

Combinatorial Therapy: Targeting CD133+ Glioma Stem-like Cells with a Polysaccharide–Prodrug Complex Functionalised Gold Nanocages
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis and Characterisation of Hollow AuNcgs
2.3. Synthesis of MR-5FU Prodrug and Functionalisation of AuNcgs with MR-5FU
2.4. Functionalisation of AuNcg-MR- 5FU Nanoparticles with Anti-CD133 Antibodies
2.5. Measurement of Au Concentration Using Microwave Plasma-Atomic Emission Spectroscopy (MP-AES)
2.6. In Vitro Drug Release Using Slide-A-Dialysis Cassette Method
2.7. Tumour Homogenate Assay for Ex Vivo Drug Release Study
2.8. In Vitro Cytotoxicity Assay
2.9. ROS Measurement Using 2′,7′-Dichlorofluorescein Diacetate (DCFDA) Assay
2.10. Cell Binding and Fluorescence Microscopy
2.11. Generic Caspase Assay
2.12. Live/Dead Assay
3. Results and Discussion
3.1. Synthesis of MR-5FU and TMFG Nanocages
3.2. Characterisation of TMFG Nanocages
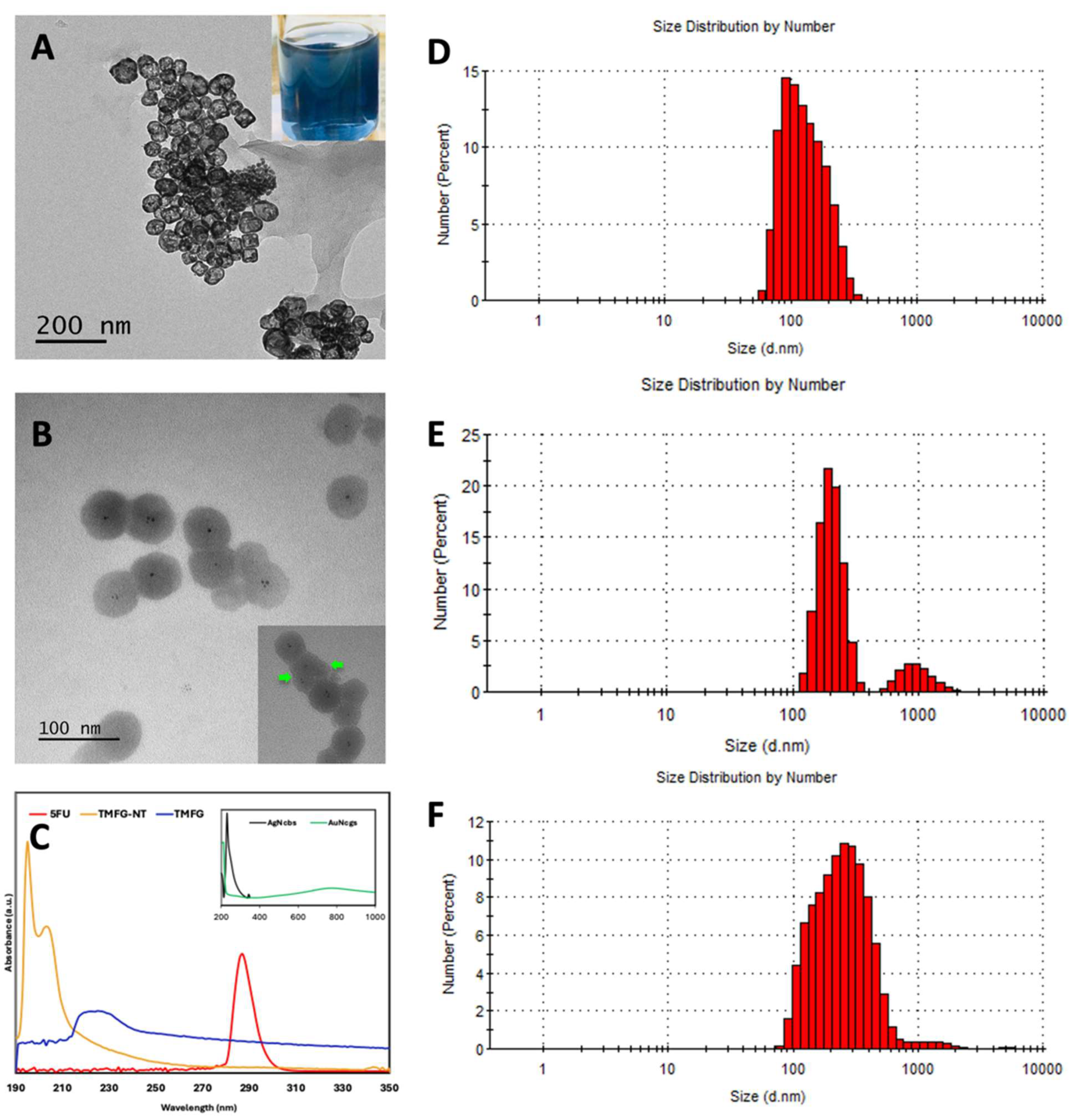

3.3. Encapsulation Efficiency TMFG Nanocages In Vitro and Ex Vivo
3.4. Cytotoxicity of TMFG Nanocages
3.5. ROS Generation via the Action of TMFG Nanocages
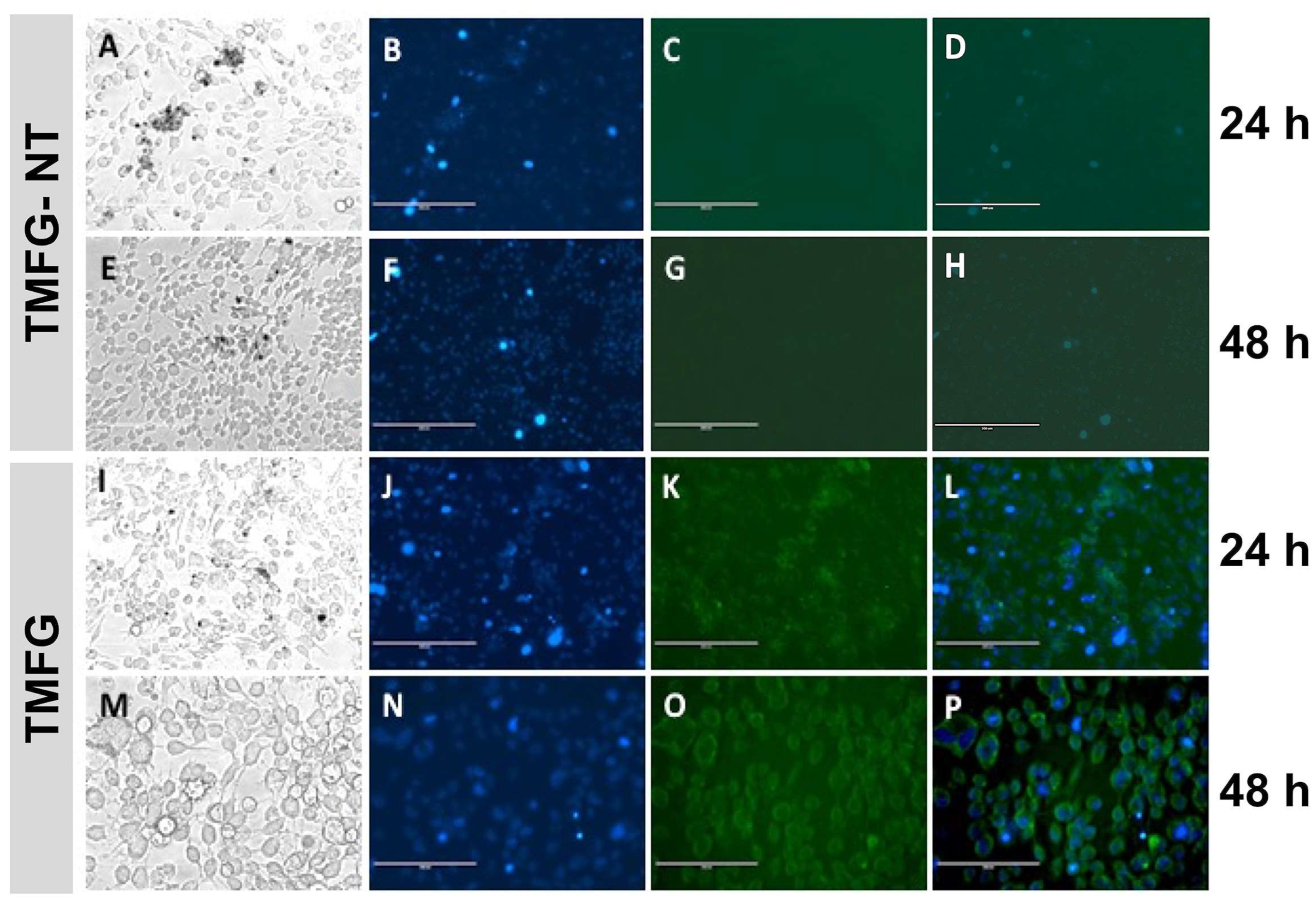
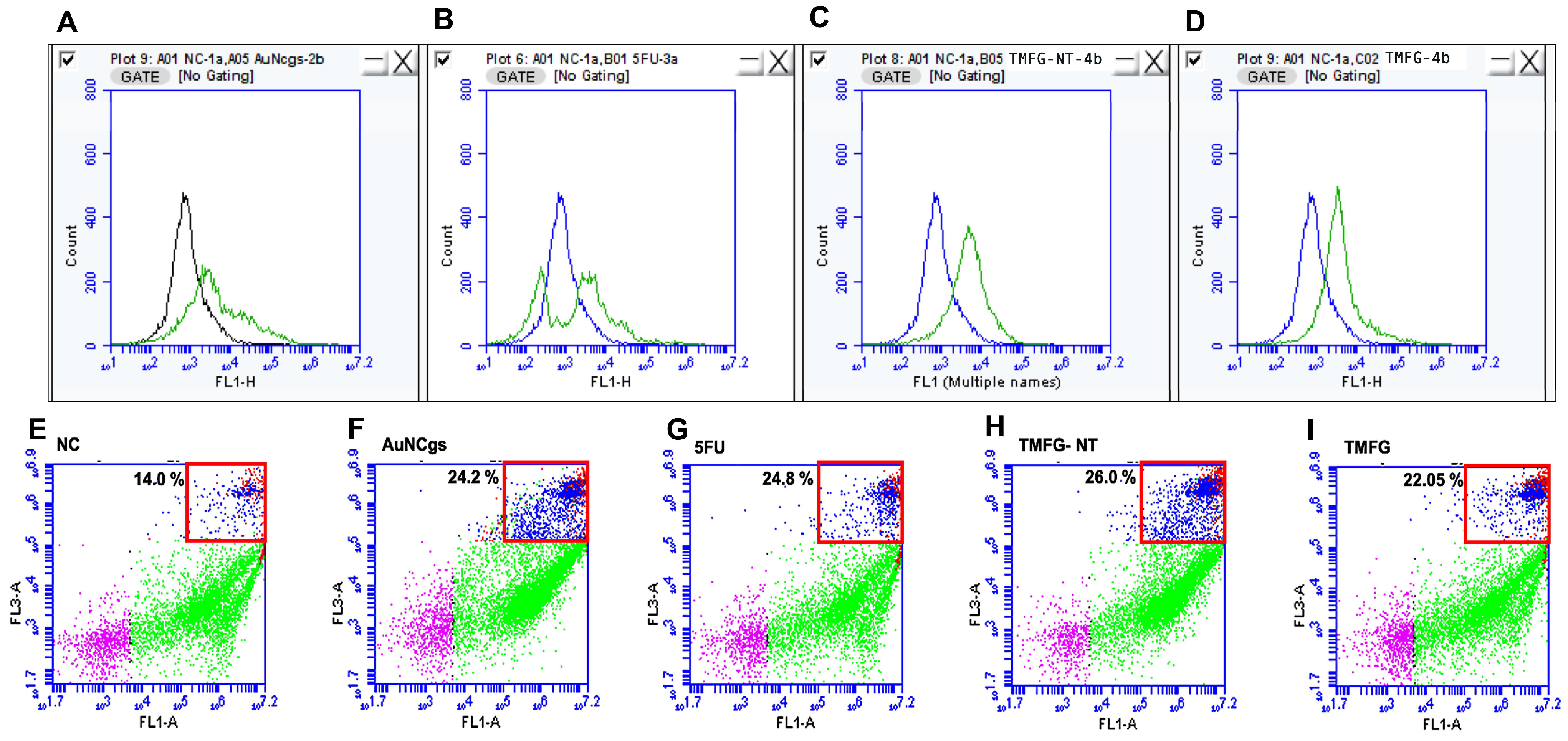
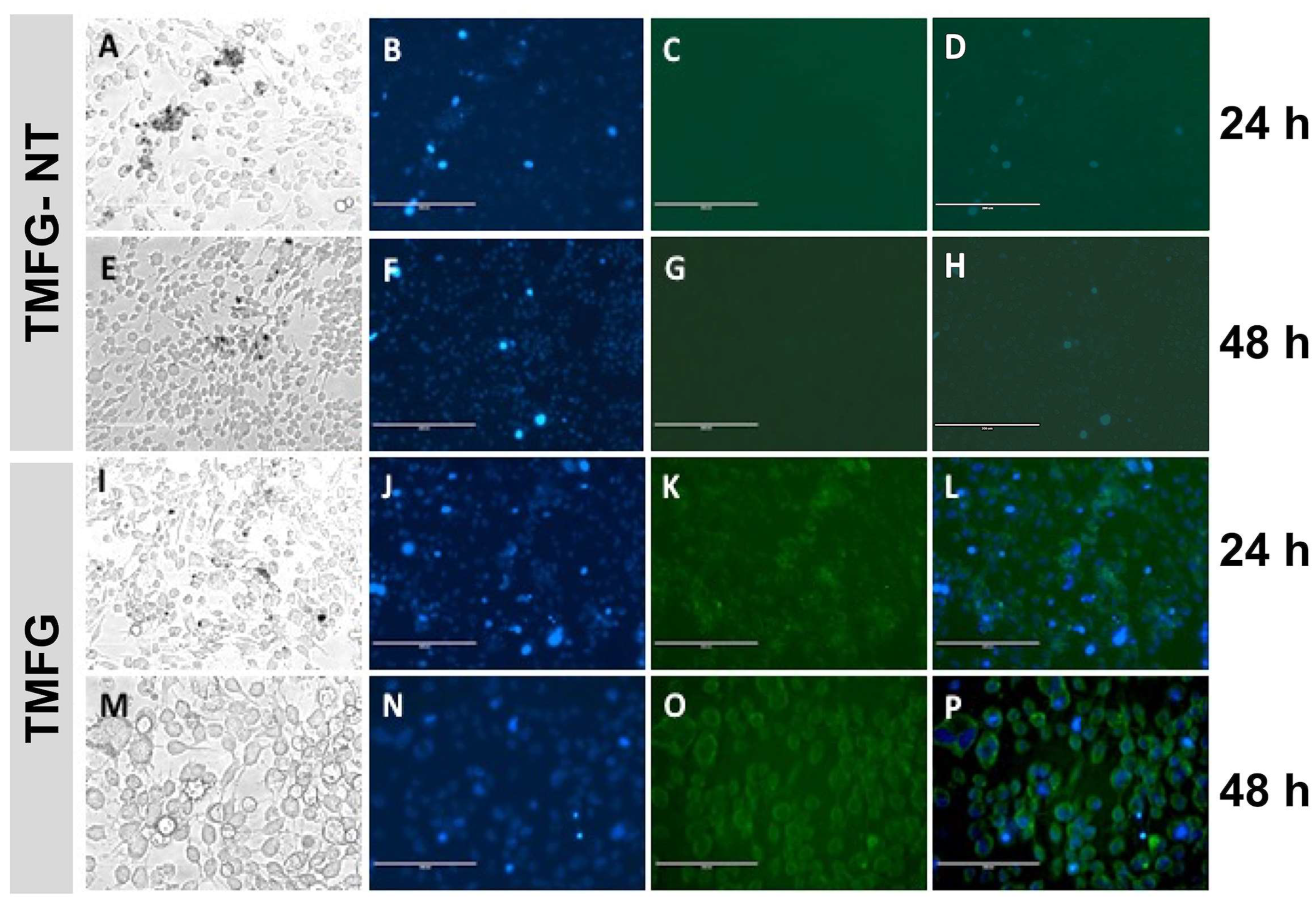
3.6. TMFG Nanocages Selectivity towards CD-133 Binding on GBM Cell Line and Caspase Assay
3.7. Live/Dead Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fang, R.H.; Zhang, L. Combinatorial Nanotherapeutics: Rewiring, Then Killing, Cancer Cells. Sci. Signal 2014, 7, pe13. [Google Scholar] [CrossRef]
- Zhang, D.; Zhao, Q.; Sun, H.; Yin, L.; Wu, J.; Xu, J.; He, T.; Yang, C.; Liang, C. Defective Autophagy Leads to the Suppression of Stem-like Features of CD271(+) Osteosarcoma Cells. J. Biomed. Sci. 2016, 23, 82. [Google Scholar] [CrossRef]
- Raveendran, S.; Sen, A.; Ito-Tanaka, H.; Kato, K.; Maekawa, T.; Kumar, D.S. Advanced Microscopic Evaluation of Parallel Type I and Type II Cell Deaths Induced by Multi-Functionalized Gold Nanocages in Breast Cancer. Nanoscale Adv. 2019, 1, 989–1001. [Google Scholar] [CrossRef]
- Marrache, S.; Dhar, S.; Siekkinen, A.R.; Mclellan, J.M.; Chen, J.; Xia, Y.; Zheng, K.; Wang, C.; Cheng, Y.; Yue, Y.; et al. Engineering of Blended Nanoparticle Platform for Delivery of Mitochondria-Acting Therapeutics. Proc. Natl. Acad. Sci. USA 2012, 109, 16288–16293. [Google Scholar] [CrossRef]
- Mohanraj, V.J.; Chen, Y. Nanoparticles—A Review. Trop. J. Pharm. Res. 2006, 5, 561–573. [Google Scholar] [CrossRef]
- Becker, J.; Trügler, A.; Jakab, A.; Hohenester, U.; Sönnichsen, C. The Optimal Aspect Ratio of Gold Nanorods for Plasmonic Bio-Sensing. Plasmonics 2010, 5, 161–167. [Google Scholar] [CrossRef]
- Shilo, M.; Sharon, A.; Baranes, K.; Motiei, M.; Lellouche, J.M.; Popovtzer, R. The Effect of Nanoparticle Size on the Probability to Cross the Blood—Brain Barrier: An in-Vitro Endothelial Cell Model. J. Nanobiotechnol. 2015, 13, 19. [Google Scholar] [CrossRef]
- Loo, C.; Lin, A.; Hirsch, L.; Lee, M.-H.; Barton, J.; Halas, N.; West, J.; Drezek, R. Nanoshell-Enabled Photonics-Based Imaging and Therapy of Cancer. Technol. Cancer Res. Treat. 2004, 3, 33–40. [Google Scholar] [CrossRef]
- Raveendran, S.; Lim, H.T.; Maekawa, T.; Vadakke Matham, M.; Sakthi Kumar, D. Gold Nanocages Entering into the Realm of High-Contrast Photoacoustic Ocular Imaging. Nanoscale 2018, 10, 13959–13968. [Google Scholar] [CrossRef]
- Cobley, C.M.; Chen, J.; Cho, E.C.; Wang, L.V.; Xia, Y. Gold Nanostructures: A Class of Multifunctional Materials for Biomedical Applications. Chem. Soc. Rev. 2011, 40, 44–56. [Google Scholar] [CrossRef]
- Yavuz, M.S.; Cheng, Y.; Chen, J.; Cobley, C.M.; Zhang, Q.; Rycenga, M.; Xie, J.; Kim, C.; Song, K.H.; Schwartz, A.G.; et al. Gold Nanocages Covered by Smart Polymers for Controlled Release with Near-Infrared Light. Nat. Mater. 2009, 8, 935–939. [Google Scholar] [CrossRef]
- Chen, J.; Glaus, C.; Laforest, R.; Zhang, Q.; Yang, M.; Gidding, M.; Welch, M.J.; Xia, Y. Gold Nanocages as Photothermal Transducers for Cancer Treatment. Small 2010, 6, 811–817. [Google Scholar] [CrossRef]
- Chen, J.; Wiley, B.; Li, Z.Y.; Campbell, D.; Saeki, F.; Cang, H.; Au, L.; Lee, J.; Li, X.; Xia, Y. Gold Nanocages: Engineering Their Structure for Biomedical Applications. Adv. Mater. 2005, 17, 2255–2261. [Google Scholar] [CrossRef]
- Brescia, P.; Ortensi, B.; Fornasari, L.; Levi, D.; Broggi, G.; Pelicci, G. CD133 Is Essential for Glioblastoma Stem Cell Maintenance. Stem Cells 2013, 31, 857–869. [Google Scholar] [CrossRef]
- Lathia, J.; Mack, S.; Mulkearns-Hubert, E.; Valentim, C.; Rich, J. Cancer Stem Cells in Glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef]
- Tilghman, J.; Wu, H.; Sang, Y.; Shi, X.; Guerrero-Cazares, H.; Quinones-Hinojosa, A.; Eberhart, C.G.; Laterra, J.; Ying, M. HMMR Maintains the Stemness and Tumorigenicity of Glioblastoma Stem-like Cells. Cancer Res. 2014, 74, 3168–3179. [Google Scholar] [CrossRef]
- Wei, G.; Jin, Q.; Giannobile, W.V.; Ma, P.X. The Enhancement of Osteogenesis by Nano-Fibrous Scaffolds Incorporating RhBMP-7 Nanospheres. Biomaterials 2007, 28, 2087–2096. [Google Scholar] [CrossRef]
- Caro, C.; Avasthi, A.; Paez-Muñoz, J.M.; Leal, M.P.; García-Martín, M.L. Passive Targeting of High-Grade Gliomas via the EPR Effect: A Closed Path for Metallic Nanoparticles? Biomater. Sci. 2021, 9, 7984–7995. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; Hjelmeland, A.B.; Rich, J.N.; Dewhirst, M.W.; Shi, Q.; Hao, Y.; McLendon, R.E.; Bao, S.; Bigner, D.D. Glioma Stem Cells Promote Radioresistance by Preferential Activation of the DNA Damage Response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Rocha, C.R.R.; Garcia, C.C.M.; Vieira, D.B.; Quinet, A.; De Andrade-Lima, L.C.; Munford, V.; Belizário, J.E.; Menck, C.F.M. Glutathione Depletion Sensitizes Cisplatin- and Temozolomide-Resistant Glioma Cells in Vitro and in Vivo. Cell Death Dis. 2014, 5, e1505. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A Restricted Cell Population Propagates Glioblastoma Growth after Chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Zustovich, F.; Lombardi, G.; Della Puppa, A.; Rotilio, A.; Scienza, R.; Pastorelli, D. A Phase II Study of Cisplatin and Temozolomide in Heavily Pre-Treated Patients with Temozolomide-Refractory High-Grade Malignant Glioma. Anticancer Res. 2009, 29, 4275–4279. [Google Scholar] [PubMed]
- Lu, B.; Morrison, R.; Schleicher, S.M.; Sun, Y.; Niermann, K.J.; Kim, S.; Spratt, D.E.; Chung, C.H. Targeting the Mechanisms of Resistance to Chemotherapy and Radiotherapy with the Cancer Stem Cell Hypothesis. J. Oncol. 2011, 2011, 941876. [Google Scholar] [CrossRef]
- Wu, A.; Oh, S.; Wiesner, S.M.; Ericson, K.; Chen, L.; Hall, W.A.; Champoux, P.E.; Low, W.C.; Ohlfest, J.R. Persistence of CD133+ Cells in Human and Mouse Glioma Cell Lines: Detailed Characterization of GL261 Glioma Cells with Cancer Stem Cell-like Properties. Stem Cells Dev. 2008, 17, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Gibson, P.; Currle, D.S.; Tong, Y.; Richardson, R.J.; Bayazitov, I.T.; Poppleton, H.; Zakharenko, S.; Ellison, D.W.; Gilbertson, R.J. Prominin 1 Marks Intestinal Stem Cells That Are Susceptible to Neoplastic Transformation. Nature 2009, 457, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Cai, G. Poly(Lactic-Co-Glycolic Acid) Nanoparticles Conjugated with CD133 Aptamers for Targeted Salinomycin Delivery to CD133+ Osteosarcoma Cancer Stem Cells. Int. J. Nanomed. 2015, 10, 2537–2554. [Google Scholar]
- Auffinger, B.; Tobias, A.L.; Han, Y.; Lee, G.; Guo, D.; Dey, M.; Lesniak, M.S.; Ahmed, A.U. Conversion of Differentiated Cancer Cells into Cancer Stem-like Cells in a Glioblastoma Model after Primary Chemotherapy. Cell Death Differ. 2014, 21, 1119–1131. [Google Scholar] [CrossRef]
- Campos, B.; Herold-Mende, C.C. Insight into the Complex Regulation of CD133 in Glioma. Int. J. Cancer 2011, 128, 501–510. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E. Targeting Brain-Tumor Stem Cells. Nat. Biotechnol. 2007, 25, 193–194. [Google Scholar] [CrossRef]
- Cobley, C.M.; Au, L.; Chen, J.; Xia, Y. Targeting gold nanocages to cancer cells for photothermal destruction and drug delivery. Expert Opin. Drug Deliv. 2010, 7, 577–587. [Google Scholar] [CrossRef]
- Shin, D.H.; Xuan, S.; Kim, W.Y.; Bae, G.U.; Kim, J.S. CD133 Antibody-Conjugated Immunoliposomes Encapsulating Gemcitabine for Targeting Glioblastoma Stem Cells. J. Mater. Chem. B 2014, 2, 3771–3781. [Google Scholar] [CrossRef]
- Diasio, R.B.; Harris, B.E. Harris Clinical Pharmacology of 5-Fluorouracil. Clin. Pharmacokinet. 1989, 16, 215–237. [Google Scholar] [CrossRef] [PubMed]
- Malet-Martino, M.; Jolimaitre, P.; Martino, R. The Prodrugs of 5-Fluorouracil. Curr. Med. Chem. Anticancer Agents 2002, 2, 267–310. [Google Scholar] [CrossRef]
- Raveendran, S.; Poulose, A.C.; Yoshida, Y.; Maekawa, T.; Kumar, D.S. Bacterial Exopolysaccharide Based Nanoparticles for Sustained Drug Delivery, Cancer Chemotherapy and Bioimaging. Carbohydr. Polym. 2013, 91, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Desaknai, S.; Lumniczky, K.; Esik, O.; Hamada, H.; Safrany, G. Local Tumour Irradiation Enhances the Anti-Tumour Effect of a Double-Suicide Gene Therapy System in a Murine Glioma Model. J. Gene Med. 2003, 5, 377–385. [Google Scholar] [CrossRef]
- Nair, K.L.; Jagadeeshan, S.; Nair, S.A.; Kumar, G.S.V. Biological Evaluation of 5-Fluorouracil Nanoparticles for Cancer Chemotherapy and Its Dependence on the Carrier, PLGA. Int. J. Nanomed. 2011, 6, 1685–1697. [Google Scholar] [CrossRef]
- Mei, Q.; Dale, K.M.; Tam, J.C. Polysacchocride Prodrug of 5-Fluorouracil (5-Fu) with Enhanced Target Specificity for Galectin-3 Expressing Cancers. U.S. Patent 11/657, 3 January 2008. [Google Scholar]
- Arias, S.; Del Moral, A.; Ferrer, M.R.; Tallon, R.; Quesada, E.; Béjar, V. Mauran, an Exopolysaccharide Produced by the Halophilic Bacterium Halomonas Maura, with a Novel Composition and Interesting Properties for Biotechnology. Extremophiles 2003, 7, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Llamas, I.; del Moral, A.; Martínez-Checa, F.; Arco, Y.; Arias, S.; Quesada, E. Halomonas Maura Is a Physiologically Versatile Bacterium of Both Ecological and Biotechnological Interest. Antonie Leeuwenhoek Int. J. Gen. Mol. Microbiol. 2006, 89, 395–403. [Google Scholar] [CrossRef]
- Raveendran, S.; Dhandayuthapani, B.; Nagaoka, Y.; Yoshida, Y.; Maekawa, T.; Sakthi Kumar, D. Biocompatible Nanofibers Based on Extremophilic Bacterial Polysaccharide, Mauran from Halomonas Maura. Carbohydr. Polym. 2013, 92, 1225–1233. [Google Scholar] [CrossRef]
- Hayashi, Y.; Jia, W.; Kidoya, H.; Muramatsu, F.; Tsukada, Y.; Takakura, N. Galectin-3 Inhibits Cancer Metastasis by Negatively Regulating Integrin Β3 Expression. Am. J. Pathol. 2019, 189, 900–910. [Google Scholar] [CrossRef]
- Zhao, T.; Fan, J.B.; Cui, J.; Liu, J.H.; Xu, X.B.; Zhu, M.Q. Microwave-Controlled Ultrafast Synthesis of Uniform Silver Nanocubes and Nanowires. Chem. Phys. Lett. 2011, 501, 414–418. [Google Scholar] [CrossRef]
- Raveendran, S.; Sen, A.; Maekawa, T.; Kumar, D.S. Ultrafast Microwave Aided Synthesis of Gold Nanocages and Structural Maneuver Studies. Nano Res. 2016, 10, 1078–1091. [Google Scholar] [CrossRef]
- Ouchi, T.; Banba, T.; Huang, T.Z.; Ohya, Y. Design of Polysaccharide-5-Fluorouracil Conjugates Exhibiting Antitumor Activities. Am. Chem. Soc. Polym. Prepr. Div. Polym. Chem. 1990, 31, 202–203. [Google Scholar]
- Kappler, K.; Hennet, T. Emergence and Significance of Carbohydrate-Specific Antibodies. Genes Immun. 2020, 21, 224–239. [Google Scholar] [CrossRef]
- McLean, G.R.; Torres, M.; Elguezabal, N.; Nakouzi, A.; Casadevall, A. Isotype Can Affect the Fine Specificity of an Antibody for a Polysaccharide Antigen. J. Immunol. 2002, 169, 1379–1386. [Google Scholar] [CrossRef]
- Broadley, K.W.R.; Hunn, M.K.; Farrand, K.J.; Price, K.M.; Grasso, C.; Miller, R.J.; Hermans, I.F.; McConnell, M.J. Side Population Is Not Necessary or Sufficient for a Cancer Stem Cell Phenotype in Glioblastoma Multiforme. Stem Cells 2011, 29, 452–461. [Google Scholar] [CrossRef]
- Promega. CellTiter 96® AQ Ueous One Solution Cell Proliferation Assay: Technical Bulletin No 169; Promega Corporation: Madison, WI, USA, 2001. [Google Scholar]
- Baskar, G.; Lalitha, K.; Garrick, B.G.; Chamundeeswari, M. Conjugation, Labeling and Characterization of Asparaginase Bound Silver Nanoparticles for Anticancer Applications. Indian J. Exp. Biol. 2017, 55, 421–426. [Google Scholar]
- Kumar, S.; Meena, V.K.; Hazari, P.P.; Sharma, R.K. FITC-Dextran Entrapped and Silica Coated Gadolinium Oxide Nanoparticles for Synchronous Optical and Magnetic Resonance Imaging Applications. Int. J. Pharm. 2016, 506, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Gan, B.K.; Rullah, K.; Yong, C.Y.; Ho, K.L.; Omar, A.R.; Alitheen, N.B.; Tan, W.S. Targeted Delivery of 5-Fluorouracil-1-Acetic Acid (5-FA) to Cancer Cells Overexpressing Epithelial Growth Factor Receptor (EGFR) Using Virus-like Nanoparticles. Sci. Rep. 2020, 10, 16867. [Google Scholar] [CrossRef]
- Attia, M.F.; Anton, N.; Wallyn, J.; Omran, Z.; Vandamme, T.F. An Overview of Active and Passive Targeting Strategies to Improve the Nanocarriers Efficiency to Tumour Sites. J. Pharm. Pharmacol. 2019, 71, 1185–1198. [Google Scholar] [CrossRef]
- Raveendran, S.; Sen, A.; Maekawa, T.; Kumar, D.S. Three-Dimensional Visualization of Subcellular Dynamics of Cancer Cell Destruction on Therapeutic Nanodrug Treatment. Small Struct. 2021, 2, 2000145. [Google Scholar] [CrossRef]



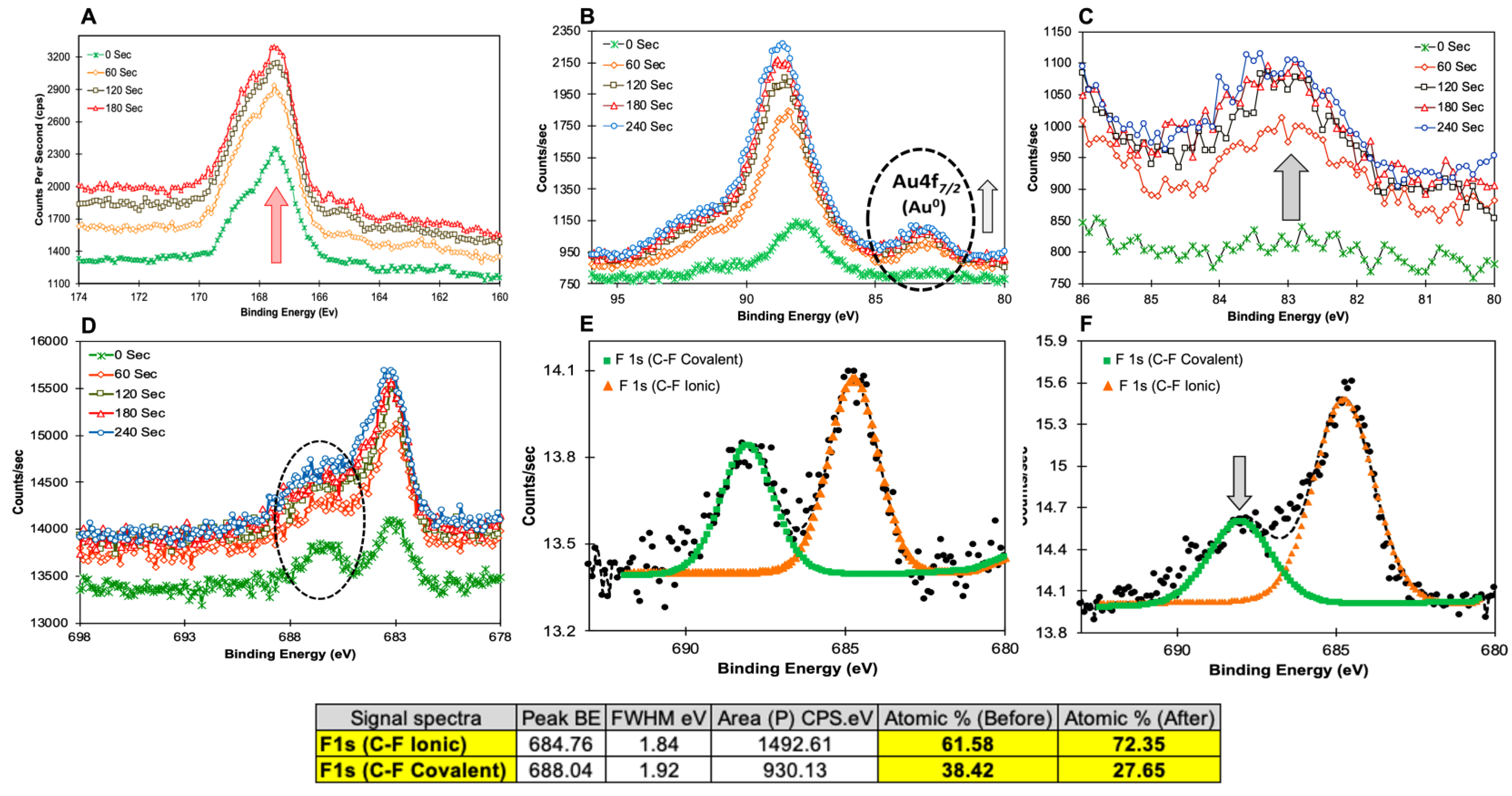
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Elements | No Sputtering | After 180 s Sputtering |
|---|---|---|
| F1s | 0.31 | 0.58 |
| O1s | 32.67 | 39.58 |
| N1s | 7.17 | 4.41 |
| C1s | 51.95 | 33.72 |
| Mg1s | 0.35 | 2.24 |
| Na1s | 3.85 | 8.74 |
| Ca2s | 1.1 | 1.56 |
| Cl2p | 0.71 | 1.03 |
| Au4f7 | 0 | 0.48 |
| Si2p | 1.14 | 6.69 |
| S2p | 0.75 | 0.97 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raveendran, S.; Giram, A.; Elmi, M.; Ray, S.; Ireson, C.; Alavijeh, M.; Savina, I.N. Combinatorial Therapy: Targeting CD133+ Glioma Stem-like Cells with a Polysaccharide–Prodrug Complex Functionalised Gold Nanocages. Biomedicines 2024, 12, 934. https://doi.org/10.3390/biomedicines12050934
Raveendran S, Giram A, Elmi M, Ray S, Ireson C, Alavijeh M, Savina IN. Combinatorial Therapy: Targeting CD133+ Glioma Stem-like Cells with a Polysaccharide–Prodrug Complex Functionalised Gold Nanocages. Biomedicines. 2024; 12(5):934. https://doi.org/10.3390/biomedicines12050934
Chicago/Turabian StyleRaveendran, Sreejith, Amit Giram, Mehrnaz Elmi, Santanu Ray, Christopher Ireson, Mo Alavijeh, and Irina N. Savina. 2024. "Combinatorial Therapy: Targeting CD133+ Glioma Stem-like Cells with a Polysaccharide–Prodrug Complex Functionalised Gold Nanocages" Biomedicines 12, no. 5: 934. https://doi.org/10.3390/biomedicines12050934