents from 2001 to 2009. JAMA 2014, 311, 1778–1786. [Google Scholar] [CrossRef]
- American Diabetes Association. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2018. Diabetes Care 2018, 41 (Suppl. 1), S13–S27. [Google Scholar] [CrossRef]
- Maric-Bilkan, C. Sex differences in micro- and macro-vascular complications of diabetes mellitus. Clin. Sci. 2017, 131, 833–846. [Google Scholar] [CrossRef]
- Dal Canto, E.; Ceriello, A.; Ryden, L.; Ferrini, M.; Hansen, T.B.; Schnell, O.; Standl, E.; Beulens, J.W. Diabetes as a cardiovascular risk factor: An overview of global trends of macro and micro vascular complications. Eur. J. Prev. Cardiol. 2019, 26 (Suppl. 2), 25–32. [Google Scholar] [CrossRef]
- Bener, A.; Kim, E.J.; Mutlu, F.; Eliyan, A.; Delghan, H.; Nofal, E.; Shalabi, L.; Wadi, N. Burden of diabetes mellitus attributable to demographic levels in Qatar: An emerging public health problem. Diabetes Metab. Syndr. 2014, 8, 216–220. [Google Scholar] [CrossRef]
- Pradeepa, R.; Mohan, V. Prevalence of type 2 diabetes and its complications in India and economic costs to the nation. Eur. J. Clin. Nutr. 2017, 71, 816–824. [Google Scholar] [CrossRef]
- Bommer, C.; Heesemann, E.; Sagalova, V.; Manne-Goehler, J.; Atun, R.; Barnighausen, T.; Vollmer, S. The global economic burden of diabetes in adults aged 20–79 years: A cost-of-illness study. Lancet Diabetes Endocrinol. 2017, 5, 423–430. [Google Scholar] [CrossRef]
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef]
- Katsarou, A.; Gudbjornsdottir, S.; Rawshani, A.; Dabelea, D.; Bonifacio, E.; Anderson, B.J.; Jacobsen, L.M.; Schatz, D.A.; Lernmark, A. Type 1 diabetes mellitus. Nat. Rev. Dis. Prim. 2017, 3, 17016. [Google Scholar] [CrossRef]
- Miller, K.M.; Foster, N.C.; Beck, R.W.; Bergenstal, R.M.; DuBose, S.N.; DiMeglio, L.A.; Maahs, D.M.; Tamborlane, W.V. Current state of type 1 diabetes treatment in the U.S.: Updated data from the T1D Exchange clinic registry. Diabetes Care 2015, 38, 971–978. [Google Scholar] [CrossRef]
- Shapiro, A.M.J.; Ricordi, C.; Hering, B.J.; Auchincloss, H.; Lindblad, R.; Robertson, R.P.; Secchi, A.; Brendel, M.D.; Berney, T.; Brennan, D.C.; et al. International trial of the Edmonton protocol for islet transplantation. N. Engl. J. Med. 2006, 355, 1318–1330. [Google Scholar] [CrossRef]
- Diane, A.; Al-Shukri, N.A.; Bin Abdul Mu, U.M.R.; Al-Siddiqi, H.H. β-cell mitochondria in diabetes mellitus: A missing puzzle piece in the generation of hPSC-derived pancreatic β-cells? J. Transl. Med. 2022, 20, 163. [Google Scholar] [CrossRef]
- Sun, Z.Y.; Yu, T.Y.; Jiang, F.X.; Wang, W. Functional maturation of immature beta cells: A roadblock for stem cell therapy for type 1 diabetes. World J. Stem Cells 2021, 13, 193–207. [Google Scholar] [CrossRef]
- McNeil, J.; Doucet, E.; Chaput, J.P. Inadequate sleep as a contributor to obesity and type 2 diabetes. Can. J. Diabetes 2013, 37, 103–108. [Google Scholar] [CrossRef]
- Marin-Penalver, J.J.; Martin-Timon, I.; Sevillano-Collantes, C.; Del Canizo-Gomez, F.J. Update on the treatment of type 2 diabetes mellitus. World J. Diabetes 2016, 7, 354–395. [Google Scholar] [CrossRef]
- Song, R. Mechanism of Metformin: A Tale of Two Sites. Diabetes Care 2016, 39, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Eldor, R.; Raz, I. Diabetes therapy--focus on Asia: Second-line therapy debate: Insulin/secretagogues. Diabetes Metab. Res. Rev. 2012, 28 (Suppl. 2), 85–89. [Google Scholar] [CrossRef] [PubMed]
- Gerich, J.; Raskin, P.; Jean-Louis, L.; Purkayastha, D.; Baron, M.A. PRESERVE-beta: Two-year efficacy and safety of initial combination therapy with nateglinide or glyburide plus metformin. Diabetes Care 2005, 28, 2093–2099. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, K.G.; Millman, J.R. Applications of iPSC-derived beta cells from patients with diabetes. Cell Rep. Med. 2021, 2, 100238. [Google Scholar] [CrossRef]
- Abdelalim, E.M.; Bonnefond, A.; Bennaceur-Griscelli, A.; Froguel, P. Pluripotent stem cells as a potential tool for disease modelling and cell therapy in diabetes. Stem Cell Rev. Rep. 2014, 10, 327–337. [Google Scholar] [CrossRef]
- Hrvatin, S.; O’donnell, C.W.; Deng, F.; Millman, J.R.; Pagliuca, F.W.; DiIorio, P.; Rezania, A.; Gifford, D.K.; Melton, D.A. Differentiated human stem cells resemble fetal, not adult, beta cells. Proc. Natl. Acad. Sci. USA 2014, 111, 3038–3043. [Google Scholar] [CrossRef]
- Klec, C.; Ziomek, G.; Pichler, M.; Malli, R.; Graier, W.F. Calcium Signaling in β-cell Physiology and Pathology: A Revisit. Int. J. Mol. Sci. 2019, 20, 6110. [Google Scholar] [CrossRef]
- Renstrom, E.; Ding, W.G.; Bokvist, K.; Rorsman, P. Neurotransmitter-induced inhibition of exocytosis in insulin-secreting beta cells by activation of calcineurin. Neuron 1996, 17, 513–522. [Google Scholar] [CrossRef]
- Lemaire, K.; Ravier, M.A.; Schraenen, A.; Creemers, J.W.M.; Van de Plas, R.; Granvik, M.; Van Lommel, L.; Waelkens, E.; Chimienti, F.; Rutter, G.A.; et al. Insulin crystallization depends on zinc transporter ZnT8 expression, but is not required for normal glucose homeostasis in mice. Proc. Natl. Acad. Sci. USA 2009, 106, 14872–14877. [Google Scholar] [CrossRef]
- Iynedjian, P.B. Molecular physiology of mammalian glucokinase. Cell. Mol. Life Sci. 2009, 66, 27–42. [Google Scholar] [CrossRef]
- Weiser, A.; Feige, J.N.; De Marchi, U. Mitochondrial Calcium Signaling in Pancreatic beta-Cell. Int. J. Mol. Sci. 2021, 22, 2515. [Google Scholar] [CrossRef]
- Prins, D.; Michalak, M. Organellar calcium buffers. Cold Spring Harb. Perspect. Biol. 2011, 3, a004069. [Google Scholar] [CrossRef]
- Berridge, M.J. The endoplasmic reticulum: A multifunctional signaling organelle. Cell Calcium 2002, 32, 235–249. [Google Scholar] [CrossRef]
- Graves, T.K.; Hinkle, P.M. Ca(2+)-induced Ca(2+) release in the pancreatic beta-cell: Direct evidence of endoplasmic reticulum Ca(2+) release. Endocrinology 2003, 144, 3565–3574. [Google Scholar] [CrossRef]
- Santulli, G.; Pagano, G.; Sardu, C.; Xie, W.; Reiken, S.; D’Ascia, S.L.; Cannone, M.; Marziliano, N.; Trimarco, B.; Guise, T.A.; et al. Calcium release channel RyR2 regulates insulin release and glucose homeostasis. J. Clin. Investig. 2015, 125, 1968–1978. [Google Scholar] [CrossRef]
- Zhang, I.X.; Raghavan, M.; Satin, L.S. The Endoplasmic Reticulum and Calcium Homeostasis in Pancreatic Beta Cells. Endocrinology 2020, 161, bqz028. [Google Scholar] [CrossRef]
- Islam, M.S. The ryanodine receptor calcium channel of beta-cells: Molecular regulation and physiological significance. Diabetes 2002, 51, 1299–1309. [Google Scholar] [CrossRef]
- Sabatini, P.V.; Speckmann, T.; Lynn, F.C. Friend and foe: Beta-cell Ca(2+) signaling and the development of diabetes. Mol. Metab. 2019, 21, 1–12. [Google Scholar] [CrossRef]
- Mitchell, K.J.; Lai, F.A.; Rutter, G.A. Ryanodine receptor type I and nicotinic acid adenine dinucleotide phosphate receptors mediate Ca2+ release from insulin-containing vesicles in living pancreatic beta-cells (MIN6). J. Biol.Chem. 2003, 278, 11057–11064. [Google Scholar] [CrossRef]
- Dixit, S.S.; Wang, T.; Manzano, E.J.; Yoo, S.; Lee, J.; Chiang, D.Y.; Ryan, N.; Respress, J.L.; Yechoor, V.K.; Wehrens, X.H. Effects of CaMKII-mediated phosphorylation of ryanodine receptor type 2 on islet calcium handling, insulin secretion, and glucose tolerance. PLoS ONE 2013, 8, e58655. [Google Scholar] [CrossRef]
- Johnson, J.D.; Kuang, S.; Misler, S.; Polonsky, K.S. Ryanodine receptors in human pancreatic beta cells: Localization and effects on insulin secretion. FASEB J. 2004, 18, 878–880. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, A.E.; Svendsen, B.; Lam, B.Y.; Yeo, G.S.; Holst, J.J.; Reimann, F.; Gribble, F.M. Transcriptomic profiling of pancreatic alpha, beta and delta cell populations identifies delta cells as a principal target for ghrelin in mouse islets. Diabetologia 2016, 59, 2156–2165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bengtsson, M.; Partridge, C.; Salehi, A.; Braun, M.; Cox, R.; Eliasson, L.; Johnson, P.R.; Renström, E.; Schneider, T.; et al. R-type Ca(2+)-channel-evoked CICR regulates glucose-induced somatostatin secretion. Nat. Cell Biol. 2007, 9, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Inositol trisphosphate and diacylglycerol as second messengers. Biochem. J. 1984, 220, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Woll, K.A.; Van Petegem, F. Calcium-release channels: Structure and function of IP3 receptors and ryanodine receptors. Physiol. Rev. 2022, 102, 209–268. [Google Scholar] [CrossRef]
- Gromada, J.; Frokjaer-Jensen, J.; Dissing, S. Glucose stimulates voltage- and calcium-dependent inositol trisphosphate production and intracellular calcium mobilization in insulin-secreting beta TC3 cells. Biochem. J. 1996, 314 Pt 1, 339–345. [Google Scholar] [CrossRef]
- Dyachok, O.; Tufveson, G.; Gylfe, E. Ca2+-induced Ca2+ release by activation of inositol 1,4,5-trisphosphate receptors in primary pancreatic beta-cells. Cell Calcium 2004, 36, 1–9. [Google Scholar] [CrossRef]
- Dyachok, O.; Gylfe, E. Ca(2+)-induced Ca(2+) release via inositol 1,4,5-trisphosphate receptors is amplified by protein kinase A and triggers exocytosis in pancreatic beta-cells. J. Biol. Chem. 2004, 279, 45455–45461. [Google Scholar] [CrossRef]
- Khan, S.; Yan-Do, R.; Duong, E.; Wu, X.; Bautista, A.; Cheley, S.; Macdonald, P.E.; Braun, M. Autocrine activation of P2Y1 receptors couples Ca(2+) influx to Ca(2+) release in human pancreatic beta cells. Diabetologia 2014, 57, 2535–2545. [Google Scholar] [CrossRef]
- Burant, C.F. Activation of GPR40 as a therapeutic target for the treatment of type 2 diabetes. Diabetes Care 2013, 36 (Suppl. 2), S175–S179. [Google Scholar] [CrossRef]
- Schnell, S.; Schaefer, M.; Schofl, C. Free fatty acids increase cytosolic free calcium and stimulate insulin secretion from beta-cells through activation of GPR40. Mol. Cell. Endocrinol. 2007, 263, 173–180. [Google Scholar] [CrossRef]
- Ferdaoussi, M.; Bergeron, V.; Zarrouki, B.; Kolic, J.; Cantley, J.; Fielitz, J.; Olson, E.N.; Prentki, M.; Biden, T.; MacDonald, P.E.; et al. G protein-coupled receptor (GPR)40-dependent potentiation of insulin secretion in mouse islets is mediated by protein kinase D1. Diabetologia 2012, 55, 2682–2692. [Google Scholar] [CrossRef]
- Fujiwara, K.; Maekawa, F.; Yada, T. Oleic acid interacts with GPR40 to induce Ca2+ signaling in rat islet beta-cells: Mediation by PLC and L-type Ca2+ channel and link to insulin release. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E670–E677. [Google Scholar] [CrossRef]
- Kiviluoto, S.; Vervliet, T.; Ivanova, H.; Decuypere, J.-P.; De Smedt, H.; Missiaen, L.; Bultynck, G.; Parys, J.B. Regulation of inositol 1,4,5-trisphosphate receptors during endoplasmic reticulum stress. Biochim. Biophys. Acta 2013, 1833, 1612–1624. [Google Scholar] [CrossRef]
- Roach, J.C.; Deutsch, K.; Li, S.; Siegel, A.F.; Bekris, L.M.; Einhaus, D.C.; Sheridan, C.M.; Glusman, G.; Hood, L.; Lernmark, Å.; et al. Genetic mapping at 3-kilobase resolution reveals inositol 1,4,5-triphosphate receptor 3 as a risk factor for type 1 diabetes in Sweden. Am. J. Hum. Genet. 2006, 79, 614–627. [Google Scholar] [CrossRef]
- Periasamy, M.; Kalyanasundaram, A. SERCA pump isoforms: Their role in calcium transport and disease. Muscle Nerve 2007, 35, 430–442. [Google Scholar] [CrossRef]
- Tong, X.; Kono, T.; Anderson-Baucum, E.K.; Yamamoto, W.; Gilon, P.; Lebeche, D.; Day, R.N.; Shull, G.E.; Evans-Molina, C. SERCA2 Deficiency Impairs Pancreatic beta-Cell Function in Response to Diet-Induced Obesity. Diabetes 2016, 65, 3039–3052. [Google Scholar] [CrossRef]
- Varadi, A.; Molnar, E.; Ashcroft, S.J. Characterisation of endoplasmic reticulum and plasma membrane Ca(2+)-ATPases in pancreatic beta-cells and in islets of Langerhans. Biochim. Biophys. Acta 1995, 1236, 119–127. [Google Scholar] [CrossRef]
- Wang, R.; McGrath, B.C.; Kopp, R.F.; Roe, M.W.; Tang, X.; Chen, G.; Cavener, D.R. Insulin secretion and Ca2+ dynamics in beta-cells are regulated by PERK (EIF2AK3) in concert with calcineurin. J. Biol. Chem. 2013, 288, 33824–33836. [Google Scholar] [CrossRef]
- Kono, T.; Ahn, G.; Moss, D.R.; Gann, L.; Zarain-Herzberg, A.; Nishiki, Y.; Fueger, P.T.; Ogihara, T.; Evans-Molina, C. PPAR-gamma activation restores pancreatic islet SERCA2 levels and prevents beta-cell dysfunction under conditions of hyperglycemic and cytokine stress. Mol. Endocrinol. 2012, 26, 257–271. [Google Scholar] [CrossRef]
- Zarain-Herzberg, A.; Garcia-Rivas, G.; Estrada-Aviles, R. Regulation of SERCA pumps expression in diabetes. Cell Calcium 2014, 56, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Aley, P.K.; Singh, N.; Brailoiu, G.C.; Brailoiu, E.; Churchill, G.C. Nicotinic acid adenine dinucleotide phosphate (NAADP) is a second messenger in muscarinic receptor-induced contraction of guinea pig trachea. J. Biol. Chem. 2013, 288, 10986–10993. [Google Scholar] [CrossRef] [PubMed]
- Masgrau, R.; Churchill, G.C.; Morgan, A.J.; Ashcroft, S.J.; Galione, A. NAADP: A new second messenger for glucose-induced Ca2+ responses in clonal pancreatic beta cells. Curr. Biol. 2003, 13, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Park, K.H.; Kim, B.J.; Shawl, A.I.; Han, M.K.; Lee, H.C.; Kim, U.H. Autocrine/paracrine function of nicotinic acid adenine dinucleotide phosphate (NAADP) for glucose homeostasis in pancreatic beta-cells and adipocytes. J. Biol. Chem. 2013, 288, 35548–35558. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-J.; Park, K.-H.; Yim, C.-Y.; Takasawa, S.; Okamoto, H.; Im, M.-J.; Kim, U.-H. Generation of nicotinic acid adenine dinucleotide phosphate and cyclic ADP-ribose by glucagon-like peptide-1 evokes Ca2+ signal that is essential for insulin secretion in mouse pancreatic islets. Diabetes 2008, 57, 868–878. [Google Scholar] [CrossRef]
- Naylor, E.; Arredouani, A.; Vasudevan, S.R.; Lewis, A.M.; Parkesh, R.; Mizote, A.; Rosen, D.; Thomas, J.M.; Izumi, M.; Ganesan, A.; et al. Identification of a chemical probe for NAADP by virtual screening. Nat. Chem. Biol. 2009, 5, 220–226. [Google Scholar] [CrossRef]
- Arredouani, A.; Ruas, M.; Collins, S.C.; Parkesh, R.; Clough, F.; Pillinger, T.; Coltart, G.; Rietdorf, K.; Royle, A.; Johnson, P.; et al. Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) and Endolysosomal Two-pore Channels Modulate Membrane Excitability and Stimulus-Secretion Coupling in Mouse Pancreatic beta Cells. J. Biol. Chem. 2015, 290, 21376–21392. [Google Scholar] [CrossRef]
- Calcraft, P.J.; Ruas, M.; Pan, Z.; Cheng, X.; Arredouani, A.; Hao, X.; Tang, J.; Rietdorf, K.; Teboul, L.; Chuang, K.-T.; et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 2009, 459, 596–600. [Google Scholar] [CrossRef]
- Cane, M.C.; Parrington, J.; Rorsman, P.; Galione, A.; Rutter, G.A. The two pore channel TPC2 is dispensable in pancreatic beta-cells for normal Ca(2+) dynamics and insulin secretion. Cell Calcium 2016, 59, 32–40. [Google Scholar] [CrossRef]
- Brailoiu, E.; Churamani, D.; Cai, X.; Schrlau, M.G.; Brailoiu, G.C.; Gao, X.; Hooper, R.; Boulware, M.J.; Dun, N.J.; Marchant, J.S.; et al. Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling. J. Cell Biol. 2009, 186, 201–209. [Google Scholar] [CrossRef]
- Guse, A.H.; Diercks, B.P. Integration of nicotinic acid adenine dinucleotide phosphate (NAADP)-dependent calcium signalling. J. Physiol. 2018, 596, 2735–2743. [Google Scholar] [CrossRef]
- Zhang, F.; Jin, S.; Yi, F.; Li, P.L. TRP-ML1 functions as a lysosomal NAADP-sensitive Ca2+ release channel in coronary arterial myocytes. J. Cell. Mol. Med. 2009, 13, 3174–3185. [Google Scholar] [CrossRef]
- Takasawa, S.; Nata, K.; Yonekura, H.; Okamoto, H. Cyclic ADP-ribose in insulin secretion from pancreatic beta cells. Science 1993, 259, 370–373. [Google Scholar] [CrossRef]
- Kato, I.; Takasawa, S.; Akabane, A.; Tanaka, O.; Abe, H.; Takamura, T.; Suzuki, Y.; Nata, K.; Yonekura, H.; Yoshimoto, T.; et al. Regulatory role of CD38 (ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase) in insulin secretion by glucose in pancreatic beta cells. Enhanced insulin secretion in CD38-expressing transgenic mice. J. Biol. Chem. 1995, 270, 30045–30050. [Google Scholar] [CrossRef]
- Kato, I.; Yamamoto, Y.; Fujimura, M.; Noguchi, N.; Takasawa, S.; Okamoto, H. CD38 disruption impairs glucose-induced increases in cyclic ADP-ribose, [Ca2+]i, and insulin secretion. J. Biol. Chem. 1999, 274, 1869–1872. [Google Scholar] [CrossRef]
- Togashi, K.; Hara, Y.; Tominaga, T.; Higashi, T.; Konishi, Y.; Mori, Y.; Tominaga, M. TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion. EMBO J. 2006, 25, 1804–1815. [Google Scholar] [CrossRef]
- Walseth, T.F.; Aarhus, R.; Kerr, J.A.; Lee, H.C. Identification of cyclic ADP-ribose-binding proteins by photoaffinity labeling. J. Biol. Chem. 1993, 268, 26686–26691. [Google Scholar] [CrossRef]
- Noguchi, N.; Takasawa, S.; Nata, K.; Tohgo, A.; Kato, I.; Ikehata, F.; Yonekura, H.; Okamoto, H. Cyclic ADP-ribose binds to FK506-binding protein 12.6 to release Ca2+ from islet microsomes. J. Biol. Chem. 1997, 272, 3133–3136. [Google Scholar] [CrossRef]
- Zhang, K.; Sun, W.; Huang, L.; Zhu, K.; Pei, F.; Zhu, L.; Wang, Q.; Lu, Y.; Zhang, H.; Jin, H.; et al. Identifying Glyceraldehyde 3-Phosphate Dehydrogenase as a Cyclic Adenosine Diphosphoribose Binding Protein by Photoaffinity Protein-Ligand Labeling Approach. J. Am. Chem. Soc. 2017, 139, 156–170. [Google Scholar] [CrossRef]
- Webb, D.L.; Islam, M.S.; Efanov, A.M.; Brown, G.; Kohler, M.; Larsson, O.; Berggren, P.O. Insulin exocytosis and glucose-mediated increase in cytoplasmic free Ca2+ concentration in the pancreatic beta-cell are independent of cyclic ADP-ribose. J. Biol. Chem. 1996, 271, 19074–19079. [Google Scholar] [CrossRef]
- Rutter, G.A.; Theler, J.M.; Li, G.; Wollheim, C.B. Ca2+ stores in insulin-secreting cells: Lack of effect of cADP ribose. Cell Calcium 1994, 16, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.S.; Berggren, P.O. Cyclic ADP-ribose and the pancreatic beta cell: Where do we stand? Diabetologia 1997, 40, 1480–1484. [Google Scholar] [CrossRef]
- Kim, U.H. Roles of cADPR and NAADP in pancreatic beta cell signaling. Cell Calcium 2022, 103, 102562. [Google Scholar] [CrossRef] [PubMed]
- Wiederkehr, A.; Wollheim, C.B. Mitochondrial signals drive insulin secretion in the pancreatic beta-cell. Mol. Cell. Endocrinol. 2012, 353, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Wiederkehr, A.; Szanda, G.; Akhmedov, D.; Mataki, C.; Heizmann, C.W.; Schoonjans, K.; Pozzan, T.; Spat, A.; Wollheim, C.B. Mitochondrial matrix calcium is an activating signal for hormone secretion. Cell Metab. 2011, 13, 601–611. [Google Scholar] [CrossRef]
- Tanaka, T.; Nagashima, K.; Inagaki, N.; Kioka, H.; Takashima, S.; Fukuoka, H.; Noji, H.; Kakizuka, A.; Imamura, H. Glucose-stimulated single pancreatic islets sustain increased cytosolic ATP levels during initial Ca2+ influx and subsequent Ca2+ oscillations. J. Biol. Chem. 2014, 289, 2205–2216. [Google Scholar] [CrossRef]
- Hansford, R.G. Relation between mitochondrial calcium transport and control of energy metabolism. Rev. Physiol. Biochem. Pharmacol. 1985, 102, 1–72. [Google Scholar]
- Denton, R.M.; McCormack, J.G. On the role of the calcium transport cycle in heart and other mammalian mitochondria. FEBS Lett. 1980, 119, 1–8. [Google Scholar] [CrossRef]
- Wiederkehr, A.; Wollheim, C.B. Impact of mitochondrial calcium on the coupling of metabolism to insulin secretion in the pancreatic beta-cell. Cell Calcium 2008, 44, 64–76. [Google Scholar] [CrossRef]
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049. [Google Scholar] [CrossRef]
- Decuypere, J.P.; Monaco, G.; Bultynck, G.; Missiaen, L.; De Smedt, H.; Parys, J.B. The IP(3) receptor-mitochondria connection in apoptosis and autophagy. Biochim. Biophys. Acta 2011, 1813, 1003–1013. [Google Scholar] [CrossRef]
- Alam, M.R.; Groschner, L.N.; Parichatikanond, W.; Kuo, L.; Bondarenko, A.I.; Rost, R.; Waldeck-Weiermair, M.; Malli, R.; Graier, W.F. Mitochondrial Ca2+ uptake 1 (MICU1) and mitochondrial Ca2+ uniporter (MCU) contribute to metabolism-secretion coupling in clonal pancreatic beta-cells. J. Biol. Chem. 2012, 287, 34445–34454. [Google Scholar] [CrossRef]
- Quan, X.; Nguyen, T.T.; Choi, S.-K.; Xu, S.; Das, R.; Cha, S.-K.; Kim, N.; Han, J.; Wiederkehr, A.; Wollheim, C.B.; et al. Essential role of mitochondrial Ca2+ uniporter in the generation of mitochondrial pH gradient and metabolism-secretion coupling in insulin-releasing cells. J. Biol. Chem. 2015, 290, 4086–4096. [Google Scholar] [CrossRef]
- Georgiadou, E.; Haythorne, E.; Dickerson, M.T.; Lopez-Noriega, L.; Pullen, T.J.; da Silva Xavier, G.; Davis, S.P.X.; Martinez-Sanchez, A.; Semplici, F.; Rizzuto, R.; et al. The pore-forming subunit MCU of the mitochondrial Ca(2+) uniporter is required for normal glucose-stimulated insulin secretion in vitro and in vivo in mice. Diabetologia 2020, 63, 1368–1381. [Google Scholar] [CrossRef]
- Gilon, P.; Shepherd, R.M.; Henquin, J.C. Oscillations of secretion driven by oscillations of cytoplasmic Ca2+ as evidences in single pancreatic islets. J. Biol. Chem. 1993, 268, 22265–22268. [Google Scholar] [CrossRef]
- Dufer, M.; Krippeit-Drews, P.; Lembert, N.; Idahl, L.A.; Drews, G. Diabetogenic effect of cyclosporin A is mediated by interference with mitochondrial function of pancreatic B-cells. Mol. Pharmacol. 2001, 60, 873–879. [Google Scholar]
- Koshkin, V.; Bikopoulos, G.; Chan, C.B.; Wheeler, M.B. The characterization of mitochondrial permeability transition in clonal pancreatic beta-cells. Multiple modes and regulation. J. Biol. Chem. 2004, 279, 41368–41376. [Google Scholar] [CrossRef]
- Lablanche, S.; Cottet-Rousselle, C.; Lamarche, F.; Benhamou, P.Y.; Halimi, S.; Leverve, X.; Fontaine, E. Protection of pancreatic INS-1 beta-cells from glucose- and fructose-induced cell death by inhibiting mitochondrial permeability transition with cyclosporin A or metformin. Cell Death Dis. 2011, 2, e134. [Google Scholar] [CrossRef]
- Bonora, M.; Patergnani, S.; Ramaccini, D.; Morciano, G.; Pedriali, G.; Kahsay, A.E.; Bouhamida, E.; Giorgi, C.; Wieckowski, M.R.; Pinton, P. Physiopathology of the Permeability Transition Pore: Molecular Mechanisms in Human Pathology. Biomolecules 2020, 10, 998. [Google Scholar] [CrossRef]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef]
- Nita, I.I.; Hershfinkel, M.; Fishman, D.; Ozeri, E.; Rutter, G.A.; Sensi, S.L.; Khananshvili, D.; Lewis, E.C.; Sekler, I. The mitochondrial Na+/Ca2+ exchanger upregulates glucose dependent Ca2+ signalling linked to insulin secretion. PLoS ONE 2012, 7, e46649. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.J.; Butler, A.E.; Saisho, Y.; Monchamp, T.; Galasso, R.; Bhushan, A.; Rizza, R.A.; Butler, P.C. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes 2008, 57, 1584–1594. [Google Scholar] [CrossRef] [PubMed]
- Finegood, D.T.; Scaglia, L.; Bonner-Weir, S. Dynamics of beta-cell mass in the growing rat pancreas. Estimation with a simple mathematical model. Diabetes 1995, 44, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Dor, Y.; Brown, J.; Martinez, O.I.; Melton, D.A. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004, 429, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Porat, S.; Weinberg-Corem, N.; Tornovsky-Babaey, S.; Schyr-Ben-Haroush, R.; Hija, A.; Stolovich-Rain, M.; Dadon, D.; Granot, Z.; Ben-Hur, V.; White, P.; et al. Control of pancreatic beta cell regeneration by glucose metabolism. Cell Metab. 2011, 13, 440–449. [Google Scholar] [CrossRef]
- Heit, J.J.; Apelqvist, A.A.; Gu, X.; Winslow, M.M.; Neilson, J.R.; Crabtree, G.R.; Kim, S.K. Calcineurin/NFAT signalling regulates pancreatic beta-cell growth and function. Nature 2006, 443, 345–349. [Google Scholar] [CrossRef]
- Lawrence, M.C.; Bhatt, H.S.; Watterson, J.M.; Easom, R.A. Regulation of insulin gene transcription by a Ca(2+)-responsive pathway involving calcineurin and nuclear factor of activated T cells. Mol. Endocrinol. 2001, 15, 1758–1767. [Google Scholar] [CrossRef]
- Miranda, J.G.; Schleicher, W.E.; Wells, K.L.; Ramirez, D.G.; Landgrave, S.P.; Benninger, R.K.P. Dynamic changes in beta-cell [Ca(2+)] regulate NFAT activation, gene transcription, and islet gap junction communication. Mol. Metab. 2022, 57, 101430. [Google Scholar] [CrossRef]
- Dai, C.; Hang, Y.; Shostak, A.; Poffenberger, G.; Hart, N.; Prasad, N.; Phillips, N.; Levy, S.E.; Greiner, D.L.; Shultz, L.D.; et al. Age-dependent human beta cell proliferation induced by glucagon-like peptide 1 and calcineurin signaling. J. Clin. Investig. 2017, 127, 3835–3844. [Google Scholar] [CrossRef]
- Goodyer, W.R.; Gu, X.; Liu, Y.; Bottino, R.; Crabtree, G.R.; Kim, S.K. Neonatal beta cell development in mice and humans is regulated by calcineurin/NFAT. Dev. Cell 2012, 23, 21–34. [Google Scholar] [CrossRef]
- Shen, W.; Taylor, B.; Jin, Q.; Nguyen-Tran, V.; Meeusen, S.; Zhang, Y.Q.; Kamireddy, A.; Swafford, A.; Powers, A.F.; Walker, J.; et al. Inhibition of DYRK1A and GSK3B induces human beta-cell proliferation. Nat. Commun. 2015, 6, 8372. [Google Scholar] [CrossRef]
- Abdolazimi, Y.; Zhao, Z.; Lee, S.; Xu, H.; Allegretti, P.; Horton, T.M.; Yeh, B.; Moeller, H.P.; Nichols, R.J.; McCutcheon, D.; et al. CC-401 Promotes beta-Cell Replication via Pleiotropic Consequences of DYRK1A/B Inhibition. Endocrinology 2018, 159, 3143–3157. [Google Scholar] [CrossRef]
- Simonett, S.P.; Shin, S.; Herring, J.A.; Bacher, R.; Smith, L.A.; Dong, C.; Rabaglia, M.E.; Stapleton, D.S.; Schueler, K.L.; Choi, J.; et al. Identification of direct transcriptional targets of NFATC2 that promote beta cell proliferation. J. Clin. Investig. 2021, 131, e144833. [Google Scholar] [CrossRef]
- Wu, X.; Liang, W.; Guan, H.; Liu, J.; Liu, L.; Li, H.; He, X.; Zheng, J.; Chen, J.; Cao, X.; et al. Exendin-4 promotes pancreatic beta-cell proliferation via inhibiting the expression of Wnt5a. Endocrine 2017, 55, 398–409. [Google Scholar] [CrossRef]
- Liu, B.; Barbosa-Sampaio, H.; Jones, P.M.; Persaud, S.J.; Muller, D.S. The CaMK4/CREB/IRS-2 cascade stimulates proliferation and inhibits apoptosis of beta-cells. PLoS ONE 2012, 7, e45711. [Google Scholar]
- Srinivasan, S.; Bernal-Mizrachi, E.; Ohsugi, M.; Permutt, M.A. Glucose promotes pancreatic islet beta-cell survival through a PI 3-kinase/Akt-signaling pathway. Am. J. Physiol. Endocrinol. Metab. 2002, 283, E784–E793. [Google Scholar] [CrossRef]
- Rachdaoui, N.; Polo-Parada, L.; Ismail-Beigi, F. Prolonged Exposure to Insulin Inactivates Akt and Erk(1/2) and Increases Pancreatic Islet and INS1E beta-Cell Apoptosis. J. Endocr. Soc. 2019, 3, 69–90. [Google Scholar] [CrossRef]
- Hara, T.; Mahadevan, J.; Kanekura, K.; Hara, M.; Lu, S.; Urano, F. Calcium efflux from the endoplasmic reticulum leads to beta-cell death. Endocrinology 2014, 155, 758–768. [Google Scholar] [CrossRef]
- Clark, A.L.; Kanekura, K.; Lavagnino, Z.; Spears, L.D.; Abreu, D.; Mahadevan, J.; Yagi, T.; Semenkovich, C.F.; Piston, D.W.; Urano, F. Targeting Cellular Calcium Homeostasis to Prevent Cytokine-Mediated Beta Cell Death. Sci. Rep. 2017, 7, 5611. [Google Scholar] [CrossRef]
- Ly, L.D.; Xu, S.; Choi, S.-K.; Ha, C.-M.; Thoudam, T.; Cha, S.-K.; Wiederkehr, A.; Wollheim, C.B.; Lee, I.-K.; Park, K.-S. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp. Mol. Med. 2017, 49, e291. [Google Scholar] [CrossRef]
- Gwiazda, K.S.; Yang, T.L.; Lin, Y.; Johnson, J.D. Effects of palmitate on ER and cytosolic Ca2+ homeostasis in beta-cells. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E690–E701. [Google Scholar] [CrossRef] [PubMed]
- Soleimanpour, S.A.; Crutchlow, M.F.; Ferrari, A.M.; Raum, J.C.; Groff, D.N.; Rankin, M.M.; Liu, C.; De Leon, D.D.; Naji, A.; Kushner, J.A.; et al. Calcineurin signaling regulates human islet beta-cell survival. J. Biol. Chem. 2010, 285, 40050–40059. [Google Scholar] [CrossRef] [PubMed]
- Marie, J.C.; Bailbe, D.; Gylfe, E.; Portha, B. Defective glucose-dependent cytosolic Ca2+ handling in islets of GK and nSTZ rat models of type 2 diabetes. J. Endocrinol. 2001, 169, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chmelova, H.; Cohrs, C.M.; Chouinard, J.A.; Jahn, S.R.; Stertmann, J.; Uphues, I.; Speier, S. Alterations in beta-Cell Calcium Dynamics and Efficacy Outweigh Islet Mass Adaptation in Compensation of Insulin Resistance and Prediabetes Onset. Diabetes 2016, 65, 2676–2685. [Google Scholar] [CrossRef]
- Rose, T.; Efendic, S.; Rupnik, M. Ca2+-secretion coupling is impaired in diabetic Goto Kakizaki rats. J. Gen. Physiol. 2007, 129, 493–508. [Google Scholar] [CrossRef]
- Li, L.; Trifunovic, A.; Kohler, M.; Wang, Y.; Petrovic Berglund, J.; Illies, C.; Juntti-Berggren, L.; Larsson, N.G.; Berggren, P.O. Defects in beta-cell Ca2+ dynamics in age-induced diabetes. Diabetes 2014, 63, 4100–4114. [Google Scholar] [CrossRef]
- Gandasi, N.; Yin, P.; Riz, M.; Chibalina, M.V.; Cortese, G.; Lund, P.-E.; Matveev, V.; Rorsman, P.; Sherman, A.; Pedersen, M.G.; et al. Ca2+ channel clustering with insulin-containing granules is disturbed in type 2 diabetes. J. Clin. Investig. 2017, 127, 2353–2364. [Google Scholar] [CrossRef]
- Bernal-Mizrachi, E.; Cras-Meneur, C.; Ye, B.R.; Johnson, J.D.; Permutt, M.A. Transgenic overexpression of active calcineurin in beta-cells results in decreased beta-cell mass and hyperglycemia. PLoS ONE 2010, 5, e11969. [Google Scholar] [CrossRef]
- Stancill, J.S.; Cartailler, J.P.; Clayton, H.W.; O’Connor, J.T.; Dickerson, M.T.; Dadi, P.K.; Osipovich, A.B.; Jacobson, D.A.; Magnuson, M.A. Chronic beta-Cell Depolarization Impairs beta-Cell Identity by Disrupting a Network of Ca(2+)-Regulated Genes. Diabetes 2017, 66, 2175–2187. [Google Scholar] [CrossRef]
- Dahan, T.; Ziv, O.; Horwitz, E.; Zemmour, H.; Lavi, J.; Swisa, A.; Leibowitz, G.; Ashcroft, F.M.; In’t Veld, P.; Glaser, B.; et al. Pancreatic beta-Cells Express the Fetal Islet Hormone Gastrin in Rodent and Human Diabetes. Diabetes 2017, 66, 426–436. [Google Scholar] [CrossRef]
- González, B.J.; Zhao, H.; Niu, J.; Williams, D.J.; Lee, J.; Goulbourne, C.N.; Xing, Y.; Wang, Y.; Oberholzer, J.; Blumenkrantz, M.H.; et al. Reduced calcium levels and accumulation of abnormal insulin granules in stem cell models of HNF1A deficiency. Commun. Biol. 2022, 5, 779. [Google Scholar] [CrossRef]
- Lee, K.; Kim, J.; Kohler, M.; Yu, J.; Shi, Y.; Yang, S.N.; Ryu, S.H.; Berggren, P.O. Blocking Ca(2+) Channel beta(3) Subunit Reverses Diabetes. Cell Rep. 2018, 24, 922–934. [Google Scholar] [CrossRef]
- Becker, A.; Wardas, B.; Salah, H.; Amini, M.; Fecher-Trost, C.; Sen, Q.; Martus, D.; Beck, A.; Philipp, S.E.; Flockerzi, V.; et al. Cavbeta3 Regulates Ca(2+) Signaling and Insulin Expression in Pancreatic beta-Cells in a Cell-Autonomous Manner. Diabetes 2021, 70, 2532–2544. [Google Scholar] [CrossRef]
- Vogel, J.; Yin, J.; Su, L.; Wang, S.X.; Zessis, R.; Fowler, S.; Chiu, C.H.; Wilson, A.C.; Chen, A.; Zecri, F.; et al. A Phenotypic Screen Identifies Calcium Overload as a Key Mechanism of beta-Cell Glucolipotoxicity. Diabetes 2020, 69, 1032–1041. [Google Scholar] [CrossRef]
- Minn, A.H.; Hafele, C.; Shalev, A. Thioredoxin-interacting protein is stimulated by glucose through a carbohydrate response element and induces beta-cell apoptosis. Endocrinology 2005, 146, 2397–2405. [Google Scholar] [CrossRef]
- Minn, A.H.; Pise-Masison, C.A.; Radonovich, M.; Brady, J.N.; Wang, P.; Kendziorski, C.; Shalev, A. Gene expression profiling in INS-1 cells overexpressing thioredoxin-interacting protein. Biochem. Biophys. Res. Commun. 2005, 336, 770–778. [Google Scholar] [CrossRef]
- Chen, J.; Saxena, G.; Mungrue, I.N.; Lusis, A.J.; Shalev, A. Thioredoxin-interacting protein: A critical link between glucose toxicity and beta-cell apoptosis. Diabetes 2008, 57, 938–944. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Xu, G.; Chen, J.; Jing, G.; Shalev, A. Preventing beta-cell loss and diabetes with calcium channel blockers. Diabetes 2012, 61, 848–856. [Google Scholar] [CrossRef]
- Ovalle, F.; Grimes, T.; Xu, G.; Patel, A.J.; Grayson, T.B.; Thielen, L.A.; Li, P.; Shalev, A. Verapamil and beta cell function in adults with recent-onset type 1 diabetes. Nat. Med. 2018, 24, 1108–1112. [Google Scholar] [CrossRef]
- Laybutt, D.R.; Preston, A.M.; Åkerfeldt, M.C.; Kench, J.G.; Busch, A.K.; Biankin, A.V.; Biden, T.J. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 2007, 50, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, P.; Bugliani, M.; Lupi, R.; Marselli, L.; Masini, M.; Boggi, U.; Filipponi, F.; Weir, G.C.; Eizirik, D.L.; Cnop, M. The endoplasmic reticulum in pancreatic beta cells of type 2 diabetes patients. Diabetologia 2007, 50, 2486–2494. [Google Scholar] [CrossRef] [PubMed]
- Cunha, D.A.; Hekerman, P.; Ladriere, L.; Bazarra-Castro, A.; Ortis, F.; Wakeham, M.C.; Moore, F.; Rasschaert, J.; Cardozo, A.K.; Bellomo, E.; et al. Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J. Cell Sci. 2008, 121 Pt 14, 2308–2318. [Google Scholar] [CrossRef] [PubMed]
- Rorsman, P.; Braun, M. Regulation of insulin secretion in human pancreatic islets. Annu. Rev. Physiol. 2013, 75, 155–179. [Google Scholar] [CrossRef]
- Zakrzewski, W.; Dobrzynski, M.; Szymonowicz, M.; Rybak, Z. Stem cells: Past, present, and future. Stem Cell Res. Ther. 2019, 10, 68. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Zeng, H.; Guo, M.; Zhou, T.; Tan, L.; Chong, C.N.; Zhang, T.; Dong, X.; Xiang, J.Z.; Yu, A.S.; Yue, L.; et al. An Isogenic Human ESC Platform for Functional Evaluation of Genome-wide-Association-Study-Identified Diabetes Genes and Drug Discovery. Cell Stem Cell 2016, 19, 326–340. [Google Scholar] [CrossRef]
- Shang, L.; Hua, H.; Foo, K.; Martinez, H.; Watanabe, K.; Zimmer, M.; Kahler, D.J.; Freeby, M.; Chung, W.; LeDuc, C.; et al. beta-cell dysfunction due to increased ER stress in a stem cell model of Wolfram syndrome. Diabetes 2014, 63, 923–933. [Google Scholar] [CrossRef]
- Hogrebe, N.J.; Maxwell, K.G.; Augsornworawat, P.; Millman, J.R. Generation of insulin-producing pancreatic beta cells from multiple human stem cell lines. Nat. Protoc. 2021, 16, 4109–4143. [Google Scholar] [CrossRef]
- Rezania, A.; Bruin, J.E.; Arora, P.; Rubin, A.; Batushansky, I.; Asadi, A.; O’Dwyer, S.; Quiskamp, N.; Mojibian, M.; Albrecht, T.; et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 1121–1133. [Google Scholar] [CrossRef]
- Pagliuca, F.W.; Millman, J.R.; Gurtler, M.; Segel, M.; Van Dervort, A.; Ryu, J.H.; Peterson, Q.P.; Greiner, D.; Melton, D.A. Generation of functional human pancreatic beta cells in vitro. Cell 2014, 159, 428–439. [Google Scholar] [CrossRef]
- Sui, L.; Danzl, N.; Campbell, S.R.; Viola, R.; Williams, D.; Xing, Y.; Wang, Y.; Phillips, N.; Poffenberger, G.; Johannesson, B.; et al. beta-Cell Replacement in Mice Using Human Type 1 Diabetes Nuclear Transfer Embryonic Stem Cells. Diabetes 2018, 67, 26–35. [Google Scholar] [CrossRef]
- Davis, J.C.; Alves, T.C.; Helman, A.; Chen, J.C.; Kenty, J.H.; Cardone, R.L.; Liu, D.R.; Kibbey, R.G.; Melton, D.A. Glucose Response by Stem Cell-Derived beta Cells In Vitro Is Inhibited by a Bottleneck in Glycolysis. Cell Rep. 2020, 31, 107623. [Google Scholar] [CrossRef]
- Veres, A.; Faust, A.L.; Bushnell, H.L.; Engquist, E.N.; Kenty, J.H.; Harb, G.; Poh, Y.C.; Sintov, E.; Gurtler, M.; Pagliuca, F.W.; et al. Charting cellular identity during human in vitro beta-cell differentiation. Nature 2019, 569, 368–373. [Google Scholar] [CrossRef]
- Sachdeva, M.M.; Claiborn, K.C.; Khoo, C.; Yang, J.; Groff, D.N.; Mirmira, R.G.; Stoffers, D.A. Pdx1 (MODY4) regulates pancreatic beta cell susceptibility to ER stress. Proc. Natl. Acad. Sci. USA 2009, 106, 19090–19095. [Google Scholar] [CrossRef]
- Johnson, J.D.; Ahmed, N.T.; Luciani, D.S.; Han, Z.; Tran, H.; Fujita, J.; Misler, S.; Edlund, H.; Polonsky, K.S. Increased islet apoptosis in Pdx1+/− mice. J. Clin. Investig. 2003, 111, 1147–1160. [Google Scholar] [CrossRef]
- Brissova, M.; Shiota, M.; Nicholson, W.E.; Gannon, M.; Knobel, S.M.; Piston, D.W.; Wright, C.V.E.; Powers, A.C. Reduction in pancreatic transcription factor PDX-1 impairs glucose-stimulated insulin secretion. J. Biol. Chem. 2002, 277, 11225–11232. [Google Scholar] [CrossRef]
- Johnson, J.S.; Kono, T.; Tong, X.; Yamamoto, W.R.; Zarain-Herzberg, A.; Merrins, M.J.; Satin, L.S.; Gilon, P.; Evans-Molina, C. Pancreatic and duodenal homeobox protein 1 (Pdx-1) maintains endoplasmic reticulum calcium levels through transcriptional regulation of sarco-endoplasmic reticulum calcium ATPase 2b (SERCA2b) in the islet beta cell. J. Biol. Chem. 2014, 289, 32798–32810. [Google Scholar] [CrossRef]
- Artner, I.; Hang, Y.; Guo, M.; Gu, G.; Stein, R. MafA is a dedicated activator of the insulin gene in vivo. J. Endocrinol. 2008, 198, 271–279. [Google Scholar] [CrossRef]
- Vanderford, N.L.; Cantrell, J.E.; Popa, G.J.; Ozcan, S. Multiple kinases regulate mafA expression in the pancreatic beta cell line MIN6. Arch. Biochem. Biophys. 2008, 480, 138–142. [Google Scholar] [CrossRef]
- Santos, G.J.; Ferreira, S.M.; Ortis, F.; Rezende, L.F.; Li, C.; Naji, A.; Carneiro, E.M.; Kaestner, K.H.; Boschero, A.C. Metabolic memory of ß-cells controls insulin secretion is mediated by CaMKII. Mol. Metab. 2014, 3, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Karagiannopoulos, A.; Eliasson, L.; Renstrom, E.; Luan, C.; Zhang, E. The Calcium Channel Subunit Gamma-4 as a Novel Regulator of MafA in Pancreatic Beta-Cell Controls Glucose Homeostasis. Biomedicines 2022, 10, 770. [Google Scholar] [CrossRef] [PubMed]
- Shimajiri, Y.; Sanke, T.; Furuta, H.; Hanabusa, T.; Nakagawa, T.; Fujitani, Y.; Kajimoto, Y.; Takasu, N.; Nanjo, K. A missense mutation of Pax4 gene (R121W) is associated with type 2 diabetes in Japanese. Diabetes 2001, 50, 2864–2869. [Google Scholar] [CrossRef] [PubMed]
- Brun, T.; Franklin, I.; St-Onge, L.; Biason-Lauber, A.; Schoenle, E.J.; Wollheim, C.B.; Gauthier, B.R. The diabetes-linked transcription factor PAX4 promotes beta-cell proliferation and survival in rat and human islets. J. Cell Biol. 2004, 167, 1123–1135. [Google Scholar] [CrossRef]
- Velazco-Cruz, L.; Song, J.; Maxwell, K.G.; Goedegebuure, M.M.; Augsornworawat, P.; Hogrebe, N.J.; Millman, J.R. Acquisition of Dynamic Function in Human Stem Cell-Derived beta Cells. Stem Cell Rep. 2019, 12, 351–365. [Google Scholar] [CrossRef]
- Nair, G.G.; Liu, J.S.; Russ, H.A.; Tran, S.; Saxton, M.S.; Chen, R.; Juang, C.; Li, M.L.; Nguyen, V.Q.; Giacometti, S.; et al. Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived beta cells. Nat. Cell Biol. 2019, 21, 263–274. [Google Scholar] [CrossRef]
- Maxwell, K.G.; Augsornworawat, P.; Velazco-Cruz, L.; Kim, M.H.; Asada, R.; Hogrebe, N.J.; Morikawa, S.; Urano, F.; Millman, J.R. Gene-edited human stem cell-derived beta cells from a patient with monogenic diabetes reverse preexisting diabetes in mice. Sci. Transl. Med. 2020, 12, eaax9106. [Google Scholar] [CrossRef]
- Cardenas-Diaz, F.L.; Osorio-Quintero, C.; Diaz-Miranda, M.A.; Kishore, S.; Leavens, K.; Jobaliya, C.; Stanescu, D.; Ortiz-Gonzalez, X.; Yoon, C.; Chen, C.S.; et al. Modeling Monogenic Diabetes using Human ESCs Reveals Developmental Metabolic Deficiencies Caused by Mutations in, H.N.F.1.A. Cell Stem Cell 2019, 25, 273–289.e5. [Google Scholar] [CrossRef]
- Lithovius, V.; Saarimäki-Vire, J.; Balboa, D.; Ibrahim, H.; Montaser, H.; Barsby, T.; Otonkoski, T. SUR1-mutant iPS cell-derived islets recapitulate the pathophysiology of congenital hyperinsulinism. Diabetologia 2021, 64, 630–640. [Google Scholar] [CrossRef]
- Wang, X.; Sterr, M.; Ansarullah Burtscher, I.; Bottcher, A.; Beckenbauer, J.; Siehler, J.; Meitinger, T.; Haring, H.U.; Staiger, H.; Cernilogar, F.M.; et al. Point mutations in the PDX1 transactivation domain impair human beta-cell development and function. Mol. Metab. 2019, 24, 80–97. [Google Scholar] [CrossRef]
- Balboa, D.; Saarimaki-Vire, J.; Borshagovski, D.; Survila, M.; Lindholm, P.; Galli, E.; Eurola, S.; Ustinov, J.; Grym, H.; Huopio, H.; et al. Insulin mutations impair beta-cell development in a patient-derived iPSC model of neonatal diabetes. Elife 2018, 7, e38519. [Google Scholar] [CrossRef]
- Balboa, D.; Barsby, T.; Lithovius, V.; Saarimäki-Vire, J.; Omar-Hmeadi, M.; Dyachok, O.; Montaser, H.; Lund, P.-E.; Yang, M.; Ibrahim, H.; et al. Functional, metabolic and transcriptional maturation of human pancreatic islets derived from stem cells. Nat. Biotechnol. 2022, 40, 1042–1055. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mu-u-min, R.B.A.; Diane, A.; Allouch, A.; Al-Siddiqi, H.H. Ca2+-Mediated Signaling Pathways: A Promising Target for the Successful Generation of Mature and Functional Stem Cell-Derived Pancreatic Beta Cells In Vitro. Biomedicines 2023, 11, 1577. https://doi.org/10.3390/biomedicines11061577
Mu-u-min RBA, Diane A, Allouch A, Al-Siddiqi HH. Ca2+-Mediated Signaling Pathways: A Promising Target for the Successful Generation of Mature and Functional Stem Cell-Derived Pancreatic Beta Cells In Vitro. Biomedicines. 2023; 11(6):1577. https://doi.org/10.3390/biomedicines11061577
Chicago/Turabian StyleMu-u-min, Razik Bin Abdul, Abdoulaye Diane, Asma Allouch, and Heba H. Al-Siddiqi. 2023. "Ca2+-Mediated Signaling Pathways: A Promising Target for the Successful Generation of Mature and Functional Stem Cell-Derived Pancreatic Beta Cells In Vitro" Biomedicines 11, no. 6: 1577. https://doi.org/10.3390/biomedicines11061577