
The Role of Cdo1 in Ferroptosis and Apoptosis in Cancer


Queen Mary School, Nanchang University, Nanchang 330047, China
*
Author to whom correspondence should be addressed.
Biomedicines 2024, 12(4), 918; https://doi.org/10.3390/biomedicines12040918
Submission received: 19 March 2024
/
Revised: 8 April 2024
/
Accepted: 16 April 2024
/
Published: 20 April 2024
(This article belongs to the Section Cancer Biology and Oncology)
Abstract
:Cysteine dioxygenase type 1 (Cdo1) is a tumor suppressor gene. It regulates the metabolism of cysteine, thereby influencing the cellular antioxidative capacity. This function puts Cdo1 in a prominent position to promote ferroptosis and apoptosis. Cdo1 promotes ferroptosis mainly by decreasing the amounts of antioxidants, leading to autoperoxidation of the cell membrane through Fenton reaction. Cdo1 promotes apoptosis mainly through the product of cysteine metabolism, taurine, and low level of antioxidants. Many cancers exhibit altered function of Cdo1, underscoring its crucial role in cancer cell survival. Genetic and epigenetic alterations have been found, with methylation of Cdo1 promoter as the most common mutation. The fact that no cancer was found to be caused by altered Cdo1 function alone indicates that the tumor suppressor role of Cdo1 is mild. By compiling the current knowledge about apoptosis, ferroptosis, and the role of Cdo1, this review suggests possibilities for how the mild anticancer role of Cdo1 could be harnessed in new cancer therapies. Here, developing drugs targeting Cdo1 is considered meaningful in neoadjuvant therapies, for example, helping against the development of anti-cancer drug resistance in tumor cells.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Cell death is an essential biological process found in all living organisms, serving various functions such as embryonic development, organ maintenance, aging, immune response coordination, and autoimmunity [1]. There exist alterations in cellular pathways either promoting or suppressing the pathways of cell death, which may result in cell immortality [1]. Cells proliferating uncontrollably and not dying are hallmarks of cancer [2]. In total, 18 million new cancer cases are diagnosed every year, and the most frequent cancer type is lung cancer followed by breast cancer, both of which amount to approximately 2.1 million cases [3]. One effector of cell death is cysteine dioxygenase type 1 (Cdo1), an enzyme that catalyzes the oxidation of cysteine. It plays roles in two different pathways of cell death, apoptosis and ferroptosis. In this review, we elaborate on how studying the anticancer role of Cdo1 in apoptosis and ferroptosis will open new avenues to treat cancer.
1.1. Apoptosis and p53
Apoptosis is a programmed cell death that results in the orderly and efficient removal of cells. In normal cells, apoptosis occurs when there is severe damage or protein misfolding, including genetic mutation and imbalance in apoptotic factors. However, in cancer cells, the genetic variations lead to the misfunction of factors that inhibit cell division or induce apoptosis, enabling unlimited cell division and evasion of cell death [4]. Apoptosis occurs when the caspases are activated by pro-apoptotic factors [5]. Caspases refer to a group of enzymes of the cysteine protease family. The mitochondrion plays a critical role in apoptosis, as it releases pro-apoptotic factors. There are three pathways involved in apoptosis: the extrinsic pathway, intrinsic pathway, and intrinsic endoplasmic reticulum pathway [5]. p53 is a prominent tumor suppressor and nuclear transcription factor that induces expression of genes of the extrinsic and intrinsic pathways [6].
The extrinsic pathway is triggered once the death ligands are bound, for example, Fas ligand [7] to the Fas cell surface receptor, which is under transcriptional control by p53. Caspase 8 is involved in this pathway [7].
The intrinsic pathway, also known as the mitochondrial pathway, is triggered by irreparable genetic damage, hypoxia, severe oxidative stress, and high cellular Ca2+ levels, which are factors causing mitochondrial intrinsic damage [8]. These factors activate the pro-apoptotic factors Bax and Bac, which form pores in the mitochondrial membrane [5,9]. Bax and a subset of pro-survival genes of the Bcl2 family contain p53-binding elements [6]. Moreover, p53 exercises this function mainly by promoting the transcription of BH3-only proteins and the APAF-1 gene [10,11,12,13]. BH3-only proteins inhibit the Bcl2 family from blocking the formation of channels on the mitochondrial membrane and promote mitochondrial outer membrane permeabilization (MOMP), which in turn promotes apoptosis [5,14]. APAF-1 acts as a scaffolding protein to allow the activation of caspase-9 [15].
1.2. Ferroptosis
Ferroptosis is a type of regulated cell death marked by the accumulation of iron as well as iron-dependent lipid peroxides [16]. In some situations, ferroptosis also displays shedding and rounding up of cells, together with increased autophagosomes [17]. Iron promotes ferroptosis as a coactivator of lipoxygenases and a generator of reactive oxygen species (ROS) [17,18]. Without abundant antioxidants, ferroptosis is triggered as the radicals produced by the reaction of phospholipid hydroperoxide (PLOOH), a product of lipid peroxidation, and iron continue with the peroxidation of membrane lipids in a process called autoperoxidation [15], a positive feedback loop. In autoperoxidation, PLOOH reacts with both ferrous (Fe2+) and ferric (Fe3+) ions, resulting in the formation of the free radicals PLO• and PLOO•, respectively [17], in the so-called Fenton reaction [19]. These free radicals then engage with polyunsaturated fatty acid–phospholipids (PUFA-PLs), promoting the continued production of PLOOH [17]. So far, three pathways with protecting roles against elevation of PLOOH have been studied.
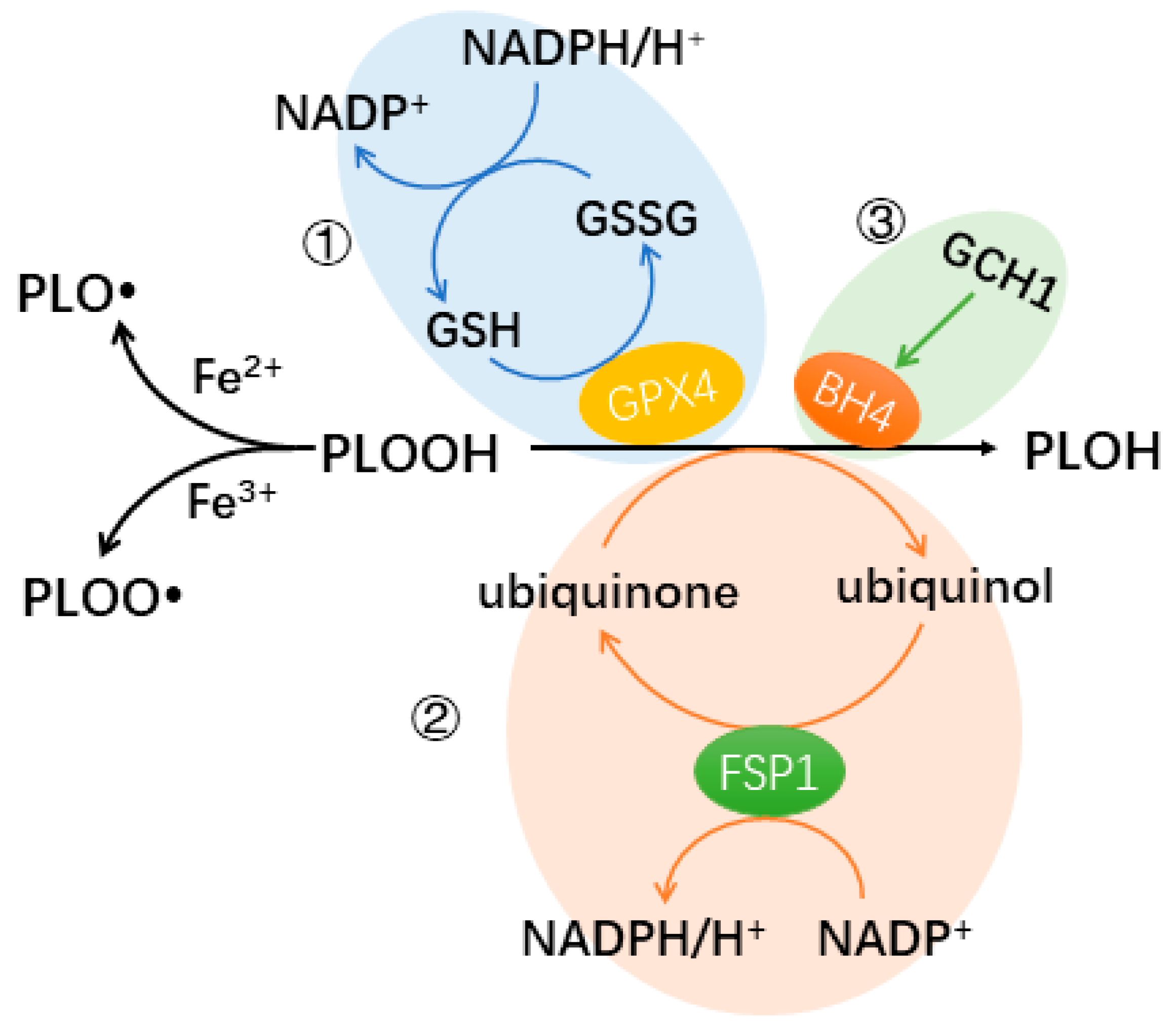
The most well-known pathway protecting cells from ferroptosis involves GPX4 as the key element [17]. The cystine/glutamate antiporters transport cystine into cells. Cystine can be reduced to cysteine, an antioxidant [20]. Additionally, cysteine is essential for the production of glutathione (GSH), a powerful antioxidant [21]. Glutathione peroxidase 4 (GPX4) inhibits the Fenton reaction, by catalyzing the reduction of PLOOH to its alcoholic form PLOH, thereby protecting cells from lipid peroxidation and ferroptosis. In this process, GSH is involved as a cofactor of GPX4 (Figure 1).
There are two other newly discovered protecting pathways. Apoptosis-inducing factor mitochondria-associated 2 (AIFM2, also known as FSP1) and GTP cyclohydrolase-1 (GCH1) participate in these two pathways, respectively. Ferroptosis suppressing protein 1 (FSP1) inhibits lipid peroxidation and ferroptosis by converting ubiquinone (CoQ10)/semihydroquinone into ubiquinol, which can directly reduce lipid radicals, similar to GPX4 [22,23]. FSP1 catalyzes the protection process with the help of NADPH [22,23]. GCH1 is an enzyme that controls the rate of tetrahydrobiopterin (BH4) synthesis, which is essential for the production of neurotransmitters such as dopamine and nitric oxide (NO) [24]. BH4 acts as a cofactor for important enzymes involved in neurotransmitter and NO production [24]. Some types of phospholipids have polyunsaturated fatty acyl tails, such as phosphatidylserine and phosphatidylcholine, which are involved in regulating the fluidity and flexibility of the membrane [25]. Through GCH1-mediated BH4 production, ferroptosis is inhibited by selectively preventing the oxidation of lipids with two polyunsaturated fatty acyl tails [24] (Figure 1).
The pathway containing GPX4 is regulated by p53 [18]. p53 plays a vital role in inhibiting cell proliferation together with promoting apoptosis and ferroptosis [26]. In ferroptosis, p53 inhibits the expression of SLC755A, a subunit of the cystine/glutamate antiporter, which reduces the synthesis of the antiporter and inhibits the uptake of cysteine by the cells, thus depleting cells of antioxidants [27]. As result of this inhibition, cells develop ferroptosis [28]. Moreover, GPX4 is inhibited by a high level of Ca2+ [28]. Consequently, any ion channel eventually increasing intracellular Ca2+ facilitates ferroptosis. Piezo1, a mechanosensitive ion channel commonly produced in cancer cells, is essentially a calcium ion channel [28,29]. Therefore, cancer cells are more likely to undergo ferroptosis than ordinary cells in response to mechanical stimuli [29]. Chloride channels have also been found to have an effect on ferroptosis [30]. Transmembrane member 16A (TMEM16A) is a component of the Ca2+-activated chloride channel [30]. This channel allows Cl− to enter the cell, and the abundance of anions in the cell encourages the cell to take up cations to maintain electric homeostasis, including Ca2+ [30]. The uptake of Ca2+ promotes ferroptosis [28].
Ferroptosis can be observed in both healthy tissues and tumors. Ferroptosis demonstrates toxic effects in healthy tissues. In the cardiovascular system, ferroptosis has been implicated in heart failure and atherosclerosis [31]. Similarly, in the nervous system, ferroptosis is observed in various neurological disorders, including neurodegenerative diseases, brain injuries, multiple sclerosis, aging, and neuroinflammation [32].
1.3. Cysteine Dioxygenase Type 1
Cysteine dioxygenase type 1 (Cdo1) is an enzyme that catalyzes the oxidation of cysteine; hence, it plays a pivotal role in cysteine metabolism by regulating the amount of cellular cysteine [20]. Cdo1 catalyzes the conversion of cysteine to its sulfinic acid, which is further metabolized in the body [33]. Cysteine is essential for the production of glutathione (GSH), a vital cellular antioxidant [34]. Since the amount of available cysteine affects the sensitivity to oxidative stress in cells, the gene Cdo1 plays a key role here [20]. Furthermore, oxidative stress is vital in inducing cell death, e.g., by apoptosis and ferroptosis. Induced cell death as an important direction in cancer chemotherapy [20], where Cdo1 has been shown to increase the vulnerability of cells [35,36]. Moreover, and importantly, a decrease in the active Cdo1 enzyme by mutation or downregulation can be seen in various types of cancers.
2. Ferroptosis and Apoptosis in Cancer
2.1. Ferroptosis in Cancer
2.1.1. Two-Sided Role of Ferroptosis in Cancer
Ferroptosis, a type of programmed cell death, is initiated by cysteine deprivation and carried out through lipid peroxidation [18,37]. In cancer cells, metabolism is heightened, leading to elevated reactive oxygen species (ROS), and subsequently, increased oxidative stress [38]. Additionally, it has been demonstrated that cancer cells have a greater need for iron, which serves as a co-factor for numerous enzymes involved in ferroptosis and impacts lipid peroxidation [16]. Consequently, cancer cells are theoretically more vulnerable to ferroptosis, and indeed, this form of cell death has been frequently observed in various cancers, including fibroblastoma, lung cancer, osteosarcoma, kidney cancer, and prostate cancer [18].
2.1.2. Inducing Ferroptosis to Combat Cancer
The human immune system is capable of inducing ferroptosis in tumor cells [39]; CD8+ T cells demonstrate their anticancer effects by releasing the cytokine IFN-γ, which triggers ferroptosis [39]. IFN-γ significantly reduces the expression of two subunits of the cystine/glutamate antiporter, SLC755A and SLC3A2, in tumor cells through activating the JAK-STAT signaling pathway [39]. This action decreases the antiporter and an inhibition of cellular uptake of cysteine, resulting in uncontrolled lipid peroxidation and ultimately ferroptosis [39]. Some tumor cells express PD-L1 on their surface, which binds to PD-1 on CD8+ T cells’ surface. As a result, these T cells fail to release cytokines, including IFN-γ, and do not exert cytotoxicity, allowing the bound tumor cell to evade the immune response [40]. Blocking this pivotal escape route has been pursued in immunotherapy; drugs that block the binding of PD-1 to PD-L1 allow CD8+ T cells not to be interfered with by PD-L1 on the tumor cell membrane, so that these CD8+ T cells can still exert cytotoxic effects to kill the cancer cell and release cytotoxic factors, including IFN-γ [41].
There are several drugs targeting ferroptosis directly. Among them, Erastin and RSL3 ((1S,3R)-RSL3) are representative [42]. Erastin has the ability to enhance the susceptibility of cancer cells to other anti-cancer drugs. Its mechanism of action involves inhibiting the expression of the subunit of the cysteine-glutamate antiporter SLC755A. This inhibition hinders the function of the antiporter, limiting the cells’ ability to uptake cysteine [43]. RSL3 binds directly to GPX4, inhibiting the action of GPX4, thus blocking the pathway that protects cancer cells from ferroptosis [44].
2.2. Apoptosis in Cancer
2.2.1. Apoptosis and Carcinogenesis
Apoptosis presents tumor-suppressing roles in cancer [45]. Under physiological conditions, the mutated cancerous cells are detected by the immune system and killed [46]. By these means, apoptosis can prevent the formation of cancer through cell death [46]. However, one crucial change among the malignant genetic transformation of cells toward cancer is the ability to evade cell death, including apoptosis [46]. Therefore, when cells evade apoptosis, carcinogenesis develops. Cancer cells evade apoptosis mainly through three kinds of pathways: losing balance between pro-apoptotic and anti-apoptotic proteins, inhibiting caspases’ functions, as well as death receptor signaling [5]. For instance, p53 is involved in death signaling as well as inhibiting cell division, and its mutation induces cancer [47]. p53 mutation is associated with over 54% of cancer types, such as melanoma [47].
2.2.2. Apoptosis in the Treatment of Cancer
Chemotherapeutic drugs induce apoptosis in cancer cells. For instance, Phikan088, a small molecule and carbazole derivative, has been proved to attach to mutated p53, thereby restoring the normal role of p53 in promoting apoptosis [48]. Therefore, developing drugs targeting molecules involved in apoptosis, such as BCL and p53, and regulating apoptosis, for example, Cdo1, is meaningful [49].
3. Ferroptosis and Cdo1
Cdo1 promotes ferroptosis. Experiments inhibiting Cdo1 production in mice indicate that Cdo1 promotes ferroptosis by increasing the oxidative stress as well as inhibiting the production of GPX4 [36,50]. Upregulation of Cdo1 in ferroptosis is thought to be regulated by c-Myb (c-Myb proto-oncogene transcription factor) by an unknown mechanism [36]. C-Myb encodes a transcription factor that directly interacts with the promoter of Cdo1 [51]. C-Myb is thought to upregulate Cdo1 in ferroptosis as cysteine enhances its DNA binding state, thereby upregulating the transcription of Cdo1 [36]. In addition, both the expression of c-Myb and Cdo1 are decreased in Erastin-induced ferroptosis in a dose-related fashion, meaning that the expressions of c-Myb and Cdo1 are positively correlated [36]. Therefore, it is reasonable to infer that c-Myb upregulated Cdo1 during ferroptosis [36].
High expression of Cdo1 leads to significant ferroptosis, reducing the number of tumor cells in many cases, and plays a role in suppressing cancer rather than targeting healthy cancer cells [18]. Cdo1 is part of the larger ferroptosis network that includes the cystine/glutamate antiporter and GPX4; therefore, the activity of each network member contributes to the activation or inhibition of ferroptosis (Figure 2). For example, the promotion of ferroptosis by Cdo1 can be influenced by ion channels that increase Ca2+ influx, thereby inhibiting GPX4. If, in cancer, the expression of these ion channels is reduced or dysfunctional, intracellular Ca2+ can be reduced, and GPX4 function is preserved [29], which counteracts GSH depletion by Cdo1 [18,20]. Moreover, dietary cystine availability should affect the concentration of cellular cysteine and thereby the amount that can be transformed by Cdo1. How nutrient availability is not only influenced by the diet, but also by the gut microbiome, is a current research question. Interestingly, it has been shown that traditional Chinese medicine (TCM) can elevate cysteine and methionine metabolism in the rat microbiome [52].
The off-target effects of Cdo1 upregulation should be considered. Cdo1 can promote ferroptosis based on the current study, but ferroptosis has negative aspects to the body other than cancer. For normal tissues, ferroptosis can easily cause diseases, for example, in the cardiovascular system, ferroptosis can lead to cardiomyopathy, myocardial infarction, and a series of cardiomyopathies, even heart failure [53,54]. In terms of metabolism, the off-target effect of Cdo1 upregulation is still unclear and needs further study. In addition to drugs that specifically target ferroptosis, there are also a variety of agents that unexpectedly cause ferroptosis and are already in clinical use, such as cisplatin, sorafenib, and statin [55,56]. These drugs can lead to an off-target effect. Cisplatin promotes ferroptosis by inhibiting GPX4, and its off-target effect is mainly reflected in its nephrotoxicity [55,56,57]. Sorafenib induces ferroptosis by increasing intracellular iron levels and inhibiting the cystine/glutamate antiporter, and its off-target effect is mainly due to skin toxicity and causes diarrhea and arterial hypertension in patients [55,57,58]. Statin induces ferroptosis mainly by inhibiting two protective pathways containing GPX4 and FSP1, and its off-target effect is mainly to increase the Hemorrhagic Stroke Risk, as well as the myopathic effect [55,57,59].
4. Apoptosis and Cdo1
4.1. Detailed Tumor-Suppressing Role of Cdo1 in Cancer
Yang et al. found that in Cdo1-overexpressing breast cancer cells, the expression levels of tumor suppressors such as PTEN and BAX were increased, whereas the expression levels of proto-oncogenes such as PI3K and AKT, which promote cell division, were decreased [60]. In general, the gene expression analysis showed that Cdo1-overexpressing breast cancer cells had lower expression of metastatic and aggressiveness-related genes [60]. In cell culture, Cdo1-overexpressing cells divide much slower and form many fewer tribes than other tumor cells [60]. In breast cancer patients, those with Cdo1 promoter hypermethylation showed a worse prognosis. This experiment illustrates the tumor-suppressing role of Cdo1 both in terms of inhibiting cell division (PTEN, p53, PI3K, AKT) and promoting apoptosis (p53, BAX) [60].
4.2. Cdo1 Influences Lipid Peroxidation during Apoptosis
With Cdo1 decreasing the cytosolic antioxidants cysteine and GSH, lipid peroxidation is more likely to happen [34]. It has been shown that lipid peroxidation is able to trigger and enhance apoptosis [18,61].
The products of lipid peroxidation affect the expression of elements involved in apoptotic signaling and cause DNA damage [62,63,64]. The inhibitor of kappa B kinase (IKK), a product of lipid peroxidation, is able to phosphorylate BCL proteins, enhancing apoptosis [65]. In addition, products of lipid peroxidation were found to form adducts with Jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK), p38, and molecules activating mitogen-activated protein kinases (MAPKs), thereby activating these enzymes in apoptosis. Activated MAPKs are required for activating caspases, the executioners of apoptosis [66,67]. Furthermore, it is speculated that the lipid peroxidation product activates protein kinase C-delta (PKCδ) [68]. When PKCδ is cleaved by caspase-3, an activated catalytic fragment can be generated, amplifying apoptosis cascades [69].
4.3. Taurine, a Product of Cysteine Oxidation, Promotes p53 Activity
Taurine is the end product of cysteine oxidation catalyzed by Cdo1 [20]. It was previously thought that taurine could only be produced in the liver, but it is now evident that taurine can also be produced in cells other than hepatocytes [70,71]. Taurine has an anti-tumor function, which promotes the production of p53, so that p53 can better perform its anti-oncogenic function [72,73].
Recent studies have found that elevated Cdo1 expression can also enhance the tumor suppressor effects of p53 [35]. Given that the production of taurine requires the catalysis of Cdo1, it is appropriate to speculate that Cdo1 enhances the tumor-suppressing effect of p53 by promoting the production of taurine. By increasing taurine expression of p53, Cdo1 indirectly exerts a tumor-suppressing effect [20,35].
5. Mild Tumor-Suppressing Role of Cdo1 in Cancer
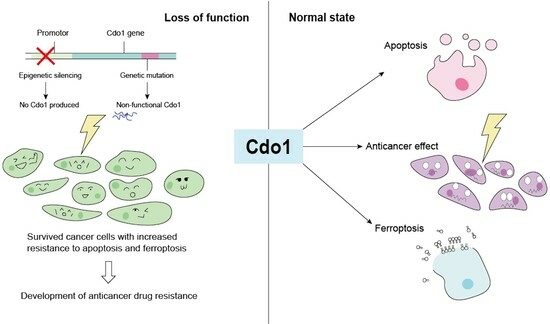
Currently, a large number of studies have confirmed that increasing the expression level of Cdo1 is able to inhibit cancer development by promoting cancer cell death, particularly by ferroptosis [60]. However, no study has found that silencing mutations in Cdo1 alone can cause cancer.
In some cancer cells, Cdo1 expression is promoted, but this promotion still fails to prevent carcinogenesis. This phenomenon implies that the anti-oncogenic effect of Cdo1 is not strong [74]. Instead, Cdo1 has a stronger effect on inhibiting malignancy than carcinogenesis [60]. The mild tumor-suppressing effect is illustrated for cells harboring mutation in the transcription factor Nrf2 [75]. Nrf2 promotes the role of Cdo1 and facilitates taurine production. Both Cdo1 and taurine have apoptosis-promoting properties, and Cdo1 also has ferroptosis-promoting properties. Therefore, from the perspective of Cdo1, this mutation is favorable for the tumor-suppressing effect of Cdo1 [75]. However, the overexpression of Nrf2 simultaneously promotes the uptake of cysteine, which is involved in the pathway that protects cells from ferroptosis [76]. This counteracts the anti-oncogenic effect of promoting Cdo1 expression. The fact that cells remained cancerous in the presence of these two opposing effects suggests that the enhancement of the tumor-suppressing effect of Cdo1 was not weaker than the promotion of cysteine uptake by Nrf2, and that cells were protected from ferroptosis [75].
6. Cdo1 Alterations in Cancer Cells
Although Cdo1 mutation is common in cancer (Figure 3, downloaded from cBioportal (April 2024)) [77], it must be clarified that mutations in the Cdo1 gene are not driver mutations of cancer [77]. Furthermore, it is worth mentioning that due to the role of Cdo1 in enhancing apoptosis and cell cycle arrest, but not inducing apoptosis or cell cycle arrest, it is suggested that Cdo1 mutation cannot be the driver mutation leading to cancer [78].
6.1. Genetic Mutations and Structural Variations
Observed genetic mutations comprise missense mutation, splicing mutation, and truncating mutation (Figure 4, downloaded from cBioportal (April 2024)) [78]. Missense mutation is the most common type, accounting for about 82% of all genetic alterations of Cdo1 (Figure 4) [78]. Splicing mutations and missense mutations have similar outcomes: the production of non-functional Cdo1 [78]. Structural variation is even less common than genetic mutation [78]. Moreover, Cdo1 mutation can be seen in every cancer type, but only in a low percentage [78], with the highest percentage in lung cancer being still less than 10% (Figure 4). Cancer types in which Cdo1 mutation and structural variation are relatively common are melanoma, breast cancer, and pancreatic cancer [77]. Since Cdo1 mutation is not the driver mutation leading to tumorigenesis, it is understandable that Cdo1 genetic alterations occur at low frequency. Figure 3 demonstrates that missense mutations have occurred at nearly every position of the Cdo1 gene. The most common splice mutation usually occurs at the beginning and the end of exon 2. There exist truncations in the middle of exon 1 and the middle of exon 2.
6.2. Epigenetic Silencing of Cdo1
Epigenetic silencing is cancer-specific, and the most obvious form is methylation of the Cdo1 promoter [78]; generally, methylation of a gene’s promoter inhibits its transcription [79]. Methylation of the Cdo1 promoter can be observed in various cancers, including colorectal cancer, breast cancer, and gastric cancer [80,81,82]. Owing to its frequent occurrence in cancers, Cdo1 methylation has become a biomarker for various types of cancers [83], and blood testing for the methylated Cdo1 promoter offers useful information in detecting cancer in patients [83]. It is reasonable to assume that Cdo1 expression can be regulated through histone modification [84]. However, the effect of histone modification on Cdo1 expression is not clear. Therefore, this section will focus on the mechanism of DNA methylation.
Currently, the mechanism of methylation of the Cdo1 promoter is not fully understood. There are two theories. ① The methylation of the targeted CpG sequence by DNA methyltransferase (DNMT) is enhanced [85]; DNA methyltransferase adds methyl groups to the nucleotides, especially the CpG nucleotide [86]. ② This specific gene methylation is just a consequence of cell proliferation, especially in acute mononuclear leukemia [87]. Two possible mechanisms supporting the first theory are introduced (Figure 5).
The first theory suggests that a methylated tumor suppressor gene makes the cell cancerous [88]. The methylation of the Cdo1 promoter is induced by Chd4/NuRD chromatin remodeling factors in tumor cells [88,89]. In normal cells, the Chd4/NuRD chromatin remodeling factors appear to cue the DNMT site of action only after DNA damage to Cdo1 [90]. Such methylation in a tumor suppressor gene can directly make the cell cancerous, leaving the tumor suppressor gene promotor methylated and non-functional [88]. The exact mechanism of this phenomenon has not been investigated [88]. However, regarding the mild tumor-suppressing effect of Cdo1, it could be possible that this epigenetic methylation is generated after carcinogenesis because cancer induced by Cdo1 silencing has not been found.
Another possible mechanism supporting the first theory is the ten-eleven translocation (TET)-mediated pathway dysregulation [88]. TET-dependent 5-hydroxymethylation is crucial in preserving the unmethylated state of normal cells’ CpG islands, thus exhibiting the opposite function to DNMT [90,91]. The most recent study found that hypermethylation is relevant to a bivalent promoter [92]. This study illustrated that some promoters showed partial methylation of the CpG island edges, resulting in the formation of smaller, unmethylated CpG islands [88]. The adjacent borders of these smaller unmethylated CpG islands are targeted by DNMT3A and TET. 5hmC (5-hydroxymethylcytosine) and DNA methylation inhibits binding, maintaining the length of the island [93,94]. The protein recruiting DNMT4 and TET in this process is H3K4me1 [92]. This protein marks the TET-mediated 5hmC at the borders of the unmethylated CpG islands [95]. In cancer cells, the activity of TET is lost, and the 5hmC level is low, indicating that there is no competition with DNMT and the binding of DNMT is allowed. This mechanism is seen in acute myeloid leukemia (AML), where CpG island methylation contributes to the upregulated proliferation of cancer cells in AML [95]. Considering the mechanism of epigenetic silencing of tumor-suppressing genes in cancer, targeting DNMTs in cancer therapy is supposed to be a powerful direction in chemotherapy [88].
Since the expression of Cdo1 is inhibited by methylation, its tumor-suppressing effects, including the promotion of apoptosis and ferroptosis, are limited [60]. Therefore, epigenetic methylation has an important inhibitory effect on the function of Cdo1. In cancer cells, methylation is the most common variant of Cdo1, and although it is not sufficient to make cells cancerous, the presence of this variant can exacerbate the progression of cancer.
7. Conclusions and Outlook
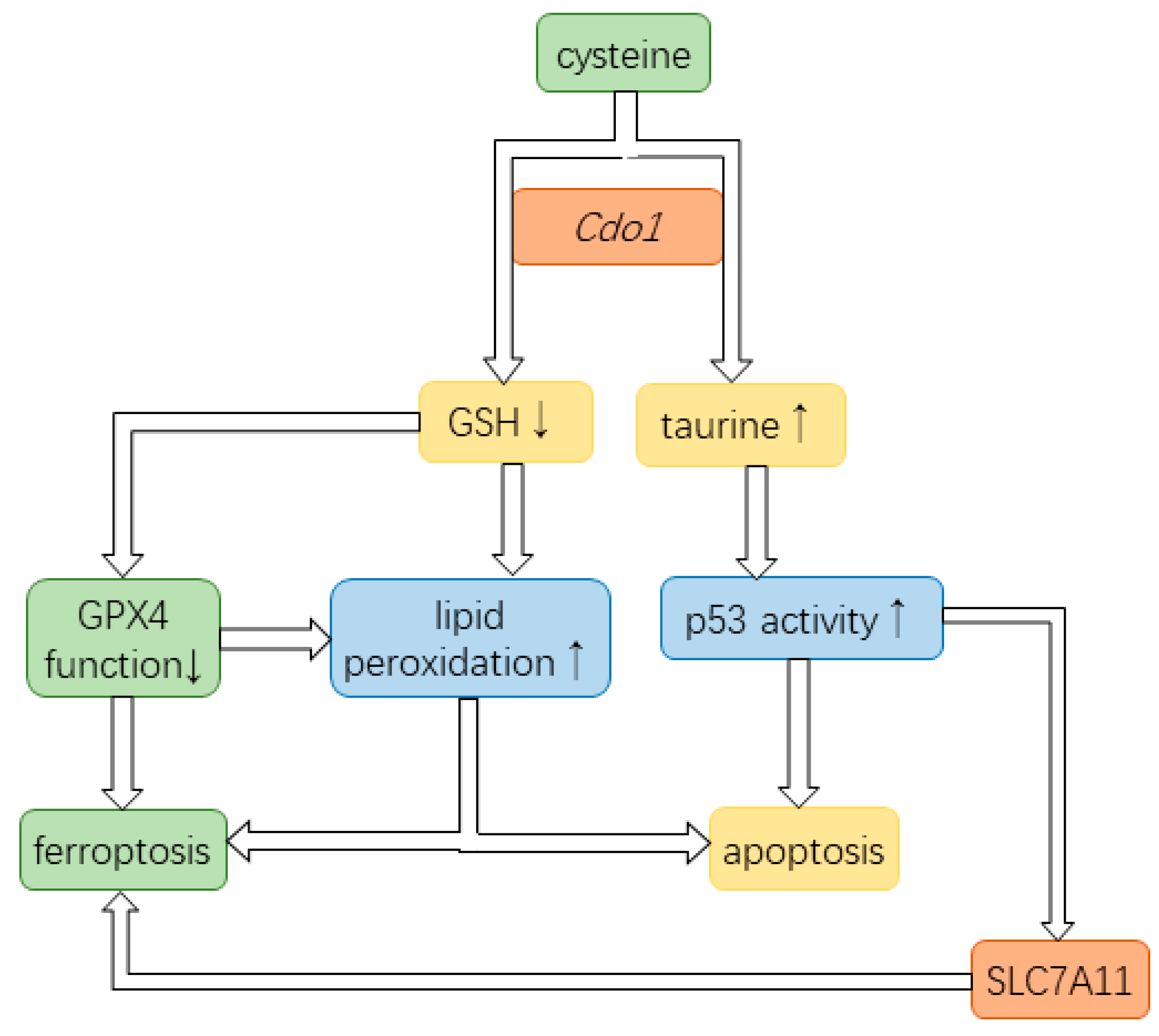
Cdo1 is considered a tumor suppressor as it enhances ferroptosis and apoptosis in cancer cells. Mechanistically, Cdo1 enhances ferroptosis by promoting cysteine starvation, thereby also reducing GSH levels [17]. GSH is the cofactor of GPX4, the enzyme protecting a cell from autoperoxidation of membranes [17] (Figure 6). In conclusion, Cdo1 conducts its tumor-suppressing role by reducing the antioxidants protecting cells from ferroptosis [20]. Moreover, Cdo1 promotes apoptosis by aggravating lipid peroxidation as well as upregulating the activity of p53 [60,61] (Figure 6).
Although the tumor-suppressing effect of Cdo1 is mild, it plays important roles in assisting anticancer agents in overcoming drug resistance. For instance, Cdo1 plays a significant role in mediating Erastin-induced ferroptosis in gastric cancer cells [36]. When the Cdo1 gene is silenced, ferroptosis does not occur even though Erastin is applied [36]. Furthermore, epigenetic silencing of Cdo1 contributes to resistance against ROS-generating chemotherapeutic drugs, including anthracycline [96]. Therefore, upregulating Cdo1 in cancer cells is likely to assist the effect of major chemotherapeutic agents.
In conclusion, Cdo1 conducts the tumor-suppressing role in cancer cells by contributing to ferroptosis as well as enhancing the anti-tumor effects of p53, thereby mediating apoptosis and cell cycle arrest. Furthermore, p53 enhances ferroptosis (Figure 6). Therefore, Cdo1 is a useful cancer drug target. Based on the current literature, combined anticancer therapy ensuring high Cdo1 activity should be a powerful approach to ensure and enhance the effect of anti-cancer drugs and to impede the development of drug resistance in cancer cells.
Author Contributions
Conceptualization, A.P. and X.C.; software, A.P. and X.C; validation, Ansgar A.P. and X.C; investigation, A.P. and X.C; writing—original draft preparation A.P. and X.C.; writing—review and editing, A.P.; supervision, A.P.; project administration A.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors thank Wangya Yang for proofreading the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Pouyssegur, J. Tumor Cell Metabolism: Cancer’s Achilles’ Heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Mattiuzzi, C. Current Cancer Epidemiology. J. Epidemiol. Glob. Health 2019, 9. [Google Scholar]
- Zhao, L.; Zhou, X.; Xie, F.; Zhang, L.; Yan, H.; Huang, J.; Zhang, C.; Zhou, F.; Chen, J.; Zhang, L. Ferroptosis in cancer and cancer immunotherapy. Cancer Commun. 2022, 42, 88–116. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S.Y. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef]
- Bates, S.; Vousden, K.H. Mechanisms of p53-mediated apoptosis. Cell. Mol. Life Sci. 1999, 55, 28–37. [Google Scholar] [CrossRef]
- Schneider, P.; Tschopp, J. Apoptosis induced by death receptors. Pharm. Acta Helv. 2000, 74, 281–286. [Google Scholar] [CrossRef]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef]
- Danial, N.N.; Korsmeyer, S.J. Cell death: Critical control points. Cell 2004, 116, 205–219. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z.; Verma, C.S. Tumor suppressor p53 and metabolism. J. Mol. Cell Biol. 2019, 1, 284–292. [Google Scholar] [CrossRef]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Cecconi, F.; Alvarez-Bolado, G.; Meyer, B.I.; Roth, K.A.; Gruss, P. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell 1998, 94, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Selvakumaran, M.; Lin, H.K.; Miyashita, T.; Wang, H.G.; Krajewski, S.; Reed, J.C.; Hoffman, B.; Liebermann, D. Immediate early up-regulation of bax expression by p53 but not TGF beta 1: A paradigm for distinct apoptotic pathways. Oncogene 1994, 9, 1791–1798. [Google Scholar] [PubMed]
- Reed, J.C. Bcl-2 family proteins: Regulators of apoptosis and chemoresistance in hematologic malignancies. Semin. Hematol. 1997, 34 (Suppl. 5), 9–19. [Google Scholar] [PubMed]
- Campioni, M.; Santini, D.; Tonini, G.; Murace, R.; Dragonetti, E.; Spugnini, E.P.; Baldi, A. Role of Apaf-1, a key regulator of apoptosis, in melanoma progression and chemoresistance. Exp. Dermatol. 2005, 14, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2020, 31, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Fenton, H.J.H. LXXIII.—Oxidation of tartaric acid in presence of iron. J. Chem. Soc. Trans. 1894, 65, 899–910. [Google Scholar] [CrossRef]
- Chen, M.; Zhu, J.-Y.; Mu, W.-J.; Guo, L. Cysteine dioxygenase type 1 (CDO1): Its functional role in physiological and pathophysiological processes. Genes Dis. 2023, 10, 877–890. [Google Scholar] [CrossRef]
- Paul, B.D.; Sbodio, J.I.; Snyder, S.H. Cysteine Metabolism in Neuronal Redox Homeostasis. Trends Pharmacol. Sci. 2018, 39, 513–524. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Grocin, A.G.; da Silva, T.N.X.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Kraft, V.A.N.; Bezjian, C.T.; Pfeiffer, S.; Ringelstetter, L.; Müller, C.; Zandkarimi, F.; Merl-Pham, J.; Bao, X.; Anastasov, N.; Kössl, J.; et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent. Sci. 2020, 6, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Lima, P.H.C.; Butera, A.P.; Cabeça, L.F.; Ribeiro-Viana, R.M. Liposome surface modification by phospholipid chemical reactions. Chem. Phys. Lipids 2021, 237, 105084. [Google Scholar] [CrossRef] [PubMed]
- Blagih, J.; Buck, M.D.; Vousden, K.H. p53, cancer and the immune response. J. Cell Sci. 2020, 133, jcs237453. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Kroemer, G.; Tang, D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic. Biol. Med. 2019, 133, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, W.; Zhang, P.; Wang, Z.; Ma, X.; Liu, C.; Vasilev, K.; Zhang, L.; Zhou, X.; Liu, L.; et al. Mechanical overloading induces GPX4-regulated chondrocyte ferroptosis in osteoarthritis via Piezo1 channel facilitated calcium influx. J. Adv. Res. 2022, 41, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Hyun, J. Mechanosensitive ion channels in apoptosis and ferroptosis: Focusing on the role of Piezo1. BMB Rep. 2023, 56, 145–152. [Google Scholar] [CrossRef]
- Guo, J.; Song, Z.; Yu, J.; Li, C.; Jin, C.; Duan, W.; Liu, X.; Liu, Y.; Huang, S.; Tuo, Y.; et al. Hepatocyte-specific TMEM16A deficiency alleviates hepatic ischemia/reperfusion injury via suppressing GPX4-mediated ferroptosis. Cell Death Dis. 2022, 13, 1072. [Google Scholar] [CrossRef]
- Fang, X.; Ardehali, H.; Min, J.; Wang, F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat. Rev. Cardiol. 2023, 20, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Parsons, R.B.; Waring, R.H.; Ramsden, D.B.; Williams, A.C. Toxicity of cysteine and cysteine sulphinic acid to human neuronal cell-lines. J. Neurol. Sci. 1997, 152 (Suppl. 1), S62–S66. [Google Scholar] [CrossRef] [PubMed]
- Turell, L.; Zeida, A.; Trujillo, M. Mechanisms and consequences of protein cysteine oxidation: The role of the initial short-lived intermediates. Essays Biochem. 2020, 64, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 3217–3266. [Google Scholar] [CrossRef] [PubMed]
- Booken, N.; Gratchev, A.; Utikal, J.; Weiß, C.; Yu, X.; Qadoumi, M.; Schmuth, M.; Sepp, N.; Nashan, D.; Rass, K.; et al. Sézary syndrome is a unique cutaneous T-cell lymphoma as identified by an expanded gene signature including diagnostic marker molecules CDO1 and DNM3. Leukemia 2008, 22, 393–399. [Google Scholar] [CrossRef]
- Hao, S.; Yu, J.; He, W.; Huang, Q.; Zhao, Y.; Liang, B.; Zhang, S.; Wen, Z.; Dong, S.; Rao, J.; et al. Cysteine Dioxygenase 1 Mediates Erastin-Induced Ferroptosis in Human Gastric Cancer Cells. Neoplasia 2017, 19, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Ren, Y.; Chen, S.; Wang, Y.; Chu, L. Ferroptosis and tumor immunotherapy: A promising combination therapy for tumors. Front. Oncol. 2023, 13, 1119369. [Google Scholar] [CrossRef]
- Lei, Q.; Wang, D.; Sun, K.; Wang, L.; Zhang, Y. Resistance Mechanisms of Anti-PD1/PDL1 Therapy in Solid Tumors. Front. Cell Dev. Biol. 2020, 8, 672. [Google Scholar] [CrossRef]
- Topalian, S.L.; Taube, J.M.; Pardoll, D.M. Neoadjuvant checkpoint blockade for cancer immunotherapy. Science 2020, 367, eaax0182. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.J.; Li, D.; Ou, Y.; Jiang, L.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell Rep. 2016, 17, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, Y.; Zhang, R.; Wang, F.; Wang, T.; Jiao, Y. The Role of Erastin in Ferroptosis and Its Prospects in Cancer Therapy. OncoTargets Ther. 2020, 13, 5429–5441. [Google Scholar] [CrossRef]
- Sui, X.; Zhang, R.; Liu, S.; Duan, T.; Zhai, L.; Zhang, M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front. Pharmacol. 2018, 9, 1371. [Google Scholar] [CrossRef]
- Kashyap, D.; Garg, V.K.; Goel, N. Intrinsic and extrinsic pathways of apoptosis: Role in cancer development and prognosis. Adv. Protein Chem. Struct. Biol. 2021, 125, 73–120. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.S.; Mhoumadi, Y.; Verma, C.S. Roles of computational modelling in understanding p53 structure, biology, and its therapeutic targeting. J. Mol. Cell Biol. 2019, 11, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Boeckler, F.M.; Joerger, A.C.; Jaggi, G.; Rutherford, T.J.; Veprintsev, D.B.; Fersht, A.R. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl. Acad. Sci. USA 2008, 105, 10360–10365. [Google Scholar] [CrossRef] [PubMed]
- Yamada, A.; Kogure, Y. I. Review of Cytotoxic Chemotherapy for Non-Small Cell Lung Cancer. Gan To Kagaku Ryoho 2020, 47, 1165–1170. [Google Scholar]
- Ma, G.; Zhao, Z.; Qu, Y.; Cai, F.; Liu, S.; Liang, H.; Zhang, R.; Deng, J. Cysteine dioxygenase 1 attenuates the proliferation via inducing oxidative stress and integrated stress response in gastric cancer cells. Cell Death Discov. 2022, 8, 493. [Google Scholar] [CrossRef]
- Guehmann, S.; Vorbrueggen, G.; Kalkbrenner, F.; Moelling, K. Reduction of a conserved Cys is essential for Myb DNA-binding. Nucleic Acids Res. 1992, 20, 2279–2286. [Google Scholar] [CrossRef] [PubMed]
- Qu, W.; Liu, S.; Zhang, W.; Zhu, H.; Tao, Q.; Wang, H.; Yan, H. Impact of traditional Chinese medicine treatment on chronic unpredictable mild stress-induced depression-like behaviors: Intestinal microbiota and gut microbiome function. Food Funct. 2019, 10, 5886–5897. [Google Scholar] [CrossRef]
- Wu, X.; Li, Y.; Zhang, S.; Zhou, X. Ferroptosis as a novel therapeutic target for cardiovascular disease. Theranostics. 2021, 11, 3052–3059. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Tian, X.M.; Li, W.; Hao, L.Y. Ferroptosis in cardiac hypertrophy and heart failure. Biomed. Pharmacother. 2023, 168, 115765. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Meng, Y.; Li, D.; Yao, L.; Le, J.; Liu, Y.; Sun, Y.; Zeng, F.; Chen, X.; Deng, G. Ferroptosis in cancer: From molecular mechanisms to therapeutic strategies. Signal Transduct. Target. Ther. 2024, 9, 55. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Tang, L.; Zhang, Y.; Ge, G.; Jiang, X.; Mo, Y.; Wu, P.; Deng, X.; Li, L.; Zuo, S.; et al. Regulatory pathways and drugs associated with ferroptosis in tumors. Cell Death Dis. 2022, 13, 544. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, A.; Dinda, A.K.; Mukhopadhyay, C.K. Effect of Cisplatin on Renal Iron Homeostasis Components: Implication in Nephropathy. ACS Omega 2022, 7, 27804–27817. [Google Scholar] [CrossRef] [PubMed]
- Di Costanzo, G.G.; de Stefano, G.; Tortora, R.; Farella, N.; Addario, L.; Lampasi, F.; Lanza, A.G.; Cordone, G.; Imparato, M.; Caporaso, N. Sorafenib off-target effects predict outcomes in patients treated for hepatocellular carcinoma. Future Oncol. 2015, 11, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Stephens, M.; Roizes, S.; von der Weid, P.Y. Off-Target Effect of Lovastatin Disrupts Dietary Lipid Uptake and Dissemination through Pro-Drug Inhibition of the Mesenteric Lymphatic Smooth Muscle Cell Contractile Apparatus. Int. J. Mol. Sci. 2021, 22, 11756. [Google Scholar] [CrossRef]
- Yang, J.; Sun, L.; Liu, X.Y.; Huang, C.; Peng, J.; Zeng, X.; Zheng, H.; Cen, W.; Xu, Y.; Zhu, W.; et al. Targeted demethylation of the CDO1 promoter based on CRISPR system inhibits the malignant potential of breast cancer cells. Clin. Transl. Med. 2023, 13, e1423. [Google Scholar] [CrossRef]
- Su, L.-J.; Zhang, J.-H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.-Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid. Med. Cell. Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef] [PubMed]
- Elkin, E.R.; Harris, S.M.; Loch-Caruso, R. Trichloroethylene metabolite S-(1,2-dichlorovinyl)-l-cysteine induces lipid peroxidation-associated apoptosis via the intrinsic and extrinsic apoptosis pathways in a first-trimester placental cell line. Toxicol. Appl. Pharmacol. 2018, 338, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.; Sharma, R.; Sharma, A.; Vatsyayan, R.; Yadav, S.; Singhal, S.S.; Rauniyar, N.; Prokai, L.; Awasthi, S.; Awasthi, Y.C. Mechanisms of 4-hydroxy-2-nonenal induced pro- and anti-apoptotic signaling. Biochemistry 2010, 49, 6263–6275. [Google Scholar] [CrossRef] [PubMed]
- Łuczaj, W.; Gęgotek, A.; Skrzydlewska, E. Antioxidants and HNE in redox homeostasis. Free Radic. Biol. Med. 2017, 111, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Bodur, C.; Kutuk, O.; Tezil, T.; Basaga, H. Inactivation of Bcl-2 through IκB kinase (IKK)-dependent phosphorylation mediates apoptosis upon exposure to 4-hydroxynonenal (HNE). J. Cell. Physiol. 2012, 227, 3556–3565. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Seo, I.; Choi, M.H.; Jeong, D. Roles of Mitogen-Activated Protein Kinases in Osteoclast Biology. Int. J. Mol. Sci. 2018, 19, 3004. [Google Scholar] [CrossRef] [PubMed]
- Preston, G.A.; Zarella, C.S.; Pendergraft, W.F., 3rd; Rudolph, E.H.; Yang, J.J.; Sekura, S.B.; Jennette, J.C.; Falk, R.J. Novel effects of neutrophil-derived proteinase 3 and elastase on the vascular endothelium involve in vivo cleavage of NF-kappaB and proapoptotic changes in JNK, ERK, and p38 MAPK signaling pathways. J. Am. Soc. Nephrol. 2002, 13, 2840–2849. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Burrington, C.M.; Davenport, S.K.; Johnson, A.K.; Horsman, M.J.; Chowdhry, S.; Greene, M.W. PKCδ regulates hepatic triglyceride accumulation and insulin signaling in Lepr(db/db) mice. Biochem. Biophys. Res. Commun. 2014, 450, 1619–1625. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Xia, L.; Chen, G.Q. Protein kinase cδ in apoptosis: A brief overview. Arch. Immunol. Ther. Exp. 2012, 60, 361–372. [Google Scholar] [CrossRef]
- Ueki, I.; Roman, H.B.; Hirschberger, L.L.; Junior, C.; Stipanuk, M.H. Extrahepatic tissues compensate for loss of hepatic taurine synthesis in mice with liver-specific knockout of cysteine dioxygenase. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1292–E1299. [Google Scholar] [CrossRef]
- He, F.; Ma, N.; Midorikawa, K.; Hiraku, Y.; Oikawa, S.; Mo, Y.; Zhang, Z.; Takeuchi, K.; Murata, M. Anti-Cancer Mechanisms of Taurine in Human Nasopharyngeal Carcinoma Cells. Adv. Exp. Med. Biol. 2019, 1155, 533–541. [Google Scholar] [PubMed]
- Guo, Y.Y.; Li, B.Y.; Xiao, G.; Liu, Y.; Guo, L.; Tang, Q.Q. Cdo1 promotes PPARγ-mediated adipose tissue lipolysis in male mice. Nat. Metab. 2022, 4, 1352–1368. [Google Scholar] [CrossRef] [PubMed]
- Ueki, I.; Roman, H.B.; Valli, A.; Fieselmann, K.; Lam, J.; Peters, R.; Hirschberger, L.L.; Stipanuk, M.H. Knockout of the murine cysteine dioxygenase gene results in severe impairment in ability to synthesize taurine and an increased catabolism of cysteine to hydrogen sulfide. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E668–E684. [Google Scholar] [CrossRef] [PubMed]
- Kubota, Y.; Tanabe, S.; Azuma, M.; Horio, K.; Fujiyama, Y.; Soeno, T.; Furue, Y.; Wada, T.; Watanabe, A.; Ishido, K.; et al. Predictive Significance of Promoter DNA Methylation of Cysteine Dioxygenase Type 1 (CDO1) in Metachronous Gastric Cancer. J. Gastric Cancer 2021, 21, 379–391. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.P.; Torrente, L.; Falzone, A.; Elkins, C.M.; Liu, M.; Asara, J.M.; Dibble, C.C.; DeNicola, G.M. Cysteine dioxygenase 1 is a metabolic liability for non-small cell lung cancer. eLife 2019, 8, e45572. [Google Scholar] [CrossRef] [PubMed]
- CBioportal (2023): Cancers Containing Cdo1 mutation [Pancancer Database]. Available online: https://www.cbioportal.org/results/cancerTypesSummary?tab_index=tab_visualize&Action=Submit&session_id=6622277083e9543d61902383&plots_horz_selection=%7B%7D&plots_vert_selection=%7B%7D&plots_coloring_selection=%7B%7D (accessed on 15 April 2024).
- CBioportal (2023): Types of Cdo1 mutation [Pancancer Database]. Available online: https://www.cbioportal.org/results/mutations?tab_index=tab_visualize&Action=Submit&session_id=6622277083e9543d61902383&plots_horz_selection=%7B%7D&plots_vert_selection=%7B%7D&plots_coloring_selection=%7B%7D (accessed on 15 April 2024).
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Harada, H.; Hosoda, K.; Moriya, H.; Mieno, H.; Ema, A.; Ushiku, H.; Washio, M.; Nishizawa, N.; Ishii, S.; Yokota, K.; et al. Cancer-specific promoter DNA methylation of Cysteine dioxygenase type 1 (CDO1) gene as an important prognostic biomarker of gastric cancer. PLoS ONE 2019, 14, e0214872. [Google Scholar] [CrossRef] [PubMed]
- Kojima, K.; Nakamura, T.; Ohbu, M.; Katoh, H.; Ooizumi, Y.; Igarashi, K.; Ishii, S.; Tanaka, T.; Yokoi, K.; Nishizawa, N.; et al. Cysteine dioxygenase type 1 (CDO1) gene promoter methylation during the adenoma-carcinoma sequence in colorectal cancer. PLoS ONE 2018, 13, e0194785. [Google Scholar] [CrossRef]
- Minatani, N.; Waraya, M.; Yamashita, K.; Kikuchi, M.; Ushiku, H.; Kojo, K.; Ema, A.; Nishimiya, H.; Kosaka, Y.; Katoh, H.; et al. Prognostic Significance of Promoter DNA Hypermethylation of cysteine dioxygenase 1 (CDO1) Gene in Primary Breast Cancer. PLoS ONE 2016, 11, e0144862. [Google Scholar] [CrossRef]
- Meller, S.; Zipfel, L.; Gevensleben, H.; Dietrich, J.; Ellinger, J.; Majores, M.; Stein, J.; Sailer, V.; Jung, M.; Kristiansen, G.; et al. CDO1 promoter methylation is associated with gene silencing and is a prognostic biomarker for biochemical recurrence-free survival in prostate cancer patients. Epigenetics 2016, 11, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, Z.; Jia, J.; Du, T.; Zhang, N.; Tang, Y.; Fang, Y.; Fang, D. Overview of Histone Modification. Adv. Exp. Med. Biol. 2021, 1283, 1–16. [Google Scholar] [PubMed]
- Simon, J.A.; Lange, C.A. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat. Res. 2008, 647, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Aguilera, O.; Depreux, P.; Halby, L.; Arimondo, P.B.; Goossens, L. DNA Methylation Targeting: The DNMT/HMT Crosstalk Challenge. Biomolecules 2017, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar] [PubMed]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA methylation landscape of cancer. Trends Genet. 2021, 37, 1012–1027. [Google Scholar] [CrossRef]
- O’Hagan, H.M.; Wang, W.; Sen, S.; Destefano Shields, C.; Lee, S.S.; Zhang, Y.W.; Clements, E.G.; Cai, Y.; Van Neste, L.; Easwaran, H.; et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell 2011, 20, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, Y.; Salz, T.; Hansen, K.D.; Feinberg, A. Whole-genome analysis of the methylome and hydroxymethylome in normal and malignant lung and liver. Genome Res. 2016, 26, 1730–1741. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Lu, Y.; Jelinek, J.; Liang, S.; Estecio, M.R.; Barton, M.C.; Issa, J.-P.J. TET1 is a maintenance DNA demethylase that prevents methylation spreading in differentiated cells. Nucleic Acids Res. 2014, 42, 6956–6971. [Google Scholar] [CrossRef]
- Skvortsova, K.; Masle-Farquhar, E.; Luu, P.L.; Song, J.Z.; Qu, W.; Zotenko, E.; Gould, C.M.; Du, Q.; Peters, T.J.; Colino-Sanguino, Y.; et al. DNA Hypermethylation Encroachment at CpG Island Borders in Cancer Is Predisposed by H3K4 Monomethylation Patterns. Cancer Cell 2019, 35, 297–314.e8. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Spencer, D.H.; Russler-Germain, D.A.; Ketkar, S.; Helton, N.M.; Lamprecht, T.L.; Fulton, R.S.; Fronick, C.C.; O’Laughlin, M.; Heath, S.E.; Shinaw, M.; et al. CpG Island Hypermethylation Mediated by DNMT3A Is a Consequence of AML Progression. Cell 2017, 168, 801–816.e13. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, J.; O’Hagan, H.M.; Zhang, W.; Vatapalli, R.; Calmon, M.F.; Danilova, L.; Nelkenbrecher, C.; Van Neste, L.; Bijsmans, I.T.G.W.; Van Engeland, M.; et al. Frequent inactivation of cysteine dioxygenase type 1 contributes to survival of breast cancer cells and resistance to anthracyclines. Clin. Cancer Res. 2013, 19, 3201–3211. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Known pathways protecting cells from ferroptosis. The 3 known pathways involve GPX4 (①), FSP1 (②), and BH4 generated from GCH1 (③), respectively. The 3 pathways have the same effect: protecting the cells from further lipid peroxidation by reducing PLOOH levels. Iron [II/III] reacts with PLOOH via the Fenton reaction to produce free radicals, which can induce the peroxidation of polyunsaturated fatty acids (PUFAs), promoting the loss of membrane integrity and cell death. (GSH: glutathione, GSSG: oxidized glutathione, GPX4: glutathione peroxidase 4, PLOOH: phospholipid hydroperoxide, PLOH: the corresponding alcohol of PLOOH, FSP1: ferroptosis suppressing protein 1, GCH1: GTP cyclohydrolase-1, BH4: tetrahydrobiopterin).
Figure 1.
Known pathways protecting cells from ferroptosis. The 3 known pathways involve GPX4 (①), FSP1 (②), and BH4 generated from GCH1 (③), respectively. The 3 pathways have the same effect: protecting the cells from further lipid peroxidation by reducing PLOOH levels. Iron [II/III] reacts with PLOOH via the Fenton reaction to produce free radicals, which can induce the peroxidation of polyunsaturated fatty acids (PUFAs), promoting the loss of membrane integrity and cell death. (GSH: glutathione, GSSG: oxidized glutathione, GPX4: glutathione peroxidase 4, PLOOH: phospholipid hydroperoxide, PLOH: the corresponding alcohol of PLOOH, FSP1: ferroptosis suppressing protein 1, GCH1: GTP cyclohydrolase-1, BH4: tetrahydrobiopterin).

Figure 2.
Function of Cdo1 in ferroptosis and targets of Erastin and RSL3 for inducing ferroptosis. Erastin inhibits the cystine/glutamate antiporter by binding to the subunit SLC755A, leading to cysteine deprivation by suppressing cystine uptake and triggering ferroptosis. Similarly, p53 inhibits the synthesis of SLC755A, the subunit of the cystine-glutamate antiporter, suppressing the uptake of cysteine. RSL3 directly binds to GPX4, inhibiting its function. Calcium ions inhibit GPX4 function, too. C-Myb is supposed to regulate Cdo1 expression in ferroptosis by an unknown mechanism. Enzymatic conversion by Cdo1 lowers the cysteine concentration, thereby depleting the pool available for the formation of GSH. Consequently, the autoperoxidation of lipids by the Fenton reaction and PLOOH cannot be inhibited, leading to ferroptosis. (GPX4: glutathione peroxidase 4, PLOOH: phospholipid hydroperoxide, PLOH: the corresponding alcohol of PLOOH, Cdo1: cysteine dioxygenase type 1, GSR: glutathione-disulfide reductase, GSSG: oxidized glutathione, GSH: glutathione, NADPH: triphosphopyridine nucleotide, c-Myb: c-Myb proto-oncogene transcription factor, RSL3: Ras-selective lethal 3).
Figure 2.
Function of Cdo1 in ferroptosis and targets of Erastin and RSL3 for inducing ferroptosis. Erastin inhibits the cystine/glutamate antiporter by binding to the subunit SLC755A, leading to cysteine deprivation by suppressing cystine uptake and triggering ferroptosis. Similarly, p53 inhibits the synthesis of SLC755A, the subunit of the cystine-glutamate antiporter, suppressing the uptake of cysteine. RSL3 directly binds to GPX4, inhibiting its function. Calcium ions inhibit GPX4 function, too. C-Myb is supposed to regulate Cdo1 expression in ferroptosis by an unknown mechanism. Enzymatic conversion by Cdo1 lowers the cysteine concentration, thereby depleting the pool available for the formation of GSH. Consequently, the autoperoxidation of lipids by the Fenton reaction and PLOOH cannot be inhibited, leading to ferroptosis. (GPX4: glutathione peroxidase 4, PLOOH: phospholipid hydroperoxide, PLOH: the corresponding alcohol of PLOOH, Cdo1: cysteine dioxygenase type 1, GSR: glutathione-disulfide reductase, GSSG: oxidized glutathione, GSH: glutathione, NADPH: triphosphopyridine nucleotide, c-Myb: c-Myb proto-oncogene transcription factor, RSL3: Ras-selective lethal 3).

Figure 3.
Percentage of Cdo1 mutations and structural variants in cancers. Overview of cancer types that contain Cdo1 mutations and structural variants. Copy number alterations means the change in the number of copies of a particular region of DNA in the genome, involving either a gain or a loss of copies of a specific DNA segment. Cdo1 mutations appear in nearly every kind of cancer; the figure displays only cancers with a relatively high percentage of Cdo1 genetic variation. Source: CBioportal (2024). Cancers with Cdo1 mutation [Pancancer database] (https://www.cbioportal.org/).
Figure 3.
Percentage of Cdo1 mutations and structural variants in cancers. Overview of cancer types that contain Cdo1 mutations and structural variants. Copy number alterations means the change in the number of copies of a particular region of DNA in the genome, involving either a gain or a loss of copies of a specific DNA segment. Cdo1 mutations appear in nearly every kind of cancer; the figure displays only cancers with a relatively high percentage of Cdo1 genetic variation. Source: CBioportal (2024). Cancers with Cdo1 mutation [Pancancer database] (https://www.cbioportal.org/).
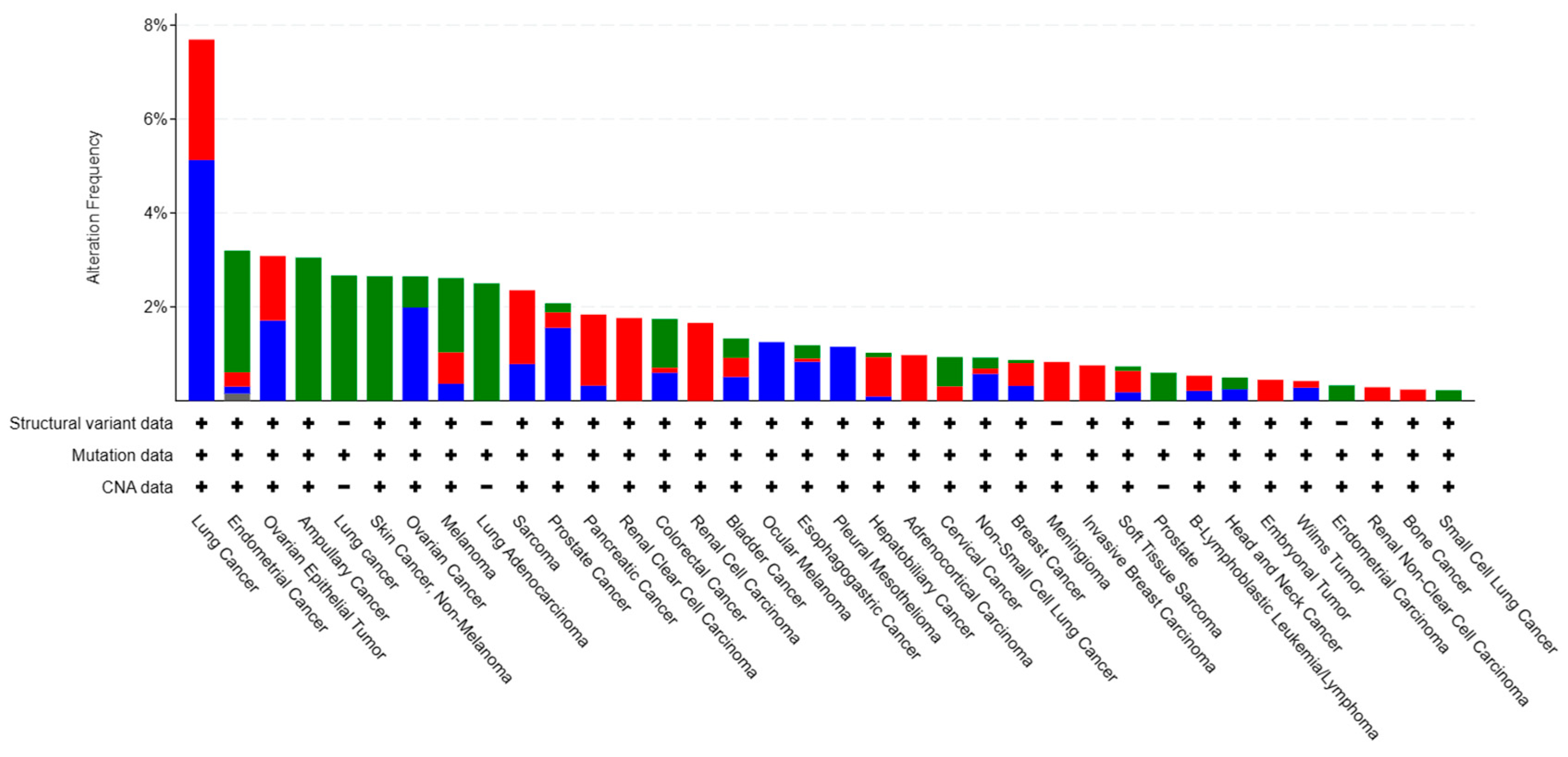
Figure 4.
Positions of genetic alterations on Cdo1. This lollipop figure shows the mutated amino acid positions on the Cdo1 protein and the frequency of observation in patients. Source: CBioportal (2024). Types of Cdo1 mutations in cancers [Pancancer database] (https://www.cbioportal.org/).
Figure 4.
Positions of genetic alterations on Cdo1. This lollipop figure shows the mutated amino acid positions on the Cdo1 protein and the frequency of observation in patients. Source: CBioportal (2024). Types of Cdo1 mutations in cancers [Pancancer database] (https://www.cbioportal.org/).

Figure 5.
Theories explaining the methylation of the Cdo1 promoter in cancer. (a) When DNA damage occurs to a tumor suppressor gene, the methylation of the promoter occurs to inhibit the expression of the damaged gene while repairing. When the damaged gene is an anticancer gene, this inhibition can be related to carcinogenesis. In this case, by an unknown mechanism, the methylation of this promoter is not re-exposed in cancer cells for removal. (b) Alternative mechanism for Cdo1 methylation in cancer cells. Cancer cells contain low levels of 5hmC, an element that inhibits the binding of TET and DNMT to the adjacent shore of the unmethylated CpG island. The binding is guided by H3K4me1. In normal cells, the binding of DNMT and TET is largely inhibited by the methylated DNA and 5hmC. Therefore, the unmethylated CpG island is maintained. However, in cancer cells, the 5hmC level is low, H3K4me1 appears at the border directing the binding of DNMT, and TET activity is lost. Only DNMT works on the shore of the island, and the shore is methylated, shortening the island. With a larger portion of the promoter methylated, the expression of Cdo1 is downregulated.
Figure 5.
Theories explaining the methylation of the Cdo1 promoter in cancer. (a) When DNA damage occurs to a tumor suppressor gene, the methylation of the promoter occurs to inhibit the expression of the damaged gene while repairing. When the damaged gene is an anticancer gene, this inhibition can be related to carcinogenesis. In this case, by an unknown mechanism, the methylation of this promoter is not re-exposed in cancer cells for removal. (b) Alternative mechanism for Cdo1 methylation in cancer cells. Cancer cells contain low levels of 5hmC, an element that inhibits the binding of TET and DNMT to the adjacent shore of the unmethylated CpG island. The binding is guided by H3K4me1. In normal cells, the binding of DNMT and TET is largely inhibited by the methylated DNA and 5hmC. Therefore, the unmethylated CpG island is maintained. However, in cancer cells, the 5hmC level is low, H3K4me1 appears at the border directing the binding of DNMT, and TET activity is lost. Only DNMT works on the shore of the island, and the shore is methylated, shortening the island. With a larger portion of the promoter methylated, the expression of Cdo1 is downregulated.

Figure 6.
Cdo1 partakes in pathways of ferroptosis and apoptosis. Cdo1 is involved in converting cysteine to taurine, thereby decreasing the cysteine pool for the production of GSH. GSH is a required cofactor of GPX4 that prevents lipid peroxidation. The reduced function of GPX4 allows lipid peroxidation on the cell membrane, ultimately leading to cell death. Upregulated taurine can increase the level of p53, a vital tumor suppressor. Additionally, p53 can induce cell cycle arrest. Lipid peroxidation increases the likelihood of apoptosis and ferroptosis. p53 contributes to ferroptosis by inhibiting the production of the SLC755A, a subunit in the cystine-glutamate antiporter that uptakes cystine.
Figure 6.
Cdo1 partakes in pathways of ferroptosis and apoptosis. Cdo1 is involved in converting cysteine to taurine, thereby decreasing the cysteine pool for the production of GSH. GSH is a required cofactor of GPX4 that prevents lipid peroxidation. The reduced function of GPX4 allows lipid peroxidation on the cell membrane, ultimately leading to cell death. Upregulated taurine can increase the level of p53, a vital tumor suppressor. Additionally, p53 can induce cell cycle arrest. Lipid peroxidation increases the likelihood of apoptosis and ferroptosis. p53 contributes to ferroptosis by inhibiting the production of the SLC755A, a subunit in the cystine-glutamate antiporter that uptakes cystine.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chen, X.; Poetsch, A. The Role of Cdo1 in Ferroptosis and Apoptosis in Cancer. Biomedicines 2024, 12, 918. https://doi.org/10.3390/biomedicines12040918
AMA Style
Chen X, Poetsch A. The Role of Cdo1 in Ferroptosis and Apoptosis in Cancer. Biomedicines. 2024; 12(4):918. https://doi.org/10.3390/biomedicines12040918
Chicago/Turabian StyleChen, Xiaoyi, and Ansgar Poetsch. 2024. "The Role of Cdo1 in Ferroptosis and Apoptosis in Cancer" Biomedicines 12, no. 4: 918. https://doi.org/10.3390/biomedicines12040918
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.