

Sleep Respiratory Disorders in Children and Adolescents with Cystic Fibrosis and Primary Ciliary Dyskinesia
 ,
,  , , and
, , and
Abstract
:1. Introduction
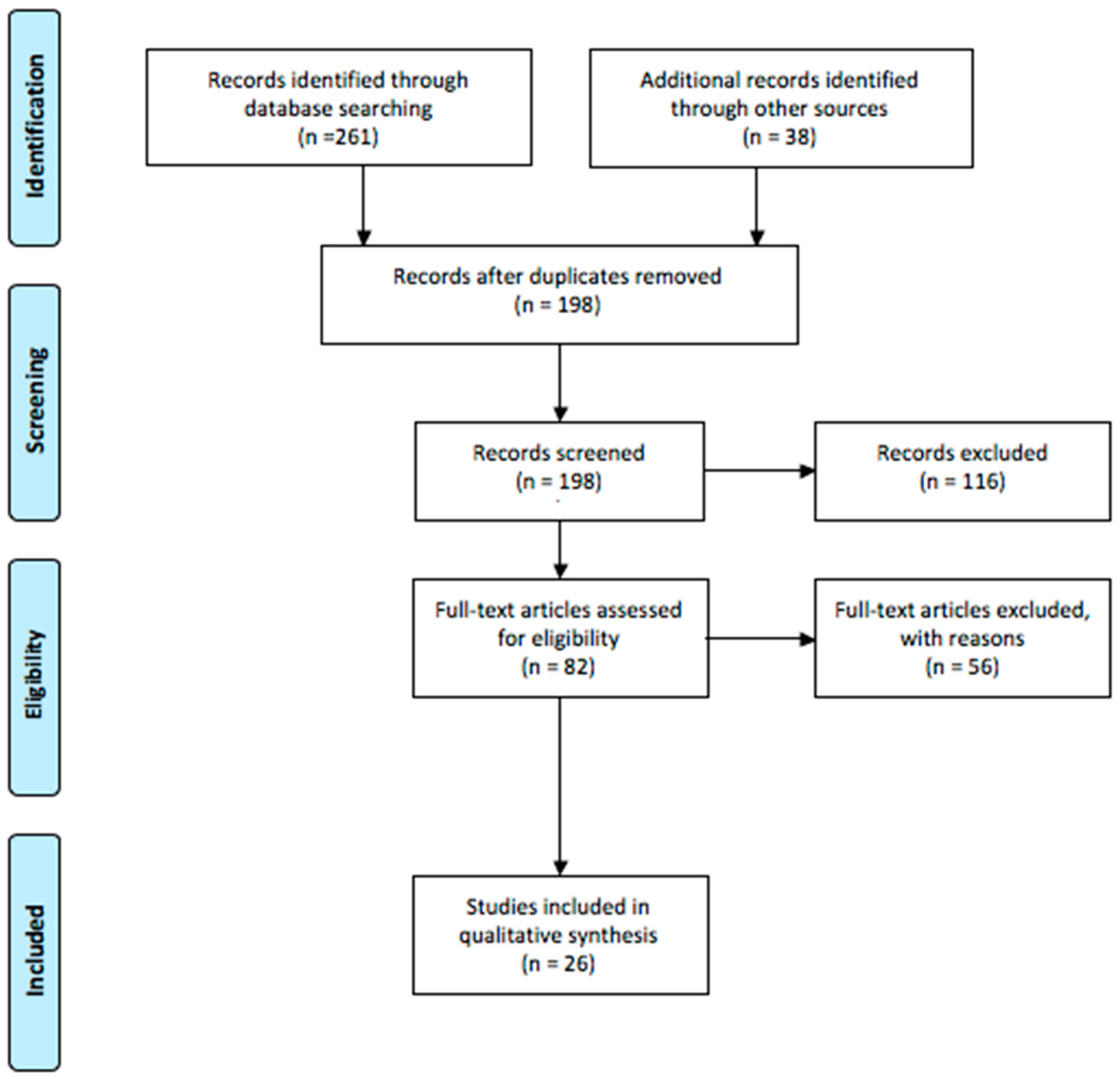
2. Materials and Methods
3. Results
3.1. SRDs in CF Patients
3.2. SRD in PCD Patients
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Cohen-Cymberknoh, M.; Simanovsky, N.; Hiller, N.; Hillel, A.G.; Shoseyov, D.; Kerem, E. Differences in disease expression between primary ciliary dyskinesia and cystic fibrosis with and without pancreatic insufficiency. Chest 2014, 145, 738–744. [Google Scholar] [CrossRef]
- Shapiro, A.J.; Davis, S.D.; Polineni, D.; Manion, M.; Rosenfeld, M.; Dell, S.D.; Chilvers, M.A.; Ferkol, T.W.; Zariwala, M.A.; Sagel, S.D.; et al. Diagnosis of Primary Ciliary Dyskinesia. An Official American Thoracic Society Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 197, e24–e39. [Google Scholar] [CrossRef]
- Farrell, P.M.; White, T.B.; Ren, C.L.; Hempstead, S.E.; Accurso, F.; Derichs, N.; Howenstine, M.; McColley, S.A.; Rock, M.; Rosenfeld, M.; et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J. Pediatr. 2017, 181S, S4–S15.e1. [Google Scholar] [CrossRef]
- Cohen-Cymberknoh, M.; Atia, O.; Gileles-Hillel, A.; Kerem, E.; Reiter, J. Sleep disorders in patients with primary ciliary dyskinesia, cystic fibrosis with and without pancreatic insufficiency. Respir. Med. 2019, 151, 96–101. [Google Scholar] [CrossRef]
- Naqvi, S.K.; Sotelo, C.; Murry, L.; Simakajornboon, N. Sleep architecture in children and adolescents with cystic fibrosis and the association with severity of lung disease. Sleep Breath. 2008, 12, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Amin, R.; Bean, J.; Burklow, K.; Jeffries, J. The relationship between sleep disturbance and pulmonary function in stable pediatric cystic fibrosis patients. Chest 2005, 128, 1357–1363. [Google Scholar] [CrossRef]
- Milross, M.A.; Piper, A.J.; Norman, M.; Dobbin, C.J.; Grunstein, R.R.; E Sullivan, C.; Bye, P.T. Subjective sleep quality in cystic fibrosis. Sleep Med. 2002, 3, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Vandeleur, M.; Walter, L.M.; Armstrong, D.S.; Robinson, P.; Nixon, G.M.; Horne, R.S. Quality of life and mood in children with cystic fibrosis: Associations with sleep quality. J. Cyst. Fibros. 2018, 17, 811–820. [Google Scholar] [CrossRef]
- Johnson, D.A.; Billings, M.E.; Hale, L. Environmental Determinants of Insufficient Sleep and Sleep Disorders: Implications for Population Health. Curr. Epidemiol. Rep. 2018, 5, 61–69. [Google Scholar] [CrossRef]
- Abbott, J.; Elborn, J.S.; Georgiopoulos, A.; Goldbeck, L.; Marshall, B.; Sabadosa, K.; Smith, B.; Quittner, A. Cystic Fibrosis Foundation and European Cystic Fibrosis Society Survey of cystic fibrosis mental health care delivery. J. Cyst. Fibros. 2015, 14, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Parisi, G.F.; Papale, M.; Tardino, L.; Nenna, R.; Midulla, F.; Leonardi, S. Biomarkers in Pediatric Lung Diseases Including Cystic Fibrosis. Curr. Respir. Med. Rev. 2019, 15, 163–173. [Google Scholar] [CrossRef]
- Manti, S.; Parisi, G.F.; Papale, M.; Marseglia, G.L.; Licari, A.; Leonardi, S. Type 2 inflammation in cystic fibrosis: New insights. Pediatr Allergy Immunol. 2022, 33 (Suppl. S27), 15–17. [Google Scholar] [CrossRef] [PubMed]
- Urquhart, D.S.; Montgomery, H.; Jaffé, A. Assessment of hypoxia in children with cystic fibrosis. Arch. Dis. Child. 2005, 90, 1138–1143. [Google Scholar] [CrossRef]
- Isaiah, A.; Daher, A.; Sharma, P.B. Predictors of sleep hypoxemia with cystic fibrosis. Pediatr. Pulmonol. 2019, 54, 273–279. [Google Scholar] [CrossRef]
- Perin, C.; Fagondes, S.C.; Casarotto, F.C.; Pinotti, A.F.F.; Barreto, S.S.M.; Dalcin, P.d.T.R. Sleep findings and predictors of sleep desaturation in adult cystic fibrosis patients. Sleep Breath. 2012, 16, 1041–1048. [Google Scholar] [CrossRef]
- Versteegh, F.G.; Bogaard, J.M.; Raatgever, J.; Stam, H.; Neijens, H.; Kerrebijn, K. Relationship between airway obstruction, desaturation during exercise and nocturnal hypoxaemia in cystic fibrosis patients. Eur. Respir. J. 1990, 3, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Poncin, W.; Lebecque, P. L’indice de clairance pulmonaire dans la mucoviscidose [Lung clearance index in cystic fibrosis]. Rev. Mal. Respir. 2019, 36, 377–395. [Google Scholar] [CrossRef]
- Papale, M.; Parisi, G.F.; Spicuzza, L.; Licari, A.; Bongiovanni, A.; Mulè, E.; Rotolo, N.; Manti, S.; Leonardi, S. Lung clearance index evaluation in detecting nocturnal hypoxemia in cystic fibrosis patients: Toward a new diagnostic tool. Respir. Med. 2020, 164, 105906. [Google Scholar] [CrossRef]
- Spicuzza, L.; Sciuto, C.; Leonardi, S.; La Rosa, M. Early occurrence of obstructive sleep apnea in infants and children with cystic fibrosis. Arch. Pediatr. Adolesc. Med. 2012, 166, 1165–1169. [Google Scholar] [CrossRef]
- American Thoracic Society. Standardization of Spirometry, 1994 Update. Am. J. Respir. Crit. Care Med. 1995, 152, 1107–1136. [Google Scholar] [CrossRef] [PubMed]
- Braggion, C.; Pradal, U.; Mastella, G. Hemoglobin desaturation during sleep and daytime in patients with cystic fibrosis and severe airway obstruction. Acta Paediatr. 1992, 81, 1002–1006. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.; Descalço, A.; Salgueiro, M.; Pereira, L.; Barreto, C.; Bandeira, T.; Ferreira, R. Respiratory sleep disturbance in children and adolescents with cystic fibrosis. Rev. Port. Pneumol. 2016, 22, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Milross, M.A.; Piper, A.J.; Norman, M.; Willson, G.N.; Grunstein, R.R.; E Sullivan, C.; Bye, P.T. Night-to-night variability in sleep in cystic fibrosis. Sleep Med. 2002, 3, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Hassett, C. Treatment of exacerbations of cystic fibrosis improves nocturnal desaturation independent of changes in FEV1. Respirology 2002, A47, 7. [Google Scholar]
- De Castro-Silva, C.; de Bruin, V.M.; Cavalcante, A.G.M.; Bittencourt, L.R.A.; de Bruin, P.F.C. Nocturnal hypoxia and sleep disturbances in cystic fibrosis. Pediatr. Pulmonol. 2009, 44, 1143–1150. [Google Scholar] [CrossRef]
- Villa, M.P.; Pagani, J.; Lucidi, V.; Palamides, S.; Ronchetti, R. Nocturnal oximetry in infants with cystic fibrosis. Arch. Dis. Child. 2001, 84, 50–54. [Google Scholar] [CrossRef]
- Paranjape, S.M.; McGinley, B.M.; Braun, A.T.; Schneider, H. Polysomnographic Markers in Children with Cystic Fibrosis Lung Disease. Pediatrics 2015, 136, 920–926. [Google Scholar] [CrossRef]
- Van der Giessen, L.; Loeve, M.; de Jongste, J.; Hop, W.; Tiddens, H. Nocturnal cough in children with stable cystic fibrosis. Pediatr. Pulmonol. 2009, 44, 859–865. [Google Scholar] [CrossRef]
- Spier, S.; Rivlin, J.; Hughes, D.; Levison, H. The effect of oxygen on sleep, blood gases, and ventilation in cystic fibrosis. Am. Rev. Respir. Dis. 1984, 129, 712–718. [Google Scholar] [CrossRef]
- Gozal, D. Nocturnal ventilatory support in patients with cystic fibrosis: Comparison with supplemental oxygen. Eur. Respir. J. 1997, 10, 1999–2003. [Google Scholar] [CrossRef]
- Ramos, R.T.; Santana, M.A.; Almeida, P.d.C.; Júnior, A.d.S.M.; Araújo-Filho, J.B.; Salles, C. Nocturnal hypoxemia in children and adolescents with cystic fibrosis. J. Bras. Pneumol. 2013, 39, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Moran, F.; Bradley, J.M.; Piper, A.J. Non-invasive ventilation for cystic fibrosis. Cochrane Database Syst. Rev. 2017, 2, CD002769. [Google Scholar] [CrossRef] [PubMed]
- Fauroux, B.; Le Roux, E.; Ravilly, S.; Bellis, G.; Clément, A. Long-term noninvasive ventilation in patients with cystic fibrosis. Respiration 2008, 76, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.R.; Daniels, L.A.; Davis, S.D.; Zariwala, M.A.; Leigh, M.W. Primary ciliary dyskinesia. Recent advances in diagnostics, genetics, and characterization of clinical disease. Am. J. Respir. Crit. Care Med. 2013, 188, 913–922. [Google Scholar] [CrossRef]
- Ullmann, N.; Santamaria, F.; Allegorico, A.; Fainardi, V.; Borrelli, M.; Ferraro, V.A.; Proietti, E.; Parisi, G.F.; Romagnoli, V.; Lucca, F.; et al. Primary ciliary dyskinesia: A multicenter survey on clinical practice and patient management in Italy. Pediatr. Pulmonol. 2023, 58, 1127–1135. [Google Scholar] [CrossRef]
- Sommer, J.U.; Schäfer, K.; Omran, H.; Olbrich, H.; Wallmeier, J.; Blum, A.; Hörmann, K.; Stuck, B.A. ENT manifestations in patients with primary ciliary dyskinesia: Prevalence and significance of otorhinolaryngologic co-morbidities. Eur. Arch. Oto-Rhino-Laryngol. 2011, 268, 383–388. [Google Scholar] [CrossRef]
- Maglione, M.; Bush, A.; Montella, S.; Mollica, C.; Manna, A.; Esposito, A.; Santamaria, F. Progression of lung disease in primary ciliary dyskinesia: Is spirometry less accurate than CT? Pediatr. Pulmonol. 2012, 47, 498–504. [Google Scholar] [CrossRef]
- Kieckhefer, G.M.; Lentz, M.J.; Tsai, S.-Y.; Ward, T.M. Parent-child agreement in report of nighttime respiratory symptoms and sleep disruptions and quality. J. Pediatr. Health Care 2009, 23, 315–326. [Google Scholar] [CrossRef]
- Oktem, S.; Karadag, B.; Erdem, E.; Gokdemir, Y.; Karakoc, F.; Dagli, E.; Ersu, R. Sleep disordered breathing in patients with primary ciliary dyskinesia. Pediatr. Pulmonol. 2013, 48, 897–903. [Google Scholar] [CrossRef]
- Santamaria, F.; Esposito, M.; Montella, S.; Cantone, E.; Mollica, C.; De Stefano, S.; Mirra, V.; Carotenuto, M. Sleep disordered breathing and airway disease in primary ciliary dyskinesia. Respirology 2014, 19, 570–575. [Google Scholar] [CrossRef]
- Goldbart, A.D.; Tal, A.; Givon-Lavi, N.; Bar-Ziv, J.; Dagan, R.; Greenberg, D. Sleep-disordered breathing is a risk factor for community-acquired alveolar pneumonia in early childhood. Chest 2012, 141, 1210–1215. [Google Scholar] [CrossRef] [PubMed]
- Goh, D.Y.; Galster, P.; Marcus, C.L. Sleep architecture and respiratory disturbances in children with obstructive sleep apnea. Am. J. Respir. Crit. Care Med. 2000, 162 Pt 1, 682–686. [Google Scholar] [CrossRef] [PubMed]
- American Academy of Pediatrics. Section on Pediatric Pulmonology, Subcommittee on Obstructive Sleep Apnea Syndrome. Clinical practice guideline: Diagnosis and management of childhood obstructive sleep apnea syndrome. Pediatrics 2002, 109, 704–712. [Google Scholar] [CrossRef]
- Helbich, T.H.; Heinz-Peer, G.; Eichler, I.; Wunderbaldinger, P.; Götz, M.; Wojnarowski, C.; Brasch, R.C.; Herold, C.J. Cystic fibrosis: CT assessment of lung involvement in children and adults. Radiology 1999, 213, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.; Payne, D.; Pike, S.; Jenkins, G.; Henke, M.O.; Rubin, B.K. Mucus properties in children with primary ciliary dyskinesia: Comparison with cystic fibrosis. Chest 2006, 129, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Green, K.; Buchvald, F.F.; Marthin, J.K.; Hanel, B.; Gustafsson, P.M.; Nielsen, K.G. Ventilation inhomogeneity in children with primary ciliary dyskinesia. Thorax 2012, 67, 49–53. [Google Scholar] [CrossRef]

{kind=link}
| CF | ||||||
|---|---|---|---|---|---|---|
| Authors | Type of Study | Number of Patients | Mean Age (yrs) | Variable Assessed | Main Findings | References |
| Cohen-Cymberknoh et al., 2019 | Observational | 39 | 11.1 | Lung function, sleep disorders and their correlation with QoL | Sleep impairment correlated with disease severity and affected QoL. | [4] |
| Isaiah et al., 2019 | Retrospective case series | 41 | 11.6 | Pulmonary function, polysomnographic variables | FEV1 was the best predictor of sleep hypoxemia in children with CF and referred for polysomnography. | [14] |
| Perin et al., 2012 | Prospective | 51 | 25.1 ± 6.7 | Cardiac and pulmonary function, and polysomnographic variables | Desaturation was common and not associated with obstructive events during sleep. It can be predicted by awake resting SpO2 < 94% | [15] |
| Papale et al., 2020 | Observational | 31 | 17.4 ± 5.2 | Lung function and sleep disorders | LCI has a higher effectiveness in predicting nocturnal hypoxemia in stable patients with CF than traditional parameter of lung function such as FEV 1 . | [18] |
| Spicuzza L. et al. | Case-control | 40 | 6.3 ± 5.6 | Polysomnographic variables and sleep quality | Early occurrence of obstructive sleep apnea in children with CF in stable condition. | [19] |
| Milross et al., 2002 | Observational | 31 | 27 ± 8 | First-night effect of polysomnographic measurements on nocturnal oxygenation | A single-night with polysomnographic measurements in patients with CF provided information on nocturnal oxygenation and respiratory disturbance. | [23] |
| De Castro Silva et al., 2009 | Prospective | 30 | 12.8 | Polysomnographic variables | Desaturation during sleep can be predicted by FEV1 < 64% | [25] |
| Paranjape et al., 2015 | Observational | 43 | 9.6 ± 3.6 | Analysis of breathing patterns, gas exchange and polysomnographic variables. | Children with CF demonstrated lower oxyhemoglobin saturation and a higher proportion of inspiratory flow limitation, compared with control group | [27] |
| PCD | ||||||
| Authors | Type of study | Number of patients | Mean age (yrs) | Variable assessed | Main findings | References |
| Cohen-Cymberknoh et al., 2019 | Observational | 39 | 11.1 | Lung function, sleep disorders and their correlation with QoL | Sleep impairment correlated with disease severity and affected QoL. | [4] |
| Oktem et al., 2013 | Observational Case control | 29 | 10.9 | Sleep quality and sleep disorders | Patients with PCD have decreased sleep quality and higher rate of OSAS compared to controls | [39] |
| Santamaria et al., 2014 | Observational Case control | 60 | 12.3 | Lung function, nasal endoscopy and sleep disorders | Nocturnal desaturation was linked with lung function and structure abnormalities | [40] |
| Clinical Parameters Predicting of Sleep Respiratory Disorders | Monitoring |
|---|---|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papale, M.; Manti, S.; Presti, S.; Mollica, F.; Parisi, G.F.; Leonardi, S. Sleep Respiratory Disorders in Children and Adolescents with Cystic Fibrosis and Primary Ciliary Dyskinesia. Children 2023, 10, 1707. https://doi.org/10.3390/children10101707
Papale M, Manti S, Presti S, Mollica F, Parisi GF, Leonardi S. Sleep Respiratory Disorders in Children and Adolescents with Cystic Fibrosis and Primary Ciliary Dyskinesia. Children. 2023; 10(10):1707. https://doi.org/10.3390/children10101707
Chicago/Turabian StylePapale, Maria, Sara Manti, Santiago Presti, Federico Mollica, Giuseppe F. Parisi, and Salvatore Leonardi. 2023. "Sleep Respiratory Disorders in Children and Adolescents with Cystic Fibrosis and Primary Ciliary Dyskinesia" Children 10, no. 10: 1707. https://doi.org/10.3390/children10101707
APA StylePapale, M., Manti, S., Presti, S., Mollica, F., Parisi, G. F., & Leonardi, S. (2023). Sleep Respiratory Disorders in Children and Adolescents with Cystic Fibrosis and Primary Ciliary Dyskinesia. Children, 10(10), 1707. https://doi.org/10.3390/children10101707