
Combined Metabolic Activators with Different NAD+ Precursors Improve Metabolic Functions in the Animal Models of Neurodegenerative Diseases
 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Study Design for AD
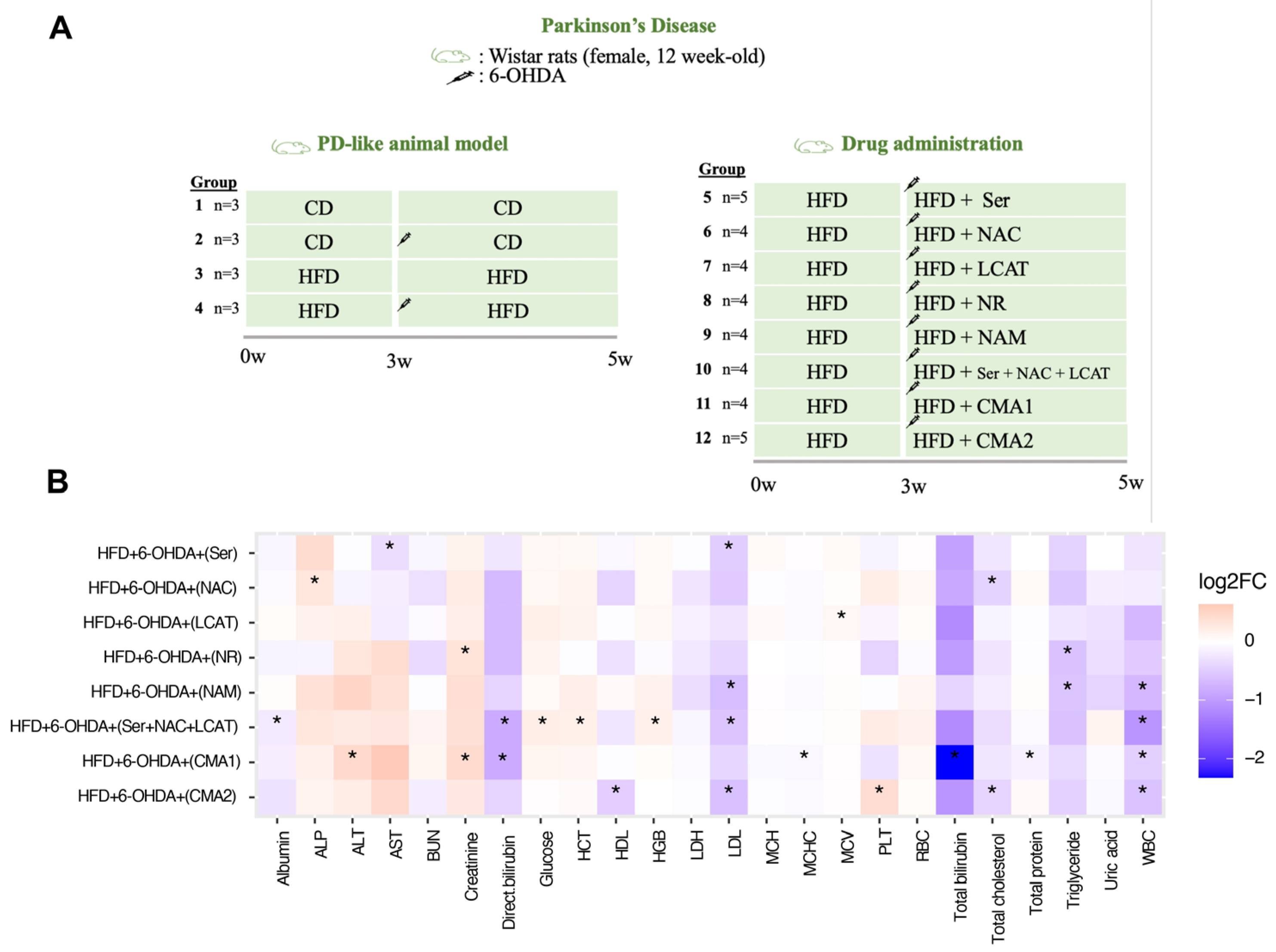
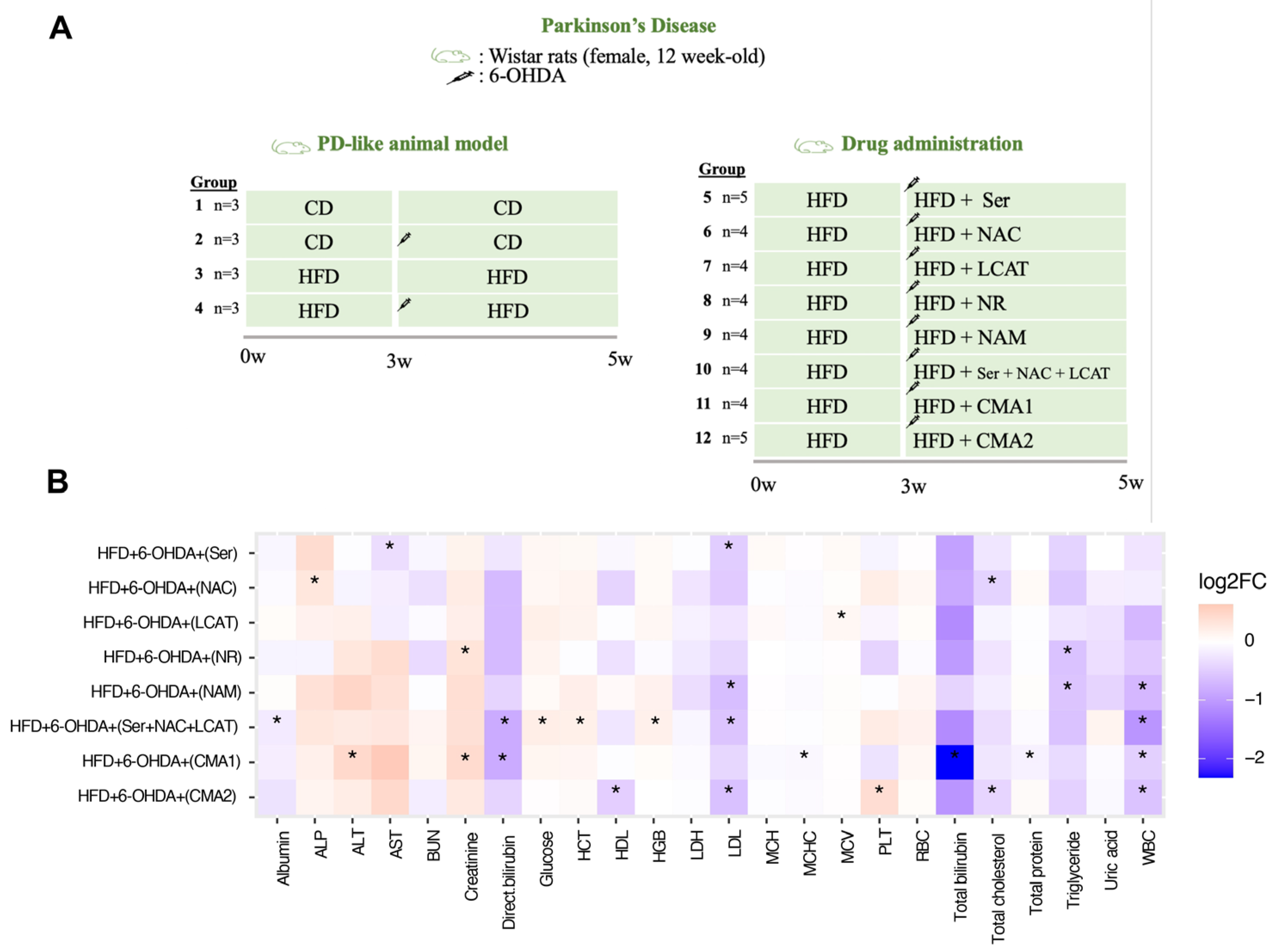
2.2. Animal Study Design for PD
2.2.1. Histopathological, Immunohistochemical and Immunofluorescence Examination
2.2.2. Behavioral Test in the PD-like Animal Model
2.3. Statistical Analysis
3. Results
3.1. Both CMA1 and CMA2 Improve Metabolic Functions in AD Animal Model
3.2. Both CMA1 and CMA2 Improve Metabolic Functions in PD Animal Model
3.3. Both CMA1 and CMA2 Shows a Beneficial Effect on Behavioural Functions in PD Rats
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Dementia 2022. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 29 October 2023).
- Azam, S.; Haque, M.E.; Balakrishnan, R.; Kim, I.-S.; Choi, D.-K. The Ageing Brain: Molecular and Cellular Basis of Neurodegeneration. Front. Cell Dev. Biol. 2021, 9, 683459. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Bassendine, M.F.; Taylor-Robinson, S.D.; Fertleman, M.; Khan, M.; Neely, D. Is Alzheimer’s Disease a Liver Disease of the Brain? J. Alzheimers Dis. 2020, 75, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Haxaire, C.; Turpin, F.R.; Potier, B.; Kervern, M.; Sinet, P.M.; Barbanel, G.; Mothet, J.P.; Dutar, P.; Billard, J.M. Reversal of age-related oxidative stress prevents hippocampal synaptic plasticity deficits by protecting D-serine-dependent NMDA receptor activation. Aging Cell 2012, 11, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Moutinho, M.; Puntambekar, S.S.; Tsai, A.P.; Coronel, I.; Lin, P.B.; Casali, B.T.; Martinez, P.; Oblak, A.L.; Lasagna-Reeves, C.A.; Lamb, B.T.; et al. The niacin receptor HCAR2 modulates microglial response and limits disease progression in a mouse model of Alzheimer’s disease. Sci. Transl. Med. 2022, 14, eabl7634. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Lautrup, S.; Cordonnier, S.; Wang, Y.; Croteau, D.L.; Zavala, E.; Zhang, Y.; Moritoh, K.; O’Connell, J.F.; Baptiste, B.A.; et al. NAD(+) supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl. Acad. Sci. USA 2018, 115, E1876–E1885. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Wei, Y.; Lautrup, S.; Yang, B.; Wang, Y.; Cordonnier, S.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. NAD(+) supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proc. Natl. Acad. Sci. USA 2021, 118, e2011226118. [Google Scholar] [CrossRef] [PubMed]
- Schöndorf, D.C.; Ivanyuk, D.; Baden, P.; Sanchez-Martinez, A.; De Cicco, S.; Yu, C.; Giunta, I.; Schwarz, L.K.; Di Napoli, G.; Panagiotakopoulou, V.; et al. The NAD+ Precursor Nicotinamide Riboside Rescues Mitochondrial Defects and Neuronal Loss in iPSC and Fly Models of Parkinson’s Disease. Cell Rep. 2018, 23, 2976–2988. [Google Scholar] [CrossRef]
- Le Douce, J.; Maugard, M.; Veran, J.; Matos, M.; Jégo, P.; Vigneron, P.A.; Faivre, E.; Toussay, X.; Vandenberghe, M.; Balbastre, Y.; et al. Impairment of Glycolysis-Derived l-Serine Production in Astrocytes Contributes to Cognitive Deficits in Alzheimer’s Disease. Cell Metab. 2020, 31, 503–517.e8. [Google Scholar] [CrossRef] [PubMed]
- Brakedal, B.; Dölle, C.; Riemer, F.; Ma, Y.; Nido, G.S.; Skeie, G.O.; Craven, A.R.; Schwarzlmüller, T.; Brekke, N.; Diab, J.; et al. The NADPARK study: A randomized phase I trial of nicotinamide riboside supplementation in Parkinson’s disease. Cell Metabolism. 2022, 34, 396–407.e6. [Google Scholar] [CrossRef]
- Caudle, W.M.; Bammler, T.K.; Lin, Y.; Pan, S.; Zhang, J. Using ‘omics’ to define pathogenesis and biomarkers of Parkinson’s disease. Expert. Rev. Neurother. 2010, 10, 925–942. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.M.; Kiser, G.L.; Kaysser-Kranich, T.M.; Lockner, R.J.; Palaniappan, C.; Federoff, H.J. Robust dysregulation of gene expression in substantia nigra and striatum in Parkinson’s disease. Neurobiol. Dis. 2006, 21, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Duke, D.C.; Moran, L.B.; Kalaitzakis, M.E.; Deprez, M.; Dexter, D.T.; Pearce, R.K.; Graeber, M.B. Transcriptome analysis reveals link between proteasomal and mitochondrial pathways in Parkinson’s disease. Neurogenetics 2006, 7, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Ruffini, N.; Klingenberg, S.; Schweiger, S.; Gerber, S. Common Factors in Neurodegeneration: A Meta-Study Revealing Shared Patterns on a Multi-Omics Scale. Cells 2020, 9, 2642. [Google Scholar] [CrossRef] [PubMed]
- Aracil-Bolaños, I.; Sampedro, F.; Marín-Lahoz, J.; Horta-Barba, A.; Martínez-Horta, S.; Botí, M.; Pérez-Pérez, J.; Bejr-Kasem, H.; Pascual-Sedano, B.; Campolongo, A.; et al. A divergent breakdown of neurocognitive networks in Parkinson’s Disease mild cognitive impairment. Hum. Brain Mapp. 2019, 40, 3233–3242. [Google Scholar] [CrossRef] [PubMed]
- Aracil-Bolaños, I.; Sampedro, F.; Pujol, J.; Soriano-Mas, C.; Gónzalez-de-Echávarri, J.M.; Kulisevsky, J.; Pagonabarraga, J. The impact of dopaminergic treatment over cognitive networks in Parkinson’s disease: Stemming the tide? Hum. Brain Mapp. 2021, 42, 5736–5746. [Google Scholar] [CrossRef] [PubMed]
- Christopher, L.; Duff-Canning, S.; Koshimori, Y.; Segura, B.; Boileau, I.; Chen, R.; Lang, A.E.; Houle, S.; Rusjan, P.; Strafella, A.P. Salience network and parahippocampal dopamine dysfunction in memory-impaired Parkinson disease. Ann. Neurol. 2015, 77, 269–280. [Google Scholar] [CrossRef]
- Saxena, U. Bioenergetics failure in neurodegenerative diseases: Back to the future. Expert. Opin. Ther. Targets 2012, 16, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Chianese, R.; Coccurello, R.; Viggiano, A.; Scafuro, M.; Fiore, M.; Coppola, G.; Operto, F.F.; Fasano, S.; Laye, S.; Pierantoni, R. Meccariello RImpact of Dietary Fats on Brain Functions. Curr. Neuropharmacol. 2018, 16, 1059–1085. [Google Scholar] [CrossRef] [PubMed]
- Janssen, C.I.F.; Jansen, D.; Mutsaers, M.P.C.; Dederen, P.J.W.C.; Geenen, B.; Mulder, M.T.; Kiliaan, A.J. The Effect of a High-Fat Diet on Brain Plasticity, Inflammation and Cognition in Female ApoE4-Knockin and ApoE-Knockout Mice. PLoS ONE 2016, 11, e0155307. [Google Scholar] [CrossRef] [PubMed]
- Del Olmo, N.; Ruiz-Gayo, M. Influence of High-Fat Diets Consumed During the Juvenile Period on Hippocampal Morphology and Function. Front. Cell. Neurosci. 2018, 12, 439. [Google Scholar] [CrossRef] [PubMed]
- Bosoi, C.R.; Vandal, M.; Tournissac, M.; Leclerc, M.; Fanet, H.; Mitchell, P.L.; Verreault, M.; Trottier, J.; Virgili, J.; Tremblay, C.; et al. High-Fat Diet Modulates Hepatic Amyloid β and Cerebrosterol Metabolism in the Triple Transgenic Mouse Model of Alzheimer’s Disease. Hepatol. Commun. 2021, 5, 446–460. [Google Scholar] [CrossRef] [PubMed]
- de Paula, G.C.; Brunetta, H.S.; Engel, D.F.; Gaspar, J.M.; Velloso, L.A.; Engblom, D.; de Oliveira, J.; de Bem, A.F. Hippocampal Function Is Impaired by a Short-Term High-Fat Diet in Mice: Increased Blood-Brain Barrier Permeability and Neuroinflammation as Triggering Events. Front. Neurosci. 2021, 15, 734158. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Nepal, P.; Odelade, A.; Freely, F.D.; Belton, D.M.; Graves, J.L., Jr.; Maldonado-Devincci, A.M. High-Fat Diet-Induced Weight Gain, Behavioral Deficits, and Dopamine Changes in Young C57BL/6J Mice. Front. Nutr. 2021, 7, 591161. [Google Scholar] [CrossRef] [PubMed]
- Cleland, N.R.W.; Al-Juboori, S.I.; Dobrinskikh, E.; Bruce, K.D. Altered substrate metabolism in neurodegenerative disease: New insights from metabolic imaging. J. Neuroinflamm. 2021, 18, 248. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Uhlen, M.; Nielsen, J. Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 3083. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Bjornson, E.; Zhang, C.; Klevstig, M.; Söderlund, S.; Ståhlman, M.; Adiels, M.; Hakkarainen, A.; Lundbom, N.; Kilicarslan, M.; et al. Personal model-assisted identification of NAD(+) and glutathione metabolism as intervention target in NAFLD. Mol. Syst. Biol. 2017, 13, 916. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Bjornson, E.; Arif, M.; Tebani, A.; Lovric, A.; Benfeitas, R.; Ozcan, M.; Juszczak, K.; Kim, W.; Kim, J.T.; et al. The acute effect of metabolic cofactor supplementation: A potential therapeutic strategy against non-alcoholic fatty liver disease. Mol. Syst. Biol. 2020, 16, e9495. [Google Scholar] [CrossRef] [PubMed]
- Mardinoglu, A.; Ural, D.; Zeybel, M.; Yuksel, H.H.; Uhlén, M.; Borén, J. The Potential Use of Metabolic Cofactors in Treatment of NAFLD. Nutrients 2019, 11, 1578. [Google Scholar] [CrossRef]
- Altay, O.; Arif, M.; Li, X.; Yang, H.; Aydın, M.; Alkurt, G.; Kim, W.; Akyol, D.; Zhang, C.; Dinler-Doganay, G.; et al. Combined Metabolic Activators Accelerates Recovery in Mild-to-Moderate COVID-19. Adv. Sci. 2021, 8, e2101222. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Wu, H.; Bjornson, E.; Zhang, C.; Hakkarainen, A.; Räsänen, S.M.; Lee, S.; Mancina, R.M.; Bergentall, M.; Pietiläinen, K.H.; et al. An Integrated Understanding of the Rapid Metabolic Benefits of a Carbohydrate-Restricted Diet on Hepatic Steatosis in Humans. Cell Metab. 2018, 27, 559–571.e5. [Google Scholar] [CrossRef] [PubMed]
- Zeybel, M.; Altay, O.; Arif, M.; Li, X.; Yang, H.; Fredolini, C.; Akyildiz, M.; Saglam, B.; Gonenli, M.G.; Ural, D.; et al. Combined metabolic activators therapy ameliorates liver fat in nonalcoholic fatty liver disease patients. Mol. Syst. Biol. 2021, 17, e10459. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Mayneris-Perxachs, J.; Boqué, N.; Del Bas, J.M.; Arola, L.; Yuan, M.; Türkez, H.; Uhlén, M.; Borén, J.; Zhang, C.; et al. Combined Metabolic Activators Decrease Liver Steatosis by Activating Mitochondrial Metabolism in Hamsters Fed with a High-Fat Diet. Biomedicines 2021, 9, 1440. [Google Scholar] [CrossRef] [PubMed]
- Turkez, H.; Altay, O.; Yildirim, S.; Li, X.; Yang, H.; Bayram, C.; Bolat, I.; Oner, S.; Tozlu, O.O.; Arslan, M.E.; et al. Combined metabolic activators improve metabolic functions in the animal models of neurodegenerative diseases. Life Sci. 2023, 314, 121325. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Li, X.; Jin, H.; Turkez, H.; Ozturk, G.; Doganay, H.L.; Zhang, C.; Nielsen, J.; Uhlén, M.; Borén, J.; et al. Longitudinal metabolomics analysis reveals the acute effect of cysteine and NAC included in the combined metabolic activators. Free Radic. Biol. Med. 2023, 204, 347–358. [Google Scholar] [CrossRef]
- Li, X.; Yang, H.; Jin, H.; Turkez, H.; Ozturk, G.; Doganay, H.L.; Zhang, C.; Nielsen, J.; Uhlén, M.; Borén, J.; et al. The acute effect of different NAD+ precursors included in the combined metabolic activators. Free Radic. Biol. Med. 2023, 205, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Hang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD(+) metabolism: Pathophysiologic mechanisms and therapeutic potential. Signal Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Lautrup, S.; Hou, Y.; Demarest, T.G.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. NAD(+) in Aging: Molecular Mechanisms and Translational Implications. Trends Mol. Med. 2017, 23, 899–916. [Google Scholar] [CrossRef] [PubMed]
- Surjana, D.; Halliday, G.M.; Damian, D.L. Role of nicotinamide in DNA damage, mutagenesis, and DNA repair. J. Nucleic Acids. 2010, 2010, 157591. [Google Scholar] [CrossRef] [PubMed]
- Klaidman, L.K.; Mukherjee, S.K.; Hutchin, T.P.; Adams, J.D. Nicotinamide as a precursor for NAD+ prevents apoptosis in the mouse brain induced by tertiary-butylhydroperoxide. Neurosci. Lett. 1996, 206, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Green, K.N.; Steffan, J.S.; Martinez-Coria, H.; Sun, X.; Schreiber, S.S.; Thompson, L.M.; LaFerla, F.M. Nicotinamide restores cognition in Alzheimer’s disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-phosphotau. J. Neurosci. 2008, 28, 11500–11510. [Google Scholar] [CrossRef]
- Liu, D.; Pitta, M.; Jiang, H.; Lee, J.H.; Zhang, G.; Chen, X.; Kawamoto, E.M.; Mattson, M.P. Nicotinamide forestalls pathology and cognitive decline in Alzheimer mice: Evidence for improved neuronal bioenergetics and autophagy procession. Neurobiol. Aging 2013, 34, 1564–1580. [Google Scholar] [CrossRef] [PubMed]
- Bayrakdar, E.T.; Armagan, G.; Uyanikgil, Y.; Kanit, L.; Koylu, E.; Yalcin, A. Ex vivo protective effects of nicotinamide and 3-aminobenzamide on rat synaptosomes treated with Aβ(1-42). Cell Biochem. Funct. 2014, 32, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.C.; Hill, L.J.; Ramsden, D.B. Nicotinamide, NAD(P)(H), and Methyl-Group Homeostasis Evolved and Became a Determinant of Ageing Diseases: Hypotheses and Lessons from Pellagra. Curr. Gerontol. Geriatr. Res. 2012, 2012, 302875. [Google Scholar] [CrossRef] [PubMed]
- Gil-Martínez, A.-L.; Cuenca, L.; Sánchez, C.; Estrada, C.; Fernández-Villalba, E.; Herrero, M.T. Effect of NAC treatment and physical activity on neuroinflammation in subchronic Parkinsonism; is physical activity essential? J. Neuroinflamm. 2018, 15, 328. [Google Scholar]
- Saiki, S.; Hatano, T.; Fujimaki, M.; Ishikawa, K.I.; Mori, A.; Oji, Y.; Okuzumi, A.; Fukuhara, T.; Koinuma, T.; Imamichi, Y.; et al. Decreased long-chain acylcarnitines from insufficient β-oxidation as potential early diagnostic markers for Parkinson’s disease. Sci. Rep. 2017, 7, 7328. [Google Scholar] [CrossRef] [PubMed]
- Virmani, M.A.; Cirulli, M. The Role of l-Carnitine in Mitochondria, Prevention of Metabolic Inflexibility and Disease Initiation. Int. J. Mol. Sci. 2022, 23, 2717. [Google Scholar] [CrossRef] [PubMed]
- Virmani, M.A.; Caso, V.; Spadoni, A.; Rossi, S.; Russo, F.; Gaetani, F. The action of acetyl-L-carnitine on the neurotoxicity evoked by amyloid fragments and peroxide on primary rat cortical neurones. Ann. N. Y. Acad. Sci. 2001, 939, 162–178. [Google Scholar] [CrossRef] [PubMed]
- Mota, S.I.; Pita, I.; Águas, R.; Tagorti, S.; Virmani, A.; Pereira, F.C.; Rego, A.C. Mechanistic perspectives on differential mitochondrial-based neuroprotective effects of several carnitine forms in Alzheimer’s disease in vitro model. Arch. Toxicol. 2021, 95, 2769–2784. [Google Scholar] [CrossRef] [PubMed]
- Abdul, H.M.; Calabrese, V.; Calvani, M.; Butterfield, D.A. Acetyl-L-carnitine-induced up-regulation of heat shock proteins protects cortical neurons against amyloid-beta peptide 1-42-mediated oxidative stress and neurotoxicity: Implications for Alzheimer’s disease. J. Neurosci. Res. 2006, 84, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Vlassenko, A.G.; Raichle, M.E. Brain aerobic glycolysis functions and Alzheimer’s disease. Clin. Transl. Imaging 2015, 3, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Sun, Y.; Jiang, Z.; Wang, G.L. Serine, an Endogenous Amino Acid, Is a Potential Neuroprotective Agent for Neurological Disease and Injury. Front. Mol. Neurosci. 2021, 14, 726665. [Google Scholar] [CrossRef] [PubMed]
- Phone Myint, S.M.M.; Sun, L.Y. L-serine: Neurological Implications and Therapeutic Potential. Biomedicines 2023, 11, 2117. [Google Scholar] [CrossRef]
- Liu, L.; Martin, R.; Chan, C. Palmitate-activated astrocytes via serine palmitoyltransferase increase BACE1 in primary neurons by sphingomyelinases. Neurobiol. Aging 2013, 34, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Qiang, R.; Yang, Y.; Jiang, Z.L.; Wang, G.H.; Zhao, G.W.; Ren, T.J.; Jiang, R.; Xu, L.H. L-serine treatment may improve neurorestoration of rats after permanent focal cerebral ischemia potentially through improvement of neurorepair. PLoS ONE 2014, 9, e93405. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Stereotypic Movement | Ambulatory Movement | Vertical Activity | Horizontal Activity | Distance Traveled (cm) | Resting (%) |
|---|---|---|---|---|---|---|
| CD | 404.25 ± 42.70 | 593.75 ± 95.05 | 50.00 ± 18.50 | 24.50 ± 6.40 | 1715.50 ± 495.90 | 50.75 ± 3.20 |
| HFD | 425.00 ± 100.50 | 643.75 ± 146.05 | 36.75 ± 17.40 | 26.00 ± 6.30 | 1784.00 ± 537.20 | 53.00 ± 5.60 |
| CD + 6-OHDA | 248.25 ± 71.50 | 236.00 ± 61.30 | 15.00 ± 2.60 | 10.25 ± 1.30 | 650.00 ± 131.30 | 73.75 ± 2.50 |
| HFD + 6-OHDA | 228.25 ± 85.00 | 224.75 ± 79.05 | 11.25 ± 2.10 | 9.75 ± 1.50 | 536.00 ± 113.90 | 85.25 ± 3.10 |
| HFD + 6-OHDA + Ser | 263.00 ± 62.70 | 260.75 ± 68.25 * | 12.75 ± 1.90 | 10.50 ± 1.20 | 616.75 ± 118.80 | 70.00 ± 3.80 * |
| HFD + 6-OHDA +NAC | 307.75 ± 96.05 | 598.00 ± 76.3 * | 23.75 ± 3.10 * | 19.75 ± 2.30 * | 1359.00 ± 159.80 * | 51.25 ± 4.70 * |
| HFD + 6-OHDA + LCAT | 370.75 ± 89.60 | 466.00 ± 64.4 5* | 27.00 ± 4.80 | 20.00 ± 2.30 * | 1199.25 ± 188.50 * | 57.50 ± 4.40 * |
| HFD + 6-OHDA +NR | 361.75 ± 108.10 | 704.75 ± 144.10 * | 37.75 ± 6.30 * | 20.50 ± 5.50 * | 1587.50 ± 429.20 * | 49.50 ± 11.40 * |
| HFD + 6-OHDA +NAM | 387.25 ± 87.05 | 587.00 ± 121.70 * | 25.25 ± 10.20 * | 23.25 ± 3.10 * | 1619.00 ± 379.10 * | 50.25 ± 4.70 * |
| HFD + 6-OHDA+Ser + NAC + LCAT | 390.50 ± 135.50 | 492.25 ± 90.05 * | 24.75 ± 8.30 * | 20.75 ± 5.30 * | 1380.25 ± 243.80 * | 55.50 ± 7.10 * |
| HFD + 6-OHDA +CMA1 | 429.75 ± 63.70 * | 453.75 ± 75.80 * | 29.50 ± 11.60 * | 21.00 ± 5.40 * | 1470.00 ± 343.80 * | 58.25 ± 6.50 * |
| HFD + 6-OHDA +CMA2 | 367.20 ± 99.40 * | 648.00 ± 173.40 * | 34.50 ± 5.30 * | 25.75 ± 7.40 * | 1873.00 ± 456.30 * | 53.50 ± 9.70 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altay, O.; Yang, H.; Yildirim, S.; Bayram, C.; Bolat, I.; Oner, S.; Tozlu, O.O.; Arslan, M.E.; Hacimuftuoglu, A.; Shoaie, S.; et al. Combined Metabolic Activators with Different NAD+ Precursors Improve Metabolic Functions in the Animal Models of Neurodegenerative Diseases. Biomedicines 2024, 12, 927. https://doi.org/10.3390/biomedicines12040927
Altay O, Yang H, Yildirim S, Bayram C, Bolat I, Oner S, Tozlu OO, Arslan ME, Hacimuftuoglu A, Shoaie S, et al. Combined Metabolic Activators with Different NAD+ Precursors Improve Metabolic Functions in the Animal Models of Neurodegenerative Diseases. Biomedicines. 2024; 12(4):927. https://doi.org/10.3390/biomedicines12040927
Chicago/Turabian StyleAltay, Ozlem, Hong Yang, Serkan Yildirim, Cemil Bayram, Ismail Bolat, Sena Oner, Ozlem Ozdemir Tozlu, Mehmet Enes Arslan, Ahmet Hacimuftuoglu, Saeed Shoaie, and et al. 2024. "Combined Metabolic Activators with Different NAD+ Precursors Improve Metabolic Functions in the Animal Models of Neurodegenerative Diseases" Biomedicines 12, no. 4: 927. https://doi.org/10.3390/biomedicines12040927