
The Role of Lipid and Lipoprotein Metabolism in Non‐Alcoholic Fatty Liver Disease

{kind=link}
{kind=link}
Abstract
:1. Introduction
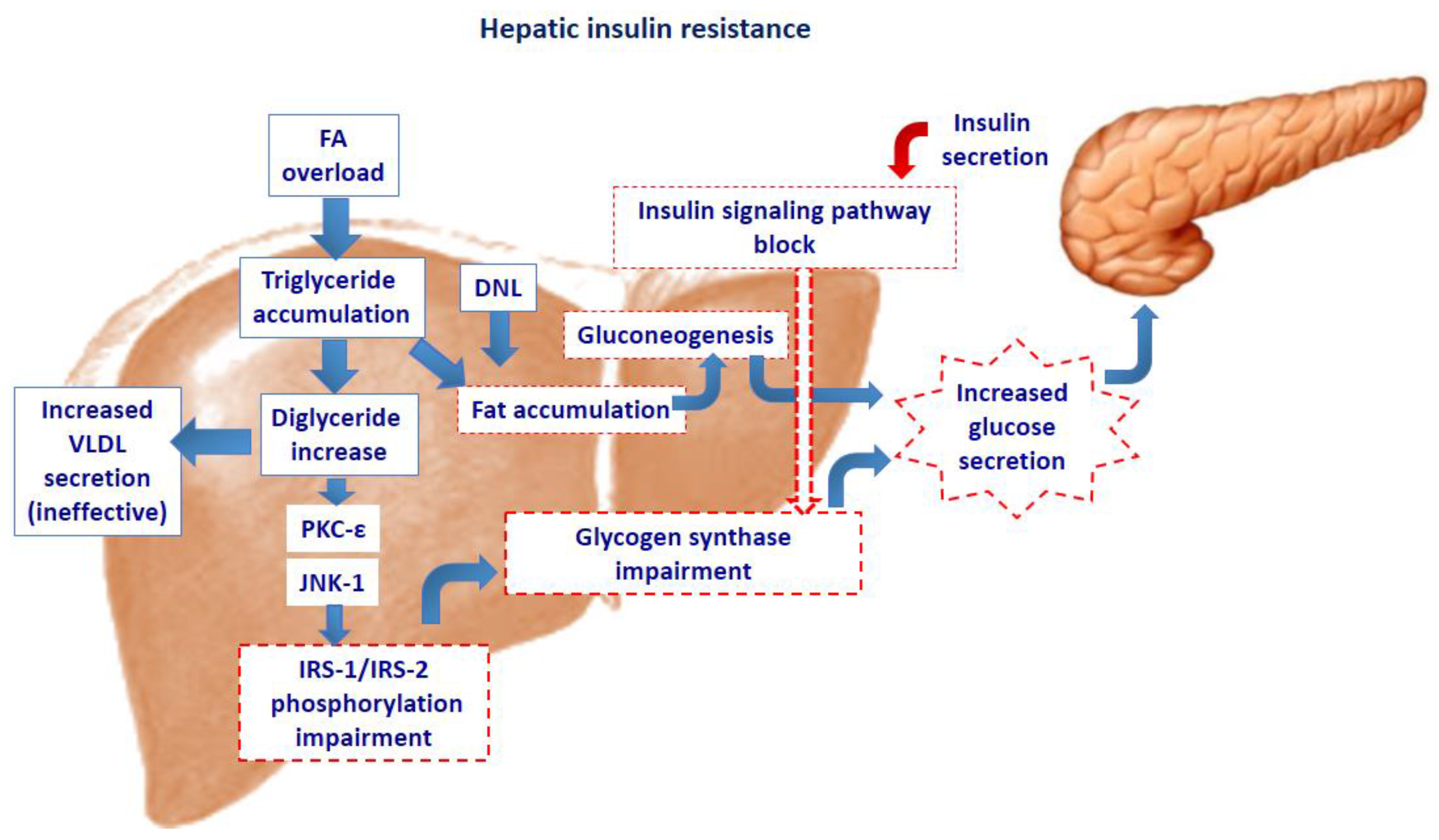
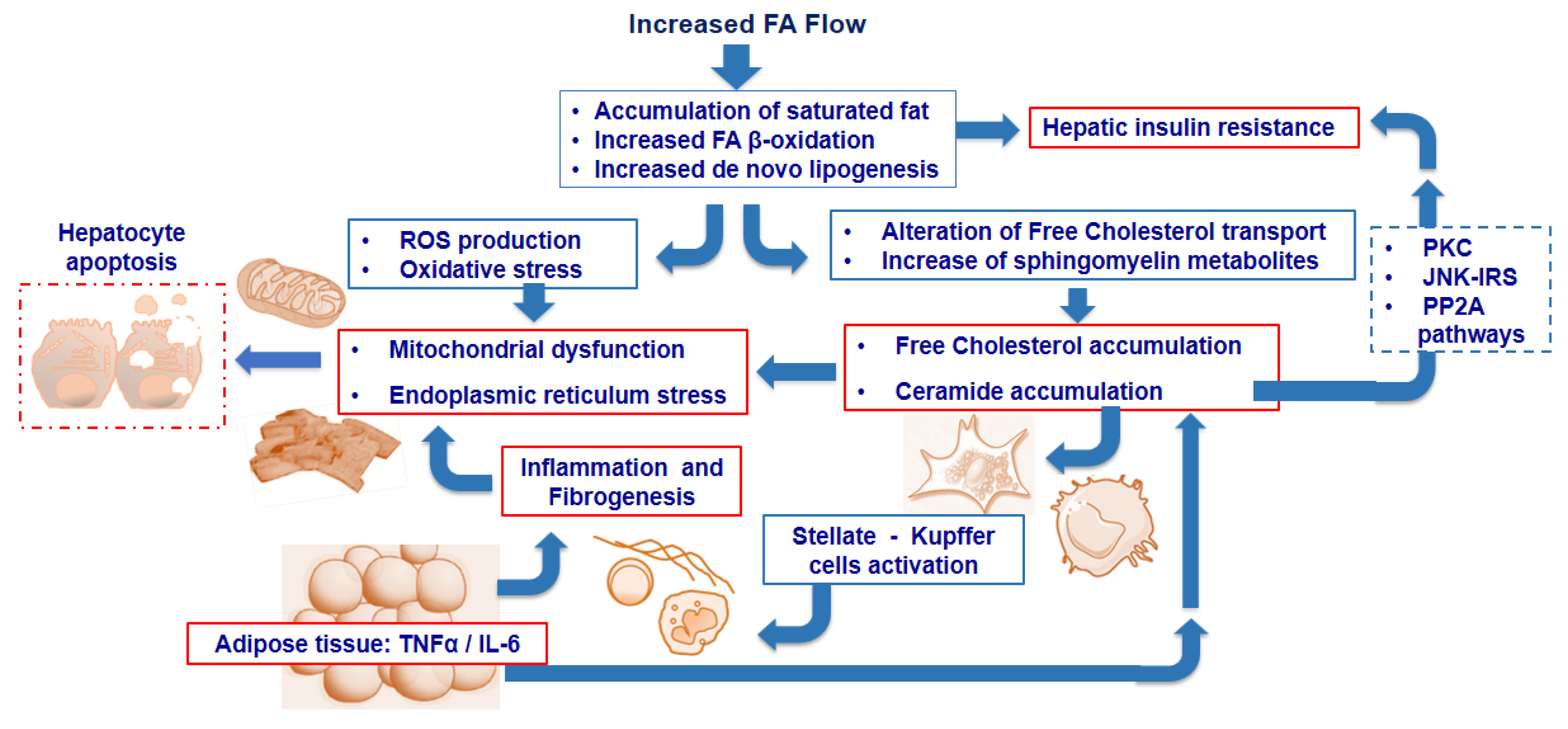
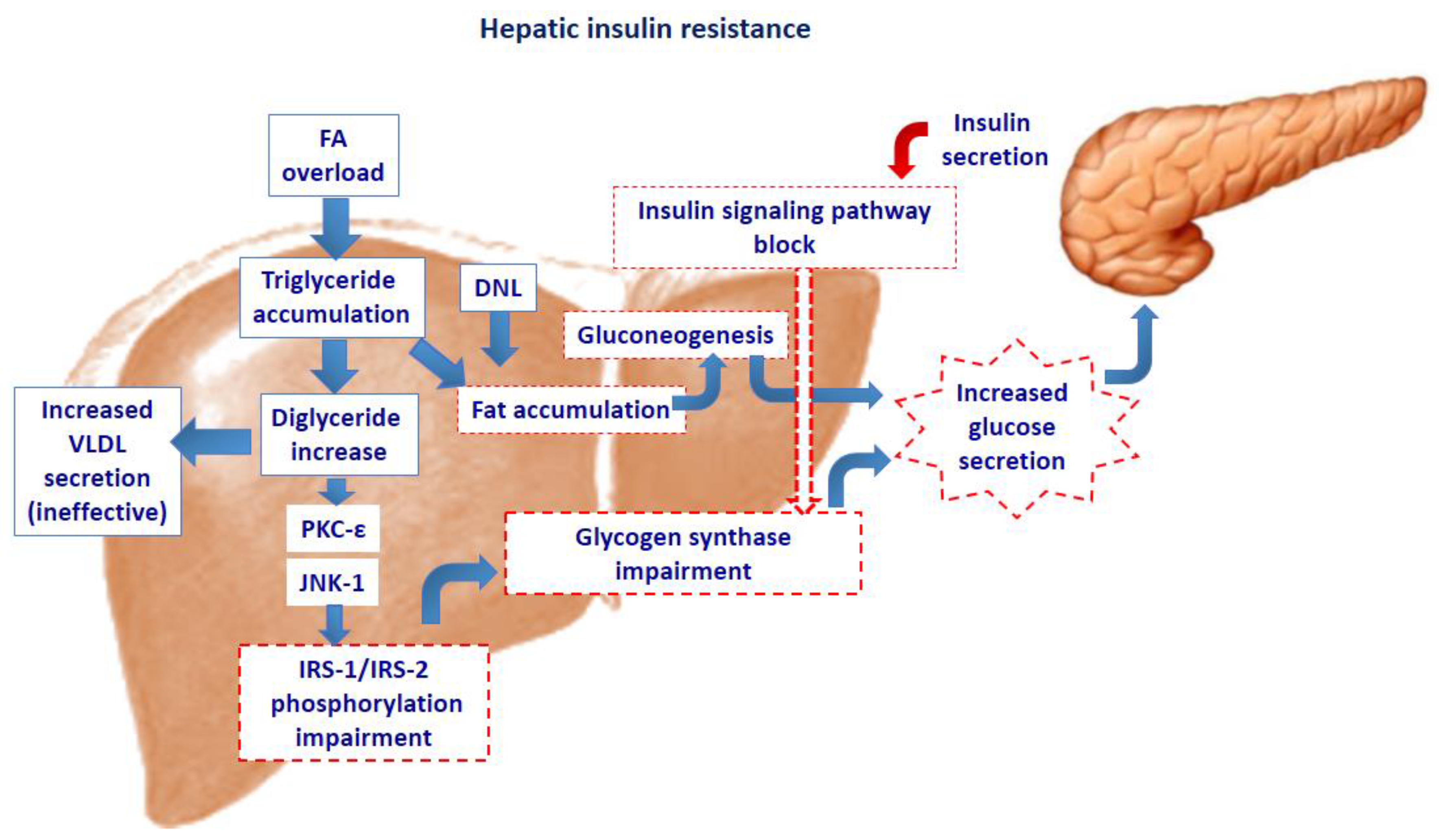
2. Intra-Cellular Lipid Accumulation and Insulin Resistance
3. Lipid and Lipoprotein Metabolism in Nonalcoholic Fatty Liver Disease
3.1. Fatty Acid Uptake
3.2. Hepatic de Novo Lipogenesis
3.3. Fatty Acid Oxidation
3.4. Very Low Density Lipoproteins Secretion
4. Role of Cholesterol in the Pathogenesis of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis
5. Role of Ceramides in Nonalcoholic Fatty Liver Disease
6. Medical Therapy
6.1. Orlistat
6.2. Omega-3 Fatty Acids
6.3. Ezetimibe
6.4. Statins
6.5. Farnesoid X Receptor (FXR) Agonists
7. Conclusions
Conflicts of Interest
References
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef] [PubMed]
- Mencin, A.A.; Lavine, J.E. Advances in pediatric nonalcoholic fatty liver disease. Pediatr. Clin. N. Am. 2011, 58, 1375–1392. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Sirlin, C.B.; Schwimmer, J.B.; Lavine, J.E. Advances in pediatric nonalcoholic fatty liver disease. Hepatology 2009, 50, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.L.; Howe, L.D.; Jones, H.E.; Higgins, J.P.; Lawlor, D.A.; Fraser, A. The Prevalence of Non-Alcoholic Fatty Liver Disease in Children and Adolescents: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0140908. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Alisi, A.; Newton, K.P.; Schwimmer, J.B. Comparison of the Phenotype and Approach to Pediatric vs Adult Patients With Nonalcoholic Fatty Liver Disease. Comparison of the Phenotype and Approach to Pediatric vs Adult Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2016, 150, 1798–1810. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, N.; Bai, C.; Fang, Y.; Srishord, M.; McCullough, A.; Gramlich, T.; Younossi, Z.M. Long-Term Follow-Up of Patients With Nonalcoholic Fatty Liver. Clin. Gastroenterol. Hepatol. 2009, 7, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.D. Ectopic fat, insulin resistance and non-alcoholic fatty liver disease. Proc. Nutr. Soc. 2013, 72, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Petersen, K.F.; Shulman, G.I. Lipid-induced insulin resistance: Unraveling the mechanism. Lancet 2010, 375, 2267–2277. [Google Scholar] [CrossRef]
- Cali, A.M.; De Oliveira, A.M.; Kim, H.; Chen, S.; Reyes-Mugica, M.; Escalera, S.; Dziura, J.; Taksali, S.E.; Kursawe, R.; Shaw, M.; et al. Glucose dysregulation and hepatic steatosis in obese adolescents: Is there a link? Hepatology 2009, 49, 1896–1903. [Google Scholar] [CrossRef] [PubMed]
- D’Adamo, E.; Cali, A.M.; Weiss, R.; Santoro, N.; Pierpont, B.; Northrup, V.; Caprio, S. Central role of fatty liver in the pathogenesis of insulin resistance in obese adolescents. Diabetes Care 2010, 33, 1817–1822. [Google Scholar] [CrossRef] [PubMed]
- Kotronen, A.; Yki-Järvinen, H. Fatty liver: A novel component of the metabolic syndrome. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Schwimmer, J.B.; Pardee, P.E.; Lavine, J.E.; Blumkin, A.K.; Cook, S. Cardiovascular risk factors and the metabolic syndrome in pediatric nonalcoholic fatty liver disease. Circulation 2008, 118, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Pacifico, L.; Nobili, V.; Anania, C.; Verdecchia, P.; Chiesa, C. Pediatric nonalcoholic fatty liver disease, metabolic syndrome and cardiovascular risk. World J. Gastroenterol. 2011, 17, 3082–3091. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Day, C.P.; Bonora, E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N. Engl. J. Med. 2010, 363, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Pacifico, L.; Anania, C.; Martino, F.; Cantisani, V.; Pascone, R.; Marcantonio, A.; Chiesa, C. Functional and morphological vascular changes in pediatric nonalcoholic fatty liver disease. Hepatology 2010, 52, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Pacifico, L.; Di Martino, M.; De Merulis, A.; Bezzi, M.; Osborn, J.F.; Catalano, C.; Chiesa, C. Left ventricular dysfunction in obese children and adolescents with nonalcoholic fatty liver disease. Hepatology 2014, 59, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Clemente, M.G.; Mandato, C.; Poeta, M.; Vajro, P. Pediatric non-alcoholic fatty liver disease: Recent solutions, unresolved issues, and future research directions. World J. Gastroenterol. 2016, 22, 8078–8093. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Liu, Z.X.; Qu, X.; Elder, B.D.; Bilz, S.; Befroy, D.; Romanelli, A.J.; Shulman, G.I. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 2004, 279, 32345–32353. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Liu, Z.X.; Wang, A.; Beddow, S.A.; Geisler, J.G.; Kahn, M.; Zhang, X.M.; Monia, B.P.; Bhanot, S.; Shulman, G.I. Inhibition of protein kinase Cε prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J. Clin. Invest. 2007, 117, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Kumashiro, N.; Erion, D.M.; Zhang, D.; Kahn, M.; Beddow, S.A.; Chu, X.; Still, C.D.; Gerhard, G.S.; Han, X.; Dziura, J.; et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2011, 108, 16381–16385. [Google Scholar] [CrossRef] [PubMed]
- Magkos, F.; Su, X.; Bradley, D.; Fabbrini, E.; Conte, C.; Eagon, J.C.; Varela, J.E.; Brunt, E.M.; Patterson, B.W.; Klein, S. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology 2012, 142, 1444–1446. [Google Scholar] [CrossRef] [PubMed]
- Birkenfeld, A.L.; Shulman, G.I. Nonalcoholic Fatty Liver Disease, Hepatic Insulin Resistance, and Type 2 Diabetes. Hepatology 2014, 9, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Turinsky, J.; Bayly, B.P.; O’Sullivan, D.M. 1,2-Diacylglycerol and ceramide levels in rat skeletal muscle and liver in vivo. Studies with insulin, exercise, muscle denervation, and vasopressin. J. Biol. Chem. 1990, 265, 7933–7938. [Google Scholar] [PubMed]
- Chavez, J.A.; Summers, S.A. A ceramide-centric view of insulin resistance. Cell Metab. 2012, 15, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Arner, P.; Caro, J.F.; Atkinson, R.L.; Spiegelman, B.M. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J. Clin. Investig. 1995, 95, 2409–2415. [Google Scholar] [CrossRef] [PubMed]
- Muoio, D.M.; Newgard, C.B. Mechanisms of disease: Molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-β and NF-κB. Nat. Med. 2005, 11, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Nishimura, T.; Ishiba, H.; Seko, Y.; Okajima, A.; Fujii, H.; Tochiki, N.; Umemura, A.; Moriguchi, M.; Sumida, Y.; et al. Blockade of interleukin 6 signalling ameliorates systemic insulin resistance through upregulation of glucose uptake in skeletal muscle and improves hepatic steatosis in high-fat diet fed mice. Liver Int. 2015, 35, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Q.; Xu, C.F.; Yu, C.H.; Chen, W.X.; Li, Y.M. Role of endoplasmic reticulum stress in the pathogenesis of nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 1768–1776. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wei, Y.; Pagliassotti, M.J. Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology 2006, 147, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Mirshahi, F.; Cheung, O.; Natarajan, R.; Maher, J.W.; Kellum, J.M.; Sanyal, A.J. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 2008, 134, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Gregor, M.F.; Yang, L.; Fabbrini, E.; Mohammed, B.S.; Eagon, J.C.; Hotamisligil, G.S.; Klein, S. Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes 2009, 58, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Görgün, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Kars, M.; Yang, L.; Gregor, M.F.; Mohammed, B.S.; Pietka, T.A.; Finck, B.N.; Patterson, B.W.; Horton, J.D.; Mittendorfer, B.; Hotamisligil, G.S.; et al. Tauroursodeoxycholic Acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes 2010, 59, 1899–1905. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Magkos, F. Hepatic steatosis as a marker of metabolic dysfunction. Nutrients 2015, 7, 4995–5019. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology 2010, 51, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.; Guo, Z.; Johnson, C.M.; Hensrud, D.D.; Jensen, M.D. Splanchnic lipolysis in human obesity. J. Clin. Investig. 2004, 113, 1582–1588. [Google Scholar] [CrossRef] [PubMed]
- Mittendorfer, B.; Magkos, F.; Fabbrini, E.; Mohammed, B.S.; Klein, S. Relationship between body fat mass and free fatty acid kinetics in men and women. Obesity 2009, 17, 1872–1877. [Google Scholar] [CrossRef] [PubMed]
- Diraison, F.; Moulin, P.; Beylot, M. Contribution of hepatic de novo lipogenesis and reesterification of plasma non esterified fatty acids to plasma triglyceride synthesis during non-alcoholic fatty liver disease. Diabetes Metab. 2003, 29, 478–485. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Dufour, S.; Savage, D.B.; Bilz, S.; Salomon, G.; Yonemitsu, S.; Cline, G.W.; Befroy, D.; Zemany, L.; Kahn, B.B.; et al. The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 12587–12594. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.; Komen, J.; Kemp, S. Fatty acid omega-oxidation as a rescue pathway for fatty acid oxidation disorders in humans. FEBS J. 2011, 278, 182–194. [Google Scholar] [CrossRef] [PubMed]
- McGarry, J.D.; Woeltje, K.F.; Kuwajima, M.; Foster, D.W. Regulation of ketogenesis and the renaissance of carnitine palmitoyltransferase. Diabetes Metab. Rev. 1989, 5, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Serra, D.; Mera, P.; Malandrino, M.I.; Mir, J.F.; Herrero, L. Mitochondrial fatty acid oxidation in obesity. Antioxid. Redox Signal. 2013, 19, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Liu, Z.X.; Choi, C.S.; Tian, L.; Kibbey, R.; Dong, J.; Cline, G.W.; Wood, P.A.; Shulman, G.I. Mitochondrial dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 17075–17080. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, R.P.; Kelly, K.B.; Leonard, K.A.; Jacobs, R.L. Creatine reduces hepatic TG accumulation in hepatocytes by stimulating fatty acid oxidation. Biochim. Biophys. Acta 2014, 1841, 1639–1646. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, S.H.; Swerdlow, R.H.; Khan, E.M.; Iezzoni, J.C.; Hespenheide, E.E.; Parks, J.K.; Parker, W.D., Jr. Mitochondrial abnormalities in non-alcoholic steatohepatitis. J. Hepatol. 1999, 31, 430–434. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Campbell-Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Perez-Carreras, M.; Del Hoyo, P.; Martin, M.A.; Rubio, J.C.; Martin, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Cortez-Pinto, H.; Chatham, J.; Chacko, V.P.; Arnold, C.; Rashid, A.; Diehl, A.M. Alterations in liver ATP homeostasis in human nonalcoholic steatohepatitis: A pilot study. JAMA 1999, 282, 1659–1664. [Google Scholar] [CrossRef] [PubMed]
- Kohjima, M.; Enjoji, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Fujino, T.; Yada, M.; Yada, R.; Harada, N.; et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2007, 20, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Shelness, G.S.; Sellers, J.A. Very-low-density lipoprotein assembly and secretion. Curr. Opin. Lipidol. 2001, 12, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Elovson, J.; Chatterton, J.E.; Bell, G.T.; Schumaker, V.N.; Reuben, M.A.; Puppione, D.L.; Reeve, J.R., Jr.; Young, N.L. Plasma very low density lipoproteins contain a single molecule of apolipoprotein B. J. Lipid Res. 1988, 29, 1461–1473. [Google Scholar] [PubMed]
- Packard, C.J. Understanding coronary heart disease as a consequence of defective regulation of apolipoprotein B metabolism. Curr. Opin. Lipidol. 1999, 10, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Mohammed, B.S.; Magkos, F.; Korenblat, K.M.; Patterson, B.W.; Klein, S. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology 2008, 134, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.; Watts, G.F.; Gan, S.; Wong, A.T.; Ooi, E.M.; Barrett, P.H. Nonalcoholic fatty liver disease as the transducer of hepatic oversecretion of very-low-density lipoprotein-apolipoprotein B-100 in obesity. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1043–1050. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Shimano, H.; Hamilton, R.L.; Brown, M.S.; Goldstein, J.L. Disruption of LDL receptor gene in transgenic SREBP-1a mice unmasks hyperlipidemia resulting from production of lipid-rich VLDL. J. Clin. Investig. 1999, 103, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Tiemann Luecking, C.; Love-Gregory, L.; Okunade, A.L.; Yoshino, M.; Fraterrigo, G.; Patterson, B.W.; Klein, S. Physiological Mechanisms of Weight Gain-Induced Steatosis in People With Obesity. Gastroenterology 2016, 150, 79–81. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N. The role of cholesterol in the pathogenesis of NASH. Trends Endocrinol. Metab. 2015, 27, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Van Rooyen, D.M.; Larter, C.Z.; Haigh, V.G.; Yeh, M.M.; Iannou, G.; Kuver, R.; Lee, S.P.; Tech, N.C.; Farrell, G.C. Hepatic free cholesterol accumulates in obese, diabetic mice and causes nonalcoholic steatohepatitis. Gastroenterology 2011, 14, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Caballero, F.; Fernández, A.; De Lacy, A.M.; Fernández-Checa, J.C.; Caballería, J.; García-Ruiz, C. Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH. J. Hepatol. 2009, 50, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Nuño-Lámbarri, N.; Domínguez-Pérez, M.; Baulies-Domenech, A.; Monte, M.J.; Marin, J.J.; Rosales-Cruz, P.; Souza, V.; Miranda, R.U.; Bucio, L.; Montalvo-Jave, E.E.; et al. Liver Cholesterol Overload Aggravates Obstructive Cholestasis by Inducing Oxidative Stress and Premature Death in Mice. Oxid. Med. Cell. Longev. 2016, 2016, 9895176. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997, 89, 331–340. [Google Scholar] [CrossRef]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Flamment, M.; Hajduch, E.; Ferré, P.; Foufelle, F. New insights into ER stress-induced insulin resistance. Trends Endocrinol. Metab. 2012, 23, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Teratani, T.; Suzuki, T.; Shimizu, M.; Sato, H.; Narimatsu, K.; Okada, Y.; Kurihara, C.; Irie, R.; Yokoyama, H.; et al. Free cholesterol accumulation in hepatic stellate cells: mechanism of liver fibrosis aggravation in nonalcoholic steatohepatitis in mice. Hepatology 2014, 59, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Mari, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernandez, A.; Enrich, C.; Fernandez-Checa, J.C.; Garcia-Ruiz, C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006, 4, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Wouters, K.; van Bilsen, M.; van Gorp, P.J.; Bieghs, V.; Lutjohann, D.; Kerksiek, A.; Staels, B.; Hofker, M.H.; Shiri-Sverdlov, R. Intrahepatic cholesterol influences progression, inhibition and reversal of non-alcoholic steatohepatitis in hyperlipidemic mice. FEBS Lett. 2010, 584, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphyngolipid metabolism: from synthesis to breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar] [PubMed]
- Reali, F.; Morine, M.J.; Kahramanoğulları, O.; Raichur, S.; Schneider, H.C.; Crowther, D.; Priami, C. Mechanistic interplay between ceramide and insulin resistance. Sci. Rep. 2017, 7, 41231. [Google Scholar] [CrossRef] [PubMed]
- Correnti, J.M.; Juskeviciute, E.; Swarup, A.; Hoek, J.B. Pharmacological ceramide reduction alleviates alcohol-induced steatosis and hepatomegaly in adiponectin knockout mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G959–G973. [Google Scholar] [CrossRef] [PubMed]
- Fucho, R.; Casals, N.; Serra, D.; Herrero, L. Ceramides and mitochondrial fatty acid oxidation in obesity. FASEB J. 2017, 31, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Pagadala, M.; Kasumov, T.; McCullough, A.J.; Zein, N.N.; Kirwan, J.P. Role of ceramides in nonalcoholic fatty liver disease. Trends Endocrinol. Metab. 2012, 23, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Barlow, S.E.; Expert Committee. Expert committee recommendations regarding the prevention, assessment, and treatment of child and adolescent overweight and obesity: summary report. Pediatrics 2007, 120 (Suppl. 4), S164–S192. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.S.; Barlow, S.E.; Quiros-Tejeira, R.E.; Scheimann, A.; Skelton, J.; Suskind, D.; Tsai, P.; Uko, V.; Warolin, J.P.; Xanthakos, S.A.; et al. Childhood obesity for pediatric gastroenterologists. J. Pediatr. Gastroenterol. Nutr. 2013, 56, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Sjöström, L.; Narbro, K.; Sjöström, C.D.; Karason, K.; Larsson, B.; Wedel, H.; Lystig, T.; Sullivan, M.; Bouchard, C.; Carlsson, B.; et al. Effects of bariatric surgery on mortality in Swedish obese subjects. N. Engl. J. Med. 2007, 357, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Kwan, T.K.; Chadban, S.J.; McKenzie, P.R.; Saunders, J.R. Acute oxalate nephropathy secondary to orlistat-induced enteric hyperoxaluria. Nephrology 2013, 18, 241–242. [Google Scholar] [CrossRef] [PubMed]
- Pacifico, L.; Giansanti, S.; Gallozzi, A.; Chiesa, C. Long chain omega-3 polyunsaturated fatty acids in pediatric metabolic syndrome. Mini Rev. Med. Chem. 2014, 14, 791–804. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.; Watts, G.F.; Mori, T.A.; Barrett, P.H.; Redgrave, T.G.; Beilin, L.J. Randomized controlled trial of the effect of n-3 fatty acid supplementation on the metabolism of apolipoprotein B-100 and chylomicron remnants in men with visceral obesity. Am. J. Clin. Nutr. 2003, 77, 300–307. [Google Scholar] [PubMed]
- Pan, M.; Maitin, V.; Parathath, S.; Andreo, U.; Lin, S.X.; St. Germain, C.; Yao, Z.; Maxfield, F.R.; Williams, K.J.; Fisher, E.A. Presecretory oxidation, aggregation, and autophagic destruction of apoprotein-B: a pathway for late-stage quality control. Proc. Natl. Acad. Sci. USA 2008, 105, 5862–5867. [Google Scholar] [CrossRef] [PubMed]
- Caviglia, J.M.; Gayet, C.; Ota, T.; Hernandez-Ono, A.; Conlon, D.M.; Jiang, H.; Fisher, E.A.; Ginsberg, H.N. Different fatty acids inhibit apoB100 secretion by different pathways: unique roles for ER stress, ceramide, and autophagy. J. Lipid Res. 2011, 52, 1636–1651. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, O.; Martinelli, N.; Sandri, M.; Bassi, A.; Guarini, P.; Trabetti, E.; Pizzolo, F.; Girelli, D.; Friso, S.; Pignatti, P.F.; et al. Apolipoprotein C-III, n-3 polyunsaturated fatty acid, and “insulin-resistant” T-455C APOC3 gene polymorphism in heart disease patients: example of gene-diet interaction. Clin. Chem. 2005, 51, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Bedogni, G.; Alisi, A.; Pietrobattista, A.; Risé, P.; Galli, C.; Agostoni, C. Docosahexaenoic acid supplementation decreases liver fat content in children with non-alcoholic fatty liver disease: Double-blind randomised controlled clinical trial. Arch. Dis. Child. 2011, 96, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Alisi, A.; Della Corte, C.; Risé, P.; Galli, C.; Agostoni, C.; Bedogni, G. Docosahexaenoic acid for the treatment of fatty liver: Randomised controlled trial in children. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 1066–1070. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Carpino, G.; Alisi, A.; De Vito, R.; Franchitto, A.; Alpini, G.; Onori, P.; Gaudio, E. Role of docosahexaenoic acid treatment in improving liver histology in pediatric nonalcoholic fatty liver disease. PLoS ONE 2014, 9, e88005. [Google Scholar] [CrossRef] [PubMed]
- Pacifico, L.; Bonci, E.; Di Martino, M.; Versacci, P.; Andreoli, G.; Silvestri, L.M.; Chiesa, C. A double-blind, placebo-controlled randomized trial to evaluate the efficacy of docosahexaenoic acid supplementation on hepatic fat and associated cardiovascular risk factors in overweight children with nonalcoholic fatty liver disease. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 734–741. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Robidoux, J. Dyslipidemia management update. Curr. Opin. Pharmacol. 2017, 33, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Averna, M. The effect of ezetimibe on NAFLD. Atheroscler. Suppl. 2015, 17, 27–34. [Google Scholar] [CrossRef]
- Wang, X.; Sugimoto, K.; Fujisawa, T.; Shindo, N.; Minato, S.; Kamada, Y.; Hamano, M.; Ohishi, M.; Ikegami, H.; Rakugi, H. Novel effect of ezetimibe to inhibit the development of non-alcoholic fatty liver disease in Fatty Liver Shionogi mouse. Hepatol. Res. 2014, 44, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Husain, N.E.; Hassan, A.T.; Elmadhoun, W.M.; Ahmed, M.H. Evaluating the safety of Liptruzet (ezetimibe and atorvastatin): What are the potential benefits beyond low-density lipoprotein cholesterol-lowering effect? Expert Opin. Drug Saf. 2015, 14, 1445–1455. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, Y.; Takamura, T.; Honda, M.; Kita, Y.; Zen, Y.; Kato, K.; Misu, H.; Ota, T.; Nakamura, M.; Yamada, K.; et al. The effects of ezetimibe on non-alcoholic fatty liver disease and glucose metabolism: A randomised controlled trial. Diabetologia 2014, 57, 878–890. [Google Scholar] [CrossRef] [PubMed]
- Baigent, C.; Keech, A.; Kearney, P.M.; Blackwell, L.; Buck, G.; Pollicino, C.; Kirby, A.; Sourjina, T.; Peto, R.; Collins, R.; et al. Efficacy and safety of cholesterol-lowering treatment: Prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005, 366, 1267–1278. [Google Scholar] [CrossRef] [PubMed]
- Chong, L.W.; Hsu, Y.C.; Lee, T.F.; Lin, Y.; Chiu, Y.T.; Yang, K.C.; Wu, J.C.; Huang, Y.T. Fluvastatin attenuates hepatic steatosis-induced fibrogenesis in rats through inhibiting paracrine effect of hepatocyte on hepatic stellate cells. BMC Gastroenterol. 2015, 15, 22. [Google Scholar] [CrossRef] [PubMed]
- Roglans, N.; Sanguino, E.; Peris, C.; Alegret, M.; Vazquez, M.; Adzet, T.; Díaz, C.; Hernández, G.; Laguna, J.C.; Sánchez, R.M. Atorvastatin treatment induced peroxisome proliferator-activated receptor α expression and decreased plasma nonesterified fatty acids and liver triglyceride in fructose-fed rats. J. Pharmacol. Exp. Ther. 2002, 302, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Egawa, T.; Toda, K.; Nemoto, Y.; Ono, M.; Akisaw, N.; Saibara, T.; Hayashi, Y.; Hiroi, M.; Enzan, H.; Onishi, S. Pitavastatin ameliorates severe hepatic steatosis in aromatase-deficient (Ar−/−) mice. Lipids 2003, 38, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef] [PubMed]
- De Ferranti, S.D.; Rodday, A.M.; Parsons, S.K.; Cull, W.L.; O’Connor, K.G.; Daniels, S.R.; Leslie, L.K. Cholesterol Screening and Treatment Practices and Preferences: A Survey of United States Pediatricians. J. Pediatr. 2017, 185, 99–105.e2. [Google Scholar] [CrossRef] [PubMed]
- Porez, G.; Prawitt, J.; Gross, B.; Staels, B. Bile acid receptors as targets for the treatment of dyslipidemia and cardiovascular disease. J. Lipid Res. 2012, 53, 1723–1737. [Google Scholar] [CrossRef] [PubMed]
- Modica, S.; Gadaleta, R.M.; Moschetta, A. Deciphering the nuclear bile acid receptor FXR paradigm. Nucl. Recept. Signal. 2010, 8, e005. [Google Scholar] [CrossRef] [PubMed]
- Calkin, A.C.; Tontonoz, P. Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat. Rev. Mol. Cell Biol. 2012, 13, 213–224. [Google Scholar] [CrossRef] [PubMed]
- De Aguiar Vallim, T.Q.; Tarling, E.J.; Edwards, P.A. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013, 17, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, S.; Mencarelli, A.; Palladino, G.; Fiorucci, S. FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J. Lipid Res. 2010, 51, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perla, F.M.; Prelati, M.; Lavorato, M.; Visicchio, D.; Anania, C. The Role of Lipid and Lipoprotein Metabolism in Non‐Alcoholic Fatty Liver Disease. Children 2017, 4, 46. https://doi.org/10.3390/children4060046
Perla FM, Prelati M, Lavorato M, Visicchio D, Anania C. The Role of Lipid and Lipoprotein Metabolism in Non‐Alcoholic Fatty Liver Disease. Children. 2017; 4(6):46. https://doi.org/10.3390/children4060046
Chicago/Turabian StylePerla, Francesco Massimo, Maurizia Prelati, Michela Lavorato, Daniele Visicchio, and Caterina Anania. 2017. "The Role of Lipid and Lipoprotein Metabolism in Non‐Alcoholic Fatty Liver Disease" Children 4, no. 6: 46. https://doi.org/10.3390/children4060046
APA StylePerla, F. M., Prelati, M., Lavorato, M., Visicchio, D., & Anania, C. (2017). The Role of Lipid and Lipoprotein Metabolism in Non‐Alcoholic Fatty Liver Disease. Children, 4(6), 46. https://doi.org/10.3390/children4060046