
Exploring the Multifaceted Landscape of MASLD: A Comprehensive Synthesis of Recent Studies, from Pathophysiology to Organoids and Beyond



Abstract
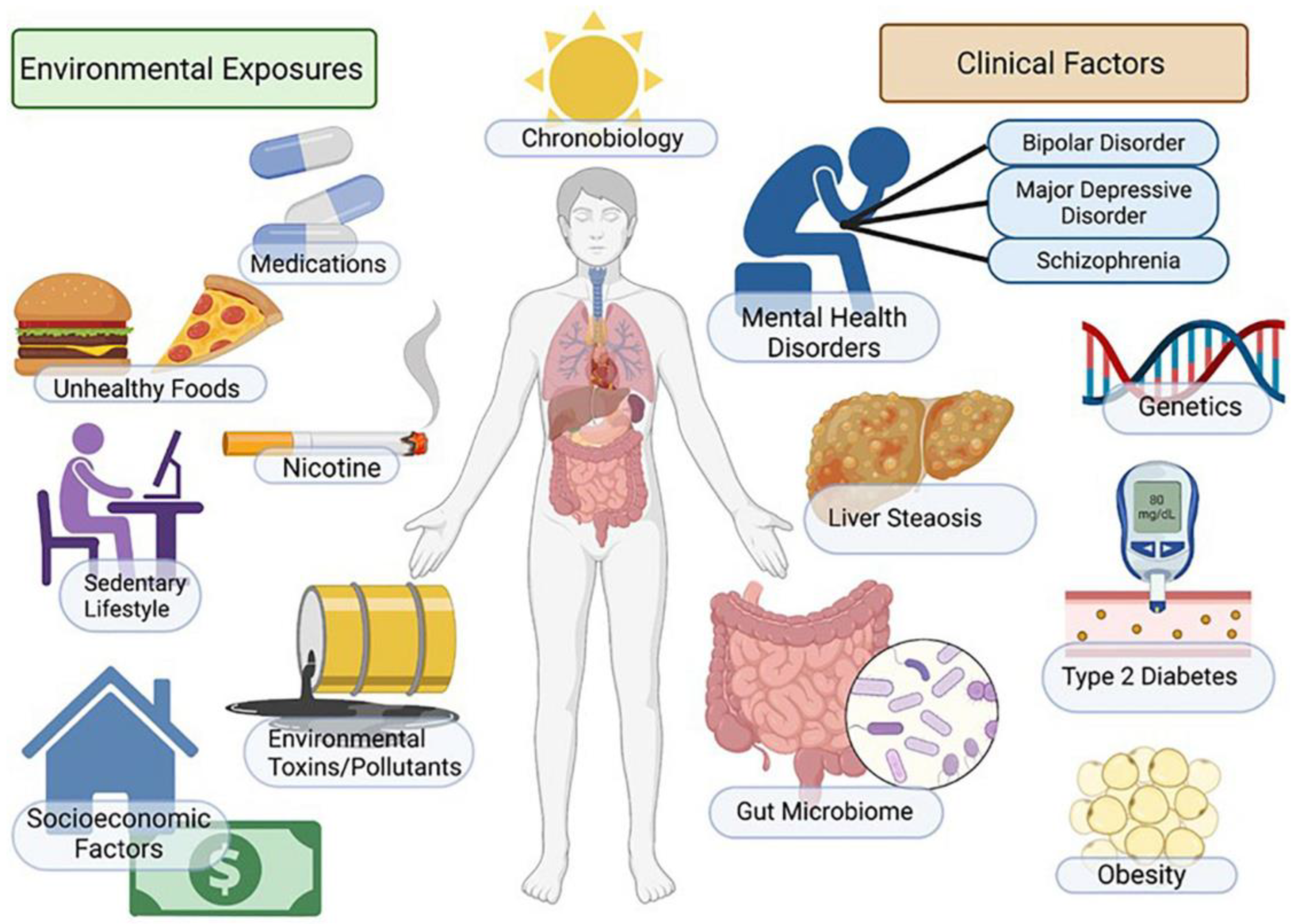
:1. Introduction
2. Pathophysiology
3. Risk Factors for MASLD
3.1. Cardiovascular Disease
3.2. Dyslipidemia
3.3. Type II Diabetes Mellitus
3.4. Obesity
3.5. Iron Overload
3.6. Galactosemia
3.7. Alpha-1-Antitrypsin
3.8. Glycogen Storage Diseases
3.9. Viral Hepatitis
3.10. Wilson’s Disease
3.11. Cystic Fibrosis
3.12. Leukocyte Telomere Length
3.13. Smoking
4. Dietary Effects on Fatty Acid Metabolism
4.1. Gut Microbiome
4.2. Gut–Liver Axis
4.3. Western Diet/Fatty Diet
4.4. Mediterranean Diet
4.5. Intermittent Fasting
5. Genetic Pathways Related to MASLD
5.1. Oxidative Stress
5.2. Genetic Mutations
5.3. Epigenetics
5.4. MicroRNA Posttranscriptional Regulation
6. The Future with Organoids
7. Therapy
7.1. Lifestyle
7.2. Bariatric Surgery
7.3. Transplant Surgery
7.4. Pharmacology
7.5. Vitamin E
7.6. Glucose Metabolism Modulators
7.7. GLP-1 Receptor Agonist Drugs
7.8. THRβ
7.9. Lipogenesis Inhibitors
7.10. ACC, FASN, SCDI, and DGAT Inhibitors, and PUFA
7.11. Bile Acid Metabolism Modulators
7.12. Fibrogenesis Inhibitors
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pouwels, S.; Sakran, N.; Graham, Y.; Leal, A.; Pintar, T.; Yang, W.; Kassir, R.; Singhal, R.; Mahawar, K.; Ramnarain, D. Non-alcoholic fatty liver disease (NAFLD): A review of pathophysiology, clinical management and effects of weight loss. BMC Endocr. Disord. 2022, 22, 63. [Google Scholar] [CrossRef]
- Jamwal, R.; Barlock, B.J. Nonalcoholic Fatty Liver Disease (NAFLD) and Hepatic Cytochrome P450 (CYP) Enzymes. Pharmaceuticals 2020, 13, 222. [Google Scholar] [CrossRef]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. Hepatology 2023, 78, 1966–1986. [Google Scholar] [CrossRef]
- Papatheodoridi, M.; Cholongitas, E. Diagnosis of Non-alcoholic Fatty Liver Disease (NAFLD): Current Concepts. Curr. Pharm. Des. 2018, 24, 4574–4586. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Golabi, P.; Paik, J.M.; Henry, A.; Van Dongen, C.; Henry, L. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): A systematic review. Hepatology 2023, 77, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Kudaravalli, P.; John, S. Nonalcoholic Fatty Liver; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Guo, X.; Yin, X.; Liu, Z.; Wang, J. Non-Alcoholic Fatty Liver Disease (NAFLD) Pathogenesis and Natural Products for Prevention and Treatment. Int. J. Mol. Sci. 2022, 23, 15489. [Google Scholar] [CrossRef] [PubMed]
- Grander, C.; Grabherr, F.; Tilg, H. Non-alcoholic fatty liver disease: Pathophysiological concepts and treatment options. Cardiovasc. Res. 2023, 119, 1787–1798. [Google Scholar] [CrossRef]
- Gill, M.G.; Majumdar, A. Metabolic associated fatty liver disease: Addressing a new era in liver transplantation. World J. Hepatol. 2020, 12, 1168–1181. [Google Scholar] [CrossRef]
- Abosheaishaa, H.; Hussein, M.; Ghallab, M.; Abdelhamid, M.; Balassiano, N.; Ahammed, R.; Baig, M.A.; Khan, J.; Elshair, M.; Soliman, M.Y.; et al. Association between non-alcoholic fatty liver disease and coronary artery disease outcomes: A systematic review and meta-analysis. Diabetes Metab. Syndr. Clin. Res. Rev. 2024, 18, 102938. [Google Scholar] [CrossRef]
- Prasad, M.; Gupta, S.; Sarin, S.K. The Independent Association of Non-alcoholic Fatty Liver Disease with Incident Cardiovascular Disease: A GRADE Evaluation of the Evidence Through a Systematic Review and Meta-analysis. J. Clin. Exp. Hepatol. 2024, 14, 101277. [Google Scholar] [CrossRef]
- Khoshbaten, M.; Maleki, S.H.; Hadad, S.; Baral, A.; Rocha, A.V.; Poudel, L.; Abdshah, A. Association of nonalcoholic fatty liver disease and carotid media-intima thickness: A systematic review and a meta-analysis. Health Sci. Rep. 2023, 6, e1554. [Google Scholar] [CrossRef]
- Mandraffino, G.; Morace, C.; Franzè, M.S.; Nassisi, V.; Sinicropi, D.; Cinquegrani, M.; Saitta, C.; Scoglio, R.; Marino, S.; Belvedere, A.; et al. Fatty Liver as Potential Biomarker of Atherosclerotic Damage in Familial Combined Hyperlipidemia. Biomedicines 2022, 10, 1770. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-Q.; Lu, L.-G. Nonalcoholic Fatty Liver Disease: Dyslipidemia, Risk for Cardiovascular Complications, and Treatment Strategy. J. Clin. Transl. Hepatol. 2015, 3, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Tanase, D.M.; Gosav, E.M.; Costea, C.F.; Ciocoiu, M.; Lacatusu, C.M.; Maranduca, M.A.; Ouatu, A.; Floria, M. The Intricate Relationship between Type 2 Diabetes Mellitus (T2DM), Insulin Resistance (IR), and Nonalcoholic Fatty Liver Disease (NAFLD). J. Diabetes Res. 2020, 2020, 3920196. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.-F.; Tsai, P.-C.; Yeh, M.-L.; Huang, C.-F.; Huang, C.-I.; Hsieh, M.-H.; Dai, C.-Y.; Yang, J.-F.; Chen, S.-C.; Yu, M.-L.; et al. Risk stratification of non-alcoholic fatty liver disease across body mass index in a community basis. Int. J. Formos. Med. Assoc. 2020, 119, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Enooku, K.; Kondo, M.; Fujiwara, N.; Sasako, T.; Shibahara, J.; Kado, A.; Okushin, K.; Fujinaga, H.; Tsutsumi, T.; Nakagomi, R.; et al. Hepatic IRS1 and ß-catenin expression is associated with histological progression and overt diabetes emergence in NAFLD patients. J. Gastroenterol. 2018, 53, 1261–1275. [Google Scholar] [CrossRef] [PubMed]
- Young, J.; Seeberg, K.A.; Aakre, K.M.; Borgeraas, H.; Nordstrand, N.; Wisløff, T.; Hjelmesæth, J.; Omland, T.; Hertel, J.K. The liver-heart axis in patients with severe obesity: The association between liver fibrosis and chronic myocardial injury may be explained by shared risk factors of cardiovascular disease. Clin. Biochem. 2024, 123, 110688. [Google Scholar] [CrossRef] [PubMed]
- Hernaez, R.; Yeung, E.; Clark, J.M.; Kowdley, K.V.; Brancati, F.L.; Kao, W.H.L. Hemochromatosis gene and nonalcoholic fatty liver disease: A systematic review and meta-analysis. J. Hepatol. 2011, 55, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Barton, J.C.; Acton, R.T. Non-alcoholic fatty liver disease in hemochromatosis probands with iron overload and HFE p.C282Y/p.C282Y. BMC Gastroenterol. 2023, 23, 137. [Google Scholar] [CrossRef]
- Yıldız, Y.; Sivri, H.S. Inborn errors of metabolism in the differential diagnosis of fatty liver disease. Turk. J. Gastroenterol. 2020, 31, 3–16. [Google Scholar] [CrossRef]
- Narayanan, P.; Mistry, P.K. Update on Alpha-1 Antitrypsin Deficiency in Liver Disease. Clin. Liver Dis. 2020, 15, 228–235. [Google Scholar] [CrossRef]
- Murali, A.R.; Prakash, S.; Sanchez, A.J. Alpha-1-Antitrypsin Pi*MZ variant increases risk of developing hepatic events in nonalcoholic fatty liver disease patients. Clin. Res. Hepatol. Gastroenterol. 2023, 47, 102066. [Google Scholar] [CrossRef]
- Allard, J.P. Other disease associations with non-alcoholic fatty liver disease (NAFLD). Best Prac. Res. Clin. Gastroenterol. 2002, 16, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Dias, C.L.; Maio, I.; Brandão, J.R.; Tomás, E.; Martins, E.; Silva, E.S. Fatty Liver Caused by Glycogen Storage Disease Type IX: A Small Series of Cases in Children. GE Port. J. Gastroenterol. 2019, 26, 430–437. [Google Scholar] [CrossRef]
- Rossi, A.; Hoogeveen, I.J.; Bastek, V.B.; de Boer, F.; Montanari, C.; Meyer, U.; Maiorana, A.; Bordugo, A.; Dianin, A.; Campana, C.; et al. Dietary lipids in glycogen storage disease type III: A systematic literature study, case studies, and future recommendations. J. Inherit. Metab. Dis. 2020, 43, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.J.; Nguyen, V.H.; Yang, H.-I.; Li, J.; Le, M.H.; Wu, W.-J.; Han, N.X.; Fong, K.Y.; Chen, E.; Wong, C.; et al. Impact of fatty liver on long-term outcomes in chronic hepatitis B: A systematic review and matched analysis of individual patient data meta-analysis. Clin. Mol. Hepatol. 2023, 29, 705–720. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cheung, K.S.; Peng, C.; Mak, L.; Cheng, H.M.; Fung, J.; Peleg, N.; Leung, H.H.; Kumar, R.; Lee, J.; et al. Steatosis, HBV-related HCC, cirrhosis, and HBsAg seroclearance: A systematic review and meta-analysis. Hepatology 2023, 77, 1735–1745. [Google Scholar] [CrossRef]
- Mahmood, S.; Inada, N.; Izumi, A.; Kawanaka, M.; Kobashi, H.; Yamada, G. Wilson’s disease masquerading as nonalcoholic steatohepatitis. N. Am. J. Med. Sci. 2009, 1, 74–76. [Google Scholar]
- Stättermayer, A.F.; Traussnigg, S.; Dienes, H.-P.; Aigner, E.; Stauber, R.; Lackner, K.; Hofer, H.; Stift, J.; Wrba, F.; Stadlmayr, A.; et al. Hepatic steatosis in Wilson disease—Role of copper and PNPLA3 mutations. J. Hepatol. 2015, 63, 156–163. [Google Scholar] [CrossRef]
- Liggi, M.; Murgia, D.; Civolani, A.; Demelia, E.; Sorbello, O.; Demelia, L. The relationship between copper and steatosis in Wilson’s disease. Clin. Res. Hepatol. Gastroenterol. 2013, 37, 36–40. [Google Scholar] [CrossRef]
- Issa, Z.; Gohy, S.; Zech, F.; Baldin, P.; Delire, B.; Dahlqvist, G. Prevalence and characteristics of cystic fibrosis liver disease: A study highlighting the lack of histological diagnosis. Clin. Res. Hepatol. Gastroenterol. 2022, 46, 101977. [Google Scholar] [CrossRef]
- Wojcicki, J.M.; Gill, R.M.; Wilson, L.; Lin, J.; Rosenthal, P. Shorter leukocyte telomere length protects against NAFLD progression in children. Sci. Rep. 2023, 13, 5446. [Google Scholar] [CrossRef]
- Jang, Y.S.; Joo, H.J.; Park, Y.S.; Park, E.-C.; Jang, S.-I. Association between smoking cessation and non-alcoholic fatty liver disease using NAFLD liver fat score. Front. Public Health 2023, 11, 1015919. [Google Scholar] [CrossRef] [PubMed]
- Gangopadhyay, A.; Ibrahim, R.; Theberge, K.; May, M.; Houseknecht, K.L. Non-alcoholic fatty liver disease (NAFLD) and mental illness: Mechanisms linking mood, metabolism and medicines. Front. Neurosci. 2022, 16, 1042442. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.-W.; Zhu, S.; Palaniappan, L.; Heshka, S.; Carnethon, M.R.; Heymsfield, S.B. The metabolic syndrome: Prevalence and associated risk factor findings in the US population from the Third National Health and Nutrition Examination Survey, 1988–1994. Arch. Intern. Med. 2003, 163, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Ruderman, N.; Chisholm, D.; Pi-Sunyer, X.; Schneider, S. The metabolically obese, normal-weight individual revisited. Diabetes 1998, 47, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Pierri, L.; Saggese, P.; Guercio Nuzio, S.; Troisi, J.; Di Stasi, M.; Poeta, M.; Savastano, R.; Marchese, G.; Tarallo, R.; Massa, G.; et al. Relations of gut liver axis components and gut microbiota in obese children with fatty liver: A pilot study. Clin. Res. Hepatol. Gastroenterol. 2018, 42, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Anania, C.; Perla, F.M.; Olivero, F.; Pacifico, L.; Chiesa, C. Mediterranean diet and nonalcoholic fatty liver disease. World J. Gastroenterol. 2018, 24, 2083–2094. [Google Scholar] [CrossRef] [PubMed]
- Kirpich, I.A.; Marsano, L.S.; McClain, C.J. Gut–liver axis, nutrition, and non-alcoholic fatty liver disease. Clin. Biochem. 2015, 48, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Ren, M.; Yang, J.; Cai, C.; Cheng, W.; Zhou, X.; Lu, D.; Ji, F. Gut microbiome and microbial metabolites in NAFLD and after bariatric surgery: Correlation and causality. Front. Microbiol. 2022, 13, 1003755. [Google Scholar] [CrossRef]
- Bäckhed, F.; Manchester, J.K.; Semenkovich, C.F.; Gordon, J.I. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. USA 2007, 104, 979–984. [Google Scholar] [CrossRef]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Mascianà, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K.; Desai, C.; Iyer, S.S.; Thorn, N.E.; Kumar, P.; Liu, Y.; Smith, T.; Neish, A.S.; Li, H.; Tan, S.; et al. Loss of Junctional Adhesion Molecule a Promotes Severe Steatohepatitis in Mice on a Diet High in Saturated Fat, Fructose, and Cholesterol. Gastroenterology 2016, 151, 733–746.e12. [Google Scholar] [CrossRef] [PubMed]
- Troisi, J.; Pierri, L.; Landolfi, A.; Marciano, F.; Bisogno, A.; Belmonte, F.; Palladino, C.; Guercio Nuzio, S.; Campiglia, P.; Vajro, P. Urinary Metabolomics in Pediatric Obesity and NAFLD Identifies Metabolic Pathways/Metabolites Related to Dietary Habits and Gut-Liver Axis Perturbations. Nutrients 2017, 9, 485. [Google Scholar] [CrossRef]
- von Schönfels, W.; Patsenker, E.; Fahrner, R.; Itzel, T.; Hinrichsen, H.; Brosch, M.; Erhart, W.; Gruodyte, A.; Vollnberg, B.; Richter, K.; et al. Metabolomic tissue signature in human non-alcoholic fatty liver disease identifies protective candidate metabolites. Liver Int. 2015, 35, 207–214. [Google Scholar] [CrossRef]
- Poeta, M.; Pierri, L.; Vajro, P. Gut–Liver Axis Derangement in Non-Alcoholic Fatty Liver Disease. Children 2017, 4, 66. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Liu, Y.; Long, J.; Chen, S.; Liao, G.; Wu, S.; Li, C.; Wang, L.; Ling, W.; Zhu, H. Trimethylamine N-Oxide Aggravates Liver Steatosis through Modulation of Bile Acid Metabolism and Inhibition of Farnesoid X Receptor Signaling in Nonalcoholic Fatty Liver Disease. Mol. Nutr. Food Res. 2019, 63, e1900257. [Google Scholar] [CrossRef]
- Barrea, L.; Annunziata, G.; Muscogiuri, G.; Di Somma, C.; Laudisio, D.; Maisto, M.; De Alteriis, G.; Tenore, G.C.; Colao, A.; Savastano, S. Trimethylamine-N-oxide (TMAO) as Novel Potential Biomarker of Early Predictors of Metabolic Syndrome. Nutrients 2018, 10, 1971. [Google Scholar] [CrossRef] [PubMed]
- León-Mimila, P.; Villamil-Ramírez, H.; Li, X.S.; Shih, D.M.; Hui, S.T.; Ocampo-Medina, E.; López-Contreras, B.; Morán-Ramos, S.; Olivares-Arevalo, M.; Grandini-Rosales, P.; et al. Trimethylamine N-oxide levels are associated with NASH in obese subjects with type 2 diabetes. Diabetes Metab. 2021, 47, 101183. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, Z.; Li, H.; Zhao, J.; Wei, X.; Lin, W.; Zhao, X.; Jiang, A.; Yuan, J. Endogenous ethanol produced by intestinal bacteria induces mitochondrial dysfunction in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2020, 35, 2009–2019. [Google Scholar] [CrossRef]
- Aljomah, G.; Baker, S.S.; Liu, W.; Kozielski, R.; Oluwole, J.; Lupu, B.; Baker, R.D.; Zhu, L. Induction of CYP2E1 in non-alcoholic fatty liver diseases. Exp. Mol. Pathol. 2015, 99, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Bovi, A.P.D.; Marciano, F.; Mandato, C.; Siano, M.A.; Savoia, M.; Vajro, P. Oxidative Stress in Non-alcoholic Fatty Liver Disease. An Updated Mini Review. Front. Med. 2021, 8, 595371. [Google Scholar] [CrossRef] [PubMed]
- Hrncir, T.; Hrncirova, L.; Kverka, M.; Hromadka, R.; Machova, V.; Trckova, E.; Kostovcikova, K.; Kralickova, P.; Krejsek, J.; Tlaskalova-Hogenova, H. Gut Microbiota and NAFLD: Pathogenetic Mechanisms, Microbiota Signatures, and Therapeutic Interventions. Microorganisms 2021, 9, 957. [Google Scholar] [CrossRef] [PubMed]
- Rakhra, V.; Galappaththy, S.L.; Bulchandani, S.; Cabandugama, P.K. Obesity and the Western Diet: How We Got Here. Momed 2020, 117, 536–538. [Google Scholar]
- Simoes, I.C.M.; Karkucinska-Wieckowska, A.; Janikiewicz, J.; Szymanska, S.; Pronicki, M.; Dobrzyn, P.; Dabrowski, M.; Do-brzyn, A.; Oliveira, P.J.; Zischka, H.; et al. Western Diet Causes Obesity-Induced Nonalcoholic Fatty Liver Disease Development by Differentially Compromising the Autophagic Response. Antioxidants 2020, 9, 995. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Long, J.; Li, W.; Yang, C.; Loughran, P.; O’doherty, R.; Billiar, T.R.; Deng, M.; Scott, M.J. Hepatocyte high-mobility group box 1 protects against steatosis and cellular stress during high fat diet feeding. Mol. Med. 2020, 26, 115. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, I.A.; Mikail, M.A.; Mustafa, M.R.; Ibrahim, M.; Othman, R. Lifestyle interventions for non-alcoholic fatty liver disease. Saudi J. Biol. Sci. 2019, 26, 1519–1524. [Google Scholar] [CrossRef]
- Torres, M.C.P.; Aghemo, A.; Lleo, A.; Bodini, G.; Furnari, M.; Marabotto, E.; Miele, L.; Giannini, E.G. Mediterranean Diet and NAFLD: What We Know and Questions That Still Need to Be Answered. Nutrients 2019, 11, 2971. [Google Scholar] [CrossRef]
- Pintó, X.; Fanlo-Maresma, M.; Corbella, E.; Corbella, X.; Mitjavila, M.T.; Moreno, J.J.; Casas, R.; Estruch, R.; Corella, D.; Bulló, M.; et al. A Mediterranean Diet Rich in Extra-Virgin Olive Oil Is Associated with a Reduced Prevalence of Nonalcoholic Fatty Liver Disease in Older Individuals at High Cardiovascular Risk. J. Nutr. 2019, 149, 1920–1929. [Google Scholar] [CrossRef]
- Samadi, M.; Moradinazar, M.; Khosravy, T.; Soleimani, D.; Jahangiri, P.; Kamari, N. A systematic review and meta-analysis of preclinical and clinical studies on the efficacy of ginger for the treatment of fatty liver disease. Phytother. Res. 2022, 36, 1182–1193. [Google Scholar] [CrossRef]
- Gillessen, A.; Schmidt, H.H.-J. Silymarin as Supportive Treatment in Liver Diseases: A Narrative Review. Adv. Ther. 2020, 37, 1279–1301. [Google Scholar] [CrossRef] [PubMed]
- Saleh, S.A.; Santos, H.O.; Găman, M.-A.; Cerqueira, H.S.; Zaher, E.A.; Alromaih, W.R.; Arafat, N.S.; Adi, A.R.; Adly, H.M.; Alyoubi, R.; et al. Effects of intermittent fasting regimens on glycemic, hepatic, anthropometric, and clinical markers in patients with non-alcoholic fatty liver disease: Systematic review and meta-analysis of randomized controlled trials. Clin. Nutr. ESPEN 2024, 59, 70–80. [Google Scholar] [CrossRef]
- Spahis, S.; Delvin, E.; Borys, J.-M.; Levy, E. Oxidative Stress as a Critical Factor in Nonalcoholic Fatty Liver Disease Pathogenesis. Antioxidants Redox Signal. 2017, 26, 519–541. [Google Scholar] [CrossRef]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef] [PubMed]
- Grohmann, M.; Wiede, F.; Dodd, G.T.; Gurzov, E.N.; Ooi, G.J.; Butt, T.; Rasmiena, A.A.; Kaur, S.; Gulati, T.; Goh, P.K.; et al. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 2018, 175, 1289–1306.e20. [Google Scholar] [CrossRef]
- Tsung, A.; Klune, J.R.; Zhang, X.; Jeyabalan, G.; Cao, Z.; Peng, X.; Stolz, D.B.; Geller, D.A.; Rosengart, M.R.; Billiar, T.R. HMGB1 release induced by liver ischemia involves Toll-like receptor 4–dependent reactive oxygen species production and calcium-mediated signaling. J. Exp. Med. 2007, 204, 2913–2923. [Google Scholar] [CrossRef]
- Xu, L.; Ge, F.; Hu, Y.; Yu, Y.; Guo, K.; Miao, C. Sevoflurane Postconditioning Attenuates Hepatic Ischemia-Reperfusion Injury by Limiting HMGB1/TLR4/NF-κB Pathway via Modulating microRNA-142 in vivo and in vitro. Front. Pharmacol. 2021, 12, 646307. [Google Scholar] [CrossRef] [PubMed]
- Rector, R.S.; Thyfault, J.P.; Uptergrove, G.M.; Morris, E.M.; Naples, S.P.; Borengasser, S.J.; Mikus, C.R.; Laye, M.J.; Laughlin, M.H.; Booth, F.W.; et al. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J. Hepatol. 2010, 52, 727–736. [Google Scholar] [CrossRef]
- Kim, J.-S.; Wang, J.-H.; Lemasters, J.J. Mitochondrial permeability transition in rat hepatocytes after anoxia/reoxygenation: Role of Ca2+-dependent mitochondrial formation of reactive oxygen species. Am. J. Physiol. Gastrointest Liver Physiol. 2012, 302, G723–G731. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ros) and ros-induced ros release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Tang, S.-P.; Mao, X.-L.; Chen, Y.-H.; Yan, L.-L.; Ye, L.-P.; Li, S.-W. Reactive Oxygen Species Induce Fatty Liver and Ischemia-Reperfusion Injury by Promoting Inflammation and Cell Death. Front. Immunol. 2022, 13, 870239. [Google Scholar] [CrossRef]
- Mirhafez, S.R.; Farimani, A.R.; Gholami, A.; Hooshmand, E.; Tavallaie, S.; Gh, B.F.N.M. The effect of curcumin with piperine supplementation on pro-oxidant and antioxidant balance in patients with non-alcoholic fatty liver disease: A randomized, double-blind, placebo-controlled trial. Drug Metab. Pers. Ther. 2019, 34, 30. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Wu, L.; Yu, M.; Chen, F.-J.; Arshad, M.; Xia, X.; Ren, H.; Yu, J.; Xu, L.; Xu, D.; et al. Differential Roles of Cell Death-inducing DNA Fragmentation Factor-α-like Effector (CIDE) Proteins in Promoting Lipid Droplet Fusion and Growth in Subpopulations of Hepatocytes. J. Biol. Chem. 2016, 291, 4282–4293. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Yin, Y.; Chua, B.T.; Li, P. CIDE family proteins control lipid homeostasis and the development of metabolic diseases. Traffic 2020, 21, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Verweij, N.; Haas, M.E.; Nielsen, J.B.; Sosina, O.A.; Kim, M.; Akbari, P.; De, T.; Hindy, G.; Bovijn, J.; Persaud, T.; et al. Germline Mutations in CIDEB and Protection against Liver Disease. N. Engl. J. Med. 2022, 387, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.C. A closer look at the mysterious HSD17B13. J. Lipid Res. 2020, 61, 1361–1362. [Google Scholar] [CrossRef] [PubMed]
- Sookoian, S.; Pirola, C.J.; Valenti, L.; Davidson, N.O. Genetic Pathways in Nonalcoholic Fatty Liver Disease: Insights From Systems Biology. Hepatology 2020, 72, 330–346. [Google Scholar] [CrossRef] [PubMed]
- Juanola, O.; Martínez-López, S.; Francés, R.; Gómez-Hurtado, I. Non-Alcoholic Fatty Liver Disease: Metabolic, Genetic, Epigenetic and Environmental Risk Factors. Int. J. Environ. Res. Public Health 2021, 18, 5227. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Romeo, S.; Valenti, L. Genetic Factors in the Pathogenesis of Nonalcoholic Fatty Liver and Steatohepatitis. BioMed Res. Int. 2015, 2015, 460190. [Google Scholar] [CrossRef]
- Luo, F.; Oldoni, F.; Das, A. TM6SF2: A Novel Genetic Player in Nonalcoholic Fatty Liver and Cardiovascular Disease. Hepatol. Commun. 2022, 6, 448–460. [Google Scholar] [CrossRef]
- Helsley, R.N.; Varadharajan, V.; Brown, A.L.; Gromovsky, A.D.; Schugar, R.C.; Ramachandiran, I.; Fung, K.; Kabbany, M.N.; Banerjee, R.; Neumann, C.K.; et al. Obesity-linked suppression of membrane-bound O-acyltransferase 7 (MBOAT7) drives non-alcoholic fatty liver disease. eLife 2019, 8, e49882. [Google Scholar] [CrossRef]
- Wang, J.; Ye, C.; Fei, S. Association between APOC3 polymorphisms and non-alcoholic fatty liver disease risk: A meta-analysis. Afr. Health Sci. 2020, 20, 1800–1808. [Google Scholar] [CrossRef]
- Harjumäki, R.; Pridgeon, C.S.; Ingelman-Sundberg, M. CYP2E1 in Alcoholic and Non-Alcoholic Liver Injury. Roles of ROS, Reactive Intermediates and Lipid Overload. Int. J. Mol. Sci. 2021, 22, 8221. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Banerjee, A.; Yoo, S.-H.; Jang, S.; Gonzalez, F.J.; Song, B.-J. Critical role of cytochrome P450 2E1 (CYP2E1) in the development of high fat-induced non-alcoholic steatohepatitis. J. Hepatol. 2012, 57, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Leung, T.-M.; Nieto, N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Kathirvel, E.; Morgan, K.; French, S.W.; Morgan, T.R. Overexpression of liver-specific cytochrome P4502E1 impairs hepatic insulin signaling in a transgenic mouse model of nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 2009, 21, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Correia, M.A.; Kwon, D. Why Hepatic CYP2E1-Elevation by Itself Is Insufficient for Inciting NAFLD/NASH: Inferences from Two Genetic Knockout Mouse Models. Biology 2020, 9, 419. [Google Scholar] [CrossRef] [PubMed]
- Bae, C.-S.; Lee, Y.; Ahn, T. Therapeutic treatments for diabetes mellitus-induced liver injury by regulating oxidative stress and inflammation. Appl. Microsc. 2023, 53, 4. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, D.; Finck, B.N. Emerging therapeutic approaches for the treatment of NAFLD and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2021, 17, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.K.; Yang, H.; Moylan, C.A.; Pang, H.; Dellinger, A.; Abdelmalek, M.F.; Garrett, M.E.; Ashley–Koch, A.; Suzuki, A.; Tillmann, H.L.; et al. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 1076–1087. [Google Scholar] [CrossRef]
- Zeybel, M.; Hardy, T.; Robinson, S.M.; Fox, C.; Anstee, Q.M.; Ness, T.; Masson, S.; Mathers, J.C.; French, J.; White, S.; et al. Differential DNA methylation of genes involved in fibrosis progression in non-alcoholic fatty liver disease and alcoholic liver disease. Clin. Epigenetics 2015, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tang, Y.; Chen, S.; Ling, W.; Wang, Q. IGFBP-2 as a biomarker in NAFLD improves hepatic steatosis: An integrated bioinformatics and experimental study. Endocr. Connect. 2021, 10, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Valenti, L.; Rametta, R.; Daly, A.K.; Nobili, V.; Mozzi, E.; Leathart, J.B.S.; Pietrobattista, A.; Burt, A.D.; Maggioni, M.; et al. Genetic variants regulating insulin receptor signalling are associated with the severity of liver damage in patients with non-alcoholic fatty liver disease. Gut 2010, 59, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Ding, R.-B.; Bao, J.; Deng, C.-X. Emerging roles of SIRT1 in fatty liver diseases. Int. J. Biol. Sci. 2017, 13, 852–867. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Dou, G.; Wang, L. MicroRNAs in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Int. J. Biol. Sci. 2021, 17, 1851–1863. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhu, Y.; Hu, S.; Pan, X.; Bawa, F.C.; Wang, H.H.; Wang, D.Q.-H.; Yin, L.; Zhang, Y. Hepatocyte miR-34a is a key regulator in the development and progression of non-alcoholic fatty liver disease. Mol. Metab. 2021, 51, 101244. [Google Scholar] [CrossRef]
- Zhang, T.; Yang, Z.; Kusumanchi, P.; Han, S.; Liangpunsakul, S. Critical Role of microRNA-21 in the Pathogenesis of Liver Diseases. Front. Med. 2020, 7, 7. [Google Scholar] [CrossRef]
- Bala, S.; Ganz, M.; Babuta, M.; Zhuang, Y.; Csak, T.; Calenda, C.D.; Szabo, G. Steatosis, inflammasome upregulation, and fibrosis are attenuated in miR-155 deficient mice in a high fat-cholesterol-sugar diet-induced model of NASH. Lab. Investig. 2021, 101, 1540–1549. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Yang, Y.-L.; Wang, P.-W.; Wang, F.-S.; Huang, Y.-H. The Emerging Role of MicroRNAs in NAFLD: Highlight of MicroRNA-29a in Modulating Oxidative Stress, Inflammation, and Beyond. Cells 2020, 9, 1041. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-L.; Wang, P.-W.; Wang, F.-S.; Lin, H.-Y.; Huang, Y.-H. miR-29a Modulates GSK3β/SIRT1-Linked Mitochondrial Proteostatic Stress to Ameliorate Mouse Non-Alcoholic Steatohepatitis. Int. J. Mol. Sci. 2020, 21, 6884. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Zhou, C.; Yang, X.; Li, L. Down-regulation of microRNA-375 regulates adipokines and inhibits inflammatory cytokines by targeting AdipoR2 in non-alcoholic fatty liver disease. Clin. Exp. Pharmacol. Physiol. 2018, 45, 819–831. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef]
- Gupta, N.; Liu, J.R.; Patel, B.; Solomon, D.E.; Vaidya, B.; Gupta, V. Microfluidics-based 3D cell culture models: Utility in novel drug discovery and delivery research. Bioeng. Transl. Med. 2016, 1, 63–81. [Google Scholar] [CrossRef] [PubMed]
- Han, D.W.; Xu, K.H.; Jin, Z.-L.; Xu, Y.-N.; Li, Y.-H.; Wang, L.; Cao, Q.; Kim, K.-P.; Ryu, D.H.; Hong, K.; et al. Customized liver organoids as an advanced in vitro modeling and drug discovery platform for non-alcoholic fatty liver diseases. Int. J. Biol. Sci. 2023, 19, 3595–3613. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Hu, H.; Kung, H.; Zou, R.; Dai, Y.; Hu, Y.; Wang, T.; Lv, T.; Yu, J.; Li, F. Organoids: The current status and biomedical applications. Medcomm 2023, 4, e274. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Chen, K.G.; Mallon, B.S.; McKay, R.D.G.; Robey, P.G. Human pluripotent stem cell culture: Considerations for maintenance, expansion, and therapeutics. Cell Stem Cell 2014, 14, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Günther, C.; Winner, B.; Neurath, M.F.; Stappenbeck, T.S. Organoids in gastrointestinal diseases: From experimental models to clinical translation. Gut 2022, 71, 1892–1908. [Google Scholar] [CrossRef]
- Schutgens, F.; Clevers, H. Human Organoids: Tools for Understanding Biology and Treating Diseases. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 211–234. [Google Scholar] [CrossRef]
- Wakamatsu, T.; Ogawa, H.; Yoshida, K.; Matsuoka, Y.; Shizuma, K.; Imura, Y.; Tamiya, H.; Nakai, S.; Yagi, T.; Nagata, S.; et al. Establishment of Organoids from Human Epithelioid Sarcoma with the Air-Liquid Interface Organoid Cultures. Front. Oncol. 2022, 12, 893592. [Google Scholar] [CrossRef]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.-H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988.e16. [Google Scholar] [CrossRef] [PubMed]
- Mittal, R.; Woo, F.W.; Castro, C.S.; Cohen, M.A.; Karanxha, J.; Mittal, J.; Chhibber, T.; Jhaveri, V.M. Organ-on-chip models: Implications in drug discovery and clinical applications. J. Cell Physiol. 2019, 234, 8352–8380. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Seiler, M.J.; Tang, W.C.; Wang, J.Y.; Delgado, J.; McLelland, B.T.; Nistor, G.; Keirstead, H.S.; Browne, A.W. Retinal organoids on-a-chip: A micro-millifluidic bioreactor for long-term organoid maintenance. Lab a Chip 2021, 21, 3361–3377. [Google Scholar] [CrossRef] [PubMed]
- Brevini, T.; Tysoe, O.C.; Sampaziotis, F. Tissue engineering of the biliary tract and modelling of cholestatic disorders. J. Hepatol. 2020, 73, 918–932. [Google Scholar] [CrossRef] [PubMed]
- Lamers, M.M.; van der Vaart, J.; Knoops, K.; Riesebosch, S.; Breugem, T.I.; Mykytyn, A.Z.; Beumer, J.; Schipper, D.; Bezstarosti, K.; Koopman, C.D.; et al. An organoid-derived bronchioalveolar model for SARS-CoV-2 infection of human alveolar type II-like cells. EMBO J. 2021, 40, e105912. [Google Scholar] [CrossRef]
- Cho, A.-N.; Jin, Y.; An, Y.; Kim, J.; Choi, Y.S.; Lee, J.S.; Kim, J.; Choi, W.-Y.; Koo, D.-J.; Yu, W.; et al. Microfluidic device with brain extracellular matrix promotes structural and functional maturation of human brain organoids. Nat. Commun. 2021, 12, 4730. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Qu, K.-Y.; Zhou, B.; Luo, Y.; Zhu, Z.; Pan, D.-J.; Cui, C.; Zhu, Y.; Chen, M.-L.; Huang, N.-P. Design and fabrication of an integrated heart-on-a-chip platform for construction of cardiac tissue from human iPSC-derived cardiomyocytes and in situ evaluation of physiological function. Biosens. Bioelectron. 2021, 179, 113080. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.C.; Shin, W.; Koh, D.; Wu, A.; Ambrosini, Y.M.; Min, S.; Eckhardt, S.G.; Fleming, R.Y.D.; Kim, S.; Park, S.; et al. Three-Dimensional Regeneration of Patient-Derived Intestinal Organoid Epithelium in a Physiodynamic Mucosal Interface-on-a-Chip. Micromachines 2020, 11, 663. [Google Scholar] [CrossRef]
- Wiedenmann, S.; Breunig, M.; Merkle, J.; von Toerne, C.; Georgiev, T.; Moussus, M.; Schulte, L.; Seufferlein, T.; Sterr, M.; Lickert, H.; et al. Single-cell-resolved differentiation of human induced pluripotent stem cells into pancreatic duct-like organoids on a microwell chip. Nat. Biomed. Eng. 2021, 5, 897–913. [Google Scholar] [CrossRef]
- Zhang, L.; Avery, J.; Yin, A.; Singh, A.M.; Cliff, T.S.; Yin, H.; Dalton, S. Generation of Functional Brown Adipocytes from Human Pluripotent Stem Cells via Progression through a Paraxial Mesoderm State. Cell Stem Cell 2020, 27, 784–797.e11. [Google Scholar] [CrossRef]
- Georgakopoulos, N.; Prior, N.; Angres, B.; Mastrogiovanni, G.; Cagan, A.; Harrison, D.; Hindley, C.J.; Arnes-Benito, R.; Liau, S.-S.; Curd, A.; et al. Long-term expansion, genomic stability and in vivo safety of adult human pancreas organoids. BMC Dev. Biol. 2020, 20, 4. [Google Scholar] [CrossRef]
- Park, Y.; Thadasina, D.; Bolujo, I.; Isidan, A.; Cross-Najafi, A.A.; Lopez, K.; Li, P.; Dahlem, A.M.; Kennedy, L.; Sato, K.; et al. Three-Dimensional Organoids as a Model to Study Nonalcoholic Fatty Liver Disease. Semin. Liver Dis. 2022, 42, 423–433. [Google Scholar] [CrossRef]
- Teng, Y.; Zhao, Z.; Tasnim, F.; Huang, X.; Yu, H. A scalable and sensitive steatosis chip with long-term perfusion of in situ differentiated HepaRG organoids. Biomaterials 2021, 275, 120904. [Google Scholar] [CrossRef] [PubMed]
- Sanders, F.W.B.; Griffin, J.L. De novo lipogenesis in the liver in health and disease: More than just a shunting yard for glucose. Biol. Rev. Camb. Philos. Soc. 2016, 91, 452–468. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Gehart, H.; van Boxtel, R.; Hamer, K.; Blokzijl, F.; Verstegen, M.M.; Ellis, E.; van Wenum, M.; Fuchs, S.A.; de Ligt, J.; et al. Long-Term culture of genome-stable bipotent stem cells from adult human liver. Cell 2015, 160, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Gehart, H.; Artegiani, B.; Löpez-Iglesias, C.; Dekkers, F.; Basak, O.; Van Es, J.; Chuva de Sousa Lopes, S.M.; Begthel, H.; Korving, J.; et al. Long-Term Expansion of Functional Mouse and Human Hepatocytes as 3D Organoids. Cell 2018, 175, 1591–1606.e19. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yan, J.; Wang, H.; Shi, M.; Zhang, M.; Yang, W.; Peng, C.; Li, H. Rapamycin ameliorates inflammation and fibrosis in the early phase of cirrhotic portal hypertension in rats through inhibition of mTORC1 but not mTORC2. PLoS ONE 2014, 9, e83908. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Wang, F.; Zhai, D.; Meng, X.; Liu, J.; Lv, X. New Drugs for Hepatic Fibrosis. Front. Pharmacol. 2022, 13, 874408. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, D.; Brouwers, J.F.; Hamer, K.; Geurts, M.H.; Luciana, L.; Massalini, S.; López-Iglesias, C.; Peters, P.J.; Rodríguez-Colman, M.J.; Lopes, S.C.d.S.; et al. Engineered human hepatocyte organoids enable CRISPR-based target discovery and drug screening for steatosis. Nat. Biotechnol. 2023, 41, 1567–1581. [Google Scholar] [CrossRef]
- Aisenbrey, E.A.; Murphy, W.L. Synthetic alternatives to Matrigel. Nat. Rev. Mater. 2020, 5, 539–551. [Google Scholar] [CrossRef]
- Lonardo, A.; Nascimbeni, F.; Mantovani, A.; Targher, G. Hypertension, diabetes, atherosclerosis and NASH: Cause or consequence? J. Hepatol. 2018, 68, 335–352. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.-S.; Kim, D. Non-alcoholic fatty liver disease and lifestyle modifications, focusing on physical activity. Korean J. Intern. Med. 2018, 33, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Zelber-Sagi, S.; Henry, L.; Gerber, L.H. Lifestyle interventions in nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 708–722. [Google Scholar] [CrossRef] [PubMed]
- Fernández, T.; Viñuela, M.; Vidal, C.; Barrera, F. Lifestyle changes in patients with non-alcoholic fatty liver disease: A systematic review and meta-analysis. PLoS ONE 2022, 17, e0263931. [Google Scholar] [CrossRef] [PubMed]
- Geerts, A.; Lefere, S. Bariatric surgery for non-alcoholic fatty liver disease: Indications and post-operative management. Clin. Mol. Hepatol. 2023, 29, S276–S285. [Google Scholar] [CrossRef] [PubMed]
- Głuszyńska, P.; Lemancewicz, D.; Dzięcioł, J.B.; Hady, H.R. Non-Alcoholic Fatty Liver Disease (NAFLD) and Bariatric/Metabolic Surgery as Its Treatment Option: A Review. J. Clin. Med. 2021, 10, 5721. [Google Scholar] [CrossRef] [PubMed]
- Battistella, S.; D’arcangelo, F.; Grasso, M.; Zanetto, A.; Gambato, M.; Germani, G.; Senzolo, M.; Russo, F.P.; Burra, P. Liver transplantation for non-alcoholic fatty liver disease: Indications and post-transplant management. Clin. Mol. Hepatol. 2023, 29, S286–S301. [Google Scholar] [CrossRef] [PubMed]
- Shetty, A.; Giron, F.; Divatia, M.K.; Ahmad, M.I.; Kodali, S.; Victor, D. Nonalcoholic Fatty Liver Disease after Liver Transplant. J. Clin. Transl. Hepatol. 2021, 9, 428–435. [Google Scholar] [CrossRef]
- Prikhodko, V.A.; Bezborodkina, N.N.; Okovityi, S.V. Pharmacotherapy for Non-Alcoholic Fatty Liver Disease: Emerging Targets and Drug Candidates. Biomedicines 2022, 10, 274. [Google Scholar] [CrossRef]
- Rong, L.; Zou, J.; Ran, W.; Qi, X.; Chen, Y.; Cui, H.; Guo, J. Advancements in the treatment of non-alcoholic fatty liver disease (NAFLD). Front. Endocrinol. 2023, 13, 1087260. [Google Scholar] [CrossRef]
- Vogli, S.; Naska, A.; Marinos, G.; Kasdagli, M.-I.; Orfanos, P. The Effect of Vitamin E Supplementation on Serum Aminotransferases in Non-Alcoholic Fatty Liver Disease (NAFLD): A Systematic Review and Meta-Analysis. Nutrients 2023, 15, 3733. [Google Scholar] [CrossRef]
- Boeckmans, J.; Natale, A.; Rombaut, M.; Buyl, K.; Rogiers, V.; De Kock, J.; Vanhaecke, T.; Rodrigues, R.M. Anti-NASH Drug Development Hitches a Lift on PPAR Agonism. Cells 2019, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Sumida, Y.; Yoneda, M.; Tokushige, K.; Kawanaka, M.; Fujii, H.; Yoneda, M.; Imajo, K.; Takahashi, H.; Ono, M.; Nozaki, Y.; et al. Hepatoprotective Effect of SGLT2 Inhibitor on Nonalcoholic Fatty Liver Disease. Diabetes Res. Open Access 2020, 2, 17–25. [Google Scholar] [CrossRef]
- Gu, Y.; Sun, L.; Zhang, W.; Kong, T.; Zhou, R.; He, Y.; Deng, C.; Yang, L.; Kong, J.; Chen, Y.; et al. Comparative efficacy of 5 sodium-glucose cotransporter protein-2 (SGLT-2) inhibitor and 4 glucagon-like peptide-1 (GLP-1) receptor agonist drugs in non-alcoholic fatty liver disease: A GRADE-assessed systematic review and network meta-analysis of randomized controlled trials. Front. Pharmacol. 2023, 14, 1102792. [Google Scholar] [CrossRef]
- Dawson, P.A.; Parini, P. Hepatic thyroid hormone receptor β1 agonism: Good for lipids, good for bile? J. Lipid Res. 2018, 59, 1551–1553. [Google Scholar] [CrossRef]
- Harrison, S.A.; Bashir, M.; Moussa, S.E.; McCarty, K.; Frias, J.P.; Taub, R.; Alkhouri, N. Effects of Resmetirom on Noninvasive Endpoints in a 36-Week Phase 2 Active Treatment Extension Study in Patients With NASH. Hepatol. Commun. 2021, 5, 573–588. [Google Scholar] [CrossRef]
- Qi, J.; Lang, W.; Geisler, J.G.; Wang, P.; Petrounia, I.; Mai, S.; Smith, C.; Askari, H.; Struble, G.T.; Williams, R.; et al. The use of stable isotope-labeled glycerol and oleic acid to differentiate the hepatic functions of DGAT1 and -2. J. Lipid Res. 2012, 53, 1106–1116. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Yang, L.; McCall, S.; Huang, J.; Yu, X.X.; Pandey, S.K.; Bhanot, S.; Monia, B.P.; Li, Y.-X.; Diehl, A.M. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007, 45, 1366–1374. [Google Scholar] [CrossRef]
- Loomba, R.; Noureddin, M.; Kowdley, K.V.; Kohli, A.; Sheikh, A.; Neff, G.; Bhandari, B.R.; Gunn, N.; Caldwell, S.H.; Goodman, Z.; et al. Combination Therapies Including Cilofexor and Firsocostat for Bridging Fibrosis and Cirrhosis Attributable to NASH. Hepatology 2021, 73, 625–643. [Google Scholar] [CrossRef]
- Calle, R.A.; Amin, N.B.; Carvajal-Gonzalez, S.; Ross, T.T.; Bergman, A.; Aggarwal, S.; Crowley, C.; Rinaldi, A.; Mancuso, J.; Aggarwal, N.; et al. ACC inhibitor alone or co-administered with a DGAT2 inhibitor in patients with non-alcoholic fatty liver disease: Two parallel, placebo-controlled, randomized phase 2a trials. Nat. Med. 2021, 27, 1836–1848. [Google Scholar] [CrossRef]
- Syed-Abdul, M.M.; Parks, E.J.; Gaballah, A.H.; Bingham, K.; Hammoud, G.M.; Kemble, G.; Buckley, D.; McCulloch, W.; Manrique-Acevedo, C. Fatty Acid Synthase Inhibitor TVB-2640 Reduces Hepatic de Novo Lipogenesis in Males with Metabolic Abnormalities. Hepatology 2020, 72, 103–118. [Google Scholar] [CrossRef]
- Ratziu, V.; de Guevara, L.; Safadi, R.; Poordad, F.; Fuster, F.; Flores-Figueroa, J.; Arrese, M.; Fracanzani, A.L.; Ben Bashat, D.; Lackner, K.; et al. Aramchol in patients with nonalcoholic steatohepatitis: A randomized, double-blind, placebo-controlled phase 2b trial. Nat. Med. 2021, 27, 1825–1835. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Morgan, E.; Watts, L.; Xia, S.; Hannan, L.A.; Geary, R.S.; Baker, B.F.; Bhanot, S. Novel antisense inhibition of diacylglycerol O-acyltransferase 2 for treatment of non-alcoholic fatty liver disease: A multicentre, double-blind, randomised, placebo-controlled phase 2 trial. Lancet Gastroenterol. Hepatol. 2020, 5, 829–838. [Google Scholar] [CrossRef]
- Dentin, R.; Benhamed, F.; Pégorier, J.-P.; Foufelle, F.; Viollet, B.; Vaulont, S.; Girard, J.; Postic, C. Polyunsaturated fatty acids suppress glycolytic and lipogenic genes through the inhibition of ChREBP nuclear protein translocation. J. Clin. Investig. 2005, 115, 2843–2854. [Google Scholar] [CrossRef]
- Climax, J.; Newsome, P.N.; Hamza, M.; Weissbach, M.; Coughlan, D.; Sattar, N.; McGuire, D.K.; Bhatt, D.L. Effects of Epeleuton, a Novel Synthetic Second-Generation n-3 Fatty Acid, on Non-Alcoholic Fatty Liver Disease, Triglycerides, Glycemic Control, and Cardiometabolic and Inflammatory Markers. J. Am. Heart Assoc. 2020, 9, e016334. [Google Scholar] [CrossRef]
- Jiang, L.; Zhang, H.; Xiao, D.; Wei, H.; Chen, Y. Farnesoid X receptor (FXR): Structures and ligands. Comput. Struct. Biotechnol. J. 2021, 19, 2148–2159. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef]
- Roth, J.D.; Feigh, M.; Veidal, S.S.; Fensholdt, L.K.; Rigbolt, K.T.; Hansen, H.H.; Chen, L.C.; Petitjean, M.; Friley, W.; Vrang, N.; et al. INT-767 improves histopathological features in a diet-induced ob/ob mouse model of biopsy-confirmed non-alcoholic steatohepatitis. World J. Gastroenterol. 2018, 24, 195–210. [Google Scholar] [CrossRef]
- Harrison, S.A.; Abdelmalek, M.F.; Neff, G.W.; Gunn, N.; Guy, C.D.; Alkhouri, N.; Bashir, M.; Freilich, B.; Almeda, J.; Knapple, W.; et al. Topline Results from the ALPINE 2/3 Study: A Randomized, Double-Blind, Placebo-Controlled, Multicenter, Phase 2b Trial Evaluating 3 Doses of the FGF19 Analogue Aldafermin on Liver Histology in Patients with Nonalcoholic Steatohepatitis and Stage 2 or 3 Fibrosis. Hepatology 2021, 74, 5A. [Google Scholar]
- Sun, M.-J.; Cao, Z.-Q.; Leng, P. The roles of galectins in hepatic diseases. J. Mol. Histol. 2020, 51, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Abdelmalek, M.F.; Garcia-Tsao, G.; Vuppalanchi, R.; Alkhouri, N.; Rinella, M.; Noureddin, M.; Pyko, M.; Shiffman, M.; Sanyal, A.; et al. Effects of Belapectin, an Inhibitor of Galectin-3, in Patients with Nonalcoholic Steatohepatitis with Cirrhosis and Portal Hypertension. Gastroenterology 2020, 158, 1334–1345.e5. [Google Scholar] [CrossRef]
- CymaBay Therapeutics CymaBay Therapeutics Reports Topline 12-Week Data from an Ongoing Phase 2b Study of Seladelpar in Patients with Nonalcoholic Steatohepatitis. 2019. Available online: https://www.globenewswire.com/news-release/2019/06/11/1866763/0/en/CymaBay-Therapeutics-Reports-Topline-12-Week-Data-from-an-Ongoing-Phase-2b-Study-of-Seladelpar-in-Patients-with-Nonalcoholic-Steatohepatitis.html. (accessed on 21 January 2024).
- Goyal, O.; Nohria, S.; Goyal, P.; Kaur, J.; Sharma, S.; Sood, A.; Chhina, R.S. Saroglitazar in patients with non-alcoholic fatty liver disease and diabetic dyslipidemia: A prospective, observational, real world study. Sci. Rep. 2020, 10, 21117. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Section | Subsection | Focus Area |
|---|---|---|
| 1. Introduction | - | Overview of MASLD |
| 2. Pathophysiology | - | Mechanisms behind MASLD |
| 3. Risk Factors | 3.1. Cardiovascular Disease | Link between MASLD and cardiovascular health |
| 3.2. Dyslipidemia | Impact of abnormal lipid levels on MASLD | |
| 3.3. Type II Diabetes Mellitus | Connection between diabetes and MASLD | |
| 3.4. Obesity | Role of obesity in MASLD development | |
| 3.5. Iron Overload | Influence of iron on MASLD | |
| 3.6. Galactosemia | Association of galactosemia with MASLD | |
| 3.7. Alpha-1-Antitrypsin | A1AT deficiency’s effect on MASLD | |
| 3.8. Glycogen Storage Diseases | GSDs and their contribution to MASLD | |
| 3.9. Viral Hepatitis | Hepatitis and MASLD relationship | |
| 3.10. Wilson’s Disease | Wilson’s Disease and its association with MASLD | |
| 3.11. Cystic Fibrosis | CFLD’s relationship with MASLD | |
| 3.12. Leukocyte Telomere Length | Leukocyte TL in relation to MASLD development | |
| 3.13. Smoking | Smoking as a risk factor for MASLD | |
| 4. Dietary Effect on Fatty Acid Metabolism | 4.1. Gut Microbiome | Gut microbiome’s impact on MASLD |
| 4.2. Gut–Liver Axis | Interaction between gut and liver in MASLD | |
| 4.3. Western Diet/Fatty Diet | Influence of diet on MASLD | |
| 4.4. Mediterranean Diet | Benefits of this diet for MASLD | |
| 4.5. Intermittent Fasting | IF’s effect on MASLD | |
| 5. Genetic Pathways Related to MASLD | 5.1. Oxidative Stress | Role of oxidative stress in MASLD |
| 5.2. Genetic Mutations | Key genetic factors in MASLD | |
| 5.3. Epigenetics | Epigenetic influences on MASLD | |
| 5.4. MicroRNA Posttranscriptional Regulation | miRNA’s role in MASLD | |
| 6. The Future with Organoids | - | Application of organoids in MASLD research |
| 7. Therapy | 7.1. Lifestyle | Lifestyle interventions for MASLD |
| 7.2. Bariatric Surgery | Surgical options for MASLD treatment | |
| 7.3. Transplant Surgery | Liver transplantation in MASLD management | |
| 7.4. Pharmacology | Drug treatments for MASLD | |
| 7.5. Vitamin E | Role of vitamin E in MASLD treatment | |
| 7.6. Glucose Metabolism Modulators | Medications affecting glucose metabolism | |
| 7.7. GLP-1 Receptor Agonist | GLP-1’s therapeutic potential in MASLD | |
| 7.8. THRβ | THRβ agonists in MASLD treatment | |
| 7.9. Lipogenesis Inhibitors | Targeting lipogenesis in MASLD | |
| 7.10. ACC, FASN, SCDI, DGAT Inhibitors and PUFA | Role of inhibitors in MASLD treatment | |
| 7.11. Bile Acid Metabolism Modulators | Bile acid’s impact on MASLD | |
| 7.12. Fibrogenesis Inhibitors | Inhibiting fibrogenesis in MASLD treatment | |
| 8. Conclusions | - | Summary of findings and future directions |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soto, A.; Spongberg, C.; Martinino, A.; Giovinazzo, F. Exploring the Multifaceted Landscape of MASLD: A Comprehensive Synthesis of Recent Studies, from Pathophysiology to Organoids and Beyond. Biomedicines 2024, 12, 397. https://doi.org/10.3390/biomedicines12020397
Soto A, Spongberg C, Martinino A, Giovinazzo F. Exploring the Multifaceted Landscape of MASLD: A Comprehensive Synthesis of Recent Studies, from Pathophysiology to Organoids and Beyond. Biomedicines. 2024; 12(2):397. https://doi.org/10.3390/biomedicines12020397
Chicago/Turabian StyleSoto, Allison, Colby Spongberg, Alessandro Martinino, and Francesco Giovinazzo. 2024. "Exploring the Multifaceted Landscape of MASLD: A Comprehensive Synthesis of Recent Studies, from Pathophysiology to Organoids and Beyond" Biomedicines 12, no. 2: 397. https://doi.org/10.3390/biomedicines12020397