
Low-Dose Metformin as a Monotherapy Does Not Reduce Non-Small-Cell Lung Cancer Tumor Burden in Mice

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Animals
2.2. Culturing Non-Small-Cell Lung Cancer Cells
2.3. Orthotopic Injection
2.4. In Vivo Imaging
2.5. Metformin Treatment
2.6. Tumor Burden
2.7. Tumor Tissue and Gastrocnemius Muscle Homogenization and mRNA Extraction
2.8. cDNA and Real-Time PCR
2.9. Gastrocnemius Tissue Protein Isolation and Quantification
2.10. Western Blotting
2.11. Statistical Analyses
3. Results
3.1. Body Mass in C57BL/6J Mice with NSCLC
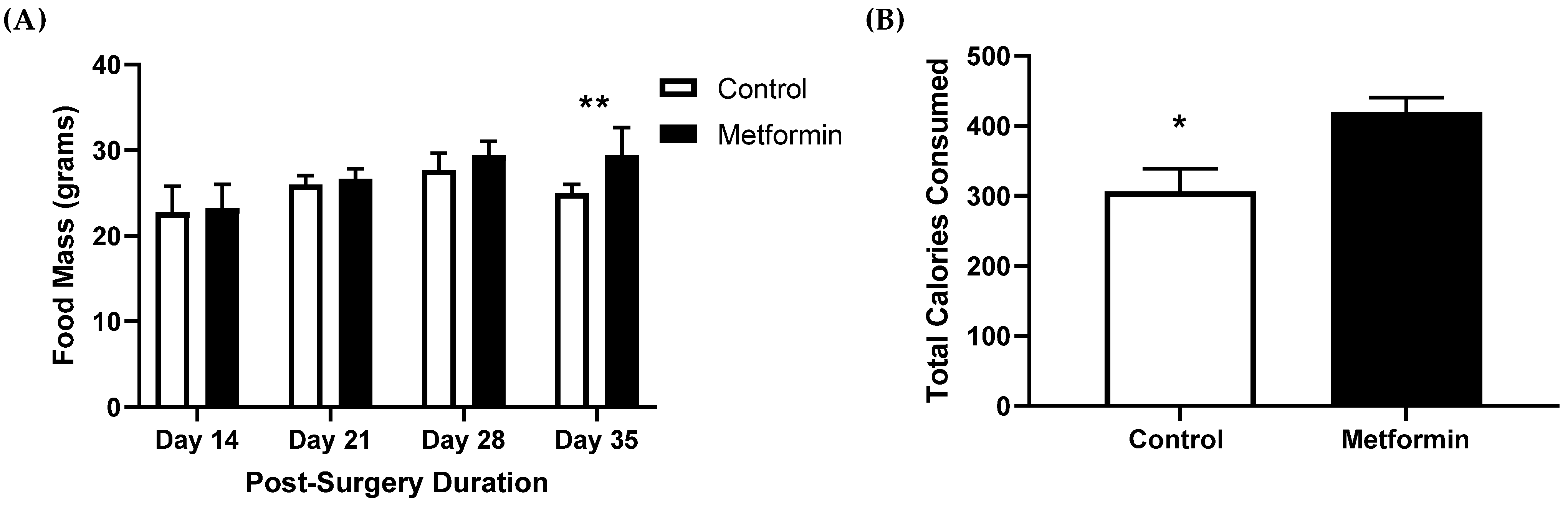
3.2. Food Consumption in C57BL/6J Mice with NSCLC
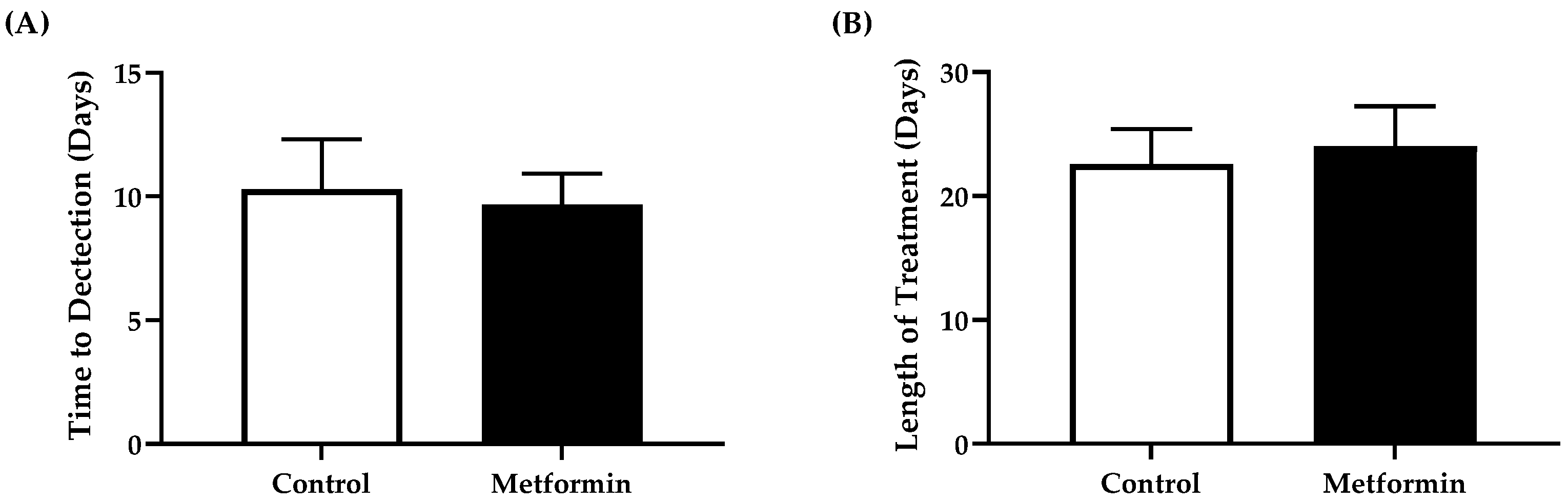
3.3. Time to Tumor Detection and Length of Treatment
3.4. NSCLC Tumor Burden and Animal Survival
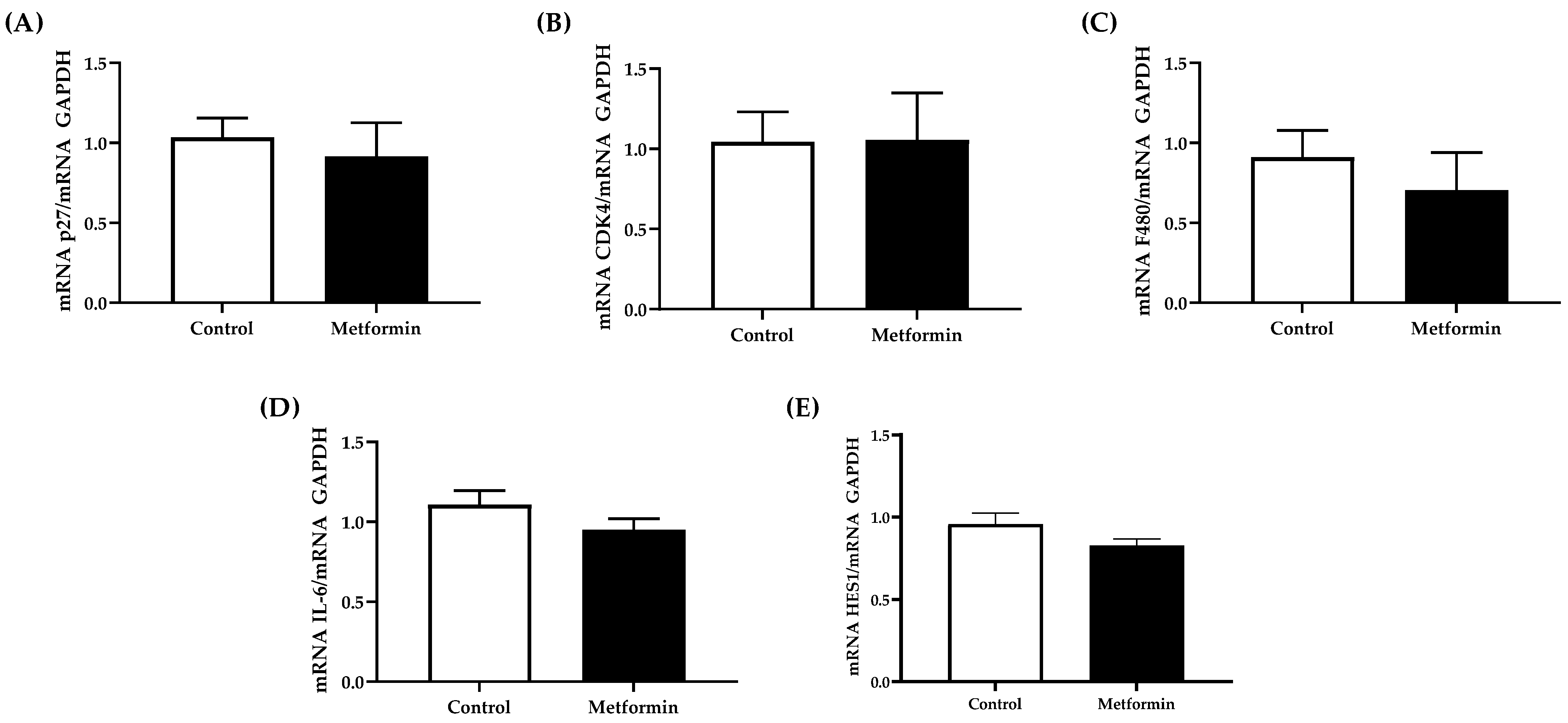
3.5. NSCLC Tumor Gene Expression
3.6. Maintenance of Skeletal Muscle Mass
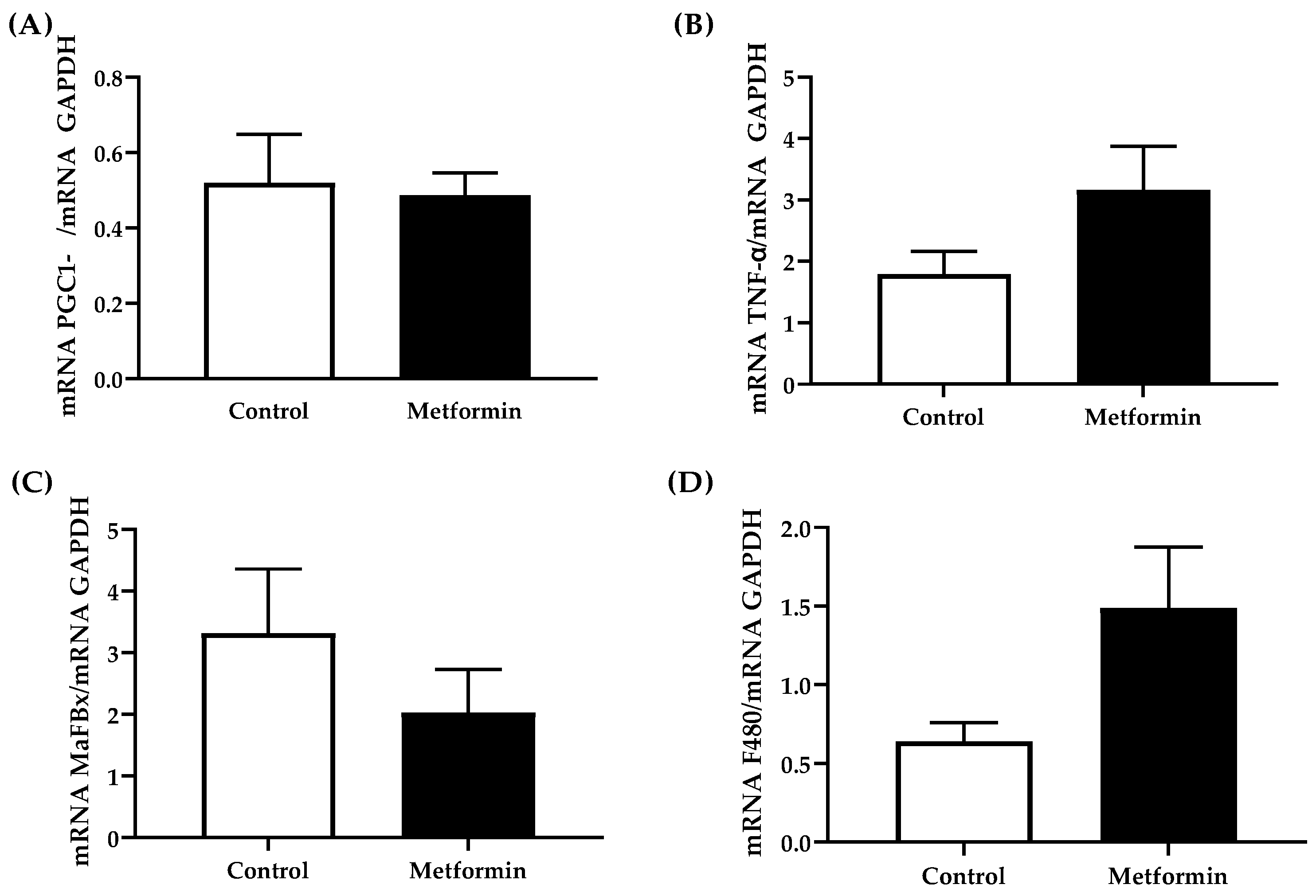
3.7. Gastrocnemius Gene Expression
3.8. Gastrocnemius Protein Expression
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Cancer Society. Cancer Facts and Figures 2019; American Cancer Society: Atlanta, GA, USA, 2019; Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2019/cancer-facts-and-figures-2019.pdf (accessed on 5 December 2020).
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Hirsch, F.R.; Suda, K.; Wiens, J.; Bunn, P.A. New and emerging targeted treatments in advanced non-small-cell lung cancer. Lancet 2016, 388, 1012–1024. [Google Scholar] [CrossRef]
- Irie, H.; Banno, K.; Yanokura, M.; Iida, M.; Adachi, M.; Nakamura, K.; Umene, K.; Nogami, Y.; Masuda, K.; Kobayashi, Y.; et al. Metformin: A candidate for the treatment of gynecological tumors based on drug repositioning. Oncol. Lett. 2016, 11, 1287–1293. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.T. Food and Drug Administration Drug Approval Process: A History and Overview. Nurs. Clin. N. Am. 2016, 51, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sleire, L.; Førde, H.E.; Netland, I.A.; Leiss, L.; Skeie, B.S.; Enger, P.Ø. Drug repurposing in cancer. Pharmacol. Res. 2017, 124, 74–91. [Google Scholar] [CrossRef]
- Queiroz, E.A.I.F.; Puukila, S.; Eichler, R.; Sampaio, S.C.; Forsyth, H.L.; Lees, S.J.; Barbosa, A.M.; Dekker, R.F.H.; Fortes, Z.B.; Khaper, N. Metformin Induces Apoptosis and Cell Cycle Arrest Mediated by Oxidative Stress, AMPK and FOXO3a in MCF-7 Breast Cancer Cells. PLoS ONE 2014, 9, e98207. [Google Scholar] [CrossRef]
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin Is an AMP Kinase–Dependent Growth Inhibitor for Breast Cancer Cells. Cancer Res. 2006, 66, 10269–10273. [Google Scholar] [CrossRef] [Green Version]
- Sarmento-Cabral, A.; L-López, F.; Gahete, M.D.; Castaño, J.P.; Luque, R.M. Metformin Reduces Prostate Tumor Growth, in a Diet-Dependent Manner, by Modulating Multiple Signaling Pathways. Mol. Cancer Res. 2017, 15, 862–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.W.; Lee, J.H.; Park, Y.H.; Park, S.J.; Cheon, J.H.; Kim, W.H.; Kim, T.I. Sex-dependent difference in the effect of metformin on colorectal cancer-specific mortality of diabetic colorectal cancer patients. World J. Gastroenterol. 2017, 23, 5196–5205. [Google Scholar] [CrossRef] [PubMed]
- Checkley, L.A.; Rho, O.; Angel, J.M.; Cho, J.; Blando, J.; Beltran, L.; Hursting, S.D.; DiGiovanni, J. Metformin Inhibits Skin Tumor Promotion in Overweight and Obese Mice. Cancer Prev. Res. 2014, 7, 54–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, N.; Zhang, Y.; Gong, Y.I.; He, J.; Chen, X. Metformin and lung cancer risk of patients with type 2 diabetes mellitus: A meta-analysis. Biomed. Rep. 2015, 3, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Tseng, C.-H. Metformin and lung cancer risk in patients with type 2 diabetes mellitus. Oncotarget 2017, 8, 41132–41142. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.-S.; Chuang, M.-C.; Chen, Y.-C.; Lee, C.-P.; Yang, T.-M.; Chen, P.-C.; Tsai, Y.-H.; Yang, Y.-H. Metformin Prolongs Survival in Type 2 Diabetes Lung Cancer Patients With EGFR-TKIs. Integr. Cancer Ther. 2019, 18, 1534735419869491. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.-J.; Yang, C.-J.; Kung, Y.-T.; Sheu, C.-C.; Shen, Y.-T.; Chang, P.-Y.; Huang, M.-S.; Chiu, H.-C. Metformin decreases lung cancer risk in diabetic patients in a dose-dependent manner. Lung Cancer 2014, 86, 137–143. [Google Scholar] [CrossRef]
- Algire, C.; Amrein, L.; Zakikhani, M.; Panasci, L.; Pollak, M. Metformin blocks the stimulative effect of a high-energy diet on colon carcinoma growth in vivo and is associated with reduced expression of fatty acid synthase. Endocr. Relat. Cancer 2010, 17, 351–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Kim, W.G.; Zhao, L.; Enomoto, K.; Willingham, M.; Cheng, S.-Y. Metformin blocks progression of obesity-activated thyroid cancer in a mouse model. Oncotarget 2016, 7, 34832–34844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Y.; Yi, Y.; Liu, Y.; Liu, X.; Keller, E.T.; Qian, C.-N.; Zhang, J.; Lu, Y. Metformin targets multiple signaling pathways in cancer. Chin. J. Cancer 2017, 36, 17. [Google Scholar] [CrossRef] [Green Version]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Prim. 2018, 4, 17105. [Google Scholar] [CrossRef]
- Evans, W.J. Skeletal muscle loss: Cachexia, sarcopenia, and inactivity. Am. J. Clin. Nutr. 2010, 91, 1123S–1127S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorensen, J. Lung Cancer Cachexia: Can Molecular Understanding Guide Clinical Management? Integr. Cancer Ther. 2018, 17, 1000–1008. [Google Scholar] [CrossRef] [Green Version]
- Aniort, J.; Stella, A.; Philipponnet, C.; Poyet, A.; Polge, C.; Claustre, A.; Combaret, L.; Béchet, D.; Attaix, D.; Boisgard, S.; et al. Muscle wasting in patients with end-stage renal disease or early-stage lung cancer: Common mechanisms at work. J. Cachexia Sarcopenia Muscle 2019, 10, 323–337. [Google Scholar] [CrossRef] [Green Version]
- Currow, D.C.; Maddocks, M.; Cella, D.; Muscaritoli, M. Efficacy of Anamorelin, a Novel Non-Peptide Ghrelin Analogue, in Patients with Advanced Non-Small Cell Lung Cancer (NSCLC) and Cachexia—Review and Expert Opinion. Int. J. Mol. Sci. 2018, 19, 3471. [Google Scholar] [CrossRef] [Green Version]
- LeBlanc, T.; Nipp, R.D.; Rushing, C.N.; Samsa, G.P.; Locke, S.; Kamal, A.H.; Cella, D.F.; Abernethy, A.P. Correlation Between the International Consensus Definition of the Cancer Anorexia-Cachexia Syndrome (CACS) and Patient-Centered Outcomes in Advanced Non-Small Cell Lung Cancer. J. Pain Symptom Manag. 2015, 49, 680–689. [Google Scholar] [CrossRef]
- Aoyagi, T.; Terracina, K.P.; Raza, A.; Matsubara, H.; Takabe, K. Cancer cachexia, mechanism and treatment. World J. Gastrointest. Oncol. 2015, 7, 17–29. [Google Scholar] [CrossRef]
- Fearon, K.C.; Voss, A.C.; Hustead, D.S. Definition of cancer cachexia: Effect of weight loss, reduced food intake, and systemic inflammation on functional status and prognosis. Am. J. Clin. Nutr. 2006, 83, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Burckart, K.; Beca, S.; Urban, R.J.; Sheffield-Moore, M. Pathogenesis of muscle wasting in cancer cachexia: Targeted anabolic and anticatabolic therapies. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 410–416. [Google Scholar] [CrossRef] [Green Version]
- Tisdale, M.J. Cancer cachexia. Curr. Opin. Gastroenterol. 2010, 26, 146–151. [Google Scholar] [CrossRef]
- Suwa, M.; Egashira, T.; Nakano, H.; Sasaki, H.; Kumagai, S. Metformin increases the PGC-1α protein and oxidative enzyme activities possibly via AMPK phosphorylation in skeletal muscle in vivo. J. Appl. Physiol. 2006, 101, 1685–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandri, M.; Lin, J.; Handschin, C.; Yang, W.; Arany, Z.P.; Lecker, S.H.; Goldberg, A.L.; Spiegelman, B.M. PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. USA 2006, 103, 16260–16265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlidou, T.; Marinkovic, M.; Rosina, M.; Fuoco, C.; Vumbaca, S.; Gargioli, C.; Castagnoli, L.; Cesareni, G. Metformin Delays Satellite Cell Activation and Maintains Quiescence. Stem Cells Int. 2019, 2019, 5980465. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Price, F.; Rudnicki, M.A. Satellite Cells and the Muscle Stem Cell Niche. Physiol. Rev. 2013, 93, 23–67. [Google Scholar] [CrossRef] [Green Version]
- Latteyer, S.; Christoph, S.; Theurer, S.; Hönes, G.S.; Schmid, K.W.; Führer, D.; Moeller, L.C. Thyroxine promotes lung cancer growth in an orthotopic mouse model. Endocr. Relat. Cancer 2019, 26, 565–574. [Google Scholar] [CrossRef]
- Mordant, P.; Loriot, Y.; Lahon, B.; Castier, Y.; Lesèche, G.; Soria, J.-C.; Vozenin, M.-C.; Decraene, C.; Deutsch, E. Bioluminescent Orthotopic Mouse Models of Human Localized Non-Small Cell Lung Cancer: Feasibility and Identification of Circulating Tumour Cells. PLoS ONE 2011, 6, e26073. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, F.; Amano, H.; Ito, Y.; Matsui, Y.; Hosono, K.; Kitasato, H.; Satoh, Y.; Majima, M. Aspirin reduces lung cancer metastasis to regional lymph nodes. Biomed. Pharmacother. 2014, 68, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E.; Litchfield, L.M.; Mitra, A.K.; Nieman, K.; Mukherjee, A.; Zhang, Y.; Johnson, A.; Bradaric, M.; Lee, W.; Romero, I.L. Metformin inhibits ovarian cancer growth and increases sensitivity to paclitaxel in mouse models. Am. J. Obstet. Gynecol. 2015, 212, 479.e1–479.e10. [Google Scholar] [CrossRef] [Green Version]
- Memmott, R.M.; Mercado, J.R.; Maier, C.R.; Kawabata, S.; Fox, S.D.; Dennis, P.A. Metformin Prevents Tobacco Carcinogen–Induced Lung Tumorigenesis. Cancer Prev. Res. 2010, 3, 1066–1076. [Google Scholar] [CrossRef] [Green Version]
- Pernicova, I.; Korbonits, M. Metformin—Mode of action and clinical implications for diabetes and cancer. Nat. Rev. Endocrinol. 2014, 10, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.J.; Turner, R.C. Metformin. N. Engl. J. Med. 1996, 334, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Panfoli, I.; Puddu, A.; Bertola, N.; Ravera, S.; Maggi, D. The Hormetic Effect of Metformin: “Less Is More”? Int. J. Mol. Sci. 2021, 22, 6297. [Google Scholar] [CrossRef] [PubMed]
- Dowling, R.J.; Lam, S.; Bassi, C.; Mouaaz, S.; Aman, A.; Kiyota, T.; Al-Awar, R.; Goodwin, P.; Stambolic, V. Metformin Pharmacokinetics in Mouse Tumors: Implications for Human Therapy. Cell Metab. 2016, 23, 567–568. [Google Scholar] [CrossRef] [Green Version]
- Heinig, K.; Bucheli, F. Fast liquid chromatographic-tandem mass spectrometric (LC–MS–MS) determination of metformin in plasma samples. J. Pharm. Biomed. Anal. 2004, 34, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Duan, J.; Liu, R.; Du, Y.; Luo, Q.; Cui, Y.; Su, Z.; Xu, J.; Xie, Y.; Lu, W. Engineered targeting tLyp-1 exosomes as gene therapy vectors for efficient delivery of siRNA into lung cancer cells. Asian J. Pharm. Sci. 2020, 15, 461–471. [Google Scholar] [CrossRef]
- Mehta, A.; Dalle Vedove, E.; Isert, L.; Merkel, O.M. Targeting KRAS Mutant Lung Cancer Cells with siRNA-Loaded Bovine Serum Albumin Nanoparticles. Pharm. Res. 2019, 36, 133. [Google Scholar] [CrossRef]
- Duncan, R.; Gaspar, R. Nanomedicine(s) under the microscope. Mol. Pharm. 2011, 8, 2101–2141. [Google Scholar] [CrossRef]
- Chaudhary, S.; Singh, A.; Kumar, P.; Kaushik, M. Strategic targeting of non-small-cell lung cancer utilizing genetic material-based delivery platforms of nanotechnology. J. Biochem. Mol. Toxicol. 2021, 35, e22784. [Google Scholar] [CrossRef]
- Knüpfer, H.; Preiss, R. Serum interleukin-6 levels in colorectal cancer patients—A summary of published results. Int. J. Color. Dis. 2009, 25, 135–140. [Google Scholar] [CrossRef]
- Zhao, Z.; Cheng, X.; Wang, Y.; Han, R.; Li, L.; Xiang, T.; He, L.; Long, H.; Zhu, B.; He, Y. Metformin Inhibits the IL-6-Induced Epithelial-Mesenchymal Transition and Lung Adenocarcinoma Growth and Metastasis. PLoS ONE 2014, 9, e95884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Liang, G.; Yao, Z.; Zhang, J.; Liu, R.; Chen, H.; Zhou, Y.; Wu, H.; Ruiyang, L.; He, Q. Metformin prevents cancer metastasis by inhibiting M2-like polarization of tumor associated macrophages. Oncotarget 2015, 6, 36441–36455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Lin, Y.; Xiong, X.; Wang, L.; Guo, Y.; Chen, Y.; Chen, S.; Wang, G.; Lin, P.; Chen, H.; et al. Low-Dose Metformin Reprograms the Tumor Immune Microenvironment in Human Esophageal Cancer: Results of a Phase II Clinical Trial. Clin. Cancer Res. 2020, 26, 4921–4932. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Tong, D.; Liu, G.; Gao, J.; Wang, L.-A.; Xu, J.; Yang, X.; Xie, Q.; Huang, Y.; Pang, J.; et al. Metformin Inhibits Prostate Cancer Progression by Targeting Tumor-Associated Inflammatory Infiltration. Clin. Cancer Res. 2018, 24, 5622–5634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.L.; Rosa-Caldwell, M.E.; Lee, D.E.; Blackwell, T.A.; Brown, L.A.; Perry, R.A.; Haynie, W.S.; Hardee, J.P.; Carson, J.; Wiggs, M.P.; et al. Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour-bearing mice. J. Cachexia Sarcopenia Muscle 2017, 8, 926–938. [Google Scholar] [CrossRef]
- Fink, L.N.; Costford, S.R.; Lee, Y.S.; Jensen, T.E.; Bilan, P.J.; Oberbach, A.; Blüher, M.; Olefsky, J.M.; Sams, A.; Klip, A. Pro-Inflammatory macrophages increase in skeletal muscle of high fat-Fed mice and correlate with metabolic risk markers in humans. Obesity 2014, 22, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Ham, D.; Murphy, K.; Chee, A.; Lynch, G.; Koopman, R. Glycine administration attenuates skeletal muscle wasting in a mouse model of cancer cachexia. Clin. Nutr. 2014, 33, 448–458. [Google Scholar] [CrossRef]
- Batista, M.L.; Neves, R.X.; Peres, S.B.; Yamashita, A.S.; Shida, C.S.; Farmer, S.R.; Seelaender, M. Heterogeneous time-dependent response of adipose tissue during the development of cancer cachexia. J. Endocrinol. 2012, 215, 363–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.-P.; Reid, M.B. Effect of tumor necrosis factor-α on skeletal muscle metabolism. Curr. Opin. Rheumatol. 2001, 13, 483–487. [Google Scholar] [CrossRef]
- Thoma, A.; Lightfoot, A.P. NF-kB and Inflammatory Cytokine Signalling: Role in Skeletal Muscle Atrophy. Adv. Exp. Med. Biol. 2018, 1088, 267–279. [Google Scholar]
- Zimmers, T.A.; Fishel, M.L.; Bonetto, A. STAT3 in the systemic inflammation of cancer cachexia. Semin. Cell Dev. Biol. 2016, 54, 28–41. [Google Scholar] [CrossRef] [Green Version]
- Guadagnin, E.; Mázala, D.; Chen, Y.-W. STAT3 in Skeletal Muscle Function and Disorders. Int. J. Mol. Sci. 2018, 19, 2265. [Google Scholar] [CrossRef] [Green Version]
- Constantinou, C.; De Oliveira, C.C.F.; Mintzopoulos, D.; Busquets, S.; He, J.; Kesarwani, M.; Mindrinos, M.; Rahme, L.G.; Argiles, J.M.; Tzika, A.A. Nuclear magnetic resonance in conjunction with functional genomics suggests mitochondrial dysfunction in a murine model of cancer cachexia. Int. J. Mol. Med. 2011, 27, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt Pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, A.G.; Gomes-Marcondes, M.C.C. Metformin treatment modulates the tumour-induced wasting effects in muscle protein metabolism minimising the cachexia in tumour-bearing rats. BMC Cancer 2016, 16, 418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Kaelin, W.G. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [Green Version]
- Britto, F.A.; Dumas, K.; Giorgetti-Peraldi, S.; Ollendorff, V.; Favier, F.B. Is REDD1 a metabolic double agent? Lessons from physiology and pathology. Am. J. Physiol. Physiol. 2020, 319, C807–C824. [Google Scholar] [CrossRef] [PubMed]
- Gordon, B.S.; Steiner, J.L.; Williamson, D.L.; Lang, C.H.; Kimball, S.R. Emerging role for regulated in development and DNA damage 1 (REDD1) in the regulation of skeletal muscle metabolism. Am. J. Physiol. Metab. 2016, 311, E157–E174. [Google Scholar] [CrossRef] [Green Version]
- Puppa, M.J.; Gao, S.; Narsale, A.A.; Carson, J.A. Skeletal muscle glycoprotein 130′s role in Lewis lung carcinoma–induced cachexia. FASEB J. 2014, 28, 998–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, M.; Li, L.; Su, Z.; Wei, L.; Pu, W.; Zhao, C.; Ding, Y.; Wazir, J.; Cao, W.; Song, S.; et al. An integrative transcriptome study reveals Ddit4/Redd1 as a key regulator of cancer cachexia in rodent models. Cell Death Dis. 2021, 12, 652. [Google Scholar] [CrossRef]
- Della Corte, C.M.; Ciaramella, V.; Di Mauro, C.; Castellone, M.; Papaccio, F.; Fasano, M.; Sasso, F.C.; Martinelli, E.; Troiani, T.; De Vita, F.; et al. Metformin increases antitumor activity of MEK inhibitors through GLI1 downregulation in LKB1 positive human NSCLC cancer cells. Oncotarget 2016, 7, 4265–4278. [Google Scholar] [CrossRef] [Green Version]
- Morgillo, F.; Fasano, M.; Della Corte, C.M.; Sasso, F.C.; Papaccio, F.; Viscardi, G.; Esposito, G.; DI Liello, R.; Normanno, N.; Capuano, A.; et al. Results of the safety run-in part of the METAL (METformin in Advanced Lung cancer) study: A multicentre, open-label phase I–II study of metformin with erlotinib in second-line therapy of patients with stage IV non-small-cell lung cancer. ESMO Open 2017, 2, e000132. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Mice Began Study, n | Mice Survived, n | Mice with Signal, n | Mice with Metastases, n |
|---|---|---|---|---|
| Males | 6 | 3 | 3 | 2 |
| Females | 6 | 4 | 4 | 1 |
| Total Control | 12 | 7 | 7 | 3 |
| Males | 6 | 3 | 3 | 0 |
| Females | 6 | 6 | 6 | 3 |
| Total Metformin | 12 | 9 | 9 | 3 |
| Primer | Sequence | |
|---|---|---|
| p27 | Forward | TCTCTTCGGCCCGGTCAAT |
| Reverse | AAATTCCACTTGCGCTGACTC | |
| F4/80 | Forward | CTTTGGCTATGGGCTTCCAGTC |
| Reverse | GCAAGGAGGACAGAGTTTATCGTG | |
| CDK4 | Forward | ATGGCTGCCACTCGATATGAA |
| Reverse | TCCTCCATTAGGAACTCTCACAC | |
| IL-6 | Forward | CTGCAAGAGCTTCCATCCAGTT |
| Reverse | GAAGTAGGGAAGGCCGTGG | |
| Hes1 | Forward | GGTCCTGGAATAGTGCTACCG |
| Reverse | CACCGGGGAGGAGGAATTTTT | |
| TNF-α | Forward | CCAGACCCTCACACTCAGATC |
| Reverse | CACTTGGTGGTTTGCTACGAC | |
| PGC-1α | Forward | TGATGTGAATGACTTGGATACAGACA |
| Reverse | GCTCATTGTTGTACTGGTTGGATATG | |
| MAFbx | Forward | CCAGGATCCGCAGCCCTCCA |
| Reverse | ATGCGGCGCGTTGGGAAGAT | |
| GAPDH | Forward | ATGTTTGTGATGGGTGTGAA |
| Reverse | ATGCCAAAGTTGTCATGGAT | |
| Left Gastrocnemius (mg) | Right Gastrocnemius (mg) | |
|---|---|---|
| Control | 100.0 ± 7.6 | 102.0 ± 8.6 |
| Metformin | 102.5 ± 6.2 | 97.5 ± 6.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bond, N.L.S.; Dréau, D.; Marriott, I.; Bennett, J.M.; Turner, M.J.; Arthur, S.T.; Marino, J.S. Low-Dose Metformin as a Monotherapy Does Not Reduce Non-Small-Cell Lung Cancer Tumor Burden in Mice. Biomedicines 2021, 9, 1685. https://doi.org/10.3390/biomedicines9111685
Bond NLS, Dréau D, Marriott I, Bennett JM, Turner MJ, Arthur ST, Marino JS. Low-Dose Metformin as a Monotherapy Does Not Reduce Non-Small-Cell Lung Cancer Tumor Burden in Mice. Biomedicines. 2021; 9(11):1685. https://doi.org/10.3390/biomedicines9111685
Chicago/Turabian StyleBond, Nicole L. Stott, Didier Dréau, Ian Marriott, Jeanette M. Bennett, Michael J. Turner, Susan T. Arthur, and Joseph S. Marino. 2021. "Low-Dose Metformin as a Monotherapy Does Not Reduce Non-Small-Cell Lung Cancer Tumor Burden in Mice" Biomedicines 9, no. 11: 1685. https://doi.org/10.3390/biomedicines9111685