
Comparing the Efficacy of Two Generations of EGFR-TKIs: An Integrated Drug–Disease Mechanistic Model Approach in EGFR-Mutated Lung Adenocarcinoma
 , , , , ,
, , , , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Improvement of ISELA-V1 Model
2.2. Modeling of Osimertinib and Gefitinib
2.2.1. Characteristics of the Two Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors and Modeling Strategy
2.2.2. Physiologically Based Pharmacokinetic Model of Osimertinib and Gefitinib
2.2.3. Modeling Tyrosine Kinase Inhibitor Mechanism of Action
2.3. Mechanisms of Resistance
2.4. Data for Model Calibration
2.5. Comparison of Model Prediction with a Retrospective Study
2.6. Effect Model
3. Results
3.1. Reproducing Pharmacokinetic Data of Gefitinib and Osimertinib
3.2. Reproducing Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Efficacy
3.3. Reproducing the Results from a Retrospective Study
3.4. Effect Model
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADME | Absorption Distribution Metabolism Elimination |
| AKT | Protein Kinase B |
| DNA | Deoxyribonucleic acid |
| EGFR | Epidermal growth factor receptor |
| ERK | Extracellular signal-regulated kinase |
| ISELA | In Silico EGFR Lung Adenocarcinoma |
| LUAD | Lung adenocarcinoma |
| MT | Metastasis |
| PK | Pharmacokinetics |
| PBPK | Physiologically based pharmacokinetics |
| PT | Primary tumor |
| RECIST | Response Evaluation Criteria In Solid Tumors |
References
- Pirlog, R.; Chiroi, P.; Rusu, I.; Jurj, A.M.; Budisan, L.; Pop-Bica, C.; Braicu, C.; Crisan, D.; Sabourin, J.C.; Berindan-Neagoe, I. Cellular and Molecular Profiling of Tumor Microenvironment and Early-Stage Lung Cancer. Int. J. Mol. Sci. 2022, 23, 5346. [Google Scholar] [CrossRef] [PubMed]
- Myers, D.J.; Wallen, J.M. Lung Adenocarcinoma. In StatPearls; StatPearls Publishing: 2023. Available online: https://pubmed.ncbi.nlm.nih.gov/30137862/ (accessed on 15 March 2024).
- Chevallier, M.; Borgeaud, M.; Addeo, A.; Friedlaender, A. Oncogenic driver mutations in non-small cell lung cancer: Past, present and future. World J. Clin. Oncol. 2021, 12, 217–237. [Google Scholar] [CrossRef] [PubMed]
- Pines, G.; Köstler, W.J.; Yarden, Y. Oncogenic mutant forms of EGFR: Lessons in signal transduction and targets for cancer therapy. FEBS Lett. 2010, 584, 2699–2706. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Huang, L.L.; Chen, J.H.; Wu, J.; Xu, Q. The emerging treatment landscape of targeted therapy in non-small-cell lung cancer. Signal Transduct. Target. Ther. 2019, 4, 61. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Lester, J.F. Tyrosine Kinase Inhibitors for the Treatment of EGFR Mutation-Positive Non–Small-Cell Lung Cancer: A Clash of the Generations. Clin. Lung Cancer 2020, 21, e216–e228. [Google Scholar] [CrossRef] [PubMed]
- Viceconti, M.; Emili, L.; Afshari, P.; Courcelles, E.; Curreli, C.; Famaey, N.; Geris, L.; Horner, M.; Jori, M.C.; Kulesza, A.; et al. Possible Contexts of Use for In Silico Trials Methodologies: A Consensus-Based Review. IEEE J. Biomed. Health Inform. 2021, 25, 3977–3982. [Google Scholar] [CrossRef] [PubMed]
- L’Hostis, A.; Palgen, J.L.; Perrillat-Mercerot, A.; Peyronnet, E.; Jacob, E.; Bosley, J.; Duruisseaux, M.; Toueg, R.; Lefèvre, L.; Kahoul, R.; et al. Knowledge-based mechanistic modeling accurately predicts disease progression with gefitinib in EGFR-mutant lung adenocarcinoma. NPJ Syst. Biol. Appl. 2023, 9, 37. [Google Scholar] [CrossRef]
- Eisenhauer, E.; Therasse, P.; Bogaerts, J.; Schwartz, L.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Nan, X.; Xie, C.; Yu, X.; Liu, J. EGFR TKI as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer. Oncotarget 2017, 8, 75712–75726. [Google Scholar] [CrossRef]
- Ishikawa, E.; Yokoyama, Y.; Chishima, H.; Kasai, H.; Kuniyoshi, O.; Kimura, M.; Hakamata, J.; Nakada, H.; Suehiro, N.; Nakaya, N.; et al. Population Pharmacokinetics, Pharmacogenomics, and Adverse Events of Osimertinib and its Two Active Metabolites, AZ5104 and AZ7550, in Japanese Patients with Advanced Non-small Cell Lung Cancer: A Prospective Observational Study. Investig. New Drugs 2023, 41, 122–133. [Google Scholar] [CrossRef]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Bremnes, R.M.; Camps, C.; Sirera, R. Angiogenesis in non-small cell lung cancer: The prognostic impact of neoangiogenesis and the cytokines VEGF and bFGF in tumours and blood. Lung Cancer 2006, 51, 143–158. [Google Scholar] [CrossRef] [PubMed]
- Schoeberl, B.; Eichler-Jonsson, C.; Gilles, E.D.; Müller, G. Computational modeling of the dynamics of the MAP kinase cascade activated by surface and internalized EGF receptors. Nat. Biotechnol. 2002, 20, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Eck, M.J.; Yun, C.H. Structural and mechanistic underpinnings of the differential drug sensitivity of EGFR mutations in non-small cell lung cancer. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2010, 1804, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Masuzawa, K.; Yasuda, H.; Hamamoto, J.; Nukaga, S.; Hirano, T.; Kawada, I.; Naoki, K.; Soejima, K.; Betsuyaku, T. Characterization of the efficacies of osimertinib and nazartinib against cells expressing clinically relevant epidermal growth factor receptor mutations. Oncotarget 2017, 8, 105479–105491. [Google Scholar] [CrossRef]
- EMA. Tagrisso Epar—Public Assessment Report—EMA/CHMP/134310/2017; Technical Report; EMA: 2017. Available online: https://www.ema.europa.eu/en/documents/product-information/tagrisso-epar-product-information_en.pdf (accessed on 15 March 2024).
- Dökümcü, K.; Farahani, R.M. Evolution of Resistance in Cancer: A Cell Cycle Perspective. Front. Oncol. 2019, 9, 376. [Google Scholar] [CrossRef] [PubMed]
- Sarmento-Ribeiro, A.B.; Scorilas, A.; Gonçalves, A.C.; Efferth, T.; Trougakos, I.P. The emergence of drug resistance to targeted cancer therapies: Clinical evidence. Drug Resist. Updat. 2019, 47, 100646. [Google Scholar] [CrossRef]
- Santoni-Rugiu, E.; Melchior, L.C.; Urbanska, E.M.; Jakobsen, J.N.; Stricker, K.d.; Grauslund, M.; Sørensen, J.B. Intrinsic Resistance to EGFR-Tyrosine Kinase Inhibitors in EGFR-Mutant Non-Small Cell Lung Cancer: Differences and Similarities with Acquired Resistance. Cancers 2019, 11, 923. [Google Scholar] [CrossRef]
- Lazzari, C.; Gregorc, V.; Karachaliou, N.; Rosell, R.; Santarpia, M. Mechanisms of resistance to osimertinib. J. Thorac. Dis. 2019, 12, 2851–2858. [Google Scholar] [CrossRef]
- Ramalingam, S.; Cheng, Y.; Zhou, C.; Ohe, Y.; Imamura, F.; Cho, B.; Lin, M.C.; Majem, M.; Shah, R.; Rukazenkov, Y.; et al. Mechanisms of acquired resistance to first-line osimertinib: Preliminary data from the phase III FLAURA study. Ann. Oncol. 2018, 29, viii740. [Google Scholar] [CrossRef]
- Morgillo, F.; Corte, C.M.D.; Fasano, M.; Ciardiello, F. Mechanisms of resistance to EGFR-targeted drugs: Lung cancer. ESMO Open 2016, 1, e000060. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, A.J.; Chan, J.M.; Kubota, D.; Sato, H.; Rizvi, H.; Daneshbod, Y.; Chang, J.C.; Paik, P.K.; Offin, M.; Arcila, M.E.; et al. Tumor Analyses Reveal Squamous Transformation and Off-Target Alterations as Early Resistance Mechanisms to First-line Osimertinib in EGFR-Mutant Lung Cancer. Clin. Cancer Res. 2020, 26, 2654–2663. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, A.; Sharma, S.; Minari, R.; Perego, P.; Giovannetti, E.; Tiseo, M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer 2019, 121, 725–737. [Google Scholar] [CrossRef]
- Kang, H.N.; Choi, J.W.; Shim, H.S.; Kim, J.; Kim, D.J.; Lee, C.Y.; Hong, M.H.; Park, S.Y.; Park, A.Y.; Shin, E.J.; et al. Establishment of a platform of non-small-cell lung cancer patient-derived xenografts with clinical and genomic annotation. Lung Cancer 2018, 124, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhou, Q.; Gallo, J.M. Demonstration of the equivalent pharmacokinetic/pharmacodynamic dosing strategy in a multiple-dose study of gefitinib. Mol. Cancer Ther. 2009, 8, 1438–1447. [Google Scholar] [CrossRef]
- Yates, J.W.; Ashton, S.; Cross, D.; Mellor, M.J.; Powell, S.J.; Ballard, P. Irreversible Inhibition of EGFR: Modeling the Combined Pharmacokinetic–Pharmacodynamic Relationship of Osimertinib and Its Active Metabolite AZ5104. Mol. Cancer Ther. 2016, 15, 2378–2387. [Google Scholar] [CrossRef] [PubMed]
- Bergman, E.; Forsell, P.; Persson, E.M.; Knutson, L.; Dickinson, P.; Smith, R.; Swaisland, H.; Farmer, M.R.; Cantarini, M.V.; Lennernäs, H. Pharmacokinetics of gefitinib in humans: The influence of gastrointestinal factors. Int. J. Pharm. 2007, 341, 134–142. [Google Scholar] [CrossRef]
- Zhao, H.; Cao, J.; Chang, J.; Zhang, Z.; Yang, L.; Wang, J.; Cantarini, M.; Zhang, L. Pharmacokinetics of Osimertinib in Chinese Patients With Advanced NSCLC: A Phase 1 Study. J. Clin. Pharmacol. 2018, 58, 504–513. [Google Scholar] [CrossRef]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in UntreatedEGFR-Mutated Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Inoue, A.; Kobayashi, K.; Maemondo, M.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Updated overall survival results from a randomized phase III trial comparing gefitinib with carboplatin–paclitaxel for chemo-naïve non-small cell lung cancer with sensitive EGFR gene mutations (NEJ002). Ann. Oncol. 2013, 24, 54–59. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum–Pemetrexed in EGFR T790M–Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Jacob, E.; Perrillat-Mercerot, A.; Palgen, J.L.; L’Hostis, A.; Ceres, N.; Boissel, J.P.; Bosley, J.; Monteiro, C.; Kahoul, R. Empirical methods for the validation of time-to-event mathematical models taking into account uncertainty and variability: Application to EGFR + lung adenocarcinoma. BMC Bioinform. 2023, 24, 331. [Google Scholar] [CrossRef] [PubMed]
- Palgen, J.L.; Perrillat-Mercerot, A.; Ceres, N.; Peyronnet, E.; Coudron, M.; Tixier, E.; Illigens, B.M.W.; Bosley, J.; L’Hostis, A.; Monteiro, C. Integration of Heterogeneous Biological Data in Multiscale Mechanistic Model Calibration: Application to Lung Adenocarcinoma. Acta Biotheor. 2022, 70, 19. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhai, Z.; Zhu, Y.; Zhou, H. Comparison of Gefitinib in the treatment of patients with non-small cell lung cancer and clinical effects of Osimertinib and EGFR Gene mutation. Pak. J. Med. Sci. 2022, 38, 1589–1594. [Google Scholar] [CrossRef] [PubMed]
- Boissel, J.P.; Kahoul, R.; Marin, D.; Boissel, F.H. Effect Model Law: An Approach for the Implementation of Personalized Medicine. J. Pers. Med. 2013, 3, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.C.; Reuben, A.; Hu, X.; McGranahan, N.; Chen, R.; Jalali, A.; Negrao, M.V.; Hubert, S.M.; Tang, C.; Wu, C.C.; et al. Multiomics profiling of primary lung cancers and distant metastases reveals immunosuppression as a common characteristic of tumor cells with metastatic plasticity. Genome Biol. 2020, 21, 271. [Google Scholar] [CrossRef] [PubMed]
- FDA. Assessing the Credibility of Computational Modeling and Simulation in Medical Device Submissions—Guidance for Industry and Food and Drug Administration Staff. 2023. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/assessing-credibility-computational-modeling-and-simulation-medical-device-submissions (accessed on 15 March 2024).
- Pappalardo, F.; Russo, G.; Tshinanu, F.M.; Viceconti, M. In silico clinical trials: Concepts and early adoptions. Briefings Bioinform. 2019, 20, 1699–1708. [Google Scholar] [CrossRef] [PubMed]
- Lameire, N.H.; Flombaum, C.D.; Moreau, D.; Ronco, C. Acute renal failure in cancer patients. Ann. Med. 2005, 37, 13–25. [Google Scholar] [CrossRef]
- Novadiscovery. Novadiscovery Announces Success of First-of-its-Kind Clinical Trial Simulation to Accurately Predict Findings of Phase III Clinical Study. Available online: https://www.novadiscovery.com/novadiscovery-announces-success-of-first-of-its-kind-clinical-trial-simulation-to-accurately-predict-findings-of-phase-iii-clinical-study/ (accessed on 12 September 2023).
- Novadiscovery. New Demonstration of the Predictive Power of an In Silico Clinical Trial in Oncology. 2024. Available online: https://www.novadiscovery.com/new-demonstration-of-the-predictive-power-of-an-in-silico-clinical-trial-in-oncology/ (accessed on 13 November 2023).
- Brøgger-Mikkelsen, M.; Zibert, J.R.; Andersen, A.D.; Lassen, U.; Hædersdal, M.; Ali, Z.; Thomsen, S.F. Changes in key recruitment performance metrics from 2008–2019 in industry-sponsored phase III clinical trials registered at ClinicalTrials.gov. PLoS ONE 2022, 17, e0271819. [Google Scholar] [CrossRef]
- Singhania, R.; Sramkoski, R.M.; Jacobberger, J.W.; Tyson, J.J. A Hybrid Model of Mammalian Cell Cycle Regulation. PLos Comput Biol. 2011, 7, e1001077. [Google Scholar] [CrossRef]
- Hnit, S.S.T.; Xie, C.; Yao, M.; Holst, J.; Bensoussan, A.; De Souza, P.; Li, Z.; Dong, Q. p27Kip1 signaling: Transcriptional and post-translational regulation. Int. J. Biochem. Cell Biol. 2015, 68, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Stein, G.S.; Medina, R.; van Wijnen, A.J.; Stein, J.L.; Lian, J.B.; Owen, T.A. Cell Cycle. ELS. John Wiley and Sons Ltd. Available online: https://onlinelibrary.wiley.com/doi/10.1002/9780470015902.a0001354.pub2 (accessed on 15 March 2024).
- Castedo, M.; Perfettini, J.-L.; Roumier, T.; Andreau, K.; Medema, R.; Kroemer, G. Cell death by mitotic catastrophe: A molecular definition. Oncogene 2004, 23, 2825–2837. [Google Scholar] [CrossRef] [PubMed]
- Ghelli Luserna di Rorà, A.; Cerchione, C.; Martinelli, G.; Simonetti, G. A WEE1 family business: Regulation of mitosis, cancer progression, and therapeutic target. J. Hematol. Oncol. 2020, 13, 126. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.-S.; Chen, I.-C.; Lin, P.-Y.; Lung, J.-H.; Li, Y.-C.; Lin, Y.-C.; Yang, C.-T.; Tsai, Y.-H. Epidermal growth factor receptor mutation enhances expression of vascular endothelial growth factor in lung cancer. Oncol. Lett. 2016, 12, 4598–4604. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, S.; Anido, U.; Lazaro, M.; Santome, L.; Afonso, J.; Fernandez, O.; Martinez de Alegria, A.; Aparicio, L.A. Angiogenesis and Lung Cancer. Oncogenesis, Inflammatory and Parasitic Tropical Diseases of the Lung. 2013, Volume 1. Available online: https://www.intechopen.com/chapters/42623 (accessed on 15 March 2024).
- Bielenberg, D.R.; Zetter, B.R. The Contribution of Angiogenesis to the Process of Metastasis. Cancer J. 2015, 21, 267–273. [Google Scholar] [CrossRef] [PubMed]
- ICRP. Report of the Task Group on Reference Man; ICRP Publication 23; Pergamon Press: Oxford, UK, 1975. [Google Scholar]
- Huang, W.; Lee, S.L.; Yu, L.X. Mechanistic approaches to predicting oral drug absorption. AAPS 2009, 11, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Mayumi, K.; Ohnishi, S.; Hasegawa, H. Successful Prediction of Human Pharmacokinetics by Improving Calculation Processes of Physiologically Based Pharmacokinetic Approach. J. Pharm. Sci. 2019, 108, 2718–2727. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, T.; Rowland, M. Physiologically based pharmacokinetic modelling 2: Predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J. Pharm. Sci. 2006, 95, 1238–1257. [Google Scholar] [CrossRef]
- Taxak, N.; Bharatam, P.V. Drug Metabolism: A Fascinating Link Between Chemistry and Biology. Reson. J. Sci. Educ. 2014, 19, 259–282. [Google Scholar] [CrossRef]
- Brown, K.; Comisar, C.; Witjes, H.; Maringwa, J.; de Greef, R.; Vishwanathan, K.; Cantarini, M.; Cox, E. Population pharmacokinetics and exposure-response of osimertinib in patients with non-small cell lung cancer. Br. J. Clin. Pharmacol. 2017, 83, 1216–1226. [Google Scholar] [CrossRef]







{kind=link}
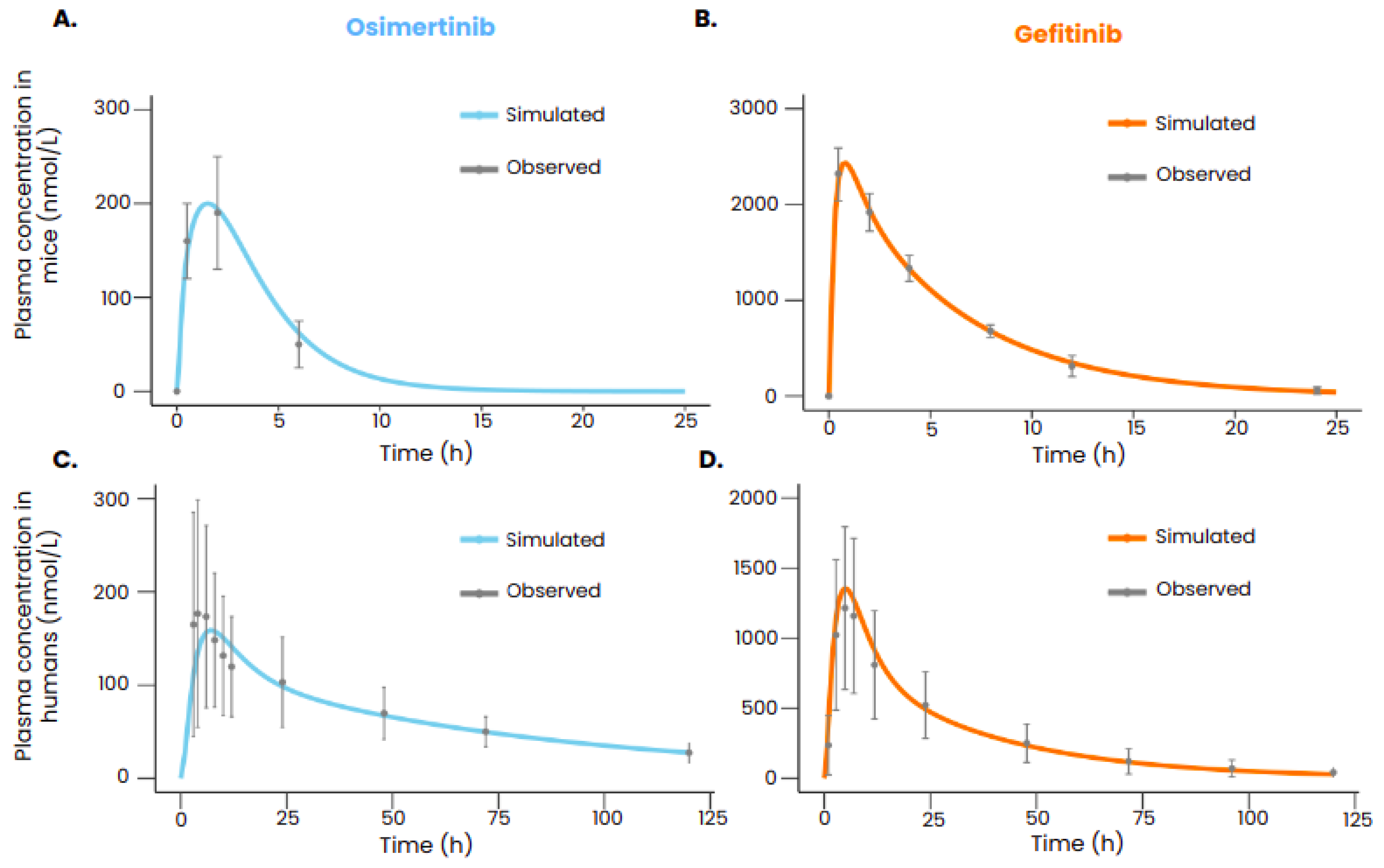{kind=link}
{kind=link}
{kind=link}
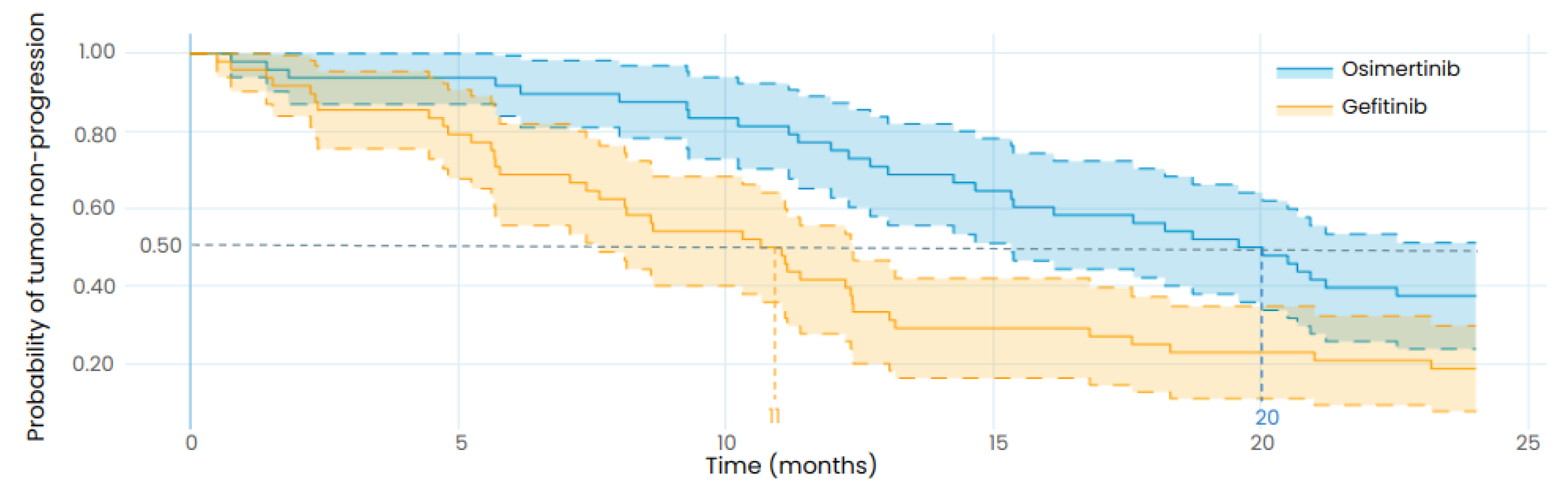{kind=link}
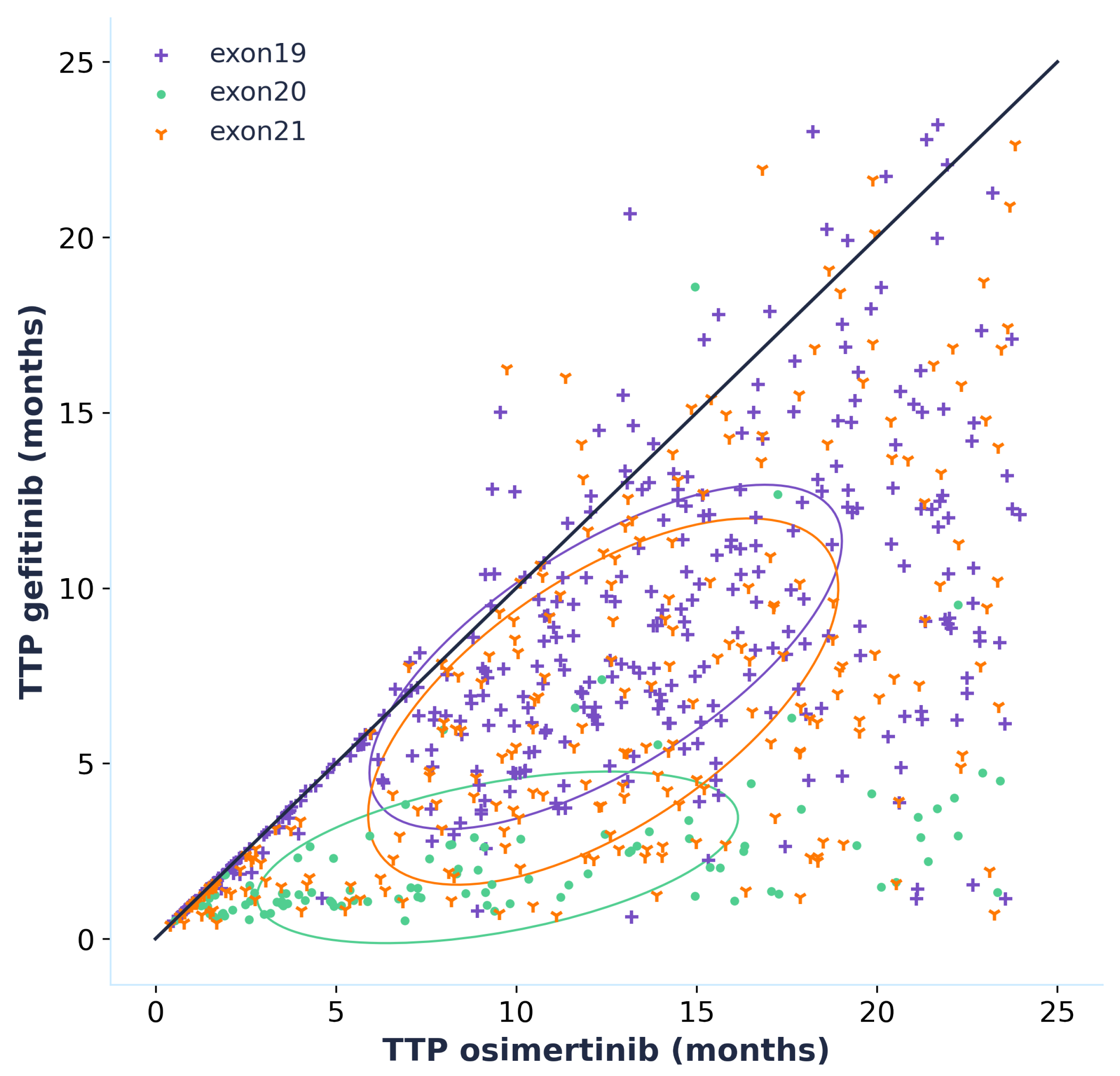{kind=link}
| Parameter Name | Description | Value |
|---|---|---|
| kaPi3kByMet | Forward speed of PI3K activation by MET | (L/mole/s) |
| kaPi3kByVegfr | Forward speed of PI3K activation by VEGFR | (L/mole/s) |
| kaPi3kByEGFR | Forward speed of PI3K activation by EGFR | (L/mole/s) |
| kaPi3kByras | Forward speed of PI3K activation by RAS | (L/mole/s) |
| kdaPi3k | Reverse speed of PI3K activation | 1 (1/s) |
| kaRasByMet | Forward speed of RAS activation by MET | (L/mole/s) |
| kaRasByVegfr | Forward speed of RAS activation by VEGFR | (L/mole/s) |
| kaRasByEGFR | Forward speed of RAS activation by EGFR | (L/mole/s) |
| kdaRas | Reverse speed of RAS activation | 1 (1/s) |
| EGFR Mutation | Tyrosine Kinase Inhibitor | (nmol/L) | |
|---|---|---|---|
| Exon 19 deletion + T790M resistance mutation | Gefitnib | 5.1 | |
| Osimertinib | |||
| Exon 21 insertion + T790M resistance mutation | Gefitinib | 3.4 | |
| Osimertinib | |||
| Exon 19 deletion mutation | Gefitnib | ||
| Osimertinib | 1.1 | ||
| Exon 20 sensitive mutation | Gefitinib | 2.5 | |
| Osimertinib | |||
| Exon 20 resistant mutation | Gefitnib | ||
| Osimertinib | 6.2 | ||
| Exon 21 insertion mutation | Gefitinib | 6.4 | |
| Osimertinib | |||
| Wild type | Gefitnib | 3.2 | |
| Osimertinib | 3.3 |
| Study | Biological Process to Reproduce | Experimental Conditions | Treatment | Type of Study |
|---|---|---|---|---|
| Kang et al. [26] | Tumor volume evolution after treatment administration in mice | Mouse PDX model YHIM-1003 harbors EGFR exon 19 deletion. Mouse PDX model YHIM-1009 harbors EGFR exon 19 deletion and PIK3CA E542K mutation. Treated with gefitinib or osimertinib | Gefitinib, osimertinib | Pre-clinical |
| Wang et al. [27] | Gefitinib plasmatic profile in mice | Tumor bearing mice that have been administered 50 mg/kg of gefitinib orally | Gefitinib | Pre-clinical |
| Yates et al. [28] | Osimertinib and AZ5104 plasmatic profile in mice | Tumor-bearing mice that have been administered 5 mg/kg of osimertinib orally | Osimertinib | Pre-clinical |
| Bergman et al. [29] | Gefitinib plasmatic profile in humans | Healthy volunteers who have been administered 250 mg of gefitinib orally | Gefitinib | Clinical |
| Zhao et al. [30] | Osimertinib and AZ5104 plasmatic profile in humans | EGFR-mutated NSCLC patients who have been administered 40 mg or 80 mg of osimertinib orally | Osimertinib | Clinical |
| FLAURA [31] 2 | Distribution of time to progression (computed from OS and PFS curves 1) in target population + distribution of progression cause and site of new lesions | Patients with an advanced stage of NSCLC harboring an EGFR mutation, treated as first line with 80 mg/day of osimertinib | Osimertinib | Clinical |
| NEJ002 [32] 2 | Distribution of time to progression (computed from OS and PFS curves 1) in target population | Patients with an advanced stage of NSCLC harboring an EGFR mutation, treated as first line with 250 mg/day of gefitinib | Gefitnib | Clinical |
| AURA3 [33] 2 | Distribution of time to progression (computed from OS and PFS curves 1) in target population + distribution of progression cause and site of new lesions | Patients with EGFR T790M-positive advanced-stage NSCLC who previously had disease progression during first-line EGFR-TKI therapy, treated with 80 mg/day of osimertinib | Osimertinib | Clinical |
| Treated with Osimertinib (n = 49) | Treated with Gefitinib (n = 53) | |
|---|---|---|
| Sex (M/F) | 24/25 | 26/27 |
| Age (<65/>65) | 27/22 | 25/28 |
| Smoking status (Y/N) | 38/11 | 35/18 |
| Cancer stage (IIIb/IV) | 30/19 | 35/18 |
| Virtual Population | |
|---|---|
| Sex (M:F ratio) | 1:2 |
| Age (mean, sd) | (67, 11) |
| Smoking status (Never, Former, Current) | (28%, 34%, 38%) |
| Ethnicity (Asian, Other) | (55%, 45%) |
| EGFR mutation (19, 20, 21) | (51.6%, 13.2%, 35.2%) |
| EGFR Mutation | Ratio of Points within the Standard Deviation | Gefitinib | Osimertinib |
|---|---|---|---|
| Exon 19 deletion mutation | 100% | 90% | 80% |
| Exon 19 deletion mutation + PI3KCA mutation | 80% | 100% | 100% |
| Retrospective Study | In Silico Clinical Trial with the Same Patient Characteristics | |
|---|---|---|
| Osimertinib | PFS: 18.1 months (95%CI: 15.4–20.7) | TTP: 20 months (95%CI: 15–24) |
| Gefitnib | PFS: 10.7 months (95%CI: 9.9–11.4) | TTP: 11 months (95%CI: 7.5–12) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Darré, H.; Masson, P.; Nativel, A.; Villain, L.; Lefaudeux, D.; Couty, C.; Martin, B.; Jacob, E.; Duruisseaux, M.; Palgen, J.-L.; et al. Comparing the Efficacy of Two Generations of EGFR-TKIs: An Integrated Drug–Disease Mechanistic Model Approach in EGFR-Mutated Lung Adenocarcinoma. Biomedicines 2024, 12, 704. https://doi.org/10.3390/biomedicines12030704
Darré H, Masson P, Nativel A, Villain L, Lefaudeux D, Couty C, Martin B, Jacob E, Duruisseaux M, Palgen J-L, et al. Comparing the Efficacy of Two Generations of EGFR-TKIs: An Integrated Drug–Disease Mechanistic Model Approach in EGFR-Mutated Lung Adenocarcinoma. Biomedicines. 2024; 12(3):704. https://doi.org/10.3390/biomedicines12030704
Chicago/Turabian StyleDarré, Hippolyte, Perrine Masson, Arnaud Nativel, Laura Villain, Diane Lefaudeux, Claire Couty, Bastien Martin, Evgueni Jacob, Michaël Duruisseaux, Jean-Louis Palgen, and et al. 2024. "Comparing the Efficacy of Two Generations of EGFR-TKIs: An Integrated Drug–Disease Mechanistic Model Approach in EGFR-Mutated Lung Adenocarcinoma" Biomedicines 12, no. 3: 704. https://doi.org/10.3390/biomedicines12030704