
Different Coactivator Recruitment to Human PPARα/δ/γ Ligand-Binding Domains by Eight PPAR Agonists to Treat Nonalcoholic Fatty Liver Disease
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Recombinant PPARα/δ/γ-LBD Expression and Purification
2.2. Coactivator Recruitment Assay
3. Results
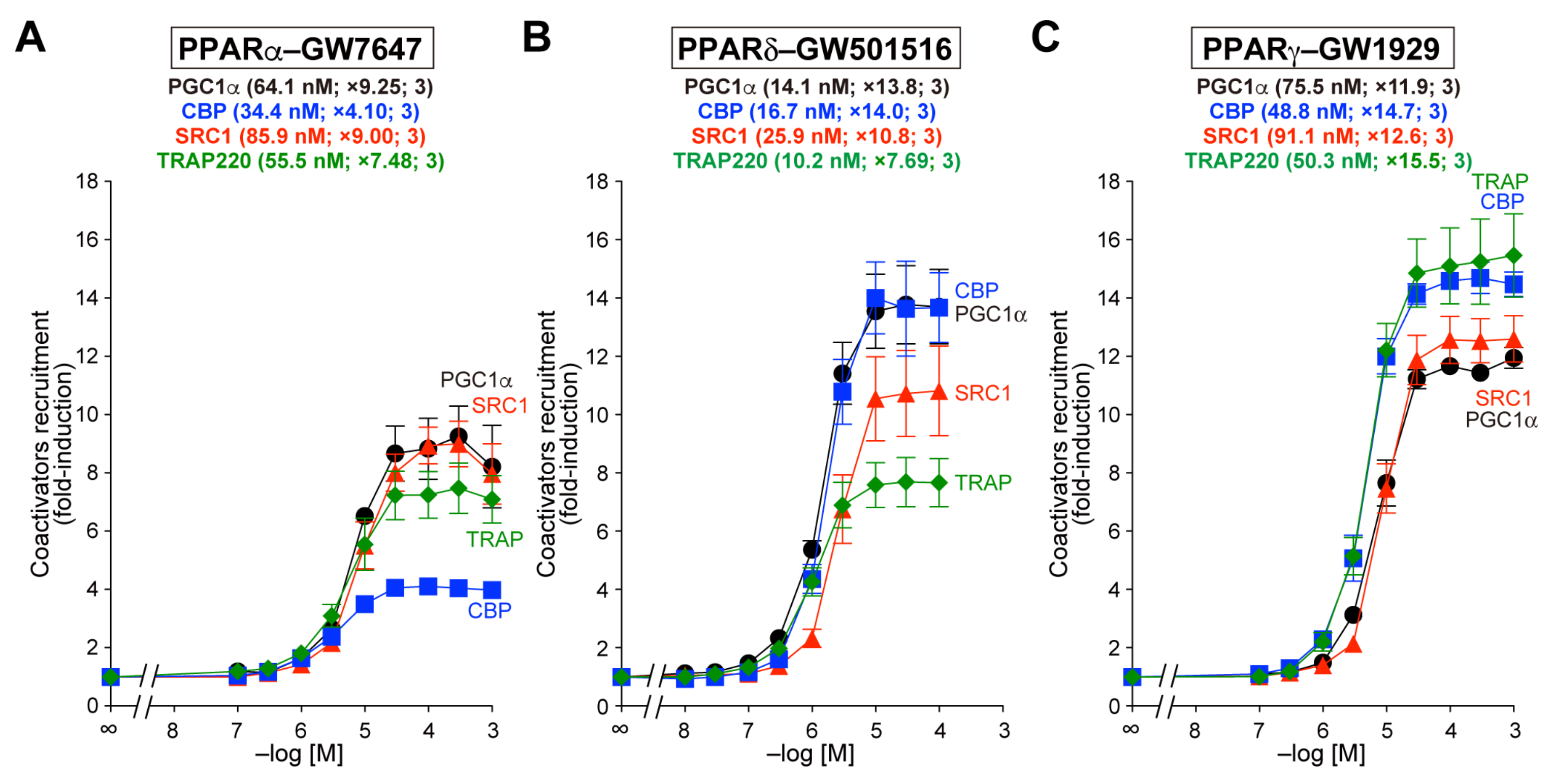
3.1. Recruitment of the Four Coactivator Species to Each of the PPARα/δ/γ-LBDs by Selective Full Agonists (as Control Experiments)
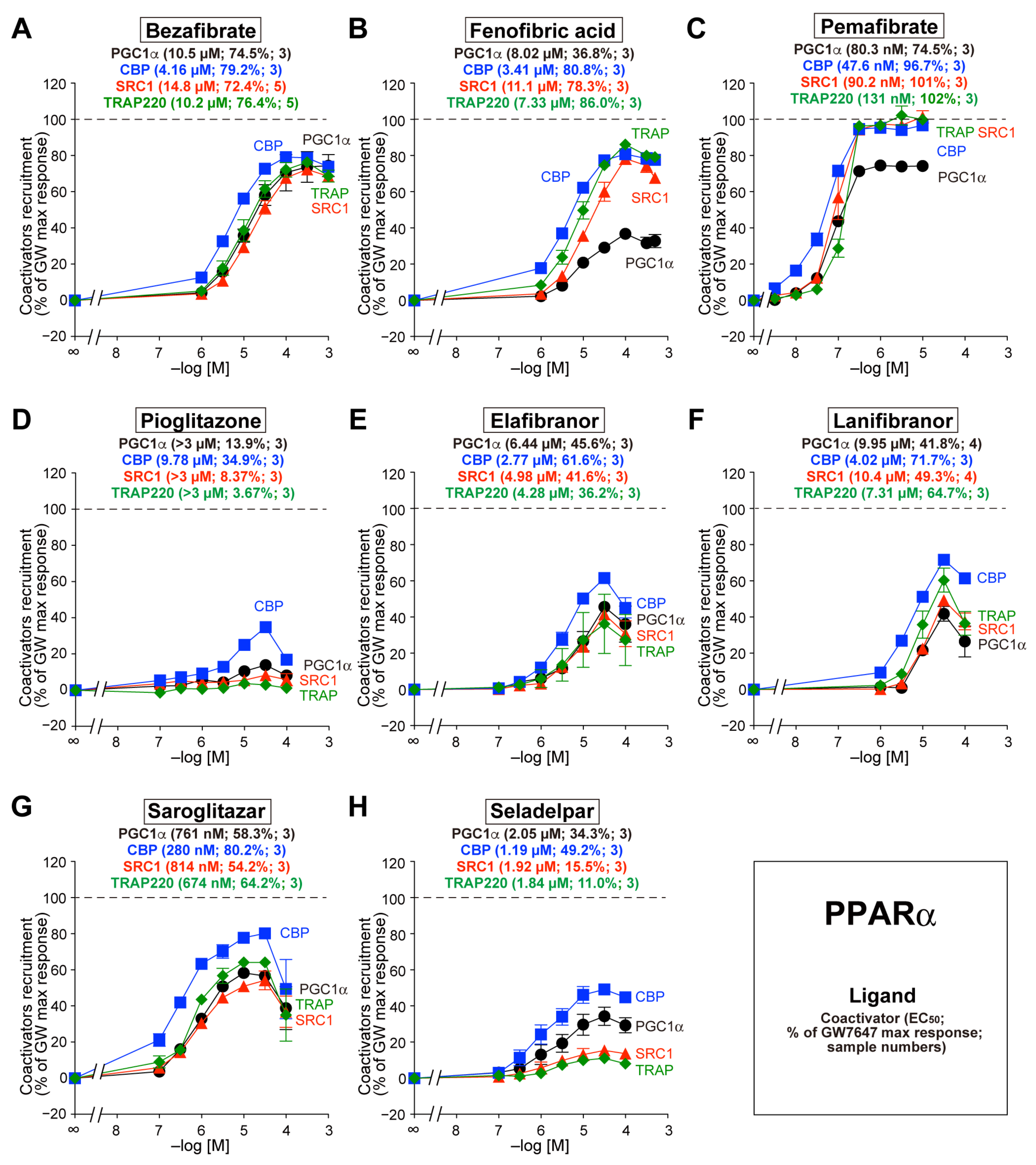
3.2. Recruitment of the Four Coactivators to PPARα-LBD by the Eight Agonists
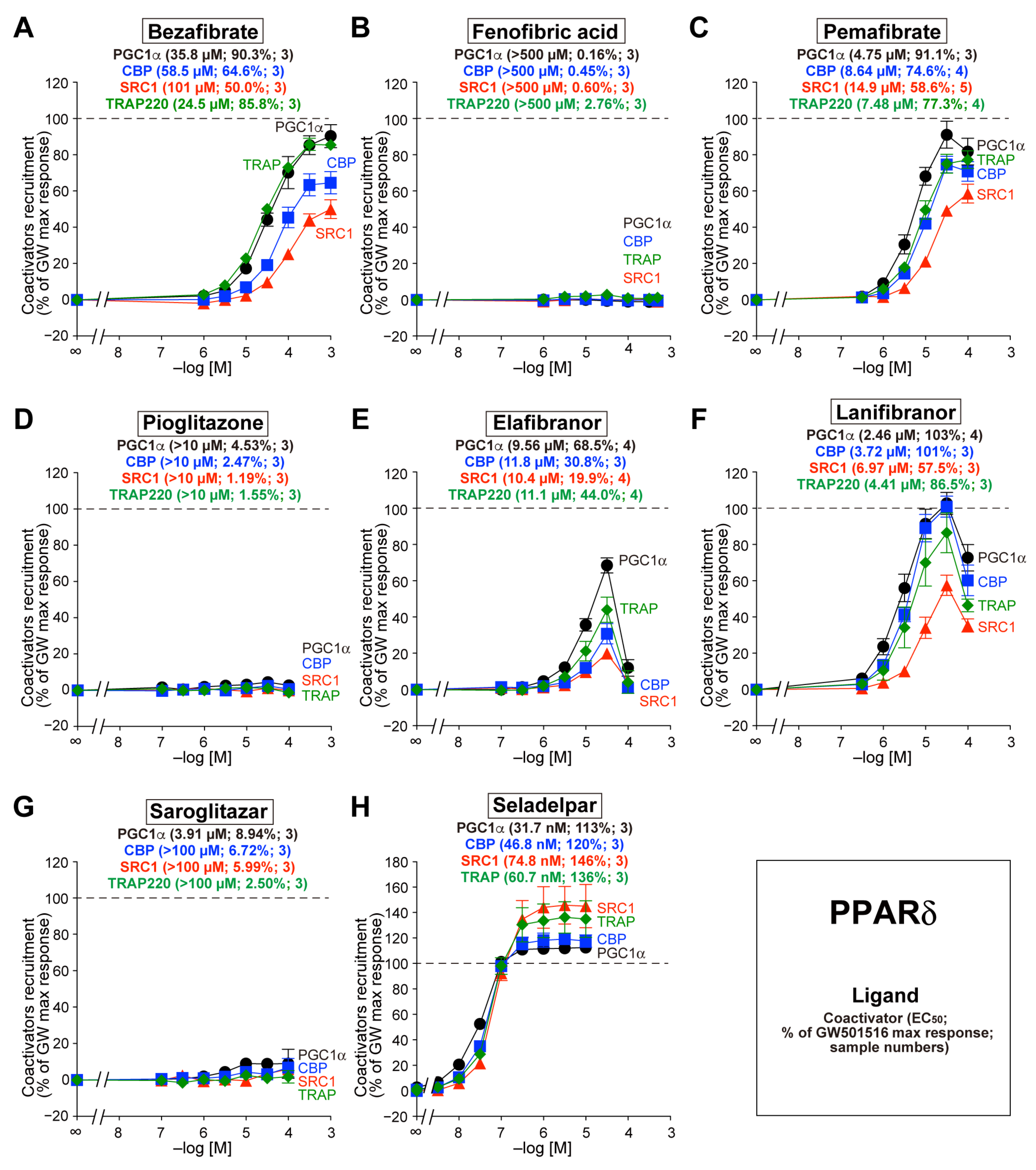
3.3. Recruitment of the Four Coactivators to PPARδ-LBD by the Eight Agonists
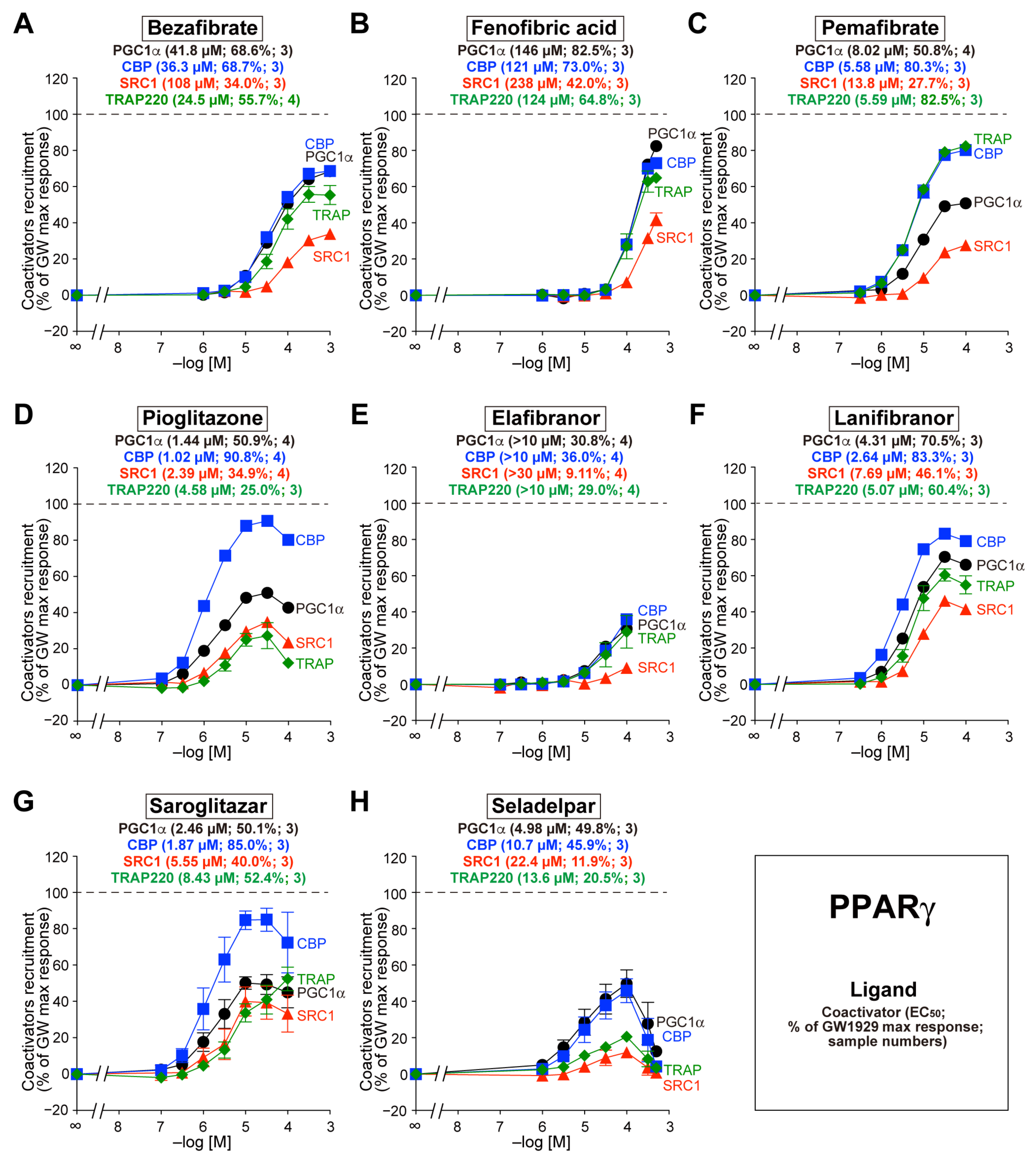
3.4. Recruitment of the Four Coactivators to PPARγ-LBD by the Eight Agonists

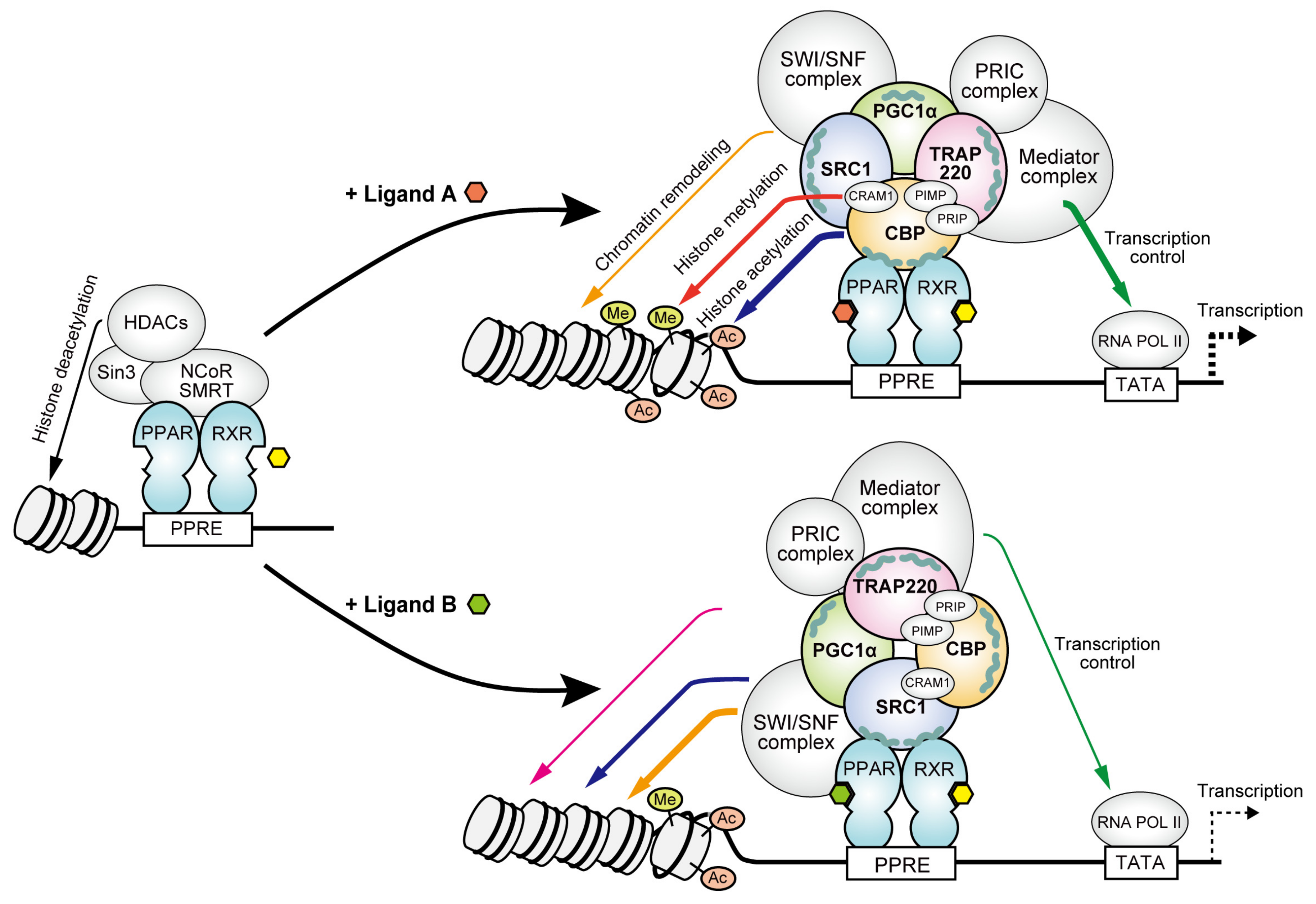
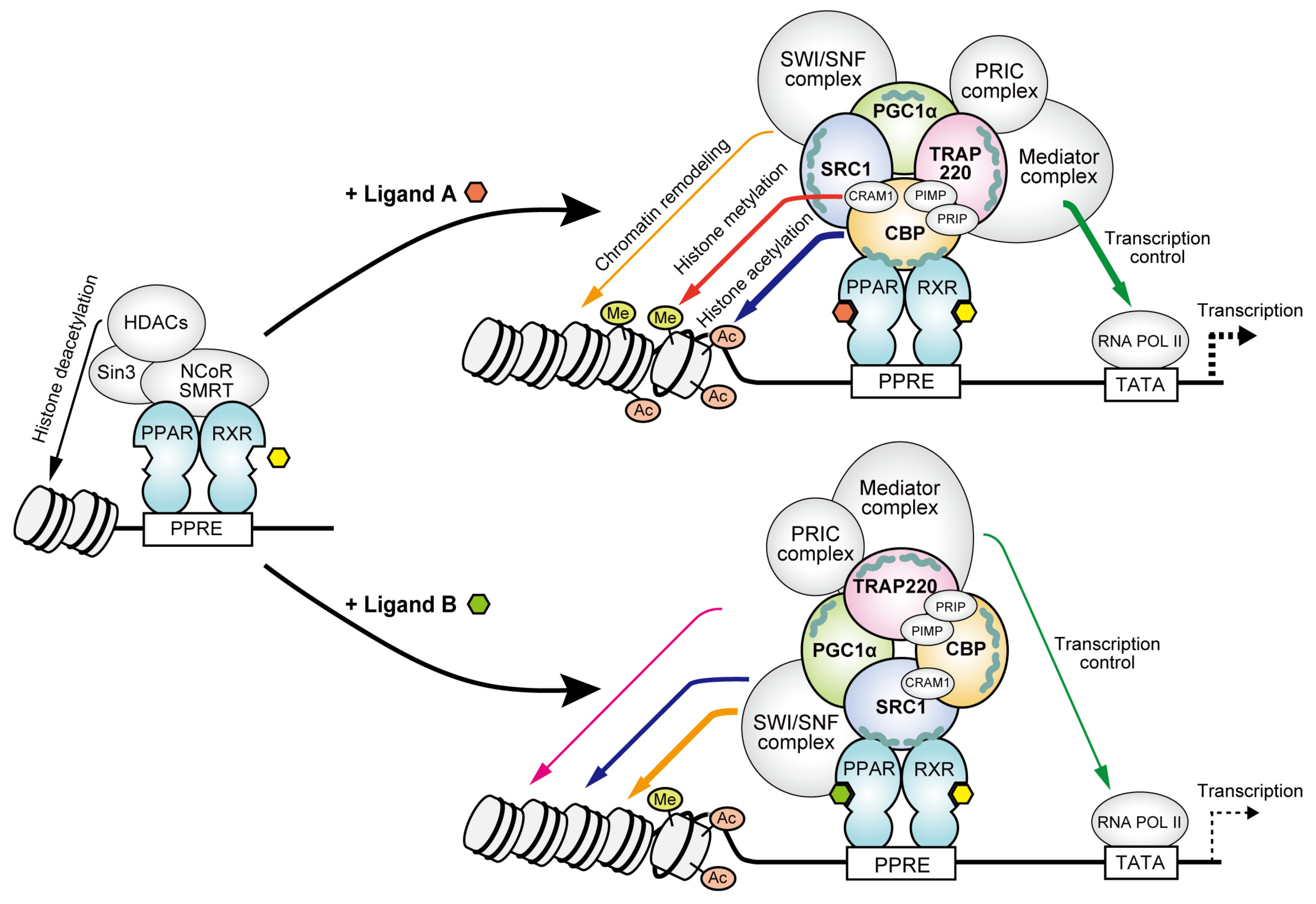
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Riazi, K.; Azhari, H.; Charette, J.H.; Underwood, F.E.; King, J.A.; Afshar, E.E.; Swain, M.G.; Congly, S.E.; Kaplan, G.G.; Shaheen, A.A. The prevalence and incidence of NAFLD worldwide: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2022, 7, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Ramai, D.; Singh, J.; Lester, J.; Khan, S.R.; Chandan, S.; Tartaglia, N.; Ambrosi, A.; Serviddio, G.; Facciorusso, A. Systematic review with meta-analysis: Bariatric surgery reduces the incidence of hepatocellular carcinoma. Aliment. Pharmacol. Ther. 2021, 53, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Pipitone, R.M.; Ciccioli, C.; Infantino, G.; La Mantia, C.; Parisi, S.; Tulone, A.; Pennisi, G.; Grimaudo, S.; Petta, S. MAFLD: A multisystem disease. Ther. Adv. Endocrinol. Metab. 2023, 14, 20420188221145549. [Google Scholar] [CrossRef]
- Vuppalanchi, R.; Noureddin, M.; Alkhouri, N.; Sanyal, A.J. Therapeutic pipeline in nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 373–392. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.S.; Tan, W.R.; Low, Z.S.; Marvalim, C.; Lee, J.Y.H.; Tan, N.S. Exploration and development of PPAR modulators in health and disease: An update of clinical evidence. Int. J. Mol. Sci. 2019, 20, 5055. [Google Scholar] [CrossRef]
- Kamata, S.; Honda, A.; Ishii, I. Current clinical trial status and future prospects of PPAR-targeted drugs for treating nonalcoholic fatty liver disease. Biomolecules 2023, 13, 1264. [Google Scholar] [CrossRef]
- Souza-Tavares, H.; Miranda, C.S.; Vasques-Monteiro, I.M.L.; Sandoval, C.; Santana-Oliveira, D.A.; Silva-Veiga, F.M.; Fernandes-da-Silva, A.; Souza-Mello, V. Peroxisome proliferator-activated receptors as targets to treat metabolic diseases: Focus on the adipose tissue, liver, and pancreas. World J. Gastroenterol. 2023, 29, 4136–4155. [Google Scholar] [CrossRef]
- Lefebvre, P.; Chinetti, G.; Fruchart, J.C.; Staels, B. Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. J. Clin. Investig. 2006, 116, 571–580. [Google Scholar] [CrossRef]
- Giordano Attianese, G.M.; Desvergne, B. Integrative and systemic approaches for evaluating PPARβ/δ (PPARD) function. Nucl. Recept. Signal. 2015, 13, e001. [Google Scholar] [CrossRef]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef]
- Dubois, V.; Eeckhoute, J.; Lefebvre, P.; Staels, B. Distinct but complementary contributions of PPAR isotypes to energy homeostasis. J. Clin. Investig. 2017, 127, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- Viswakarma, N.; Jia, Y.; Bai, L.; Vluggens, A.; Borensztajn, J.; Xu, J.; Reddy, J.K. Coactivators in PPAR-regulated gene expression. PPAR Res. 2010, 2010, 250126. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, J.; Byun, J.; Park, J.Y.; Yamamoto, T.; Schesing, K.; Tian, B.; Sadoshima, J.; Oka, S. An ideal PPAR response element bound to and activated by PPARα. PLoS ONE 2015, 10, e0134996. [Google Scholar] [CrossRef] [PubMed]
- Stallcup, M.R.; Poulard, C. Gene-specific actions of transcriptional coregulators facilitate physiological plasticity: Evidence for a physiological coregulator code. Trends Biochem. Sci. 2020, 45, 497–510. [Google Scholar] [CrossRef]
- Yu, S.; Reddy, J.K. Transcription coactivators for peroxisome proliferator-activated receptors. Biochim. Biophys. Acta 2007, 1771, 936–951. [Google Scholar] [CrossRef]
- Kamata, S.; Oyama, T.; Saito, K.; Honda, A.; Yamamoto, Y.; Suda, K.; Ishikawa, R.; Itoh, T.; Watanabe, Y.; Shibata, T.; et al. PPARα ligand-binding domain structures with endogenous fatty acids and fibrates. iScience 2020, 23, 101727. [Google Scholar] [CrossRef]
- Honda, A.; Kamata, S.; Satta, C.; Machida, Y.; Uchii, K.; Terasawa, K.; Nemoto, A.; Oyama, T.; Ishii, I. Structural basis for anti-non-alcoholic fatty liver disease and diabetic dyslipidemia drug saroglitazar as a PPAR α/γ dual agonist. Biol. Pharm. Bull. 2021, 44, 1210–1219. [Google Scholar] [CrossRef]
- Honda, A.; Kamata, S.; Akahane, M.; Machida, Y.; Uchii, K.; Shiiyama, Y.; Habu, Y.; Miyawaki, S.; Kaneko, C.; Oyama, T.; et al. Functional and structural insights into human PPARα/δ/γ subtype selectivity of bezafibrate, fenofibric acid, and pemafibrate. Int. J. Mol. Sci. 2022, 23, 4726. [Google Scholar] [CrossRef]
- Kamata, S.; Honda, A.; Ishikawa, R.; Akahane, M.; Fujita, A.; Kaneko, C.; Miyawaki, S.; Habu, Y.; Shiiyama, Y.; Uchii, K.; et al. Functional and structural insights into the human PPARα/δ/γ targeting preferences of anti-NASH investigational drugs, lanifibranor, seladelpar, and elafibranor. Antioxidants 2023, 12, 1523. [Google Scholar] [CrossRef]
- Kamata, S.; Oyama, T.; Ishii, I. Preparation of co-crystals of human PPARα-LBD and ligand for high-resolution X-ray crystallography. STAR Protoc. 2021, 2, 100364. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics 2014, 13, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Drake, K.A.; Zhang, J.H.; Harrison, R.K.; McGeehan, G.M. Development of a homogeneous, fluorescence resonance energy transfer-based in vitro recruitment assay for peroxisome proliferator-activated receptor delta via selection of active LXXLL coactivator peptides. Anal. Biochem. 2002, 304, 63–69. [Google Scholar] [CrossRef]
- Guo, L.; Fang, H.; Collins, J.; Fan, X.H.; Dial, S.; Wong, A.; Mehta, K.; Blann, E.; Shi, L.; Tong, W.; et al. Differential gene expression in mouse primary hepatocytes exposed to the peroxisome proliferator-activated receptor alpha agonists. BMC Bioinform. 2006, 7 (Suppl. S2), S18. [Google Scholar] [CrossRef]
- Toyoshiba, H.; Sawada, H.; Naeshiro, I.; Horinouchi, A. Similar compounds searching system by using the gene expression microarray database. Toxicol. Lett. 2009, 186, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Raza-Iqbal, S.; Tanaka, T.; Anai, M.; Inagaki, T.; Matsumura, Y.; Ikeda, K.; Taguchi, A.; Gonzalez, F.J.; Sakai, J.; Kodama, T. Transcriptome analysis of K-877 (a novel selective PPARα modulator (SPPARMα))-regulated genes in primary human hepatocytes and the mouse liver. J. Atheroscler. Thromb. 2015, 22, 754–772. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Raza-Iqbal, S.; Tanaka, T.; Murakami, K.; Anai, M.; Osawa, T.; Matsumura, Y.; Sakai, J.; Kodama, T. Gene expression profiles induced by a novel selective peroxisome proliferator-activated receptor α modulator (SPPARMα) pemafibrate. Int. J. Mol. Sci. 2019, 20, 5682. [Google Scholar] [CrossRef]
- Khodadadi, I.; Griffin, B.; Thumser, A. Differential effects of long-chain fatty acids and clofibrate on gene expression profiles in cardiomyocytes. Arch. Iran Med. 2008, 11, 42–49. [Google Scholar]
- Zoete, V.; Grosdidier, A.; Michielin, O. Peroxisome proliferator-activated receptor structures: Ligand specificity, molecular switch and interactions with regulators. Biochim. Biophys. Acta 2007, 1771, 915–925. [Google Scholar] [CrossRef]
- Bruning, J.B.; Chalmers, M.J.; Prasad, S.; Busby, S.A.; Kamenecka, T.M.; He, Y.; Nettles, K.W.; Griffin, P.R. Partial agonists activate PPARgamma using a helix 12 independent mechanism. Structure 2007, 15, 1258–1271. [Google Scholar] [CrossRef]
- Nolte, R.T.; Wisely, G.B.; Westin, S.; Cobb, J.E.; Lambert, M.H.; Kurokawa, R.; Rosenfeld, M.G.; Willson, T.M.; Glass, C.K.; Milburn, M.V. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 1998, 395, 137–143. [Google Scholar] [CrossRef]
- Kalkhoven, E. CBP and p300: HATs for different occasions. Biochem. Pharmacol. 2004, 68, 1145–1155. [Google Scholar] [CrossRef]
- Kung, A.L.; Rebel, V.I.; Bronson, R.T.; Ch’ng, L.E.; Sieff, C.A.; Livingston, D.M.; Yao, T.P. Gene dose-dependent control of hematopoiesis and hematologic tumor suppression by CBP. Genes Dev. 2000, 14, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.P.; Oh, S.P.; Fuchs, M.; Zhou, N.D.; Ch’ng, L.E.; Newsome, D.; Bronson, R.T.; Li, E.; Livingston, D.M.; Eckner, R. Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell 1998, 93, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Surapureddi, S.; Yu, S.; Bu, H.; Hashimoto, T.; Yeldandi, A.V.; Kashireddy, P.; Cherkaoui-Malki, M.; Qi, C.; Zhu, Y.J.; Rao, M.S.; et al. Identification of a transcriptionally active peroxisome proliferator-activated receptor alpha -interacting cofactor complex in rat liver and characterization of PRIC285 as a coactivator. Proc. Natl. Acad. Sci. USA 2002, 99, 11836–11841. [Google Scholar] [CrossRef] [PubMed]
- Dowell, P.; Ishmael, J.E.; Avram, D.; Peterson, V.J.; Nevrivy, D.J.; Leid, M. p300 functions as a coactivator for the peroxisome proliferator-activated receptor alpha. J. Biol. Chem. 1997, 272, 33435–33443. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Kawada, T.; Yamamoto, T.; Goto, T.; Taimatsu, A.; Aoki, N.; Kawasaki, H.; Taira, K.; Yokoyama, K.K.; Kamei, Y.; et al. Overexpression and ribozyme-mediated targeting of transcriptional coactivators CREB-binding protein and p300 revealed their indispensable roles in adipocyte differentiation through the regulation of peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 2002, 277, 16906–16912. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Oike, Y.; Kamon, J.; Waki, H.; Komeda, K.; Tsuchida, A.; Date, Y.; Li, M.X.; Miki, H.; Akanuma, Y.; et al. Increased insulin sensitivity despite lipodystrophy in Crebbp heterozygous mice. Nat. Genet. 2002, 30, 221–226. [Google Scholar] [CrossRef]
- Li, M.; Luo, R.Z.; Chen, J.W.; Cao, Y.; Lu, J.B.; He, J.H.; Wu, Q.L.; Cai, M.Y. High expression of transcriptional coactivator p300 correlates with aggressive features and poor prognosis of hepatocellular carcinoma. J. Transl. Med. 2011, 9, 5. [Google Scholar] [CrossRef]
- Cai, L.Y.; Chen, S.J.; Xiao, S.H.; Sun, Q.J.; Ding, C.H.; Zheng, B.N.; Zhu, X.Y.; Liu, S.Q.; Yang, F.; Yang, Y.X.; et al. Targeting p300/CBP attenuates hepatocellular carcinoma progression through epigenetic regulation of metabolism. Cancer Res. 2021, 81, 860–872. [Google Scholar] [CrossRef]
- Halachmi, S.; Marden, E.; Martin, G.; MacKay, H.; Abbondanza, C.; Brown, M. Estrogen receptor-associated proteins: Possible mediators of hormone-induced transcription. Science 1994, 264, 1455–1458. [Google Scholar] [CrossRef]
- Mouchiroud, L.; Eichner, L.J.; Shaw, R.J.; Auwerx, J. Transcriptional coregulators: Fine-tuning metabolism. Cell Metab. 2014, 20, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Stashi, E.; York, B.; O’Malley, B.W. Steroid receptor coactivators: Servants and masters for control of systems metabolism. Trends Endocrinol. Metab. 2014, 25, 337–347. [Google Scholar] [CrossRef]
- Tong, Z.; Li, M.; Wang, W.; Mo, P.; Yu, L.; Liu, K.; Ren, W.; Li, W.; Zhang, H.; Xu, J.; et al. Steroid receptor coactivator 1 promotes human hepatocellular carcinoma progression by enhancing Wnt/β-catenin signaling. J. Biol. Chem. 2015, 290, 18596–18608. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, H.; Yu, Y.; Liu, M.; Guo, D.; Zhang, X.; Zhang, J. Imbalanced expression pattern of steroid receptor coactivator-1 and -3 in liver cancer compared with normal liver: An immunohistochemical study with tissue microarray. Oncol. Lett. 2018, 16, 6339–6348. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Spiegelman, B.M. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): Transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003, 24, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Villena, J.A. New insights into PGC-1 coactivators: Redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2014, 282, 647–672. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Adelmant, G.; Wu, Z.; Fan, M.; Xu, J.; O’Malley, B.; Spiegelman, B.M. Activation of PPARgamma coactivator-1 through transcription factor docking. Science 1999, 286, 1368–1371. [Google Scholar] [CrossRef]
- Lin, J.; Puigserver, P.; Donovan, J.; Tarr, P.; Spiegelman, B.M. Peroxisome proliferator-activated receptor gamma coactivator 1beta (PGC-1beta), a novel PGC-1-related transcription coactivator associated with host cell factor. J. Biol. Chem. 2002, 277, 1645–1648. [Google Scholar] [CrossRef]
- Feige, J.N.; Auwerx, J. Transcriptional coregulators in the control of energy homeostasis. Trends Cell Biol. 2007, 17, 292–301. [Google Scholar] [CrossRef]
- Tai, C.M.; Huang, C.K.; Tu, H.P.; Hwang, J.C.; Yeh, M.L.; Huang, C.F.; Huang, J.F.; Dai, C.Y.; Chuang, W.L.; Yu, M.L. Interactions of a PPARGC1A variant and a PNPLA3 variant affect nonalcoholic steatohepatitis in severely obese Taiwanese patients. Medicine 2016, 95, e3120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.N.; Shen, F.; Pan, Q.; Cao, H.X.; Chen, G.Y.; Fan, J.G. PPARGC1A rs8192678 G>A polymorphism affects the severity of hepatic histological features and nonalcoholic steatohepatitis in patients with nonalcoholic fatty liver disease. World J. Gastroenterol. 2021, 27, 3863–3876. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.J.; Moon, I.; Han, K. Transcriptional cofactors exhibit differential preference toward peroxisome proliferator-activated receptors alpha and delta in uterine cells. Endocrinology 2004, 145, 2886–2895. [Google Scholar] [CrossRef]
- Gao, J.; Bao, M.; Xing, Y.; Ding, Y.; Han, T.; Wen, E.; Liu, J.; Yue, S.; Wang, R.; Wang, L.; et al. Mediator subunit MED1 deficiency prevents carbon tetrachloride-induced hepatic fibrosis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2023, 325, G418–G428. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Zhao, B.; Wei, D.; Wang, W.; Cui, Y.; Qian, L.; Liu, G. miR-146a improves hepatic lipid and glucose metabolism by targeting MED1. Int. J. Mol. Med. 2020, 45, 543–555. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PPAR Transcripts | Coactivator Transcripts | ||||||
|---|---|---|---|---|---|---|---|
| PPARA | PPARD | PPARG | PGC1A | CBP | SRC1 | TRAP220 | |
| Gene ID 1 | 5465 | 5467 | 5468 | 10891 | 1387 | 8648 | 5469 |
| FPKM values | |||||||
| adrenal | 3.35 | 4.34 | 0.349 | 0.985 | 4.09 | 8.79 | 4.35 |
| appendix | 2.20 | 7.55 | 0.797 | 0.181 | 7.15 | 8.66 | 7.69 |
| bone marrow | 0.492 | 2.04 | 0.0962 | 0.00309 | 11.6 | 6.44 | 5.34 |
| brain | 2.53 | 7.43 | 0.165 | 1.85 | 6.46 | 17.4 | 5.10 |
| colon | 5.88 | 7.77 | 3.61 | 2.11 | 5.63 | 8.12 | 5.62 |
| duodenum | 7.53 | 3.85 | 1.75 | 2.26 | 4.36 | 10.8 | 4.14 |
| endometrium | 2.15 | 6.20 | 0.490 | 0.307 | 10.3 | 10.0 | 7.21 |
| esophagus | 3.39 | 9.75 | 0.559 | 0.944 | 5.21 | 10.3 | 5.33 |
| fat | 3.91 | 5.05 | 18.9 | 0.696 | 6.87 | 8.08 | 5.53 |
| gall bladder | 2.57 | 6.57 | 1.11 | 0.822 | 6.55 | 9.84 | 5.80 |
| heart | 8.09 | 3.91 | 0.557 | 5.82 | 3.27 | 6.17 | 3.50 |
| kidney | 11.9 | 3.55 | 1.14 | 7.60 | 5.82 | 9.72 | 5.23 |
| liver | 6.43 | 1.35 | 1.00 | 5.97 | 3.38 | 4.28 | 2.71 |
| lung | 1.80 | 6.07 | 2.03 | 1.35 | 6.51 | 9.06 | 5.56 |
| lymph node | 1.53 | 5.37 | 0.370 | 0.0698 | 6.65 | 8.50 | 8.09 |
| ovary | 4.89 | 7.48 | 0.966 | 0.343 | 12.1 | 13.4 | 6.58 |
| pancreas | 0.893 | 0.845 | 0.0542 | 0.269 | 1.96 | 2.15 | 1.04 |
| placenta | 1.79 | 13.0 | 3.48 | 0.0421 | 6.31 | 7.72 | 5.84 |
| prostate | 2.91 | 6.53 | 0.419 | 0.983 | 6.03 | 8.15 | 4.96 |
| salivary gland | 1.56 | 2.28 | 0.193 | 3.66 | 2.84 | 3.75 | 1.98 |
| skin | 3.22 | 7.33 | 0.0981 | 0.173 | 6.47 | 7.69 | 4.80 |
| small intestine | 7.96 | 5.07 | 1.22 | 1.94 | 4.81 | 10.0 | 4.26 |
| spleen | 1.98 | 6.75 | 1.07 | 0.200 | 9.28 | 8.99 | 6.61 |
| stomach | 1.98 | 7.95 | 2.89 | 0.862 | 5.12 | 5.47 | 4.28 |
| testis | 0.989 | 5.43 | 0.801 | 1.68 | 12.7 | 14.6 | 7.22 |
| thyroid | 3.97 | 10.7 | 1.16 | 5.52 | 8.11 | 7.65 | 6.88 |
| urinary bladder | 2.76 | 5.30 | 4.24 | 0.279 | 5.75 | 8.39 | 6.14 |
| PPARα | PPARδ | PPARγ | |
|---|---|---|---|
| Bezafibrate | CBP>PGC=TRAP>SRC | TRAP>PGC>CBP>SRC | PGC=CBP=TRAP>SRC |
| Fenofibric acid | CBP>TRAP>PGC>SRC | No activation | PGC=CBP=TRAP>SRC |
| Pemafibrate | CBP>PGC=SRC>TRAP | PGC>CBP=TRAP>SRC | CBP=TRAP>PGC>SRC |
| Pioglitazone | CBP>>>PGC=SRC=TRAP | No activation | CBP>PGC>SRC>TRAP |
| Elafibranor | CBP>PGC=SRC=TRAP | PGC>TRAP>CBP>SRC | PGC=CBP=TRAP>SRC |
| Lanifibranor | CBP>TRAP>PGC=SRC | PGC>CBP>TRAP>SRC | CBP>PGC>TRAP>SRC |
| Saroglitazar | CBP>TRAP>PGC=SRC | Faint activation | CBP>PGC>TRAP=SRC |
| Seladelpar | CBP>PGC>SRC=TRAP | PGC>CBP>SRC=TRAP | PGC=CBP>TRAP>SRC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamata, S.; Honda, A.; Kashiwagi, N.; Shimamura, A.; Yashiro, S.; Komori, Y.; Hosoda, A.; Akahoshi, N.; Ishii, I. Different Coactivator Recruitment to Human PPARα/δ/γ Ligand-Binding Domains by Eight PPAR Agonists to Treat Nonalcoholic Fatty Liver Disease. Biomedicines 2024, 12, 624. https://doi.org/10.3390/biomedicines12030624
Kamata S, Honda A, Kashiwagi N, Shimamura A, Yashiro S, Komori Y, Hosoda A, Akahoshi N, Ishii I. Different Coactivator Recruitment to Human PPARα/δ/γ Ligand-Binding Domains by Eight PPAR Agonists to Treat Nonalcoholic Fatty Liver Disease. Biomedicines. 2024; 12(3):624. https://doi.org/10.3390/biomedicines12030624
Chicago/Turabian StyleKamata, Shotaro, Akihiro Honda, Nonoka Kashiwagi, Ayumi Shimamura, Sayaka Yashiro, Yuna Komori, Aoi Hosoda, Noriyuki Akahoshi, and Isao Ishii. 2024. "Different Coactivator Recruitment to Human PPARα/δ/γ Ligand-Binding Domains by Eight PPAR Agonists to Treat Nonalcoholic Fatty Liver Disease" Biomedicines 12, no. 3: 624. https://doi.org/10.3390/biomedicines12030624