
Capitalizing on Cancer Specific Replication: Oncolytic Viruses as a Versatile Platform for the Enhancement of Cancer Immunotherapy Strategies

Abstract
:
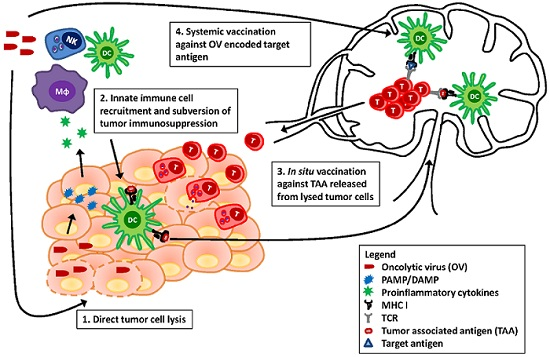{kind=link}
{kind=link}
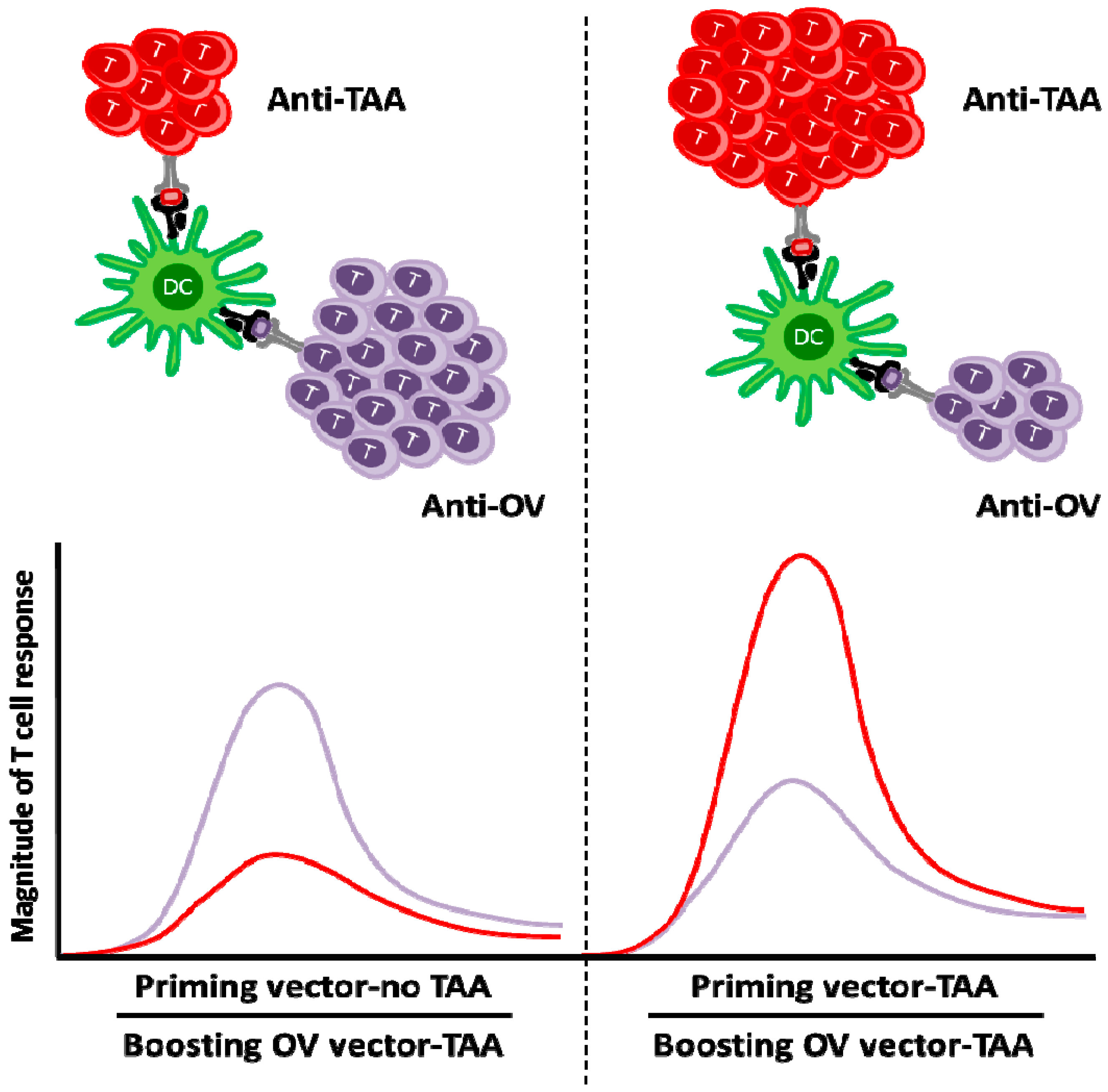{kind=link}
{kind=link}
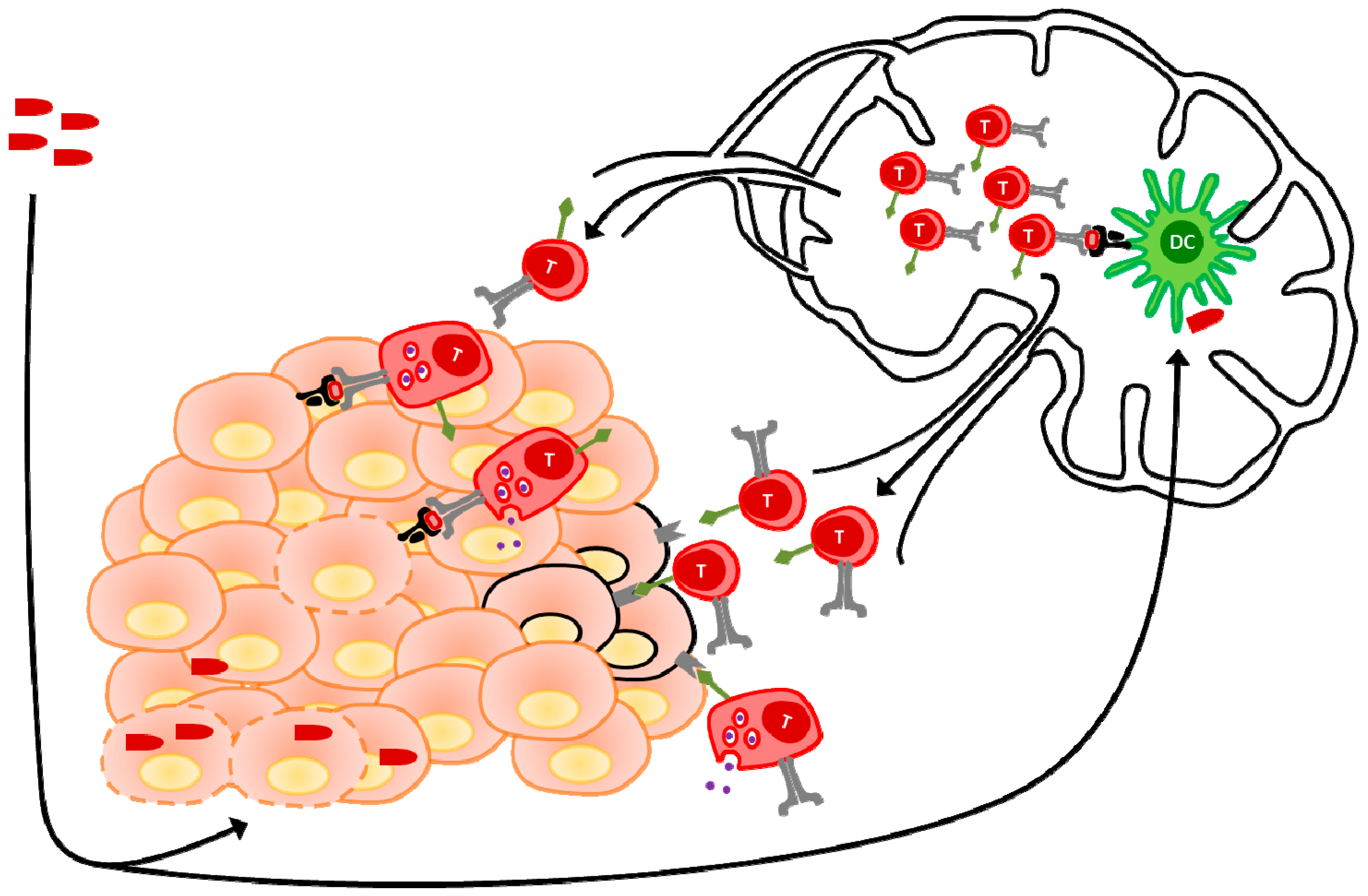{kind=link}
1. Introduction
2. Oncolytic Viruses: Cancer Specific Killers
3. Revisiting the Paradigm: The Immune Response Is Paramount
3.1. Viral Detection and Immunogenic Cell Death in Innate Activation and Adaptive Priming
3.2. Provision of Antigens
3.3. Enhanced Recruitment
4. Engineering Oncolytic Viruses to Enhance Their Immunostimulatory Potential
5. Oncolytic Viruses in Cancer Vaccination
6. Adoptive Cell Transfer and Oncolytic Viruses: Orchestrating the Attack
7. Taking off the Breaks with Checkpoint Inhibitors
8. Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gerlinger, M.; Swanton, C. How Darwinian models inform therapeutic failure initiated by clonal heterogeneity in cancer medicine. Br. J. Cancer 2010, 103, 1139–1143. [Google Scholar] [CrossRef] [PubMed]
- Couzin-Frankel, J. Cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef] [PubMed]
- Parato, K.A.; Senger, D.; Forsyth, P.A.J.; Bell, J.C. Recent progress in the battle between oncolytic viruses and tumours. Nat. Rev. Cancer 2005, 5, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.I.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H. Cancer-fighting viruses win approval. Nature 2015, 526, 622–623. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.-T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, R.J.; Harrington, K.J.; Pandha, H.S.; Vile, R.G.; Melcher, A.A.; Errington, F. Oncolytic viruses: A novel form of immunotherapy. Expert Rev. Anticancer Ther. 2008, 8, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Woller, N.; Gürlevik, E.; Ureche, C.-I.; Schumacher, A.; Kühnel, F. Oncolytic viruses as anticancer vaccines. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Lichty, B.D.; Breitbach, C.J.; Stojdl, D.F.; Bell, J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Dock, G. The influence of complicating diseases upon leukemia. Am. J. Med. Sci. 1904, 127, 563–592. [Google Scholar] [CrossRef]
- Kelly, E.; Russell, S.J. History of oncolytic viruses: Genesis to genetic engineering. Mol. Ther. 2007, 15, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Ilkow, C.S.; Swift, S.L.; Bell, J.C.; Diallo, J.S. From Scourge to Cure: Tumour-Selective Viral Pathogenesis as a New Strategy against Cancer. PLoS Pathog. 2014, 10, e1003836. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.; Rabkin, S.D. Designing herpes viruses as oncolytics. Mol. Ther. Oncolytics 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.; Wang, P.; Alusi, G.; Shi, H.; Chu, Y.; Wang, J.; Bhakta, V.; McNeish, I.; McCart, A.; Lemoine, N.R.; et al. Lister strain vaccinia virus with thymidine kinase gene deletion is a tractable platform for development of a new generation of oncolytic virus. Gene Ther. 2015, 22, 476–484. [Google Scholar] [CrossRef] [PubMed]
- McCart, J.A.; Ward, J.M.; Lee, J.; Hu, Y.; Alexander, H.R.; Libutti, S.K.; Moss, B.; Bartlett, D.L. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res. 2001, 61, 8751–8757. [Google Scholar] [PubMed]
- Stark, G.R.; Kerr, I.M.; Williams, B.R.; Silverman, R.H.; Schreiber, R.D. How cells respond to interferons. Annu. Rev. Biochem. 1998, 67, 227–264. [Google Scholar] [CrossRef] [PubMed]
- Stojdl, D.F.; Lichty, B.D.; TenOever, B.R.; Paterson, J.M.; Power, A.T.; Knowles, S.; Marius, R.; Reynard, J.; Poliquin, L.; Atkins, H.; et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 2003, 4, 263–275. [Google Scholar] [CrossRef]
- Stojdl, D.; Lichty, B.; Knowles, S.; Marius, R.; Atkins, H.; Sonenberg, N.; Bell, J.C. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med. 2000, 6, 821–825. [Google Scholar] [PubMed]
- Ilkow, C.S.; Marguerie, M.; Batenchuk, C.; Mayer, J.; Ben Neriah, D.; Cousineau, S.; Falls, T.; Jennings, V.A.; Boileau, M.; Bellamy, D.; et al. Reciprocal cellular cross-talk within the tumor microenvironment promotes oncolytic virus activity. Nat. Med. 2015, 21, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Brun, J.; McManus, D.; Lefebvre, C.; Hu, K.; Falls, T.; Atkins, H.; Bell, J.C.; McCart, J.A.; Mahoney, D.; Stojdl, D.F. Identification of genetically modified Maraba virus as an oncolytic rhabdovirus. Mol. Ther. 2010, 18, 1440–1449. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.-W.; Bell, J.C. Oncolytic Virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.-C.; Galanis, E.; Kirn, D. Clinical trial results with oncolytic virotherapy: A century of promise, a decade of progress. Nat. Clin. Pract. Oncol. 2007, 4, 101–117. [Google Scholar] [CrossRef] [PubMed]
- Khuri, F.R.; Nemunaitis, J.; Ganly, I.; Arseneau, J.; Tannock, I.F.; Romel, L.; Gore, M.; Ironside, J.; MacDougall, R.H.; Heise, C.; et al. A controlled trial of intratumoral ONYX-015, a selectively-replicating adenovirus, in combination with cisplatin and 5-fluorouracil in patients with recurrent head and neck cancer. Nat. Med. 2000, 6, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Tuve, S.; Liu, Y.; Tragoolpua, K.; Jacobs, J.D.; Yumul, R.C.; Li, Z.Y.; Strauss, R.; Hellström, K.E.; Disis, M.L.; Roffler, S.; et al. In situ adenovirus vaccination engages T effector cells against cancer. Vaccine 2009, 27, 4225–4239. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, R.J.; Ilett, E.J.; Errington, F.; Diaz, R.M.; Steele, L.P.; Kottke, T.; Thompson, J.; Galivo, F.; Harrington, K.J.; Pandha, H.S.; et al. Immune-mediated antitumor activity of reovirus is required for therapy and is independent of direct viral oncolysis and replication. Clin. Cancer Res. 2009, 15, 4374–4381. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.L.; Fraser, N.W. HSV-1 therapy of primary tumors reduces the number of metastases in an immune-competent model of metastatic breast cancer. Mol. Ther. 2003, 8, 543–551. [Google Scholar] [CrossRef]
- Diaz, R.M.; Galivo, F.; Kottke, T.; Wongthida, P.; Qiao, J.; Thompson, J.; Valdes, M.; Barber, G.; Vile, R.G. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 2007, 67, 2840–2848. [Google Scholar] [CrossRef] [PubMed]
- Naik, J.D.; Twelves, C.J.; Selby, P.J.; Vile, R.G.; Chester, J.D. Immune recruitment and therapeutic synergy: Keys to optimizing oncolytic viral therapy? Clin. Cancer Res. 2011, 17, 4214–4224. [Google Scholar] [CrossRef] [PubMed]
- Rouse, B.T.; Sehrawat, S. Immunity and immunopathology to viruses: What decides the outcome? Nat. Rev. Immunol. 2010, 10, 514–526. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Tesniere, A.; Kroemer, G. Cancer despite immunosurveillance: Immunoselection and immunosubversion. Nat. Rev. Immunol. 2006, 6, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Schreiber, H.; Fu, Y. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Janeway, C.A. Decoding the patterns of self and nonself by the innate immune system. Science 2002, 296, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.S.; Liu, Z.; Bartlett, D.L. Oncolytic Immunotherapy: Dying the Right Way is a Key to Eliciting Potent Antitumor Immunity. Front. Oncol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Kono, H.; Rock, K.L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 2008, 8, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Dalod, M.; Chelbi, R.; Malissen, B.; Lawrence, T. Dendritic cell maturation: Functional specialization through signaling specificity and transcriptional programming. EMBO J. 2014, 33, 1104–1116. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern recognition receptors and the innate immune response to viral infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, N.; Ho, S.; Antonenko, S.; Malefyt, R.W.; Kastelein, R.A.; Bazan, F.; Liu, Y.J. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J. Exp. Med. 2001, 194, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Wagner, H. Toll meets bacterial CpG-DNA. Immunity 2001, 14, 499–502. [Google Scholar] [CrossRef]
- Sieben, M.; Schäfer, P.; Dinsart, C.; Galle, P.R.; Moehler, M. Activation of the human immune system via toll-like receptors by the oncolytic parvovirus H-1. Int. J. Cancer 2013, 132, 2548–2556. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.B.; Sikorski, R.R.; Kirn, D.H.D.; Thorne, S.H.S. Synergistic anti-tumor effects between oncolytic vaccinia virus and paclitaxel are mediated by the IFN response and HMGB1. Gene Ther. 2011, 18, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Sakai, R.; Ouchi, M.; Onimatsu, H.; Hioki, M.; Kagawa, S.; Uno, F.; Watanabe, Y.; Urata, Y.; Tanaka, N.; et al. Virus-mediated oncolysis induces danger signal and stimulates cytotoxic T-lymphocyte activity via proteasome activator upregulation. Oncogene 2008, 27, 2375–2381. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, R.J.; Errington, F.; Ilett, E.J.; Morgan, R.S.M.; Scott, K.J.; Kottke, T.; Thompson, J.; Morrison, E.E.; Harrington, K.J.; Pandha, H.S.; et al. Tumor infection by oncolytic reovirus primes adaptive antitumor immunity. Clin. Cancer Res. 2008, 14, 7358–7366. [Google Scholar] [CrossRef] [PubMed]
- Bridle, B.W.; Boudreau, J.E.; Lichty, B.D.; Brunelliere, J.; Stephenson, K.; Koshy, S.; Bramson, J.L.; Wan, Y. Vesicular stomatitis virus as a novel cancer vaccine vector to prime antitumor immunity amenable to rapid boosting with adenovirus. Mol. Ther. 2009, 17, 1814–1821. [Google Scholar] [CrossRef] [PubMed]
- Gauvrit, A.; Brandler, S.; Sapede-Peroz, C.; Boisgerault, N.; Tangy, F.; Gregoire, M. Measles virus induces oncolysis of mesothelioma cells and allows dendritic cells to cross-prime tumor-specific CD8 response. Cancer Res. 2008, 68, 4882–4892. [Google Scholar] [CrossRef] [PubMed]
- Errington, F.; White, C.L.; Twigger, K.R.; Rose, A.; Scott, K.; Steele, L.; Ilett, L.J.; Prestwich, R.; Pandha, H.S.; Coffey, M.; et al. Inflammatory tumour cell killing by oncolytic reovirus for the treatment of melanoma. Gene Ther. 2008, 15, 1257–1270. [Google Scholar] [CrossRef] [PubMed]
- Schulz, O.; Diebold, S.S.; Chen, M.; Näslund, T.I.; Nolte, M.A.; Alexopoulou, L.; Azuma, Y.-T.; Flavell, R.A.; Liljeström, P.; Reis e Sousa, C. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature 2005, 433, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Le Bon, A.; Etchart, N.; Rossmann, C.; Ashton, M.; Hou, S.; Gewert, D.; Borrow, P.; Tough, D.F. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat. Immunol. 2003, 4, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Waithman, J.; Lata, R.; Mifsud, N.A.; Cebon, J.; Kay, T.; Smyth, M.J.; Sadler, A.J.; Chen, W. Influenza A infection enhances cross-priming of CD8+ T cells to cell-associated antigens in a TLR7- and type I IFN-dependent fashion. J. Immunol. 2010, 185, 6013–6022. [Google Scholar] [CrossRef] [PubMed]
- Woller, N.; Gürlevik, E.; Fleischmann-Mundt, B.; Schumacher, A.; Knocke, S.; Kloos, A.M.; Saborowski, M.; Geffers, R.; Manns, M.P.; Wirth, T.C.; et al. Viral Infection of Tumors Overcomes Resistance to PD-1-immunotherapy by Broadening Neoantigenome-directed T-cell Responses. Mol. Ther. 2015, 23, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Breitbach, C.J.; Paterson, J.M.; Lemay, C.G.; Falls, T.J.; McGuire, A.; Parato, K.A.; Stojdl, D.F.; Daneshmand, M.; Speth, K.; Kirn, D.; et al. Targeted inflammation during oncolytic virus therapy severely compromises tumor blood flow. Mol. Ther. 2007, 15, 1686–1693. [Google Scholar] [CrossRef] [PubMed]
- Gujar, S.A.; Pan, D.; Marcato, P.; Garant, K.A.; Lee, P.W. Oncolytic Virus-initiated Protective Immunity Against Prostate Cancer. Mol. Ther. 2011, 19, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Holmgaard, R.B.; Subudhi, S.K.; Park, J.S.; Mansour, M.; Palese, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-Y.; Huang, Q.; Kung, H.-F. Cytokine and immuno-gene therapy for solid tumors. Cell. Mol. Immunol. 2005, 2, 81–91. [Google Scholar] [PubMed]
- White, R.L., Jr.; Schwartzentruber, D.J.; Guleria, A.; MacFarlane, M.P.; White, D.E.; Tucker, E.; Rosenberg, S.A. Cardiopulmonary Toxicity of Treatment With High-Dose Interleukin-2 in 199 Consecutive Patients With Metastatic Melanoma or Renal-Cell Carcinoma. Cancer 1994, 74, 3212–3222. [Google Scholar] [CrossRef]
- Dranoff, G.; Jaffee, E.; Lazenby, A.; Golumbek, P.; Levitsky, H.; Brose, K.; Jackson, V.; Hamada, H.; Pardoll, D.; Mulligan, R.C. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc. Natl. Acad. Sci. USA 1993, 90, 3539–3543. [Google Scholar] [CrossRef] [PubMed]
- Mach, N.; Gillessen, S.; Wilson, S.B.; Sheehan, C.; Mihm, M.; Dranoff, G. Differences in dendritic cells stimulated in vivo by tumors engineered to secrete granulocyte-macrophage colony-stimulating factor or Flt3-ligand. Cancer Res. 2000, 60, 3239–3246. [Google Scholar]
- Dranoff, G. GM-CSF-based cancer vaccines. Immunol. Rev. 2002, 188, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kim, D.W.; DeRaffele, G.; Mitcham, J.; Coffin, R.S.; Kim-Schulze, S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann. Surg. Oncol. 2010, 17, 718–730. [Google Scholar] [CrossRef] [PubMed]
- Heol, J.; Reid, T.; Ruo, L.; Breitbach, C.J.; Rose, S.; Bloomston, M.; Cho, M.; Lim, H.Y.; Chung, H.C.; Chang, W.K.; et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 2013, 19, 329–336. [Google Scholar]
- Cerullo, V.; Pesonen, S.; Diaconu, I.; Escutenaire, S.; Arstila, P.T.; Ugolini, M.; Nokisalmi, P.; Raki, M.; Laasonen, L.; Särkioja, M.; et al. Oncolytic adenovirus coding for granulocyte macrophage colony-stimulating factor induces antitumoral immunity in cancer patients. Cancer Res. 2010, 70, 4297–4309. [Google Scholar] [CrossRef] [PubMed]
- Grote, D.; Cattaneo, R.; Fielding, A.K. Neutrophils contribute to the measles virus-induced antitumor effect: Enhancement by granulocyte macrophage colony-stimulating factor expression. Cancer Res. 2003, 63, 6463–6468. [Google Scholar] [PubMed]
- Bergman, I.; Griffin, J.A.; Gao, Y.; Whitaker-Dowling, P. Treatment of implanted mammary tumors with recombinant vesicular stomatitis virus targeted to Her2/neu. Int. J. Cancer 2007, 121, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Pachella, L.A.; Madsen, L.T.; Dains, J.E. The Toxicity and Benefit of Various Dosing Strategies for Interleukin-2 in Metastatic Melanoma and Renal Cell Carcinoma. J. Adv. Pract. Oncol. 2015, 6, 212–221. [Google Scholar] [PubMed]
- Vigil, A.; Park, M.S.; Martinez, O.; Chua, M.A.; Xiao, S.; Cros, J.F.; Martínez-Sobrido, L.; Woo, S.L.C.; García-Sastre, A. Use of reverse genetics to enhance the oncolytic properties of newcastle disease virus. Cancer Res. 2007, 67, 8285–8292. [Google Scholar] [CrossRef] [PubMed]
- Ferrantini, M.; Capone, I.; Belardelli, F. Interferon-alpha and cancer: Mechanisms of action and new perspectives of clinical use. Biochimie 2007, 89, 884–893. [Google Scholar] [CrossRef] [PubMed]
- Jonasch, E.; Haluska, F.G. Interferon in oncological practice: Review of interferon biology, clinical applications, and toxicities. Oncologist 2001, 6, 34–55. [Google Scholar] [CrossRef] [PubMed]
- LaRocca, C.J.; Han, J.; Gavrikova, T.; Armstrong, L.; Oliveira, A.R.; Shanley, R.; Vickers, S.M.; Yamamoto, M.; Davydova, J. Oncolytic adenovirus expressing interferon alpha in a syngeneic Syrian hamster model for the treatment of pancreatic cancer. Surgery 2015, 157, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Willmon, C.L.; Saloura, V.; Fridlender, Z.G.; Wongthida, P.; Diaz, R.M.; Thompson, J.; Kottke, T.; Federspiel, M.; Barber, G.; Albelda, S.M.; et al. Expression of IFN-beta enhances both efficacy and safety of oncolytic vesicular stomatitis virus for therapy of mesothelioma. Cancer Res. 2009, 69, 7713–7720. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois-Daigneault, M.-C.; Roy, D.G.; Falls, T.; Twumasi-Boateng, K.; St-Germain, L.E.; Marguerie, M.; Garcia, V.; Selman, M.; Jennings, V.A.; Pettigrew, J.; et al. Oncolytic vesicular stomatitis virus expressing interferon-γ has enhanced therapeutic activity. Mol. Ther. Oncolytics 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Peng, L.; Sham, J.; Wang, X.; Zhang, Q.; Chua, D.; Liu, C.; Cui, Z.; Xue, H.; Wu, H.; et al. Immune Gene-Viral Therapy with Triplex Efficacy Mediated by Oncolytic Adenovirus Carrying an Interferon-Gamma Gene Yields Efficient Antitumor Activity in Immunodeficient and Immunocompetent Mice. Mol. Ther. 2006, 13, 918–927. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; O’Malley, M.; Urban, J.; Sampath, P.; Guo, Z.S.; Kalinski, P.; Thorne, S.H.; Bartlett, D.L. Chemokine expression from oncolytic vaccinia virus enhances vaccine therapies of cancer. Mol. Ther. 2011, 19, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Lapteva, N.; Aldrich, M.; Weksberg, D.; Rollins, L.; Goltsova, T.; Chen, S.-Y.; Huang, X.F. Targeting the intratumoral dendritic cells by the oncolytic adenoviral vaccine expressing RANTES elicits potent antitumor immunity. J. Immunother. 2009, 32, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishna, E.; Woller, N.; Mundt, B.; Knocke, S.; Gurlevik, E.; Saborowski, M.; Malek, N.; Manns, M.P.; Wirth, T.; Kuhnel, F.; et al. Antitumoral immune response by recruitment and expansion of dendritic cells in tumors infected with telomerase-dependent oncolytic viruses. Cancer Res. 2009, 69, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Margolin, K. Cytokines in cancer immunotherapy. Cancers 2011, 3, 3856–3893. [Google Scholar] [CrossRef] [PubMed]
- Huehls, A.M.; Coupet, T.A.; Sentman, C.L. Bispecific T-cell engagers for cancer immunotherapy. Immunol. Cell Biol. 2015, 93, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Wang, X.; Guo, Z.S.; Bartlett, D.L.; Gottschalk, S.M.; Song, X.-T. T-cell engager-armed oncolytic vaccinia virus significantly enhances antitumor therapy. Mol. Ther. 2014, 22, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Klein, G.; Sjogren, H.O.; Klein, E.; Hellstrom, K.E. Demonstration of resistance against methylcholanthrene-induced sarcomas. Cancer Res. 1960, 20, 1561–1572. [Google Scholar]
- Vergati, M.; Intrivici, C.; Huen, N.-Y.; Schlom, J.; Tsang, K.Y. Strategies for cancer vaccine development. J. Biomed. Biotechnol. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Restifo, N.P. Cancer immunotherapy: Moving beyond current vaccines. Nat. Med. 2004, 10, 909–915. [Google Scholar] [CrossRef]
- Melero, I.; Gaudernack, G.; Gerritsen, W.; Huber, C.; Parmiani, G.; Scholl, S.; Thatcher, N.; Wagstaff, J.; Zielinski, C.; Faulkner, I.; et al. Therapeutic vaccines for cancer: An overview of clinical trials. Nat. Rev. Clin. Oncol. 2014, 11, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Moehler, M.H.; Zeidler, M.; Wilsberg, V.; Cornelis, J.J.; Woelfel, T.; Rommelaere, J.; Galle, P.R.; Heike, M. Parvovirus H-1-induced tumor cell death enhances human immune response in vitro via increased phagocytosis, maturation, and cross-presentation by dendritic cells. Hum. Gene Ther. 2005, 16, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Toda, M.; Rabkin, S.D.; Kojima, H.; Martuza, R.L. Herpes simplex virus as an in situ cancer vaccine for the induction of specific anti-tumor immunity. Hum. Gene Ther. 1999, 10, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Lemay, C.G.; Rintoul, J.L.; Kus, A.; Paterson, J.M.; Garcia, V.; Falls, T.J.; Ferreira, L.; Bridle, B.W.; Conrad, D.P.; Tang, V.A.; et al. Harnessing oncolytic virus-mediated antitumor immunity in an infected cell vaccine. Mol. Ther. 2012, 20, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- Kottke, T.; Errington, F.; Pulido, J.; Galivo, F.; Thompson, J.; Wongthida, P.; Diaz, R.M.; Chong, H.; Ilett, E.; Chester, J.; et al. Broad antigenic coverage induced by vaccination with virus-based cDNA libraries cures established tumors. Nat. Med. 2011, 17, 854–859. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.S.; Monken, C.E.; Lattime, E.C. Intratumoral Vaccination with Vaccinia-Expressed Tumor Antigen and Granulocyte Macrophage Colony-Stimulating Factor Overcomes Immunological Ignorance to Tumor Antigen. Cancer Res. 2003, 63, 6956–6961. [Google Scholar] [PubMed]
- Harrop, R.; John, J.; Carroll, M.W. Recombinant viral vectors: Cancer vaccines. Adv. Drug Deliv. Rev. 2006, 58, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Bridle, B.W.; Stephenson, K.B.; Boudreau, J.E.; Koshy, S.; Kazdhan, N.; Pullenayegum, E.; Brunellière, J.; Bramson, J.L.; Lichty, B.D.; Wan, Y. Potentiating cancer immunotherapy using an oncolytic virus. Mol. Ther. 2010, 18, 1430–1439. [Google Scholar] [CrossRef] [PubMed]
- Bridle, B.W.; Clouthier, D.; Zhang, L.; Pol, J.; Chen, L.; Lichty, B.D.; Bramson, J.L.; Wan, Y. Oncolytic vesicular stomatitis virus quantitatively and qualitatively improves primary CD8+ T-cell responses to anticancer vaccines. Oncoimmunology 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Pol, J.G.; Zhang, L.; Bridle, B.W.; Stephenson, K.B.; Rességuier, J.; Hanson, S.; Chen, L.; Kazdhan, N.; Bramson, J.L.; Stojdl, D.F.; et al. Maraba virus as a potent oncolytic vaccine vector. Mol. Ther. 2014, 22, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.; Pamer, E.G. Feedback regulation of pathogen-specific T cell priming. Immunity 2003, 18, 499–511. [Google Scholar] [CrossRef]
- Bridle, B.W.; Nguyen, A.; Salem, O.; Zhang, L.; Koshy, S.; Clouthier, D.; Chen, L.; Pol, J.; Swift, S.; Bowdish, D.M.E.; et al. Privileged Antigen Presentation in Splenic B Cell Follicles Maximizes T Cell Responses in Prime-Boost Vaccination. J. Immunol. 2016, 196, 4587–4595. [Google Scholar] [CrossRef]
- Canadian Cancer Trials Group; Ottawa Hospital Research Institute. MG1 Maraba/MAGE-A3, With and Without Adenovirus Vaccine, With Transgenic MAGE-A3 Insertion in Patients With Incurable MAGE-A3-Expressing Solid Tumours (I214). Available online: https://clinicaltrials.gov/ct2/show/NCT02285816 (accessed on 6 May 2016).
- Chuang, C.M.; Monie, A.; Wu, A.; Pai, S.I.; Hung, C.F. Combination of viral oncolysis and tumor-specific immunity to control established tumors. Clin. Cancer Res. 2009, 15, 4581–4588. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L.; Wallace, D.R.; Gooley, T.A.; Dang, Y.; Slota, M.; Lu, H.; Coveler, A.L.; Childs, J.S.; Higgins, D.M.; Fintak, P.A.; et al. Concurrent trastuzumab and HER2/neu-specific vaccination in patients with metastatic breast cancer. J. Clin. Oncol. 2009, 27, 4685–4692. [Google Scholar] [CrossRef] [PubMed]
- Bridle, B.W.; Hanson, S.; Lichty, B.D. Combining oncolytic virotherapy and tumour vaccination. Cytokine Growth Factor Rev. 2010, 21, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Yee, C. Adoptive T-cell therapy for cancer: Boutique therapy or treatment modality? Clin. Cancer Res. 2013, 19, 4550–4552. [Google Scholar] [CrossRef] [PubMed]
- Hinrichs, C.S.; Rosenberg, S.A. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol. Rev. 2014, 257, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Teachey, D.T.; Porter, D.L.; Grupp, S.A. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 2015, 125, 4017–4023. [Google Scholar] [CrossRef] [PubMed]
- Fousek, K.; Ahmed, N. The evolution of T-cell therapies for solid malignancies. Clin. Cancer Res. 2015, 21, 3384–3392. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Slaney, C.Y.; Kershaw, M.H.; Darcy, P.K. Trafficking of T cells into tumors. Cancer Res. 2014, 74, 7168–7174. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Rivera, A.; Tao, L.; Zhang, X. An HSV-2 based oncolytic virus can function as an attractant to guide migration of adoptively transferred T cells to tumor sites. Oncotarget 2015, 6, 902–914. [Google Scholar] [CrossRef] [PubMed]
- Park, J.R.; Digiusto, D.L.; Slovak, M.; Wright, C.; Naranjo, A.; Wagner, J.; Meechoovet, H.B.; Bautista, C.; Chang, W.-C.; Ostberg, J.R.; et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol. Ther. 2007, 15, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, C.A.; Gattinoni, L.; Restifo, N.P. Sorting through subsets: Which T-cell populations mediate highly effective adoptive immunotherapy? J. Immunother. 2012, 35, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Heemskerk, M.H.M.; Hoogeboom, M.; Hagedoorn, R.; Kester, M.G.D.; Willemze, R.; Falkenburg, J.H.F. Reprogramming of virus-specific T cells into leukemia-reactive T cells using T cell receptor gene transfer. J. Exp. Med. 2004, 199, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.; Westwood, J.A.; Brown, L.E.; Teng, M.W.L.; Moeller, M.; Xu, Y.; Smyth, M.J.; Hwu, P.; Darcy, P.K.; Kershaw, M.H. Antitumor activity of dual-specific T cells and influenza virus. Cancer Gene Ther. 2007, 14, 499–508. [Google Scholar] [CrossRef]
- Ludewig, B.; Bonilla, W.V.; Dumrese, T.; Odermatt, B.; Zinkernagel, R.M.; Hengartner, H. Perforin-independent regulation of dendritic cell homeostasis by CD8+ T cells in vivo: Implications for adaptive immunotherapy. Eur. J. Immunol. 2001, 31, 1772–1779. [Google Scholar] [CrossRef]
- Willmon, C.; Harrington, K.; Kottke, T.; Prestwich, R.; Melcher, A.; Vile, R. Cell carriers for oncolytic viruses: Fed Ex for cancer therapy. Mol. Ther. 2009, 17, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Wang, H.; Kottke, T.; Diaz, R.M.; Willmon, C.; Hudacek, A.; Thompson, J.; Parato, K.; Bell, J.; Naik, J.; et al. Loading of oncolytic vesicular stomatitis virus onto antigen-specific T cells enhances the efficacy of adoptive T-cell therapy of tumors. Gene Ther. 2008, 15, 604–616. [Google Scholar] [CrossRef] [PubMed]
- VanSeggelen, H.; Tantalo, D.G.; Afsahi, A.; Hammill, J.A.; Bramson, J.L. Chimeric antigen receptor–engineered T cells as oncolytic virus carriers. Mol. Ther. Oncolytics 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Marchini, A.; Scott, E.M.; Rommelaere, J. Overcoming barriers in oncolytic virotherapy with HDAC inhibitors and immune checkpoint blockade. Viruses 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Bauzon, M.; Hermiston, T. Armed therapeutic viruses—A disruptive therapy on the horizon of cancer immunotherapy. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Tarhini, A.; Lo, E.; Minor, D.R. Releasing the brake on the immune system: Ipilimumab in melanoma and other tumors. Cancer Biother. Radiopharm. 2010, 25, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Van Elsas, A.; Hurwitz, A.A.; Allison, J.P. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied. J. Exp. Med. 1999, 190, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.R.; Chasalow, S.D.; Wang, L.; Hamid, O.; Schmidt, H.; Cogswell, J.; Alaparthy, S.; Berman, D.; Jure-Kunkel, M.; Siemers, N.O.; et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol. Immunother. 2012, 61, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Winograd, R.; Byrne, K.T.; Evans, R.A.; Odorizzi, P.M.; Meyer, A.R.L.; Bajor, D.L.; Clendenin, C.; Stanger, B.Z.; Furth, E.E.; Wherry, E.J.; et al. Induction of T-cell Immunity Overcomes Complete Resistance to PD-1 and CTLA-4 Blockade and Improves Survival in Pancreatic Carcinoma. Cancer Immunol. Res. 2015, 3, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.J.; Sampath, P.; Hou, W.; Thorne, S.H. Defining effective combinations of immune checkpoint blockade and oncolytic virotherapy. Clin. Cancer Res. 2015, 21, 5543–5551. [Google Scholar] [CrossRef] [PubMed]
- Engeland, C.E.; Grossardt, C.; Veinalde, R.; Bossow, S.; Lutz, D.; Kaufmann, J.K.; Shevchenko, I.; Umansky, V.; Nettelbeck, D.M.; Weichert, W.; et al. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol. Ther. 2014, 22, 1949–1959. [Google Scholar] [CrossRef] [PubMed]
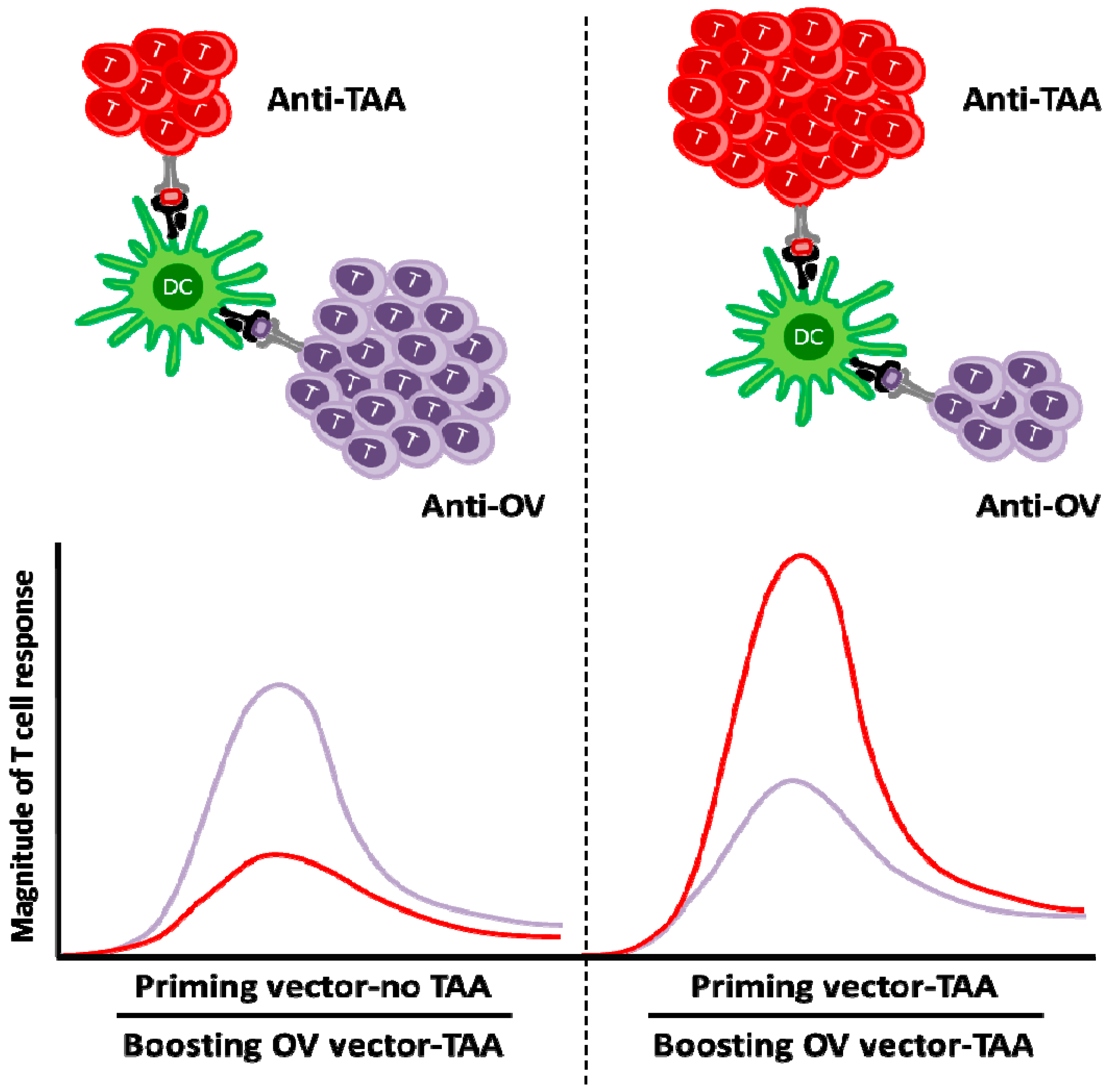
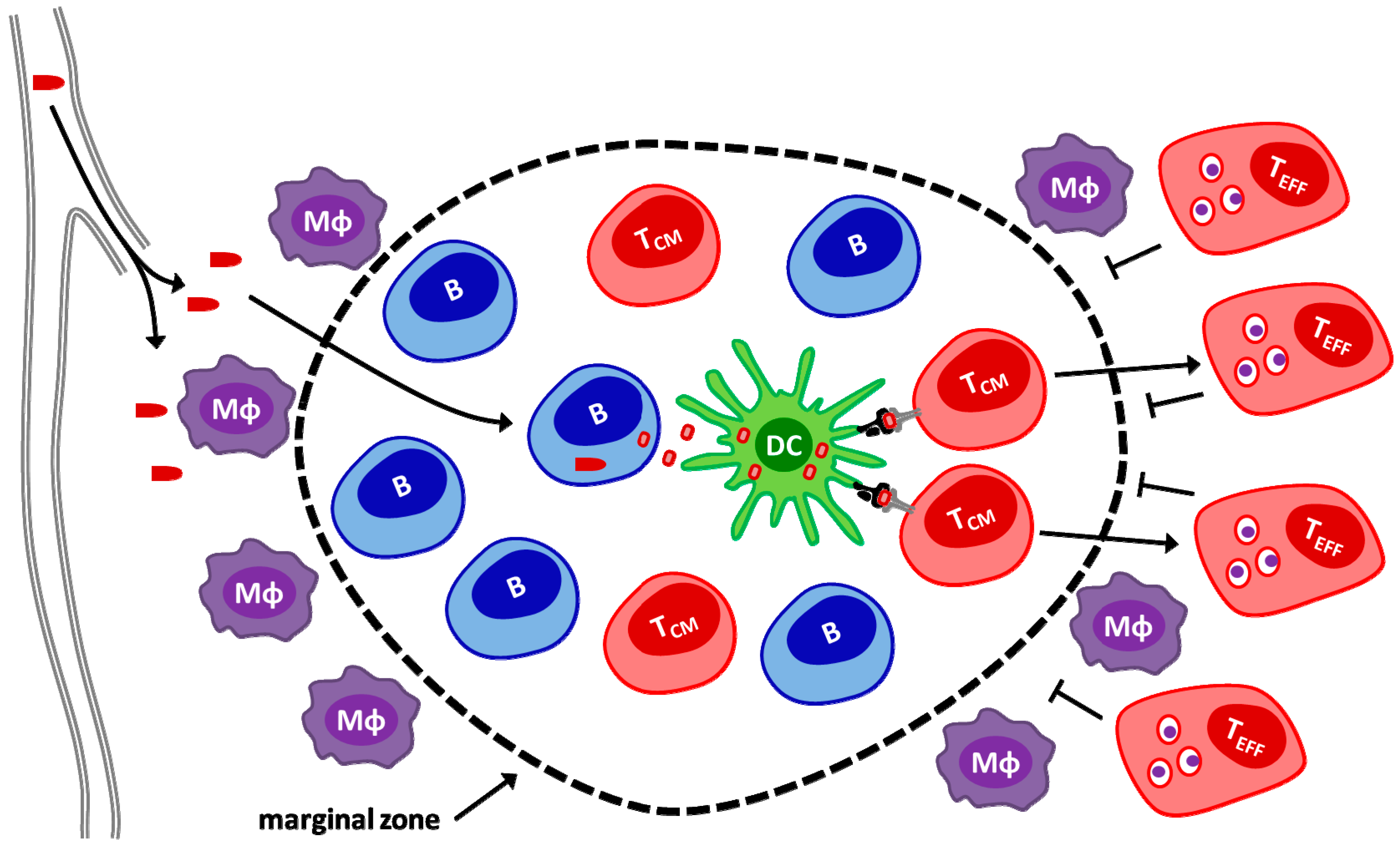

© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bastin, D.; Walsh, S.R.; Al Saigh, M.; Wan, Y. Capitalizing on Cancer Specific Replication: Oncolytic Viruses as a Versatile Platform for the Enhancement of Cancer Immunotherapy Strategies. Biomedicines 2016, 4, 21. https://doi.org/10.3390/biomedicines4030021
Bastin D, Walsh SR, Al Saigh M, Wan Y. Capitalizing on Cancer Specific Replication: Oncolytic Viruses as a Versatile Platform for the Enhancement of Cancer Immunotherapy Strategies. Biomedicines. 2016; 4(3):21. https://doi.org/10.3390/biomedicines4030021
Chicago/Turabian StyleBastin, Donald, Scott R. Walsh, Meena Al Saigh, and Yonghong Wan. 2016. "Capitalizing on Cancer Specific Replication: Oncolytic Viruses as a Versatile Platform for the Enhancement of Cancer Immunotherapy Strategies" Biomedicines 4, no. 3: 21. https://doi.org/10.3390/biomedicines4030021
APA StyleBastin, D., Walsh, S. R., Al Saigh, M., & Wan, Y. (2016). Capitalizing on Cancer Specific Replication: Oncolytic Viruses as a Versatile Platform for the Enhancement of Cancer Immunotherapy Strategies. Biomedicines, 4(3), 21. https://doi.org/10.3390/biomedicines4030021