Diagnosis, Evaluation and Treatment of Pulmonary Arterial Hypertension in Children


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
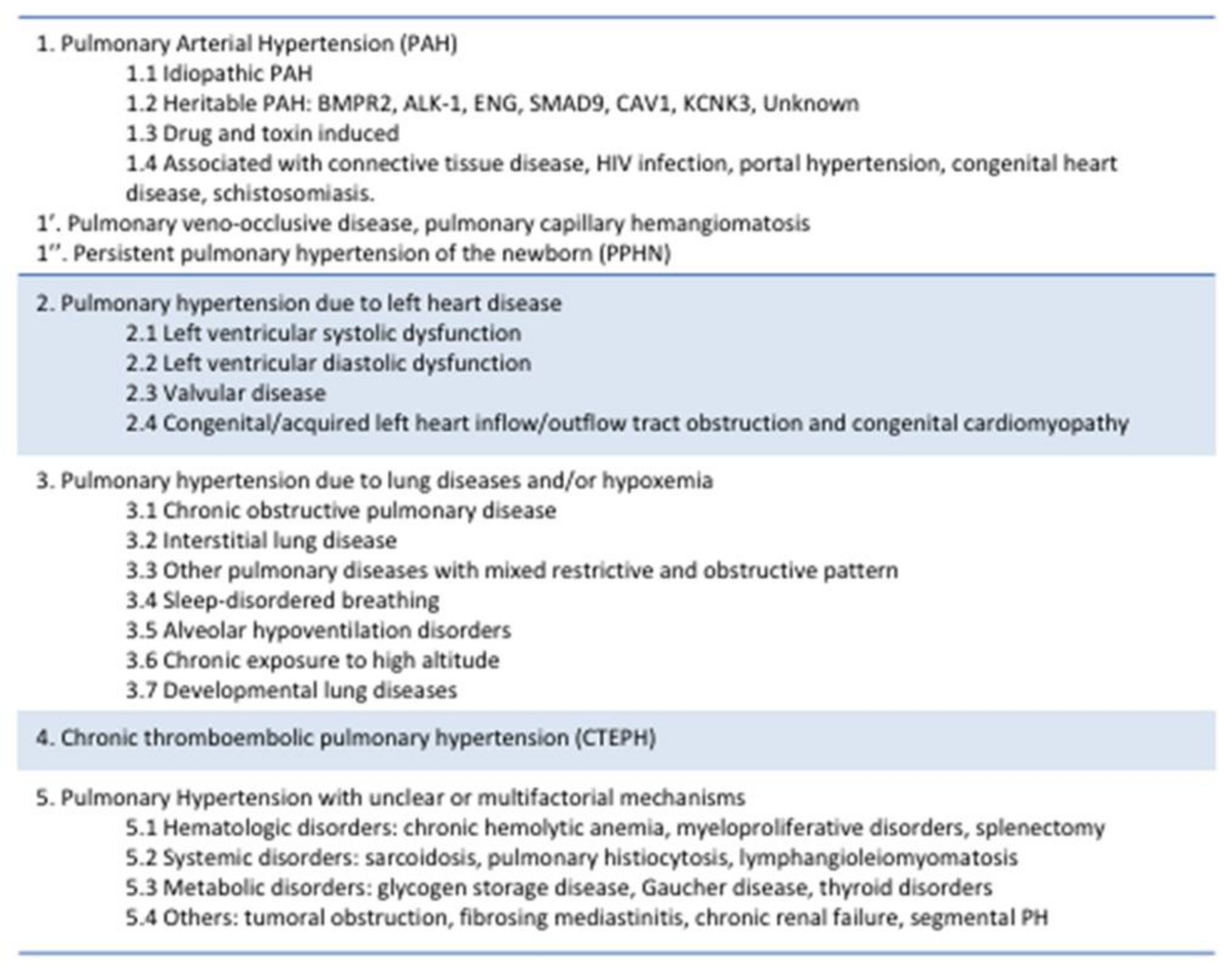
2. Definition
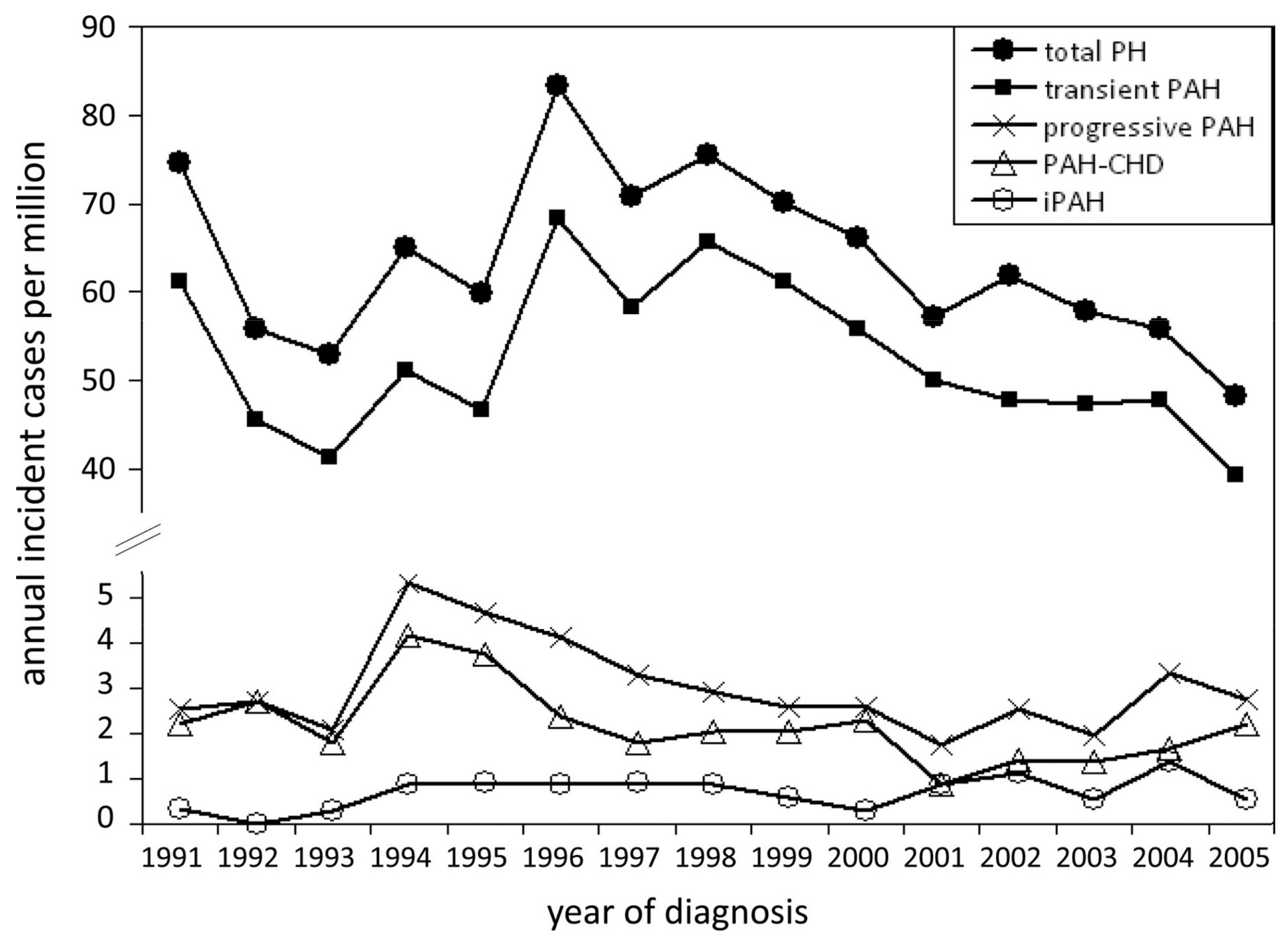
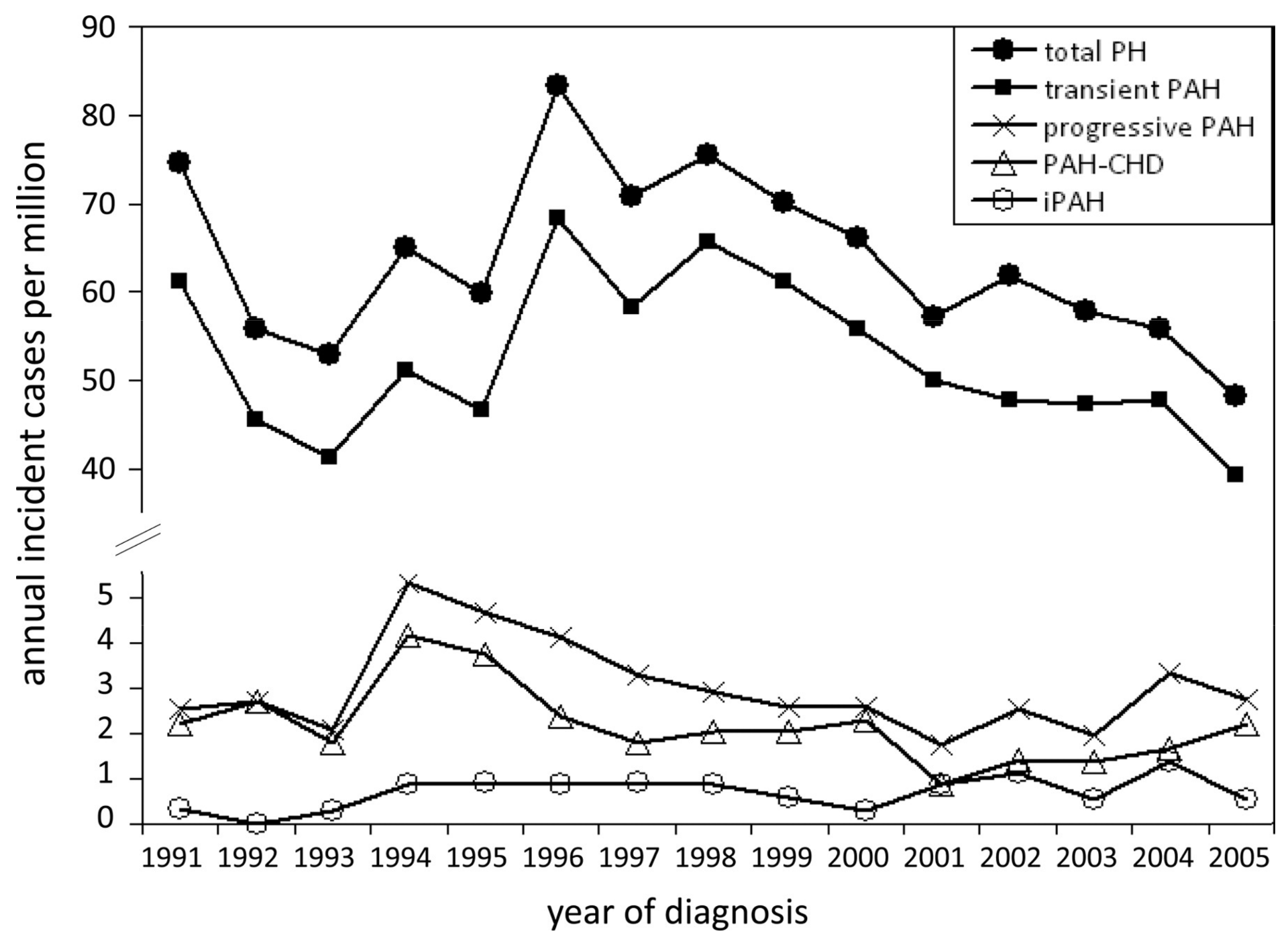
3. Epidemiology
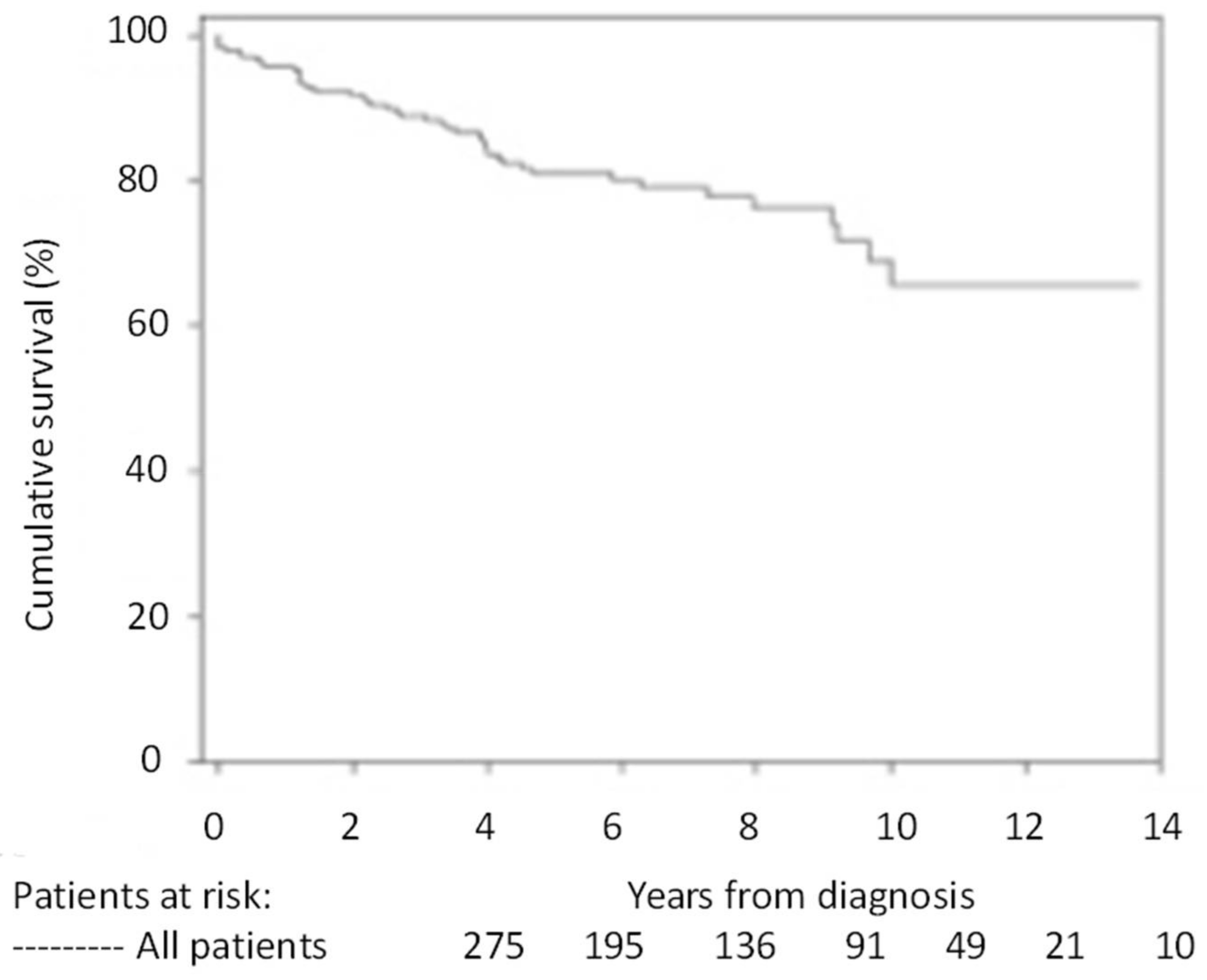
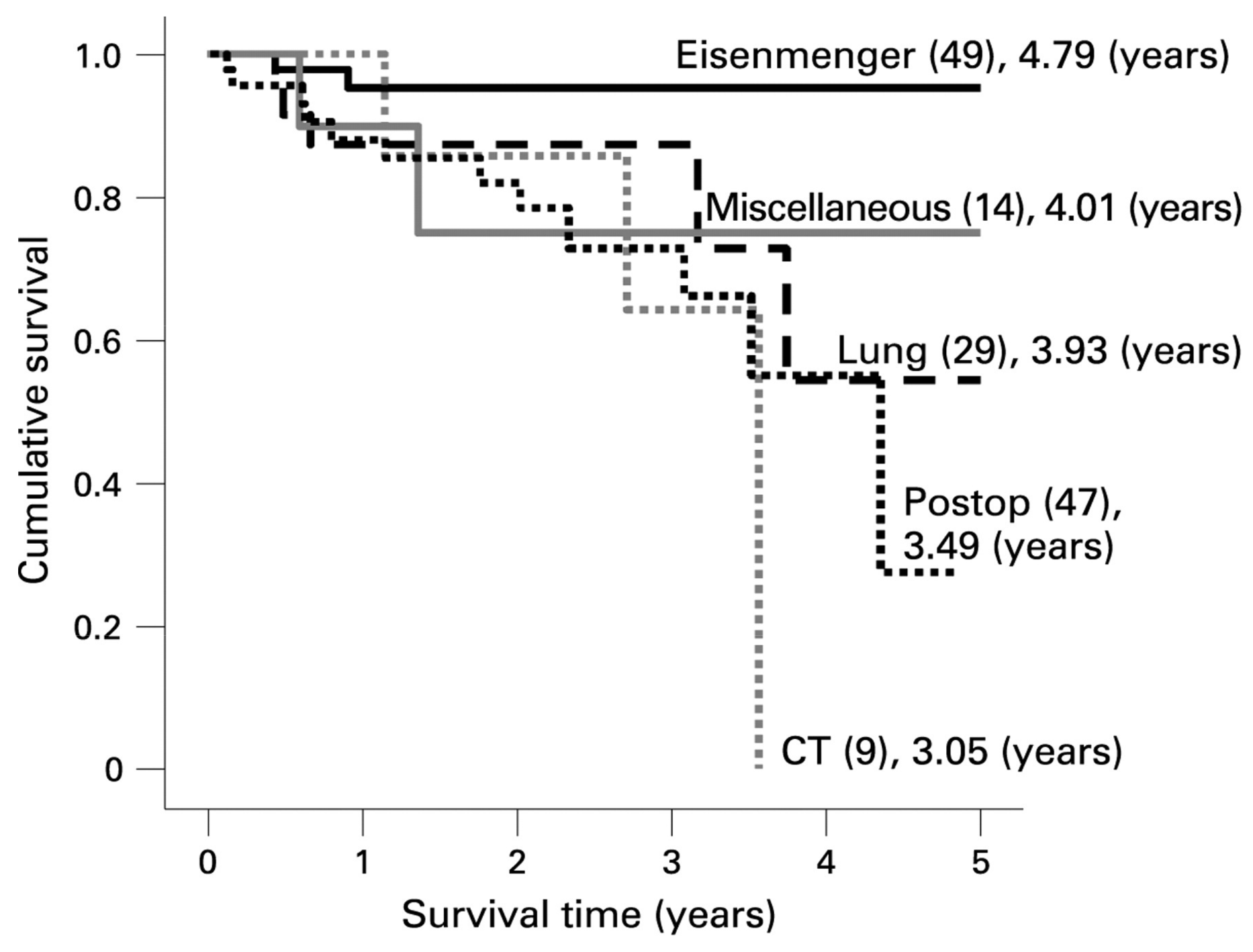
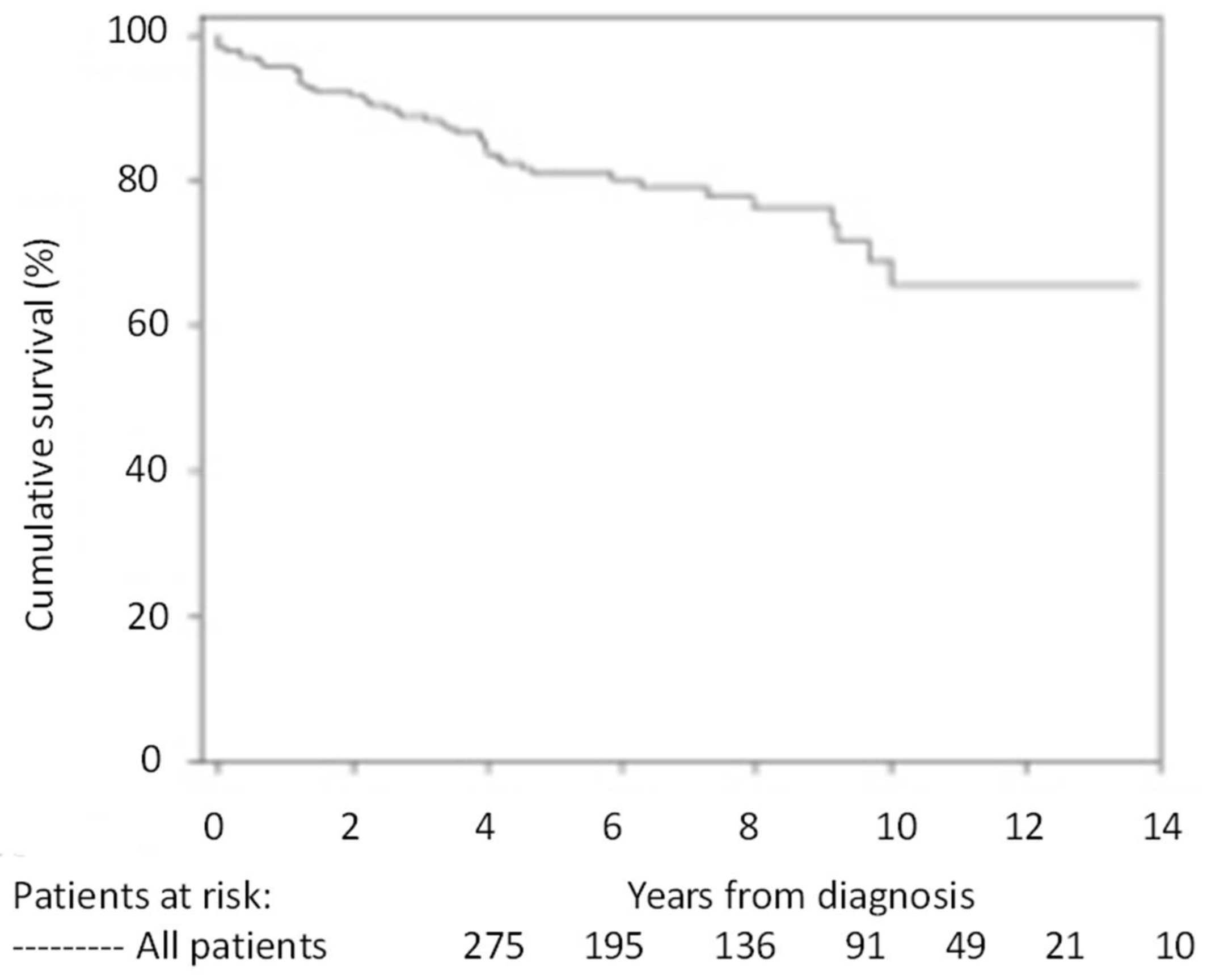
4. Prognosis
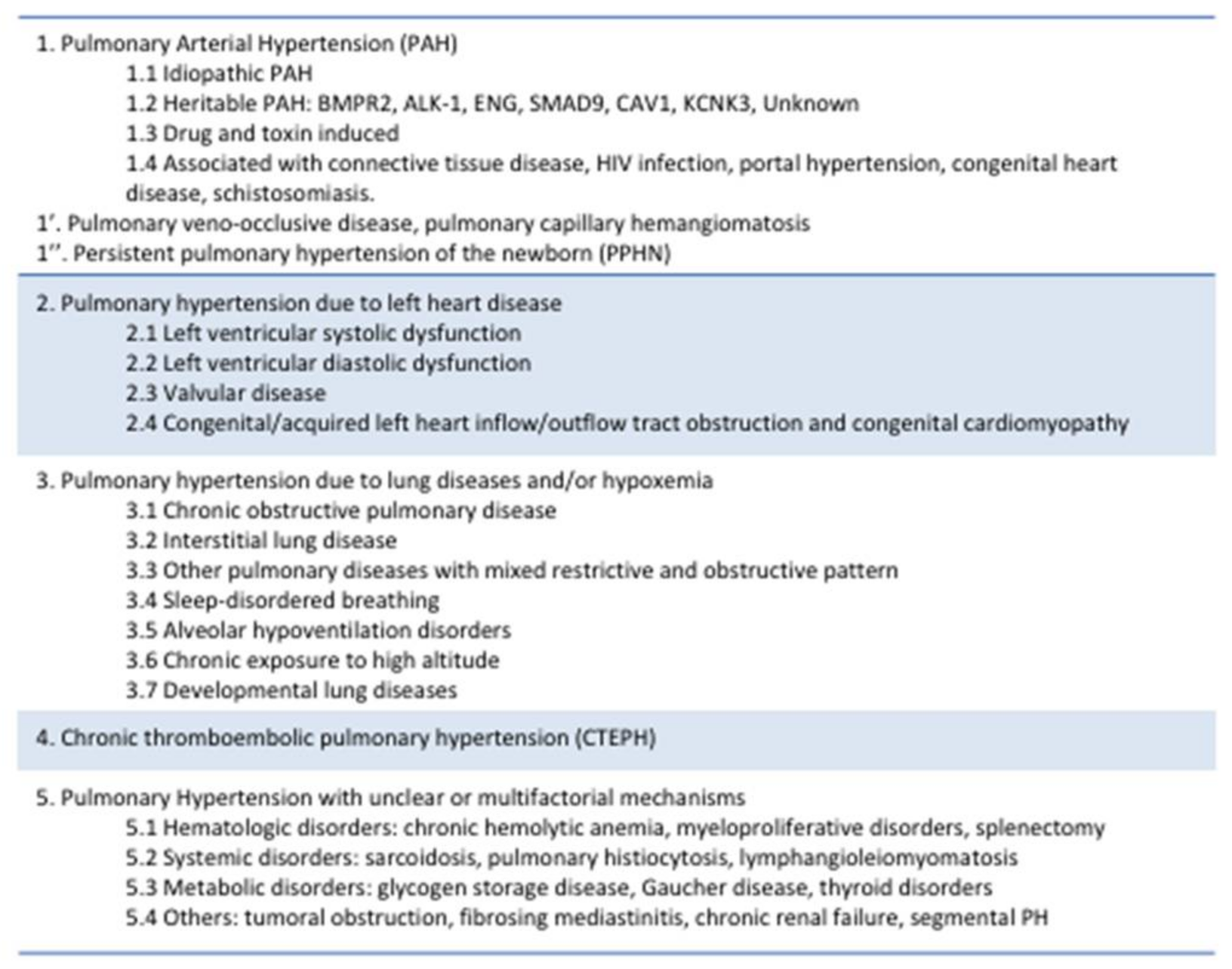
5. Highlighted Causes of Pulmonary Hypertension (PH)
5.1. Heritable Pulmonary Arterial Hypertension (PAH)
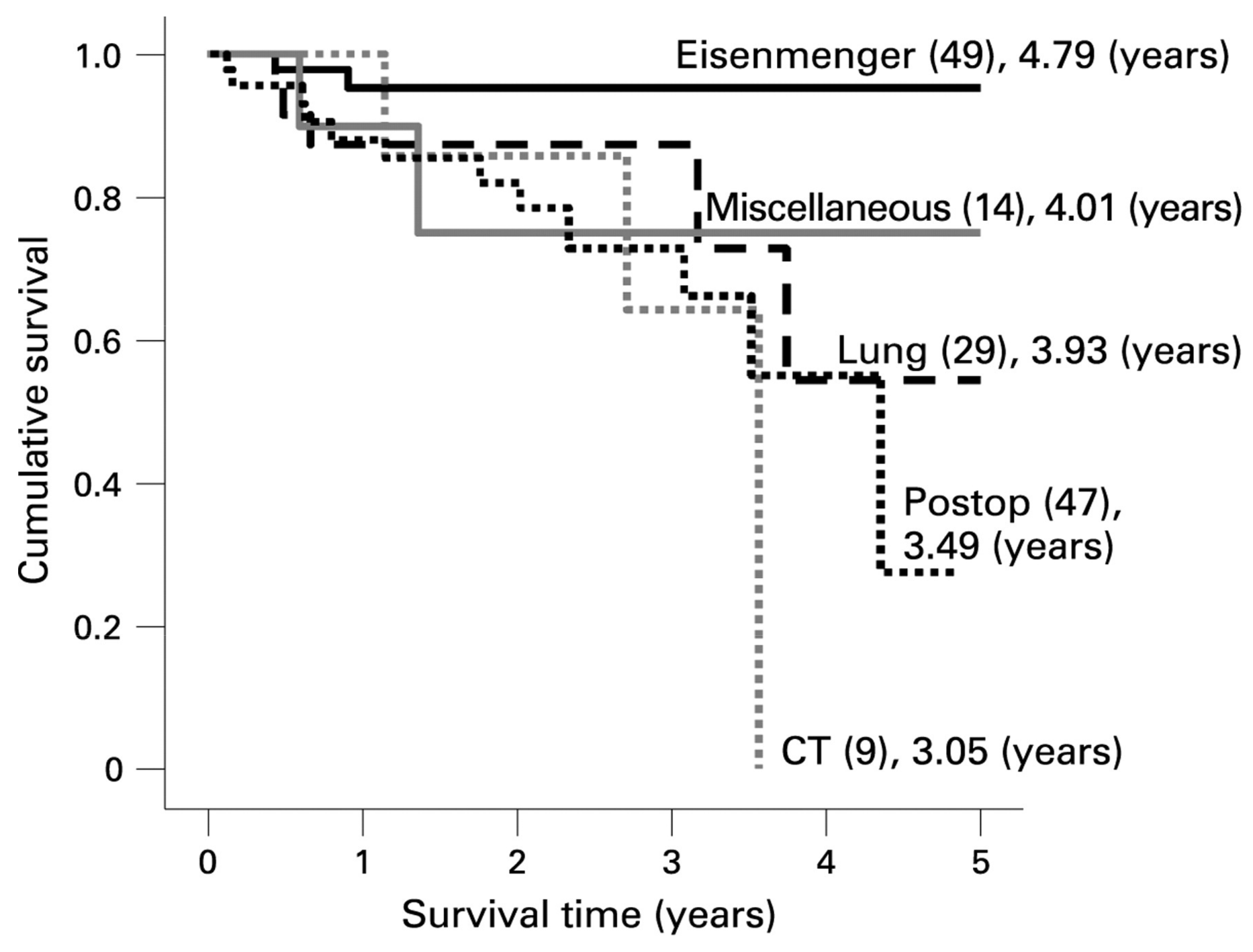
5.2. Single-Ventricle Circulation
5.3. Bronchopulmonary Dysplasia
6. Standard Evaluation
7. Emerging Evaluation Techniques
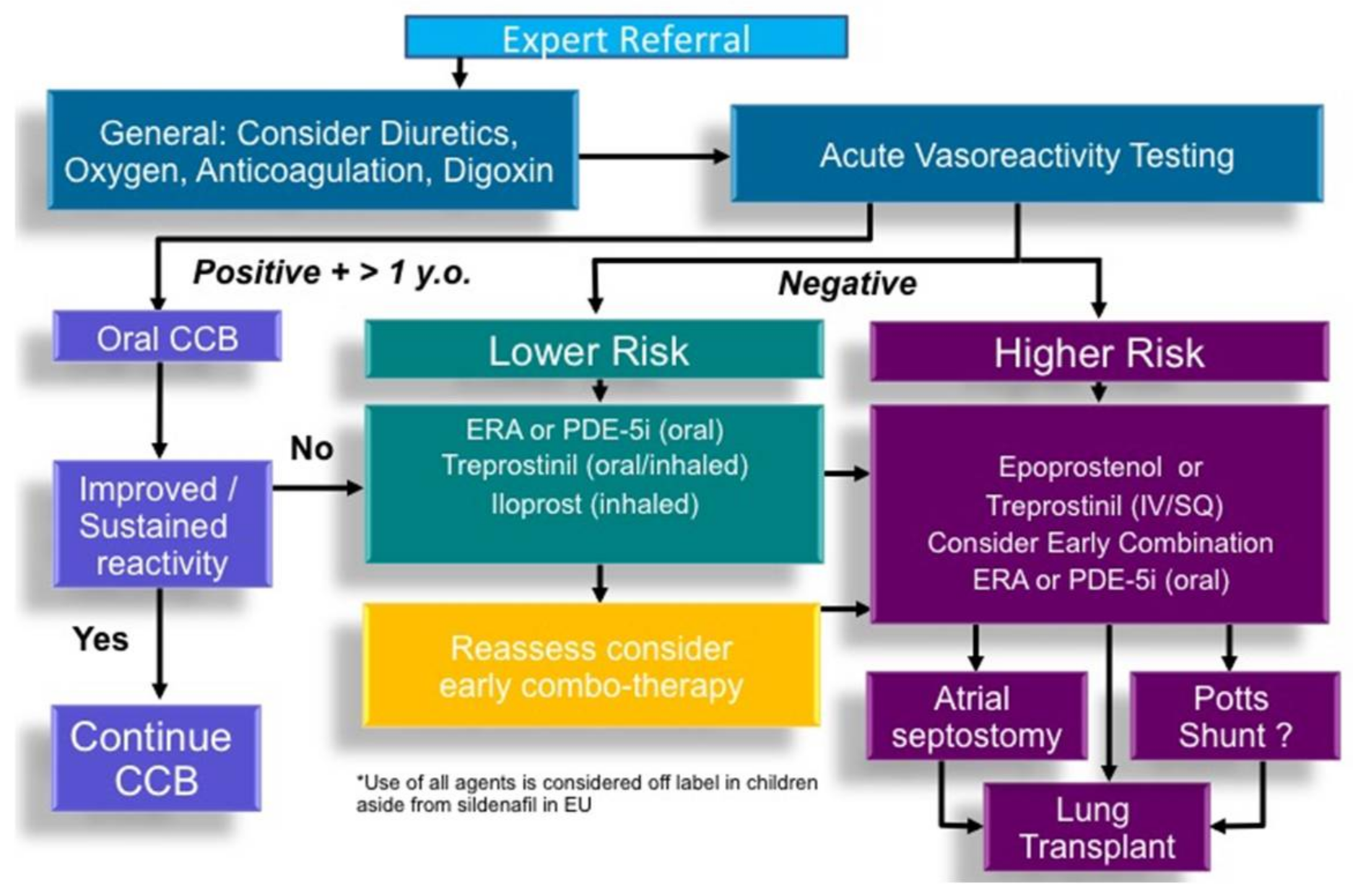
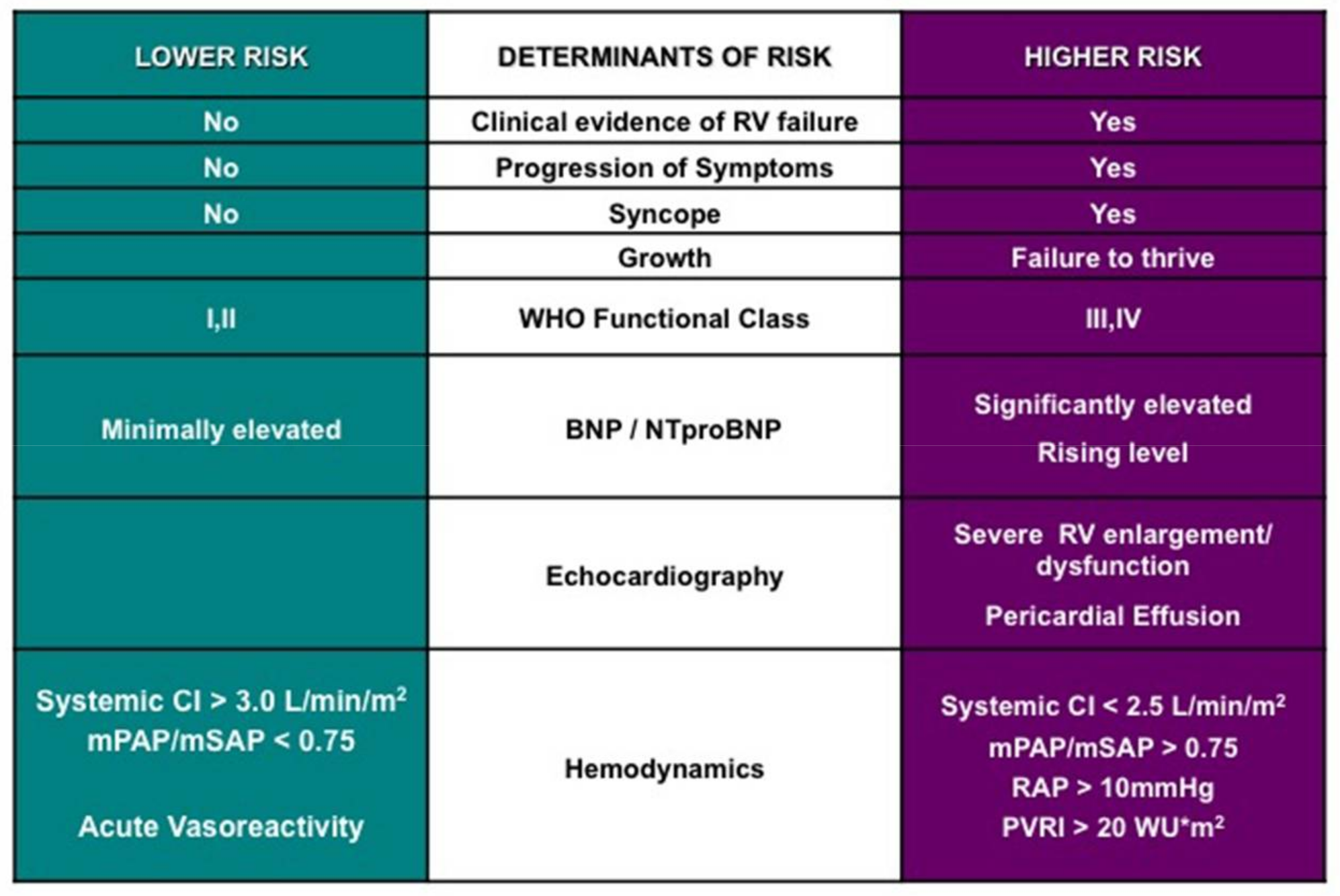
8. Cardiac Catheterization and Acute Vasoreactivity Testing
9. Targeted Pharmacological Therapy for PAH
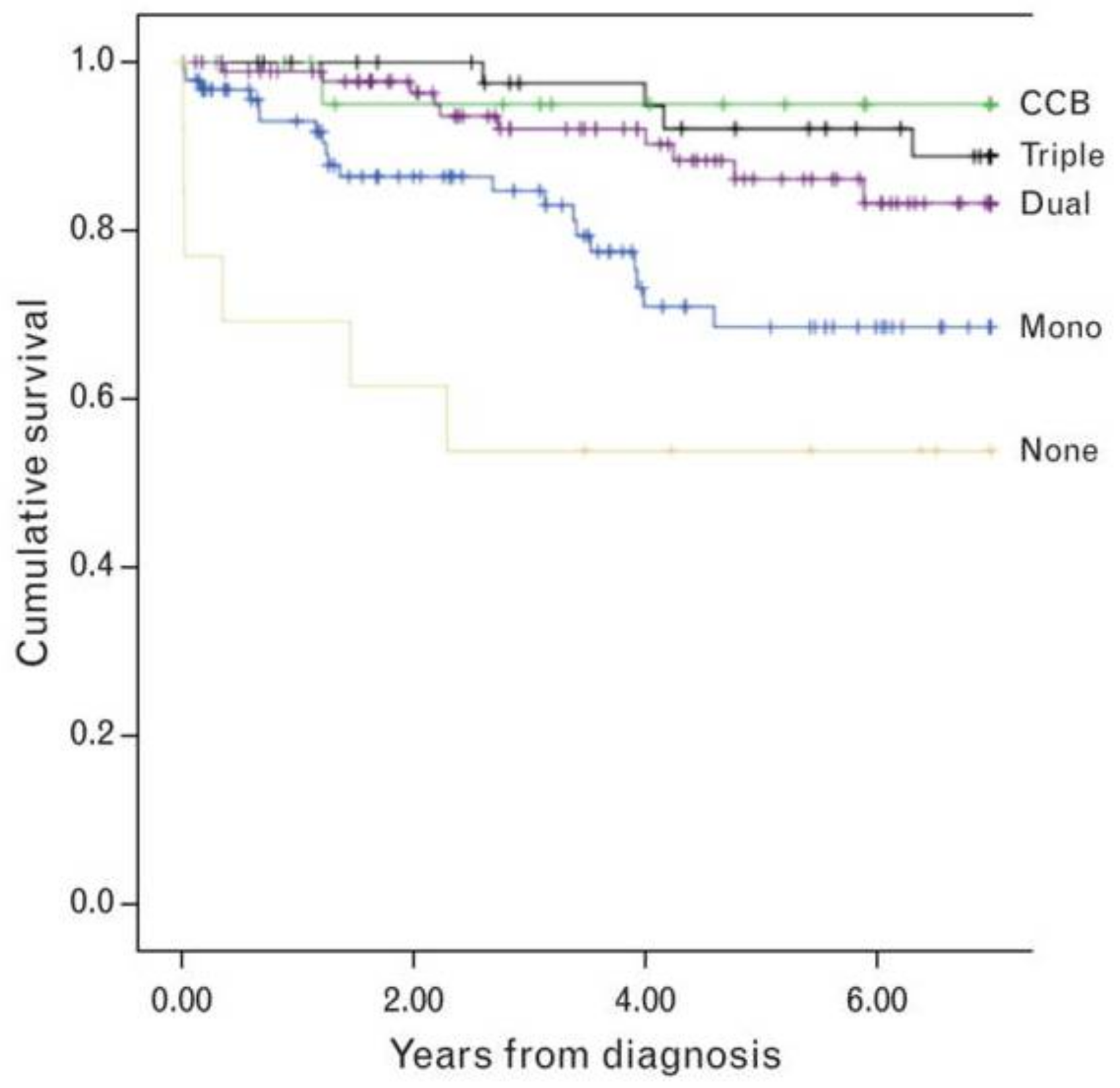
9.1. Calcium Channel Blockers
9.2. Prostacyclins
9.3. Endothelin Receptor Antagonists
9.4. Phosphodiesterase-5 Inhibitors and Soluble Guanylate Cyclase Stimulators
9.5. Combination Therapy
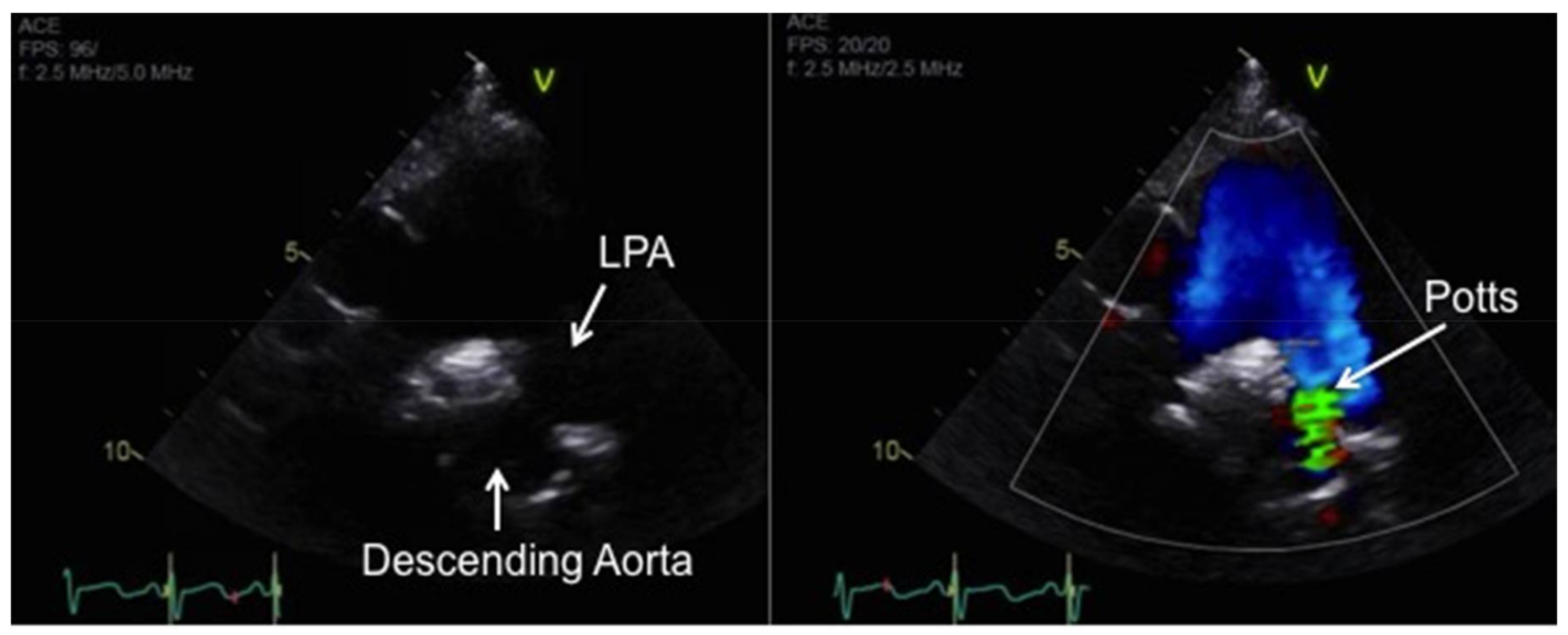
10. Atrial Septostomy and Potts Shunt for Refractory PAH
11. Transplantation
12. Adjunctive Therapy
13. Summary
Acknowledgments
Conflicts of Interest
References
- D’Alonzo, G.E.; Barst, R.J.; Ayres, S.M.; Bergofsky, E.H.; Brundage, B.H.; Detre, K.M.; Fishman, A.P.; Goldring, R.M.; Groves, B.M.; Kernis, J.T.; et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann. Intern. Med. 1991, 115, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Barst, R.J. Recent advances in the treatment of pediatric pulmonary artery hypertension. Pediatr. Clin. N. Am. 1999, 46, 331–345. [Google Scholar] [CrossRef]
- Ivy, D.D.; Abman, S.H.; Barst, R.J.; Berger, R.M.F.; Bonnet, D.; Fleming, T.R.; Haworth, S.G.; Raj, J.U.; Rosenzweig, E.B.; Schulze Neick, I.; et al. Pediatric pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D117–D126. [Google Scholar] [CrossRef] [PubMed]
- Zijlstra, W.M.; Douwes, J.M.; Rosenzweig, E.B.; Schokker, S.; Krishnan, U.; Roofthooft, M.T.R.; Miller-Reed, K.; Hillege, H.L.; Ivy, D.D.; Berger, R.M.F. Survival differences in pediatric pulmonary arterial hypertension: Clues to a better understanding of outcome and optimal treatment strategies. J. Am. Coll. Cardiol. 2014, 63, 2159–2169. [Google Scholar] [CrossRef] [PubMed]
- Barst, R.J.; Ertel, S.I.; Beghetti, M.; Ivy, D.D. Pulmonary arterial hypertension: A comparison between children and adults. Eur. Respir. J. 2011, 37, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Cerro, M.J.; Abman, S.; Diaz, G.; Freudenthal, F.; Harikrishnan, S.; Haworth, S.G.; Ivy, D.; Lopes, A.A.; Raj, J.U.; Sandoval, J.; et al. A consensus approach to the classification of pediatric pulmonary hypertensive vascular disease: Report from the PVRI Pediatric Taskforce, Panama 2011. Pulm. Circ. 2011, 1, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Corris, P.A.; Frost, A.; Girgis, R.E.; Granton, J.; Jing, Z.C.; Klepetko, W.; McGoon, M.D.; McLaughlin, V.V.; Preston, L.R.; et al. Updated treatment algorithm of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2013, 62, D60–D72. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Respir. J. 2015, 46, 1855–1856. [Google Scholar]
- Potts, W.J.; Smith, S.; Gibson, S. Anastomosis of the aorta to a pulmonary artery; certain types in congenital heart disease. J. Am. Med. Assoc. 1946, 132, 627–631. [Google Scholar] [CrossRef] [PubMed]
- Baruteau, A.E.; Belli, E.; Boudjemline, Y.; Laux, D.; Lévy, M.; Simonneau, G.; Carotti, A.; Humbert, M.; Bonnet, D. Palliative Potts shunt for the treatment of children with drug-refractory pulmonary arterial hypertension: Updated data from the first 24 patients. Eur. J. Cardiothorac. Surg. 2015, 47, e105–e110. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Barst, R.J.; Bourge, R.C.; Feldman, J.; Frost, A.E.; Galié, N.; Gómez-Sánchez, M.A.; Grimminger, F.; Grünig, E.; Hassoun, P.M.; et al. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: Results of the randomized IMPRES study. Circulation 2013, 127, 1128–1138. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Gatzoulis, M.A.; Adatia, I.; Celermajer, D.; Denton, C.; Ghofrani, A.; Gomez Sanchez, M.A.; Krishna Kumar, R.; Landzberg, M.; Machado, R.; et al. Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D34–D41. [Google Scholar] [CrossRef] [PubMed]
- Moledina, S.; Hislop, A.A.; Foster, H.; Schulze-Neick, I.; Haworth, S.G. Childhood idiopathic pulmonary arterial hypertension: A national cohort study. Heart 2010, 96, 1401–1406. [Google Scholar] [CrossRef] [PubMed]
- Van Loon, R.L.; Roofthooft, M.T.; Hillege, H.L.; ten Harkel, A.D.; van Osch-Gevers, M.; Delhaas, T.; Kapusta, L.; Strengers, J.L.; Rammeloo, L.; Clur, S.A.; et al. Pediatric pulmonary hypertension in the Netherlands: Epidemiology and characterization during the period 1991 to 2005. Circulation 2011, 124, 1755–1764. [Google Scholar] [CrossRef] [PubMed]
- Cerro Marin, M.J.; Sabate Rotes, A.; Rodriguez Ogando, A.; Mendoza Soto, A.; Quero Jiménez, M.; Gavilán Camacho, J.L.; Raposo Sonnenfeld, I.; Moya Bonora, A.; Albert Brotons, D.C.; REHIPED Investigators. Assessing Pulmonary Hypertensive Vascular Disease in Childhood: Data from the Spanish Registry. Am. J. Respir. Crit. Care Med. 2014, 190, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.B.; Crystal, M.A.; Morales, D.L.; Gerald, K.; Hanna, B.D.; Mallory, G.B., Jr.; Rossano, J.W. Trends in pediatric pulmonary hypertension-related hospitalizations in the United States from 2000–2009. Pulm. Circ. 2015, 5, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, B.G.; Nies, M.K.; Ajuba-Iwuji, C.C.; Coulson, J.D.; Romer, L.H. Trends in Hospitalization for Pediatric Pulmonary Hypertension. Pediatrics 2015, 136, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Haworth, S.G.; Hislop, A.A. Treatment and survival in children with pulmonary arterial hypertension: The UK Pulmonary Hypertension Service for Children 2001–2006. Heart 2009, 95, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Berger, R.M.; Beghetti, M.; Humpl, T.; Ivy, D.D.; Jing, Z.C.; Bonnet, D.; Schulze-Neick, I.; Barst, R.J. Clinical features of paediatric pulmonary hypertension: A registry study. Lancet 2012, 379, 537–546. [Google Scholar] [CrossRef]
- Zijlstra, W.M.; Douwes, J.M.; Ploegstra, M.J.; Krishnan, U.; Roofthooft, M.T.; Hillege, H.L.; Ivy, D.D.; Rosenzweig, E.B.; Berger, R.M. Clinical classification in pediatric pulmonary arterial hypertension associated with congenital heart disease. Pulm. Circ. 2016, 6, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Austin, E.D.; Loyd, J.E. The genetics of pulmonary arterial hypertension. Circ. Res. 2014, 115, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Haghighi, F.; Helleby, L.; Vanterpool, K.; Horn, E.M.; Barst, R.J.; Hodge, S.E.; Morse, J.H.; Knowles, J.A. Fine mapping of PPH1, a gene for familial primary pulmonary hypertension, to a 3-cM region on chromosome 2q33. Am. J. Respir. Crit. Care Med. 2000, 161, 1055–1059. [Google Scholar] [CrossRef] [PubMed]
- Grunig, E.; Koehler, R.; Miltenberger-Miltenyi, G.; Zimmermann, R.; Gorenflo, M.; Mereles, D.; Arnold, K.; Naust, B.; Wilkens, H.; Benz, A.; et al. Primary pulmonary hypertension in children may have a different genetic background than in adults. Pediatr. Res. 2004, 56, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.E.; Berger, R.; Haworth, S.G.; Tulloh, R.; Mache, C.J.; Morrell, N.W.; Aldred, M.A.; Trembath, R.C. Transforming growth factor-beta receptor mutations and pulmonary arterial hypertension in childhood. Circulation 2005, 111, 435–441. [Google Scholar] [CrossRef] [PubMed]
- International PPH Consortium; Lane, K.B.; Machado, R.D.; Pauciulo, M.W.; Thomson, J.R.; Phillips, J.A.; Loyd, J.E.; Nichols, W.C.; Trembath, R.C. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat. Genet. 2000, 26, 81–84. [Google Scholar] [PubMed]
- Rosenzweig, E.B.; Morse, J.H.; Knowles, J.A.; Chada, K.K.; Khan, A.M.; Roberts, K.E.; McElroy, J.J.; Juskiw, N.K.; Mallory, N.C.; Rich, S.; et al. Clinical implications of determining BMPR2 mutation status in a large cohort of children and adults with pulmonary arterial hypertension. J. Heart Lung Transplant. 2008, 27, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Larkin, E.K.; Newman, J.H.; Austin, E.D.; Hemnes, A.R.; Wheeler, L.; Robbins, I.M.; West, J.D.; Phillips, J.A.; Hamid, R.; Loyd, J.E. Longitudinal analysis casts doubt on the presence of genetic anticipation in heritable pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Eyries, M.; Szezepanski, I.; Ladouceur, M.; Nadaud, S.; Bonnet, D.; Soubrier, F. Genetic analyses in a cohort of children with pulmonary hypertension. Eur. Respir. J. 2016, 48, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, M.; Yagi, H.; Matsuoka, R.; Akimoto, K.; Furutani, M.; Imamura, S.; Uehara, R.; Nakayama, T.; Takao, A.; Nakazawa, M. Implications of mutations of activin receptor-like kinase 1 gene (ALK1) in addition to bone morphogenetic protein receptor II gene (BMPR2) in children with pulmonary arterial hypertension. Circ. J. 2008, 72, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Best, D.H.; Austin, E.D.; Chung, W.K.; Elliott, C.G. Genetics of pulmonary hypertension. Curr. Opin. Cardiol. 2014, 29, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Best, D.H.; Sumner, K.L.; Austin, E.D.; Chung, W.K.; Brown, L.M.; Borczuk, A.C.; Rosenzweig, E.B.; Bayrak-Toydemir, P.; Mao, R.; Cahill, B.C.; et al. EIF2AK4 mutations in pulmonary capillary hemangiomatosis. Chest 2014, 145, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Kerstjens-Frederikse, W.S.; Bongers, E.M.; Roofthooft, M.T.; Leter, E.M.; Douwes, J.M.; Van Dijk, A.; Vonk-Noordegraaf, A.; Dijk-Bos, K.K.; Hoefsloot, L.H.; Hoendermis, E.S.; et al. TBX4 mutations (small patella syndrome) are associated with childhood-onset pulmonary arterial hypertension. J. Med. Genet. 2013, 50, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Chung, W.K. The genetic basis of pulmonary arterial hypertension. Hum. Genet. 2014, 133, 471–479. [Google Scholar] [CrossRef] [PubMed]
- La Gerche, A.; Gewillig, M. What Limits Cardiac Performance during Exercise in Normal Subjects and in Healthy Fontan Patients? Int. J. Pediatr. 2010, 2010, 791291. [Google Scholar] [CrossRef] [PubMed]
- Haseyama, K.; Satomi, G.; Yasukochi, S.; Matsui, H.; Harada, Y.; Uchita, S. Pulmonary vasodilation therapy with sildenafil citrate in a patient with plastic bronchitis after the Fontan procedure for hypoplastic left heart syndrome. J. Thorac. Cardiovasc. Surg. 2006, 132, 1232–1233. [Google Scholar] [CrossRef] [PubMed]
- John, A.S.; Johnson, J.A.; Khan, M.; Driscoll, D.J.; Warnes, C.A.; Cetta, F. Clinical outcomes and improved survival in patients with protein-losing enteropathy after the Fontan operation. J. Am. Coll. Cardiol. 2014, 64, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, S.P.; Ivy, D.D.; Mitchell, M.B.; Campbell, D.N.; Dines, M.L.; Miyamoto, S.; Kay, J.; Clarke, D.R.; Lacour-Gayet, F. Performance of cavopulmonary palliation at elevated altitude: Midterm outcomes and risk factors for failure. Circulation 2008, 118, S177–S181. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Danel, C.; Laval, A.M.; Leca, F.; Vouhe, P.R.; Israel-Biet, D. Nitric oxide synthase expression by pulmonary arteries: A predictive marker of Fontan procedure outcome? J. Thorac. Cardiovasc. Surg. 2003, 125, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, D.J.; French, B.; Szwast, A.L.; McBride, M.G.; Marino, B.S.; Mirarchi, N.; Hanna, B.D.; Wernovsky, G.; Paridon, S.M.; Rychik, J. Impact of sildenafil on echocardiographic indices of myocardial performance after the Fontan operation. Pediatr. Cardiol. 2012, 33, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Morchi, G.S.; Ivy, D.D.; Duster, M.C.; Claussen, L.; Chan, K.C.; Kay, J. Sildenafil Increases Systemic Saturation and Reduces Pulmonary Artery Pressure in Patients with Failing Fontan Physiology. Congenit. Heart Dis. 2009, 4, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Van De Bruaene, A.; La Gerche, A.; Claessen, G.; De Meester, P.; Devroe, S.; Gillijns, H.; Bogaert, J.; Claus, P.; Heidbuchel, H.; Gewillig, M.; et al. Sildenafil improves exercise hemodynamics in Fontan patients. Circ. Cardiovasc. Imaging 2014, 7, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, M.; Kurosawa, H.; Hashimoto, K.; Nomura, K.; Kitamura, N. The role of plasma endothelin in the Fontan circulation. J. Cardiovasc. Surg. 2002, 43, 793–797. [Google Scholar]
- Bowater, S.E.; Weaver, R.A.; Thorne, S.A.; Clift, P.F. The safety and effects of bosentan in patients with a Fontan circulation. Congenit. Heart Dis. 2012, 7, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Derk, G.; Houser, L.; Miner, P.; Williams, R.; Moriarty, J.; Finn, P.; Alejos, J.; Aboulhosn, J. Efficacy of endothelin blockade in adults with fontan physiology. Congenit. Heart Dis. 2014, 10, E11–E16. [Google Scholar] [CrossRef] [PubMed]
- Hebert, A.; Mikkelsen, U.R.; Thilen, U.; Idorn, L.; Jensen, A.S.; Nagy, E.; Hanseus, K.; Sørensen, K.E.; Søndergaard, L. Bosentan improves exercise capacity in adolescents and adults after Fontan operation: The TEMPO (Treatment With Endothelin Receptor Antagonist in Fontan Patients, a Randomized, Placebo-Controlled, Double-Blind Study Measuring Peak Oxygen Consumption) study. Circulation 2014, 130, 2021–2030. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, U.; Rosenzweig, E.B. Pulmonary hypertension in chronic lung disease of infancy. Curr. Opin. Pediatr. 2015, 27, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Mourani, P.M.; Sontag, M.K.; Younoszai, A.; Miller, J.I.; Kinsella, J.P.; Baker, C.D.; Poindexter, B.B.; Ingram, D.A.; Abman, S.H. Early pulmonary vascular disease in preterm infants at risk for bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2015, 191, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Abman, S.H.; Ivy, D.D. Hospitalizations of Children With Pulmonary Hypertension: Implications for Improving Care. Pediatrics 2015, 136, 392–393. [Google Scholar] [CrossRef] [PubMed]
- Del Cerro, M.J.; Sabate Rotes, A.; Carton, A.; Deiros, L.; Bret, M.; Cordeiro, M.; Verdú, C.; Barrios, M.I.; Albajara, L.; Gutierrez-Larraya, F. Pulmonary hypertension in bronchopulmonary dysplasia: Clinical findings, cardiovascular anomalies and outcomes. Pediatr. Pulmonol. 2014, 49, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Khemani, E.; McElhinney, D.B.; Rhein, L.; Andrade, O.; Lacro, R.V.; Thomas, K.C.; Mullen, M.P. Pulmonary artery hypertension in formerly premature infants with bronchopulmonary dysplasia: Clinical features and outcomes in the surfactant era. Pediatrics 2007, 120, 1260–1269. [Google Scholar] [CrossRef] [PubMed]
- Augustine, S.; Avey, M.T.; Harrison, B.; Locke, T.; Ghannad, M.; Moher, D.; Thébaud, B. Mesenchymal Stromal Cell Therapy in Bronchopulmonary Dysplasia: Systematic Review and Meta-Analysis of Preclinical Studies. Stem Cells Transl. Med. 2017, 6, 2079–2093. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, U.; Feinstein, J.A.; Adatia, I.; Austin, E.D.; Mullen, M.P.; Hopper, R.K.; Hanna, B.; Romer, L.; Keller, R.L.; Fineman, J.; et al. Evaluation and management of pulmonary hypertension in children with bronchopulmonary dysplasia. J. Pediatr. 2017, 188, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Abman, S.H.; Hansmann, G.; Archer, S.L.; Ivy, D.D.; Adatia, I.; Chung, W.K.; Hanna, B.D.; Rosenzweig, E.B.; Raj, J.U.; Cornfield, D.; et al. Pediatric pulmonary hypertension: Guidelines from the American Heart Association and American Thoracic Society. Circulation 2015, 132, 2037–2099. [Google Scholar] [CrossRef] [PubMed]
- Beghetti, M.; Berger, R.M.; Schulze-Neick, I.; Day, R.W.; Pulido, T.; Feinstein, J.; Barst, R.J.; Humpl, T.; TOPP Registry Investigators. Diagnostic evaluation of paediatric pulmonary hypertension in current clinical practice. Eur. Respir. J. 2013, 42, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Jone, P.N.; Ivy, D.D. Echocardiography in pediatric pulmonary hypertension. Front. Pediatr. 2014, 2, 124. [Google Scholar] [CrossRef] [PubMed]
- Masuyama, T.; Kodama, K.; Kitabatake, A.; Sato, H.; Nanto, S.; Inoue, M. Continuous-wave Doppler echocardiographic detection of pulmonary regurgitation and its application to noninvasive estimation of pulmonary artery pressure. Circulation 1986, 74, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Forfia, P.R.; Fisher, M.R.; Mathai, S.C.; Housten-Harris, T.; Hemnes, A.R.; Borlaug, B.A.; Chamera, E.; Corretti, M.C.; Champion, H.C.; Abraham, T.P.; et al. Tricuspid annular displacement predicts survival in pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2006, 174, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Friesen, R.M.; Schafer, M.; Burkett, D.A.; Cassidy, C.J.; Ivy, D.D.; Jone, P.N. Right Ventricular Tissue Doppler Myocardial Performance Index in Children with Pulmonary Hypertension: Relation to Invasive Hemodynamics. Pediatr. Cardiol. 2017, 39, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Hinderliter, A.L.; Torbicki, A.; Fourme, T.; Simonneau, G.; Pulido, T.; Espinola-Zavaleta, N.; Rocchi, G.; Manes, A.; Frantz, R.; et al. Effects of the oral endothelin-receptor antagonist bosentan on echocardiographic and doppler measures in patients with pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2003, 41, 1380–1386. [Google Scholar] [CrossRef]
- Hinderliter, A.L.; Willis, P.W.; Barst, R.J.; Rich, S.; Rubin, L.J.; Badesch, D.B.; Groves, B.M.; McGoon, M.D.; Tapson, V.F.; Bourge, R.C.; et al. Effects of long-term infusion of prostacyclin (epoprostenol) on echocardiographic measures of right ventricular structure and function in primary pulmonary hypertension. Primary Pulmonary Hypertension Study Group. Circulation 1997, 95, 1479–1486. [Google Scholar] [CrossRef] [PubMed]
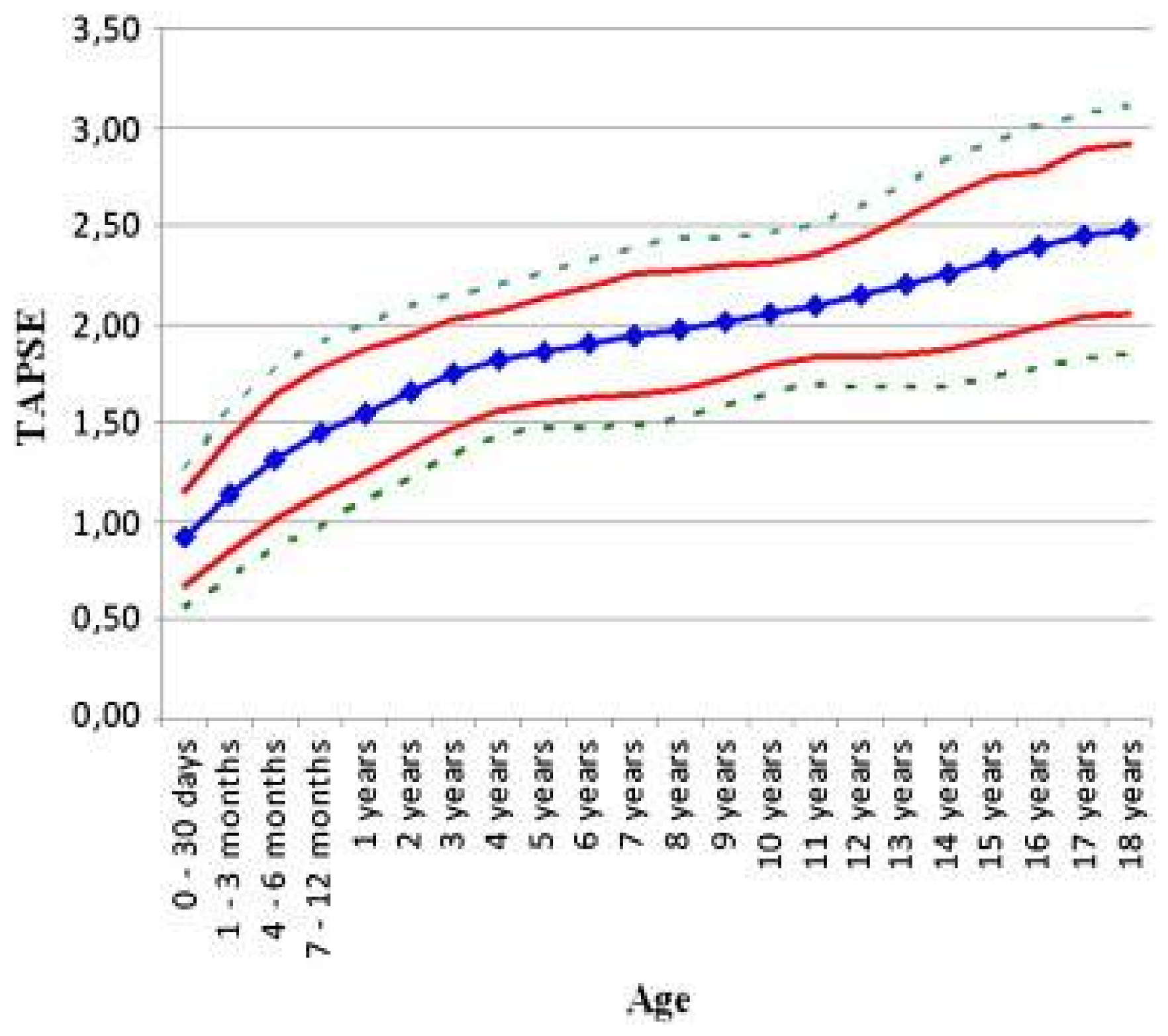
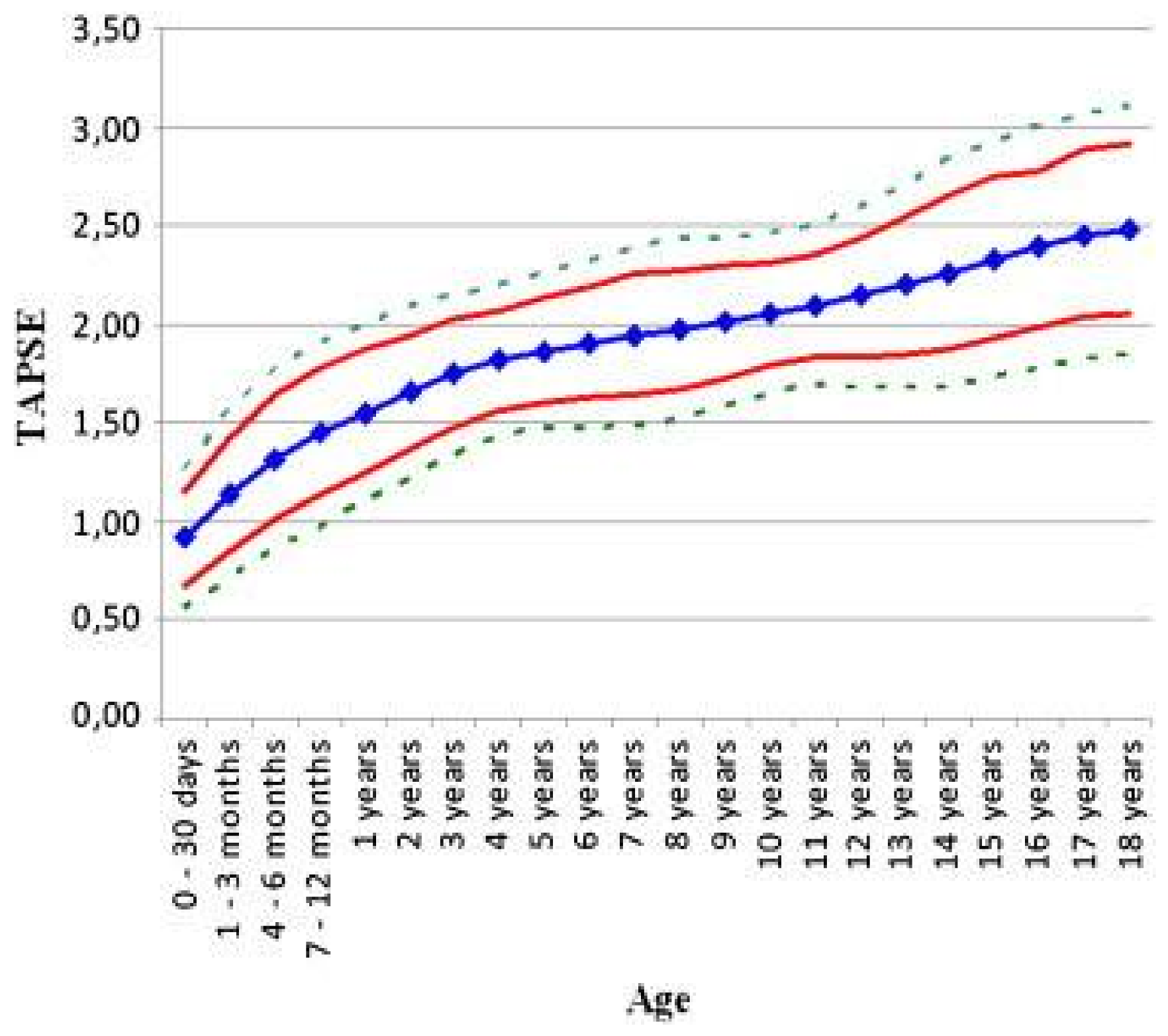
- Koestenberger, M.; Ravekes, W.; Everett, A.D.; Stueger, H.P.; Heinzl, B.; Gamillscheg, A.; Cvirn, G.; Boysen, A.; Fandl, A.; Nagel, B. Right ventricular function in infants, children and adolescents: Reference values of the tricuspid annular plane systolic excursion (TAPSE) in 640 healthy patients and calculation of z score values. J. Am. Soc. Echocardiogr. 2009, 22, 715–719. [Google Scholar] [CrossRef] [PubMed]
- Raymond, R.J.; Hinderliter, A.L.; Willis, P.W.; Ralph, D.; Caldwell, E.J.; Williams, W.; Ettinger, N.A.; Hill, N.S.; Summer, W.R.; de Boisblanc, B.; et al. Echocardiographic predictors of adverse outcomes in primary pulmonary hypertension. J. Am. Coll. Cardiol. 2002, 39, 1214–1219. [Google Scholar] [CrossRef]
- Tei, C.; Dujardin, K.S.; Hodge, D.O.; Bailey, K.R.; McGoon, M.D.; Tajik, A.J.; Seward, S.B. Doppler echocardiographic index for assessment of global right ventricular function. J. Am. Soc. Echocardiogr. 1996, 9, 838–847. [Google Scholar] [CrossRef]
- Jone, P.N.; Patel, S.S.; Cassidy, C.; Ivy, D.D. Three-dimensional Echocardiography of Right Ventricular Function Correlates with Severity of Pediatric Pulmonary Hypertension. Congenit. Heart Dis. 2016, 11, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Ploegstra, M.J.; Douwes, J.M.; Roofthooft, M.T.; Zijlstra, W.M.; Hillege, H.L.; Berger, R.M. Identification of treatment goals in paediatric pulmonary arterial hypertension. Eur. Respir. J. 2014, 44, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
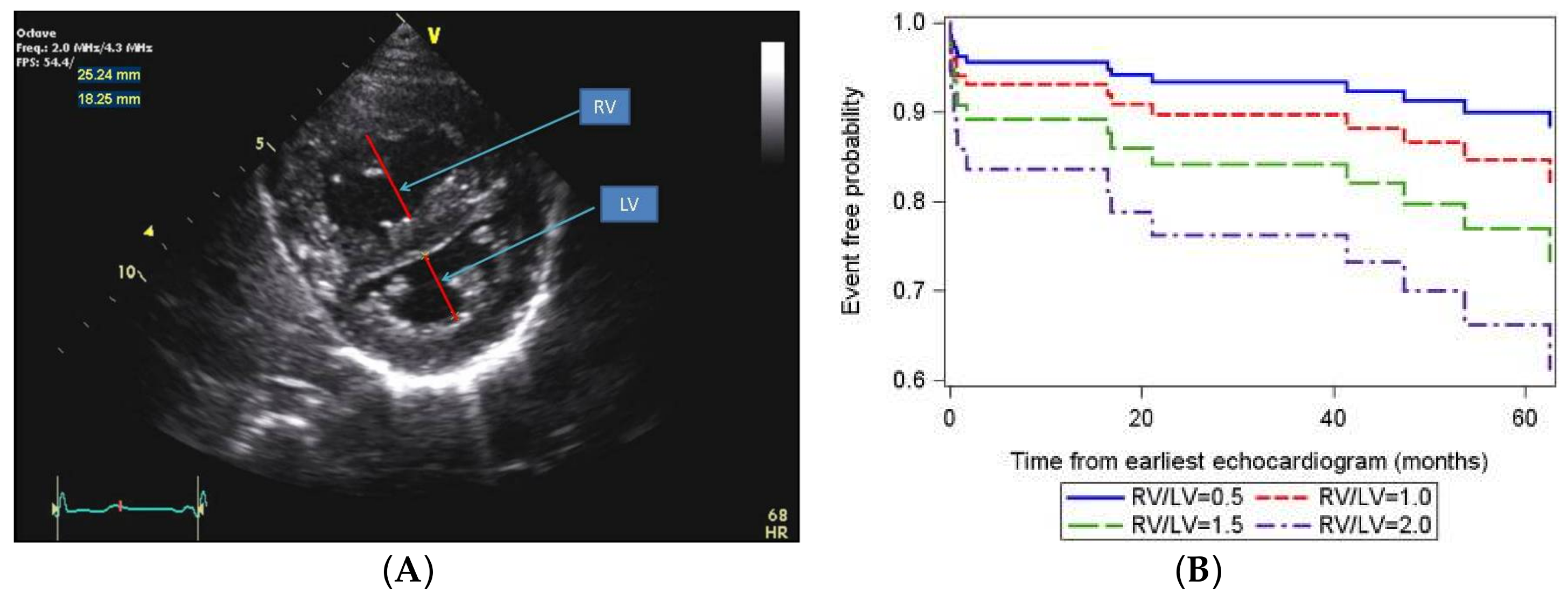
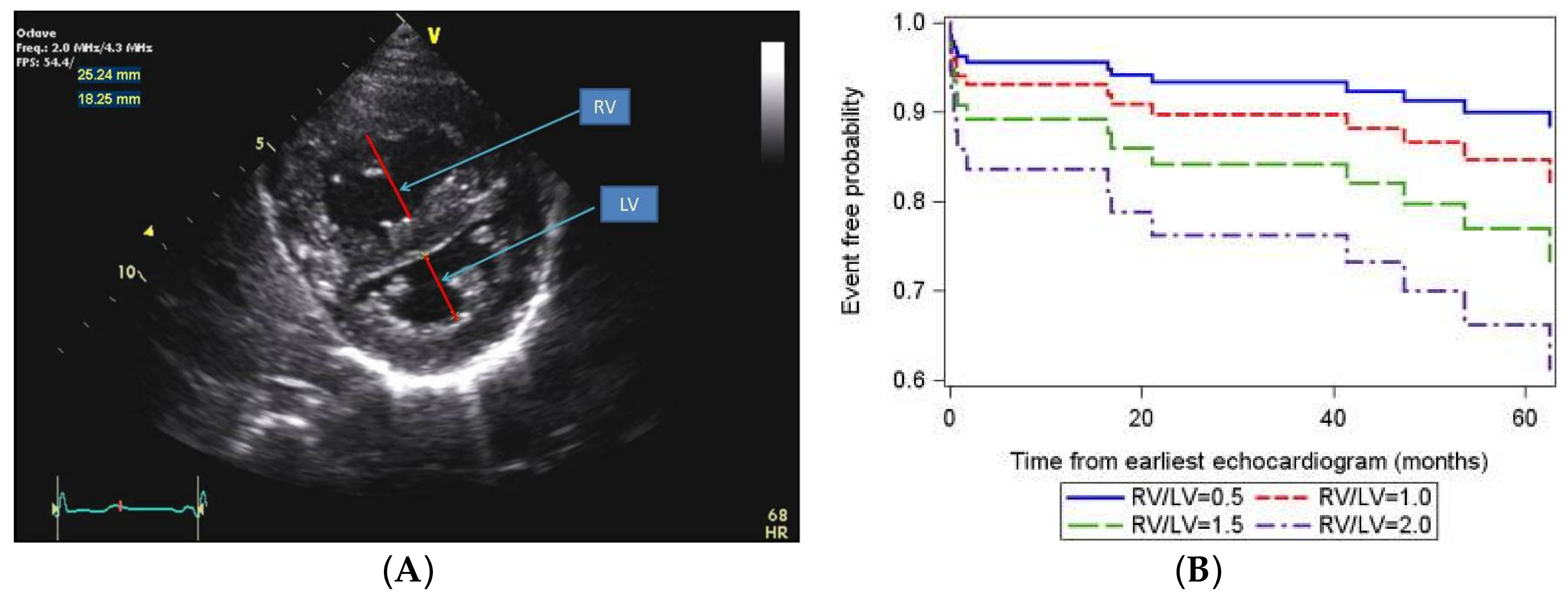
- Jone, P.N.; Hinzman, J.; Wagner, B.D.; Ivy, D.D.; Younoszai, A. Right ventricular to left ventricular diameter ratio at end-systole in evaluating outcomes in children with pulmonary hypertension. J. Am. Soc. Echocardiogr. 2014, 27, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Benza, R.L.; Miller, D.P.; Gomberg-Maitland, M.; Frantz, R.P.; Foreman, A.J.; Coffey, C.S.; Frost, A.; Barst, R.J.; Badesch, D.B.; Elliott, C.G.; et al. Predicting survival in pulmonary arterial hypertension: Insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation 2010, 122, 164–172. [Google Scholar] [CrossRef] [PubMed]
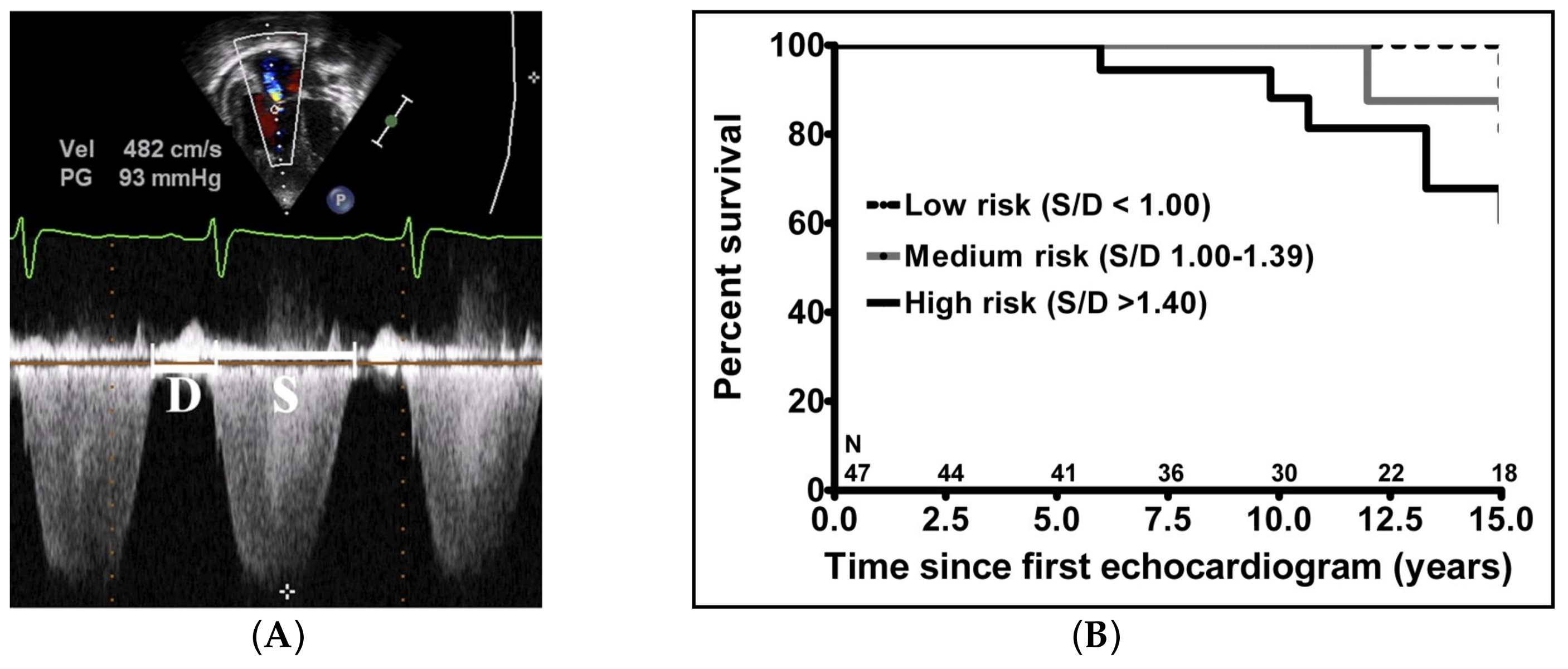
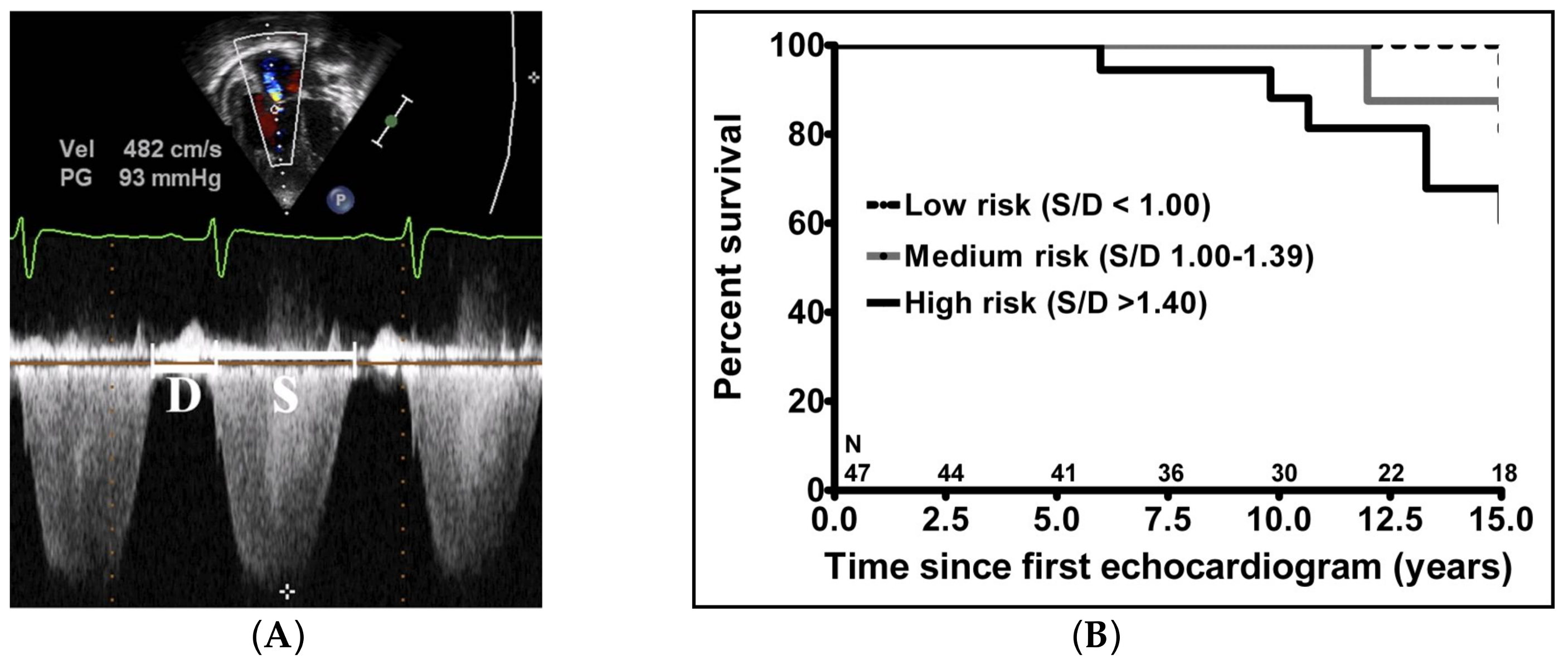
- Alkon, J.; Humpl, T.; Manlhiot, C.; McCrindle, B.W.; Reyes, J.T.; Friedberg, M.K. Usefulness of the right ventricular systolic to diastolic duration ratio to predict functional capacity and survival in children with pulmonary arterial hypertension. Am. J. Cardiol. 2010, 106, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, M.K.; Redington, A.N. Right versus left ventricular failure: Differences, similarities, and interactions. Circulation 2014, 129, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Kassem, E.; Humpl, T.; Friedberg, M.K. Prognostic significance of 2-dimensional, M-mode, and Doppler echo indices of right ventricular function in children with pulmonary arterial hypertension. Am. Heart J. 2013, 165, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
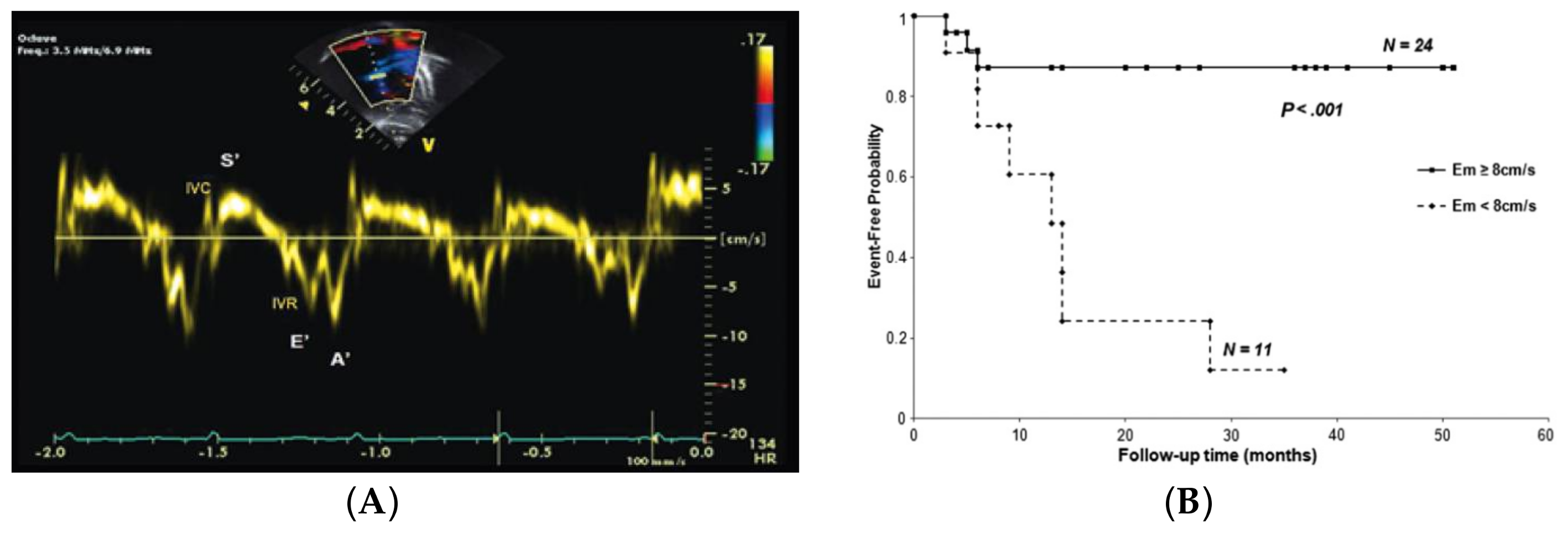
- Lammers, A.E.; Haworth, S.G.; Riley, G.; Maslin, K.; Diller, G.P.; Marek, J. Value of tissue Doppler echocardiography in children with pulmonary hypertension. J. Am. Soc. Echocardiogr. 2012, 25, 504–510. [Google Scholar] [CrossRef] [PubMed]
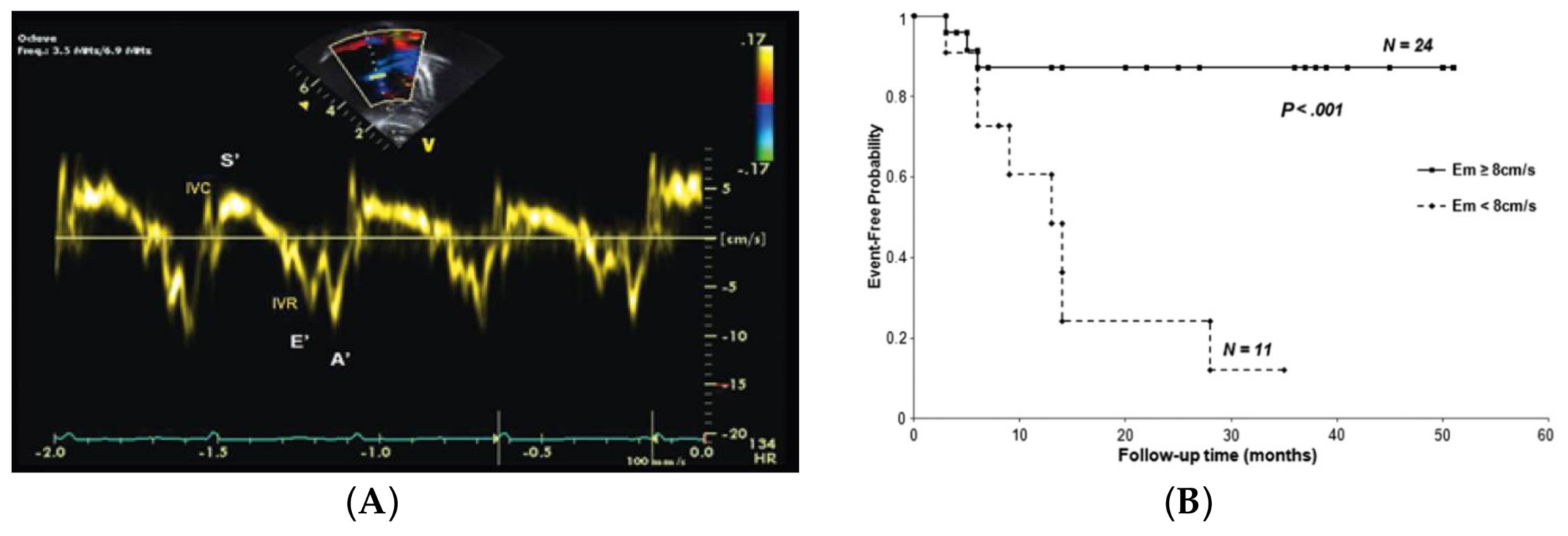
- Takatsuki, S.; Nakayama, T.; Jone, P.N.; Wagner, B.D.; Naoi, K.; Ivy, D.D.; Saji, T. Tissue Doppler imaging predicts adverse outcome in children with idiopathic pulmonary arterial hypertension. J. Pediatr. 2012, 161, 1126–1131. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, K.; Funabashi, N.; Takaoka, H.; Tanabe, N.; Yanagawa, N.; Tatsumi, K.; Kobayashi, Y. Utility of three-dimensional global longitudinal strain of the right ventricle using transthoracic echocardiography for right ventricular systolic function in pulmonary hypertension. Int. J. Cardiol. 2014, 174, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Nadvoretskiy, V.; Bu, L.; Stolpen, A.; Ayres, N.; Pignatelli, R.H.; Kovalchin, J.P.; Grenier, M.; Klas, B.; Ge, S. Accuracy and reproducibility of real-time three-dimensional echocardiography for assessment of right ventricular volumes and ejection fraction in children. J. Am. Soc. Echocardiogr. 2008, 21, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Geiger, R.; Strasak, A.; Treml, B.; Gasser, K.; Kleinsasser, A.; Fischer, V.; Geiger, H.; Loeckinger, A.; Stein, J.I. Six-minute walk test in children and adolescents. J. Pediatr. 2007, 150, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Lammers, A.E.; Hislop, A.A.; Flynn, Y.; Haworth, S.G. The 6-minute walk test: Normal values for children of 4-11 years of age. Arch. Dis. Child. 2008, 93, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Lesser, D.J.; Fleming, M.M.; Maher, C.A.; Kim, S.B.; Woo, M.S.; Keens, T.G. Does the 6-min walk test correlate with the exercise stress test in children? Pediatr. Pulmonol. 2010, 45, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Li, A.M.; Yin, J.; Au, J.T.; So, H.K.; Tsang, T.; Wong, E.; Fok, T.F.; Ng, P.C. Standard reference for the six-minute-walk test in healthy children aged 7 to 16 years. Am. J. Respir. Crit. Care Med. 2007, 176, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Van Loon, R.L.; Roofthooft, M.T.; Delhaas, T.; van Osch-Gevers, M.; ten Harkel, A.D.; Strengers, J.L.; Backx, A.; Hillege, H.L.; Berger, R.M. Outcome of pediatric patients with pulmonary arterial hypertension in the era of new medical therapies. Am. J. Cardiol. 2010, 106, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Douwes, J.M.; Hegeman, A.K.; van der Krieke, M.B.; Roofthooft, M.T.; Hillege, H.L.; Berger, R.M. Six-minute walking distance and decrease in oxygen saturation during the six-minute walk test in pediatric pulmonary arterial hypertension. Int. J. Cardiol. 2015, 202, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Garofano, R.P.; Barst, R.J. Exercise testing in children with primary pulmonary hypertension. Pediatr. Cardiol. 1999, 20, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Yetman, A.T.; Taylor, A.L.; Doran, A.; Ivy, D.D. Utility of cardiopulmonary stress testing in assessing disease severity in children with pulmonary arterial hypertension. Am. J. Cardiol. 2005, 95, 697–699. [Google Scholar] [CrossRef] [PubMed]
- Rausch, C.M.; Taylor, A.L.; Ross, H.; Sillau, S.; Ivy, D.D. Ventilatory efficiency slope correlates with functional capacity, outcomes, and disease severity in pediatric patients with pulmonary hypertension. Int. J. Cardiol. 2013, 169, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Nagaya, N.; Nishikimi, T.; Uematsu, M.; Satoh, T.; Kyotani, S.; Sakamaki, F.; Kakishita, M.; Fukushima, K.; Okano, Y.; Nakanishi, N.; et al. Plasma brain natriuretic peptide as a prognostic indicator in patients with primary pulmonary hypertension. Circulation 2000, 102, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Bernus, A.; Wagner, B.D.; Accurso, F.; Doran, A.; Kaess, H.; Ivy, D.D. Brain natriuretic peptide levels in managing pediatric patients with pulmonary arterial hypertension. Chest 2009, 135, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Lammers, A.E.; Hislop, A.A.; Haworth, S.G. Prognostic value of B-type natriuretic peptide in children with pulmonary hypertension. Int. J. Cardiol. 2009, 135, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Van Albada, M.E.; Loot, F.G.; Fokkema, R.; Roofthooft, M.T.; Berger, R.M. Biological serum markers in the management of pediatric pulmonary arterial hypertension. Pediatr. Res. 2008, 63, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Ploegstra, M.J.; Arjaans, S.; Zijlstra, W.M.; Douwes, J.M.; Vissia-Kazemier, T.R.; Roofthooft, M.T.R.; Hillege, H.L.; Berger, R.M.F. Clinical worsening as composite study end point in pediatric pulmonary arterial hypertension. Chest 2015, 148, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Hunter, K.S.; Feinstein, J.A.; Ivy, D.D.; Shandas, R. Computational simulation of the pulmonary arteries and its role in the study of pediatric pulmonary hypertension. Prog. Pediatr. Cardiol. 2010, 30, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Hunter, K.S.; Shandas, R. Impact of pulmonary vascular stiffness and vasodilator treatment in pediatric pulmonary hypertension: 21 patient-specific fluid-structure interaction studies. Comput. Methods Progr. Biomed. 2012, 108, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Tan, W.; Shandas, R.; Hunter, K.S. Influence of distal resistance and proximal stiffness on hemodynamics and RV afterload in progression and treatments of pulmonary hypertension: A computational study with validation using animal models. Comput. Math. Methods Med. 2013, 2013, 618326. [Google Scholar] [CrossRef] [PubMed]
- Douwes, J.M.; Roofthooft, M.T.; Bartelds, B.; Talsma, M.D.; Hillege, H.L.; Berger, R.M. Pulsatile haemodynamic parameters are predictors of survival in paediatric pulmonary arterial hypertension. Int. J. Cardiol. 2013, 168, 1370–1377. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, M.K.; Feinstein, J.A.; Rosenthal, D.N. Noninvasive assessment of pulmonary arterial capacitance by echocardiography. J. Am. Soc. Echocardiogr. 2007, 20, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Takatsuki, S.; Nakayama, T.; Ikehara, S.; Matsuura, H.; Ivy, D.D.; Saji, T. Pulmonary Arterial Capacitance Index Is a Strong Predictor for Adverse Outcome in Children with Idiopathic and Heritable Pulmonary Arterial Hypertension. J. Pediatr. 2017, 180, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Di Maria, M.V.; Burkett, D.A.; Younoszai, A.K.; Landeck, B.F.; Mertens, L.; Ivy, D.D.; Friedberg, M.K.; Hunter, K.S. Echocardiographic estimation of right ventricular stroke work in children with pulmonary arterial hypertension: Comparison with invasive measurements. J. Am. Soc. Echocardiogr. 2015, 28, 1350–1357. [Google Scholar] [CrossRef] [PubMed]
- Di Maria, M.V.; Younoszai, A.K.; Mertens, L.; Landeck, B.F.; Ivy, D.D.; Hunter, K.S.; Friedberg, M.K. RV stroke work in children with pulmonary arterial hypertension: Estimation based on invasive haemodynamic assessment and correlation with outcomes. Heart 2014, 100, 1342–1347. [Google Scholar] [CrossRef] [PubMed]
- Moledina, S.; Pandya, B.; Bartsota, M.; Mortensen, K.H.; McMillan, M.; Quyam, S.; Taylor, A.M.; Haworth, S.G.; Schulze-Neick, I.; Muthurangu, V. Prognostic significance of cardiac magnetic resonance imaging in children with pulmonary hypertension. Circ. Cardiovasc. Imaging 2013, 6, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, S.; Fischler, M.; Speich, R.; Bloch, K.E. Wrist actigraphy predicts outcome in patients with pulmonary hypertension. Respiration 2013, 86, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zijlstra, W.M.H.; Ploegstra, M.J.; Vissia-Kazemier, T.; Roofthooft, M.T.R.; Sarvaas, G.D.M.; Bartelds, B.; Rackowitz, A.; van den Heuvel, F.; Hillege, H.L.; Plasqui, G.; et al. Physical activity in pediatric pulmonary arterial hypertension measured by accelerometry. A candidate clinical endpoint. Am. J. Respir. Crit. Care Med. 2017, 196, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Hall, S.; Brogan, P.; Haworth, S.G.; Klein, N. Contribution of inflammation to the pathology of idiopathic pulmonary arterial hypertension in children. Thorax 2009, 64, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Nies, M.K.; Ivy, D.D.; Everett, A.D. The untapped potential of proteomic analysis in pediatric pulmonary hypertension. Proteom. Clin. Appl. 2014, 8, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Yeager, M.E.; Colvin, K.L.; Everett, A.D.; Stenmark, K.R.; Ivy, D.D. Plasma proteomics of differential outcome to long-term therapy in children with idiopathic pulmonary arterial hypertension. Proteom. Clin. Appl. 2012, 6, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Duncan, M.; Wagner, B.D.; Murray, K.; Allen, J.; Colvin, K.; Accurso, F.J.; Ivy, D.D. Circulating cytokines and growth factors in pediatric pulmonary hypertension. Mediat. Inflamm. 2012, 2012, 143428. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Umar, S.; Potus, F.; Iorga, A.; Wong, G.; Meriwether, D.; Breuils-Bonnet, S.; Mai, D.; Navab, K.; Ross, D.; et al. Apolipoprotein A-I mimetic peptide 4F rescues pulmonary hypertension by inducing microRNA-193-3p. Circulation 2014, 130, 776–785. [Google Scholar] [CrossRef] [PubMed]
- Wagner, B.D.; Takatsuki, S.; Accurso, F.J.; Ivy, D.D. Evaluation of circulating proteins and hemodynamics towards predicting mortality in children with pulmonary arterial hypertension. PLoS ONE 2013, 8, e80235. [Google Scholar] [CrossRef] [PubMed]
- Carmosino, M.J.; Friesen, R.H.; Doran, A.; Ivy, D.D. Perioperative complications in children with pulmonary hypertension undergoing noncardiac surgery or cardiac catheterization. Anesth. Analg. 2007, 104, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Friesen, R.H.; Williams, G.D. Anesthetic management of children with pulmonary arterial hypertension. Paediatr. Anaesth. 2008, 18, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Beghetti, M.; Schulze-Neick, I.; Berger, R.M.; Ivy, D.D.; Bonnet, D.; Weintraub, R.G.; Saji, T.; Yung, D.; Mallory, G.B.; Geiger, R.; et al. Haemodynamic characterisation and heart catheterisation complications in children with pulmonary hypertension: Insights from the Global TOPP Registry (tracking outcomes and practice in paediatric pulmonary hypertension). Int. J. Cardiol. 2015, 203, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Del Cerro, M.J.; Moledina, S.; Haworth, S.G.; Ivy, D.; Al Dabbagh, M.; Banjar, H.; Diaz, G.; Heath-Freudenthal, A.; Galal, A.N.; Humpl, T.; et al. Cardiac catheterization in children with pulmonary hypertensive vascular disease: Consensus statement from the Pulmonary Vascular Research Institute, Pediatric and Congenital Heart Disease Task Forces. Pulm. Circ. 2016, 6, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Barst, R.J.; Maislin, G.; Fishman, A.P. Vasodilator therapy for primary pulmonary hypertension in children. Circulation 1999, 99, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Beghetti, M. Current treatment options in children with pulmonary arterial hypertension and experiences with oral bosentan. Eur. J. Clin. Investig. 2006, 36, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Berman Rosenzweig, E.; Barst, R.J. Pulmonary arterial hypertension: A comprehensive review of pharmacological treatment. Treat. Respir. Med. 2006, 5, 117–127. [Google Scholar] [CrossRef] [PubMed]
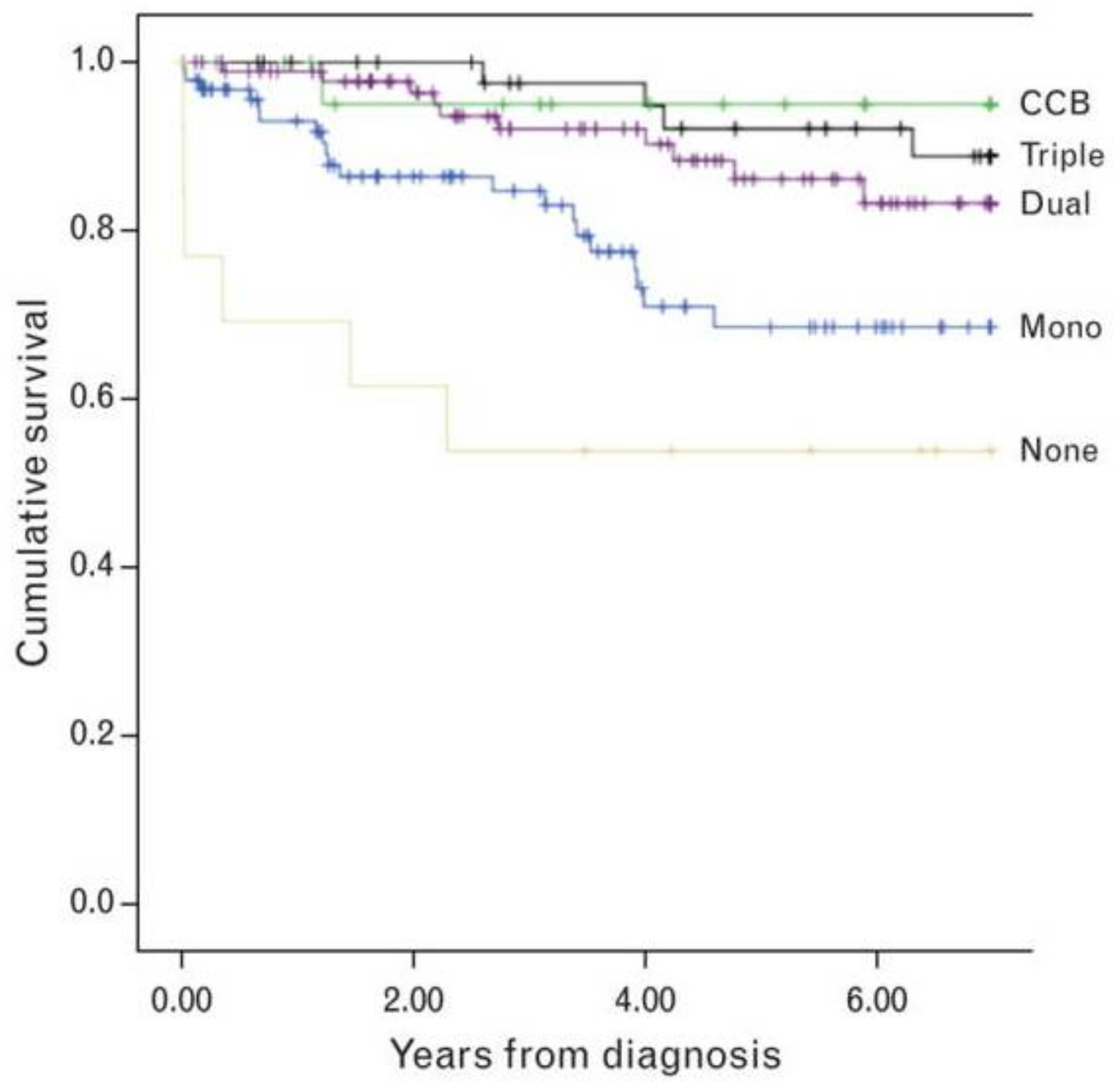
- Sitbon, O.; Humbert, M.; Jais, X.; Ioos, V.; Hamid, A.M.; Provencher, S.; Garcia, G.; Parent, F.; Hervé, P.; Simonneau, G. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 2005, 111, 3105–3111. [Google Scholar] [CrossRef] [PubMed]
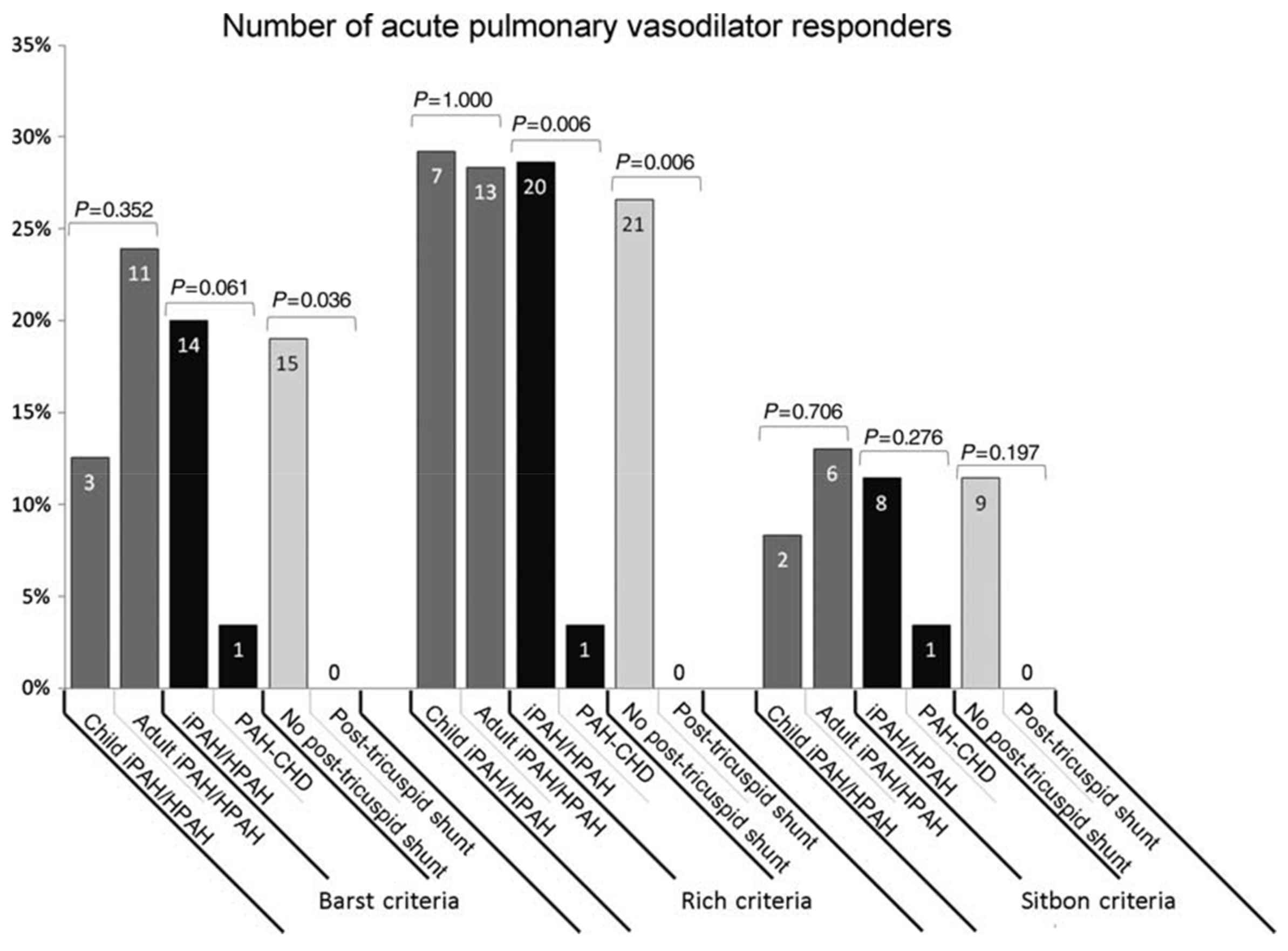
- Douwes, J.M.; van Loon, R.L.; Hoendermis, E.S.; Vonk-Noordegraaf, A.; Roofthooft, M.T.; Talsma, M.D.; Hillege, H.L.; Berger, R.M. Acute pulmonary vasodilator response in paediatric and adult pulmonary arterial hypertension: Occurrence and prognostic value when comparing three response criteria. Eur. Heart J. 2011, 32, 3137–3146. [Google Scholar] [CrossRef] [PubMed]
- Barst, R.J.; McGoon, M.D.; Elliott, C.G.; Foreman, A.J.; Miller, D.P.; Ivy, D.D. Survival in childhood pulmonary arterial hypertension: Insights from the registry to evaluate early and long-term pulmonary arterial hypertension disease management. Circulation 2012, 125, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Douwes, J.M.; Humpl, T.; Bonnet, D.; Beghetti, M.; Ivy, D.D.; Berger, R.M.; TOPP Investigators. Acute Vasodilator Response in Pediatric Pulmonary Arterial Hypertension: Current Clinical Practice From the TOPP Registry. J. Am. Coll. Cardiol. 2016, 67, 1312–1323. [Google Scholar] [CrossRef] [PubMed]
- Yung, D.; Widlitz, A.C.; Rosenzweig, E.B.; Kerstein, D.; Maislin, G.; Barst, R.J. Outcomes in children with idiopathic pulmonary arterial hypertension. Circulation 2004, 110, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Adatia, I.; Barrow, S.E.; Stratton, P.D.; Miall-Allen, V.M.; Ritter, J.M.; Haworth, S.G. Thromboxane A2 and prostacyclin biosynthesis in children and adolescents with pulmonary vascular disease. Circulation 1993, 88, 2117–2122. [Google Scholar] [CrossRef] [PubMed]
- Christman, B.W.; McPherson, C.D.; Newman, J.H.; King, G.A.; Bernard, G.R.; Groves, B.M.; Loyd, J.E. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N. Engl. J. Med. 1992, 327, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Cool, C.D.; Geraci, M.W.; Wang, J.; Abman, S.H.; Wright, L.; Badesch, D.; Voelkel, N.F. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1999, 159, 1925–1932. [Google Scholar] [CrossRef] [PubMed]
- Ivy, D.D.; Doran, A.; Claussen, L.; Bingaman, D.; Yetman, A. Weaning and discontinuation of epoprostenol in children with idiopathic pulmonary arterial hypertension receiving concomitant bosentan. Am. J. Cardiol. 2004, 93, 943–946. [Google Scholar] [CrossRef] [PubMed]
- Lammers, A.E.; Hislop, A.A.; Flynn, Y.; Haworth, S.G. Epoprostenol treatment in children with severe pulmonary hypertension. Heart 2007, 93, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Siehr, S.L.; Ivy, D.D.; Miller-Reed, K.; Ogawa, M.; Rosenthal, D.N.; Feinstein, J.A. Children with pulmonary arterial hypertension and prostanoid therapy: Long-term hemodynamics. J. Heart Lung Transplant. 2013, 32, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Barst, R.J.; Galie, N.; Naeije, R.; Simonneau, G.; Jeffs, R.; Arneson, C.; Rubin, L.J. Long-term outcome in pulmonary arterial hypertension patients treated with subcutaneous treprostinil. Eur. Respir. J. 2006, 28, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Gomberg-Maitland, M.; Tapson, V.F.; Benza, R.L.; McLaughlin, V.V.; Krichman, A.; Widlitz, A.C.; Barst, R.J. Transition from intravenous epoprostenol to intravenous treprostinil in pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2005, 172, 1586–1589. [Google Scholar] [CrossRef] [PubMed]
- Ivy, D.D.; Claussen, L.; Doran, A. Transition of stable pediatric patients with pulmonary arterial hypertension from intravenous epoprostenol to intravenous treprostinil. Am. J. Cardiol. 2007, 99, 696–698. [Google Scholar] [CrossRef] [PubMed]
- Doran, A.K.; Ivy, D.D.; Barst, R.J.; Hill, N.; Murali, S.; Benza, R.L. Guidelines for the prevention of central venous catheter-related blood stream infections with prostanoid therapy for pulmonary arterial hypertension. Int. J. Clin. Pract. Suppl. 2008, 62, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Ferdman, D.J.; Rosenzweig, E.B.; Zuckerman, W.A.; Krishnan, U. Subcutaneous treprostinil for pulmonary hypertension in chronic lung disease of infancy. Pediatrics 2014, 134, e274–e278. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Celermajer, D.S.; Bourges-Petit, E.; Del Cerro, M.J.; Bajolle, F.; Bonnet, D. Add-on therapy with subcutaneous treprostinil for refractory pediatric pulmonary hypertension. J. Pediatr. 2011, 158, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, U.; Takatsuki, S.; Ivy, D.D.; Kerstein, J.; Calderbank, M.; Coleman, E.; Rosenzweig, E.B. Effectiveness and safety of inhaled treprostinil for the treatment of pulmonary arterial hypertension in children. Am. J. Cardiol. 2012, 110, 1704–1709. [Google Scholar] [CrossRef] [PubMed]
- Takatsuki, S.; Rosenzweig, E.B.; Zuckerman, W.; Brady, D.; Calderbank, M.; Ivy, D.D. Clinical safety, pharmacokinetics, and efficacy of ambrisentan therapy in children with pulmonary arterial hypertension. Pediatr. Pulmonol. 2013, 48, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Voswinckel, R.; Enke, B.; Reichenberger, F.; Kohstall, M.; Kreckel, A.; Krick, S.; Gall, H.; Gessler, T.; Schmehl, T.; Ghofrani, H.A.; et al. Favorable effects of inhaled treprostinil in severe pulmonary hypertension: Results from randomized controlled pilot studies. J. Am. Coll. Cardiol. 2006, 48, 1672–1681. [Google Scholar] [CrossRef] [PubMed]
- Jing, Z.C.; Parikh, K.; Pulido, T.; Jerjes-Sanchez, C.; White, R.J.; Allen, R.; Torbicki, A.; Xu, K.F.; Yehle, D.; Laliberte, K.; et al. Efficacy and safety of oral treprostinil monotherapy for the treatment of pulmonary arterial hypertension: A randomized, controlled trial. Circulation 2013, 127, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Tapson, V.F.; Jing, Z.C.; Xu, K.F.; Pan, L.; Feldman, J.; Kiely, D.G.; Kotlyar, E.; McSwain, C.S.; Laliberte, K.; Arneson, C.; et al. Oral treprostinil for the treatment of pulmonary arterial hypertension in patients receiving background endothelin receptor antagonist and phosphodiesterase type 5 inhibitor therapy (the FREEDOM-C2 study): A randomized controlled trial. Chest 2013, 144, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, N.; Ikeda, S.; Tahara, N.; Fukuda, K.; Hatano, M.; Ito, H.; Nakayama, T.; Anzai, T.; Hashimoto, A.; Inoue, T.; et al. Efficacy and Safety of an Orally Administered Selective Prostacyclin Receptor Agonist, Selexipag, in Japanese Patients With Pulmonary Arterial Hypertension. Circ. J. 2017, 81, 1360–1607. [Google Scholar] [CrossRef] [PubMed]
- Gallotti, R.; Drogalis-Kim, D.E.; Satou, G.; Alejos, J. Single-center experience using selexipag in a pediatric population. Pediatr. Cardiol. 2017, 38, 1405–1409. [Google Scholar] [CrossRef] [PubMed]
- Olschewski, H.; Simonneau, G.; Galie, N.; Higenbottam, T.; Naeije, R.; Rubin, L.J.; Nikkho, S.; Speich, R.; Hoeper, M.M.; Behr, J.; et al. Inhaled iloprost for severe pulmonary hypertension. N. Engl. J. Med. 2002, 347, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Ivy, D.D.; Doran, A.K.; Smith, K.J.; Mallory, G.B., Jr.; Beghetti, M.; Barst, R.J.; Brady, D.; Law, Y.; Parker, D.; Claussen, L.; et al. Short- and long-term effects of inhaled iloprost therapy in children with pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2008, 51, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Limsuwan, A.; Wanitkul, S.; Khosithset, A.; Attanavanich, S.; Samankatiwat, P. Aerosolized iloprost for postoperative pulmonary hypertensive crisis in children with congenital heart disease. Int. J. Cardiol. 2008, 129, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.J.; Badesch, D.B.; Barst, R.J.; Galiè, N.; Black, C.M.; Keogh, A.; Pulido, T.; Frost, A.; Roux, S.; Leconte, I.; et al. Bosentan therapy for pulmonary arterial hypertension. N. Engl. J. Med. 2002, 346, 896–903. [Google Scholar] [CrossRef] [PubMed]
- Vignon-Zellweger, N.; Heiden, S.; Miyauchi, T.; Emoto, N. Endothelin and endothelin receptors in the renal and cardiovascular systems. Life Sci. 2012, 91, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.W.; Chatfield, B.A.; Koppenhafer, S.A.; Schaffer, M.S.; Wolfe, R.R.; Abman, S. Circulating Immunoreactive Endothelin-1 in Children with Pulmonary Hypertension. Am. Rev. Respir. Dis. 1993, 148, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Rubin, L.; Hoeper, M.; Jansa, P.; Al-Hiti, H.; Meyer, G.; Chiossi, E.; Kusic-Pajic, A.; Simonneau, G. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): A double-blind, randomised controlled trial. Lancet 2008, 371, 2093–2100. [Google Scholar] [CrossRef]
- Barst, R.J.; Ivy, D.; Dingemanse, J.; Widlitz, A.; Schmitt, K.; Doran, A.; Bingaman, D.; Nguyen, N.; Gaitonde, M.; van Giersbergen, P.L. Pharmacokinetics, safety, and efficacy of bosentan in pediatric patients with pulmonary arterial hypertension. Clin. Pharmacol. Ther. 2003, 73, 372–382. [Google Scholar] [CrossRef]
- Beghetti, M. Bosentan in pediatric patients with pulmonary arterial hypertension. Curr. Vasc. Pharmacol. 2009, 7, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Beghetti, M.; Haworth, S.G.; Bonnet, D.; Barst, R.J.; Acar, P.; Fraisse, A.; Ivy, D.D.; Jais, X.; Schulze-Neick, I.; Galiè, N.; et al. Pharmacokinetic and clinical profile of a novel formulation of bosentan in children with pulmonary arterial hypertension: The FUTURE-1 study. Br. J. Clin. Pharmacol. 2009, 68, 948–955. [Google Scholar] [CrossRef] [PubMed]
- Beghetti, M.; Hoeper, M.M.; Kiely, D.G.; Carlsen, J.; Schwierin, B.; Segal, E.S.; Humbert, M. Safety experience with bosentan in 146 children 2–11 years old with pulmonary arterial hypertension: Results from the European Postmarketing Surveillance program. Pediatr. Res. 2008, 64, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Hislop, A.A.; Moledina, S.; Foster, H.; Schulze-Neick, I.; Haworth, S.G. Long-term efficacy of bosentan in treatment of pulmonary arterial hypertension in children. Eur. Respir. J. 2011, 38, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Ivy, D.D.; Lee, D.S.; Rairigh, R.L.; Parker, T.A.; Abman, S.H. Endothelin B receptor blockade attenuates pulmonary vasodilation in oxygen-ventilated fetal lambs. Biol. Neonate 2004, 86, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Maiya, S.; Hislop, A.A.; Flynn, Y.; Haworth, S.G. Response to bosentan in children with pulmonary hypertension. Heart 2006, 92, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, E.B.; Ivy, D.D.; Widlitz, A.; Doran, A.; Claussen, L.R.; Yung, D.; Abman, S.H.; Morganti, A.; Nguyen, N.; Barst, R.J. Effects of long-term bosentan in children with pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2005, 46, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, M.; Ichida, F.; Hirono, K.; Miyawaki, T.; Yoshimura, N.; Nakamura, T.; Akita, C.; Nakayama, T.; Saji, T.; Kato, Y.; et al. Pharmacokinetics of bosentan in routinely treated Japanese pediatric patients with pulmonary arterial hypertension. Drug. Metab. Pharmacokinet. 2011, 26, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Berger, R.M.F.; Gehin, M.; Beghetti, M.; Ivy, D.; Kusic-Pajic, A.; Cornelisse, P.; Grill, S.; Bonnet, D.; The FUTURE-3 Investigators. A bosentan pharmacokinetic study to investigate dosing regimens in paediatric patients with pulmonary arterial hypertension: FUTURE-3. Br. J. Clin. Pharmacol. 2017, 83, 1734–1744. [Google Scholar] [CrossRef] [PubMed]
- Pulido, T.; Adzerikho, I.; Channick, R.N.; Delcroix, M.; Galiè, N.; Ghofrani, H.A.; Jansa, P.; Jing, Z.C.; Le Brun, F.O.; Mehta, S.; et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Olschewski, H.; Oudiz, R.J.; Torres, F.; Frost, A.; Ghofrani, H.A.; Badesch, D.B.; McGoon, M.D.; McLaughlin, V.V.; Roecker, E.B.; et al. Ambrisentan for the treatment of pulmonary arterial hypertension: Results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 2008, 117, 3010–3019. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, W.A.; Leaderer, D.; Rowan, C.A.; Mituniewicz, J.D.; Rosenzweig, E.B. Ambrisentan for pulmonary arterial hypertension due to congenital heart disease. Am. J. Cardiol. 2011, 107, 1381–1385. [Google Scholar] [CrossRef] [PubMed]
- Hanson, K.A.; Ziegler, J.W.; Rybalkin, S.D.; Miller, J.W.; Abman, S.H.; Clarke, W.R. Chronic pulmonary hypertension increases fetal lung cGMP phosphodiesterase activity. Am. J. Physiol. 1998, 275, L931–L941. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Ghofrani, H.A.; Torbicki, A.; Barst, R.J.; Rubin, L.J.; Badesch, D.; Fleming, T.; Parpia, T.; Burgess, G.; Branzi, A.; et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N. Engl. J. Med. 2005, 353, 2148–2157. [Google Scholar] [CrossRef] [PubMed]
- Humpl, T.; Reyes, J.T.; Holtby, H.; Stephens, D.; Adatia, I. Beneficial effect of oral sildenafil therapy on childhood pulmonary arterial hypertension: Twelve-month clinical trial of a single-drug, open-label, pilot study. Circulation 2005, 111, 3274–3280. [Google Scholar] [CrossRef] [PubMed]
- Pettit, R.S.; Johnson, C.E.; Caruthers, R.L. Stability of an extemporaneously prepared tadalafil suspension. Am. J. Health-Syst. Pharm. 2012, 69, 592–594. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, E.B. Tadalafil for the treatment of pulmonary arterial hypertension. Expert. Opin. Pharmacother. 2010, 11, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Takatsuki, S.; Calderbank, M.; Ivy, D.D. Initial experience with tadalafil in pediatric pulmonary arterial hypertension. Pediatr. Cardiol. 2012, 33, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Neick, I.; Hartenstein, P.; Li, J.; Stiller, B.; Nagdyman, N.; Hübler, M.; Butrous, G.; Petros, A.; Lange, P.; Redington, A.N. Intravenous sildenafil is a potent pulmonary vasodilator in children with congenital heart disease. Circulation 2003, 108, II167–II173. [Google Scholar] [CrossRef] [PubMed]
- Stocker, C.; Penny, D.J.; Brizard, C.P.; Cochrane, A.D.; Soto, R.; Shekerdemian, L.S. Intravenous sildenafil and inhaled nitric oxide: A randomised trial in infants after cardiac surgery. Intensive Care Med. 2003, 29, 1996–2003. [Google Scholar] [CrossRef] [PubMed]
- Atz, A.M.; Wessel, D.L. Sildenafil ameliorates effects of inhaled nitric oxide withdrawal. Anesthesiology 1999, 91, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Namachivayam, P.; Theilen, U.; Butt, W.W.; Cooper, S.M.; Penny, D.J.; Shekerdemian, L.S. Sildenafil prevents rebound pulmonary hypertension after withdrawal of nitric oxide in children. Am. J. Respir. Crit. Care Med. 2006, 174, 1042–1047. [Google Scholar] [CrossRef] [PubMed]
- Fasnacht, M.S.; Tolsa, J.F.; Beghetti, M. The Swiss registry for pulmonary arterial hypertension: The paediatric experience. Swiss Med. Wkly. 2007, 137, 510–513. [Google Scholar] [PubMed]
- Haworth, S.G. The management of pulmonary hypertension in children. Arch. Dis. Child. 2008, 93, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Karatza, A.A.; Bush, A.; Magee, A.G. Safety and efficacy of sildenafil therapy in children with pulmonary hypertension. Int. J. Cardiol. 2005, 100, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Mourani, P.M.; Sontag, M.K.; Ivy, D.D.; Abman, S.H. Effects of long-term sildenafil treatment for pulmonary hypertension in infants with chronic lung disease. J. Pediatr. 2009, 154, 379–384. [Google Scholar] [CrossRef] [PubMed]
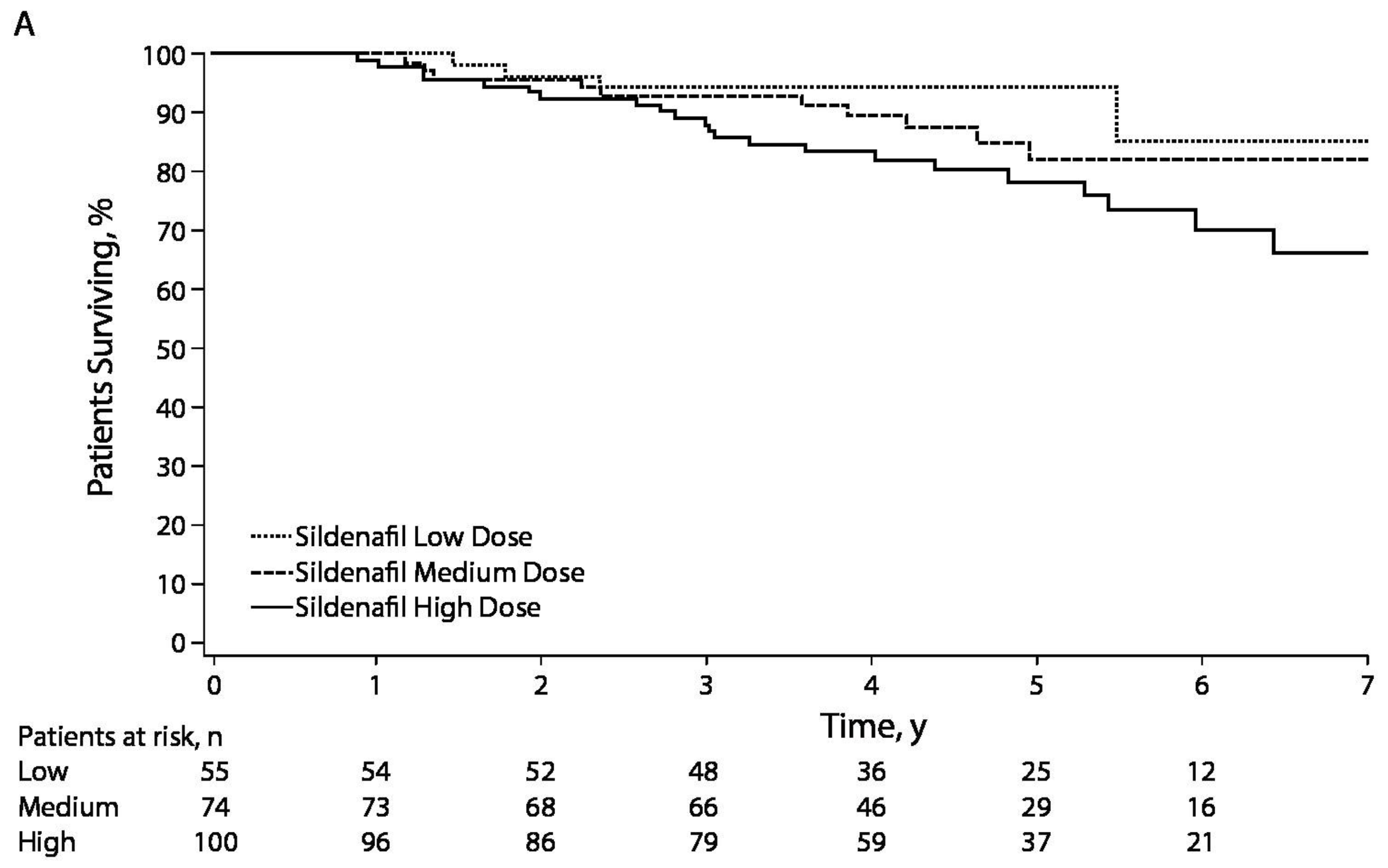
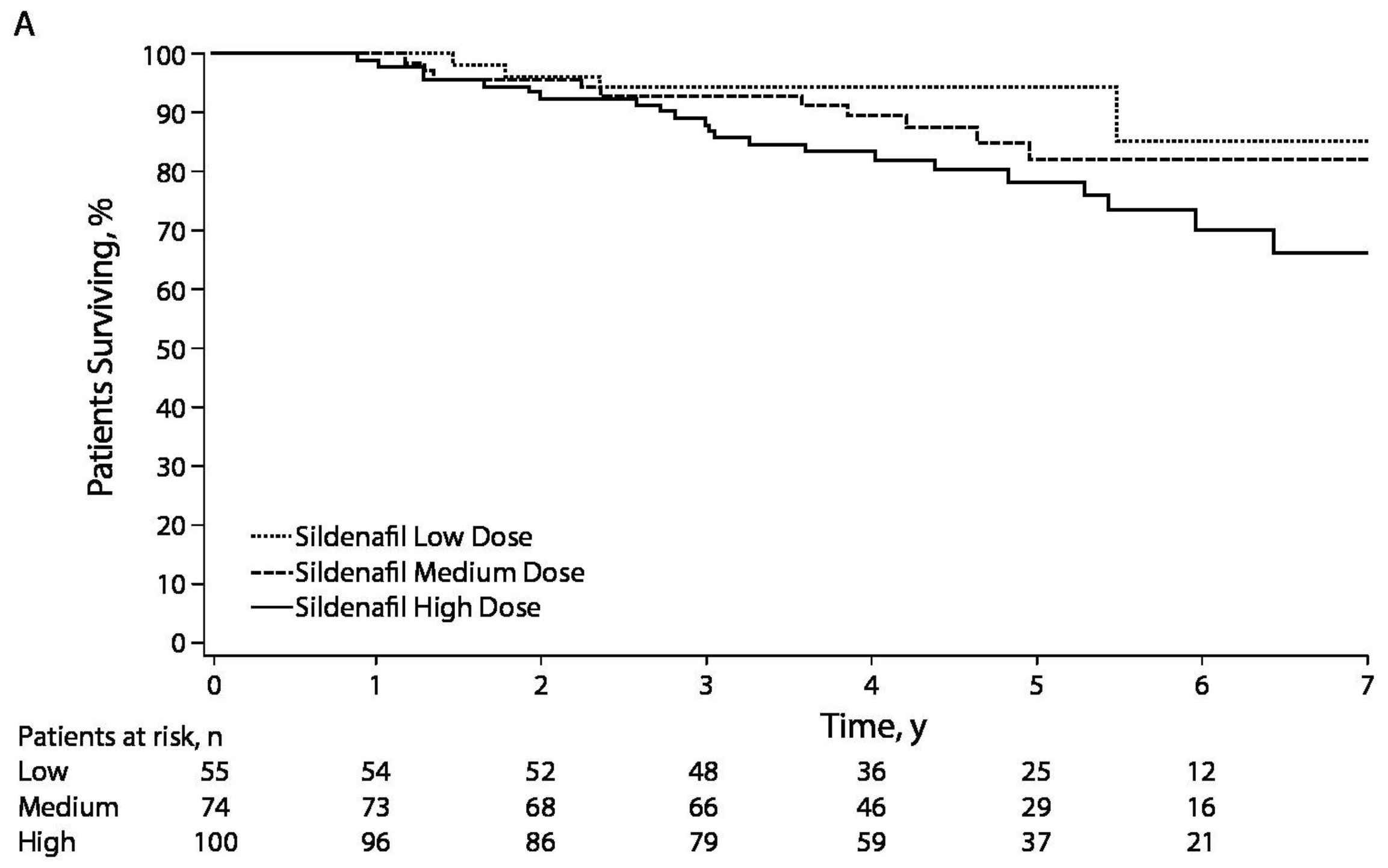
- Barst, R.J.; Ivy, D.D.; Gaitan, G.; Szatmari, A.; Rudzinski, A.; Garcia, A.E.; Sastry, B.K.; Pulido, T.; Layton, G.R.; Serdarevic-Pehar, M.; et al. A randomized, double-blind, placebo-controlled, dose-ranging study of oral sildenafil citrate in treatment-naive children with pulmonary arterial hypertension. Circulation 2012, 125, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Barst, R.J.; Beghetti, M.; Pulido, T.; Layton, G.; Konourina, I.; Zhang, M.; Ivy, D.D.; STARTS-2 Investigators. STARTS-2: Long-term survival with oral sildenafil monotherapy in treatment-naive pediatric pulmonary arterial hypertension. Circulation 2014, 129, 1914–1923. [Google Scholar] [CrossRef] [PubMed]
- Schermuly, R.T.; Janssen, W.; Weissmann, N.; Stasch, J.P.; Grimminger, F.; Ghofrani, H.A. Riociguat for the treatment of pulmonary hypertension. Expert. Opin. Investig. Drugs 2011, 20, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Ghofrani, H.A.; D’Armini, A.M.; Grimminger, F.; Hoeper, M.M.; Jansa, P.; Kim, N.H.; Mayer, E.; Simonneau, G.; Wilkins, M.R.; Fritsch, A.; et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N. Engl. J. Med. 2013, 369, 319–329. [Google Scholar] [CrossRef] [PubMed]
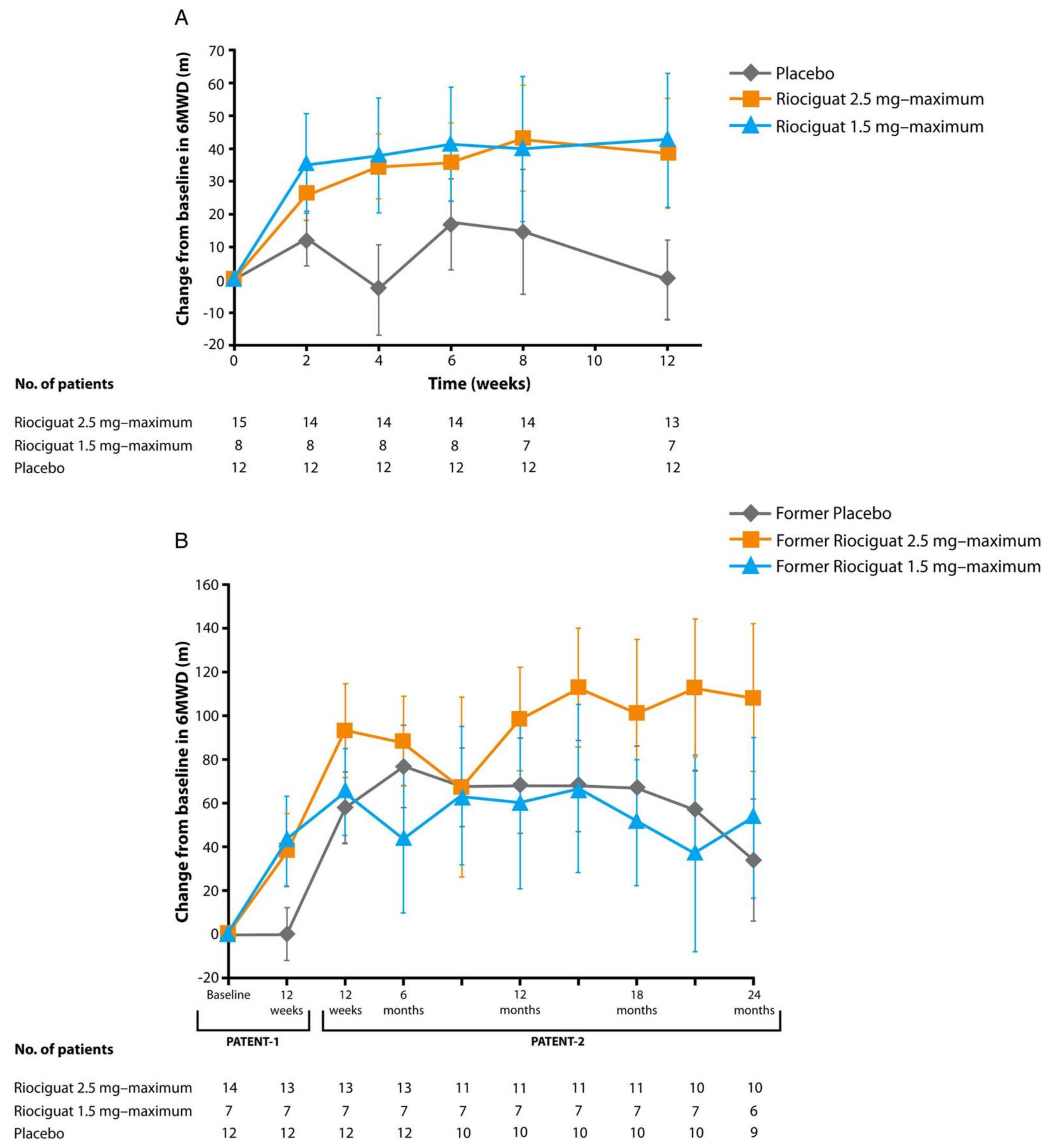
- Rosenkranz, S.; Ghofrani, H.A.; Beghetti, M.; Ivy, D.; Frey, R.; Fritsch, A.; Weimann, G.; Saleh, S.; Apitz, C. Riociguat for pulmonary arterial hypertension associated with congenital heart disease. Heart 2015, 101, 1792–1799. [Google Scholar] [CrossRef] [PubMed]
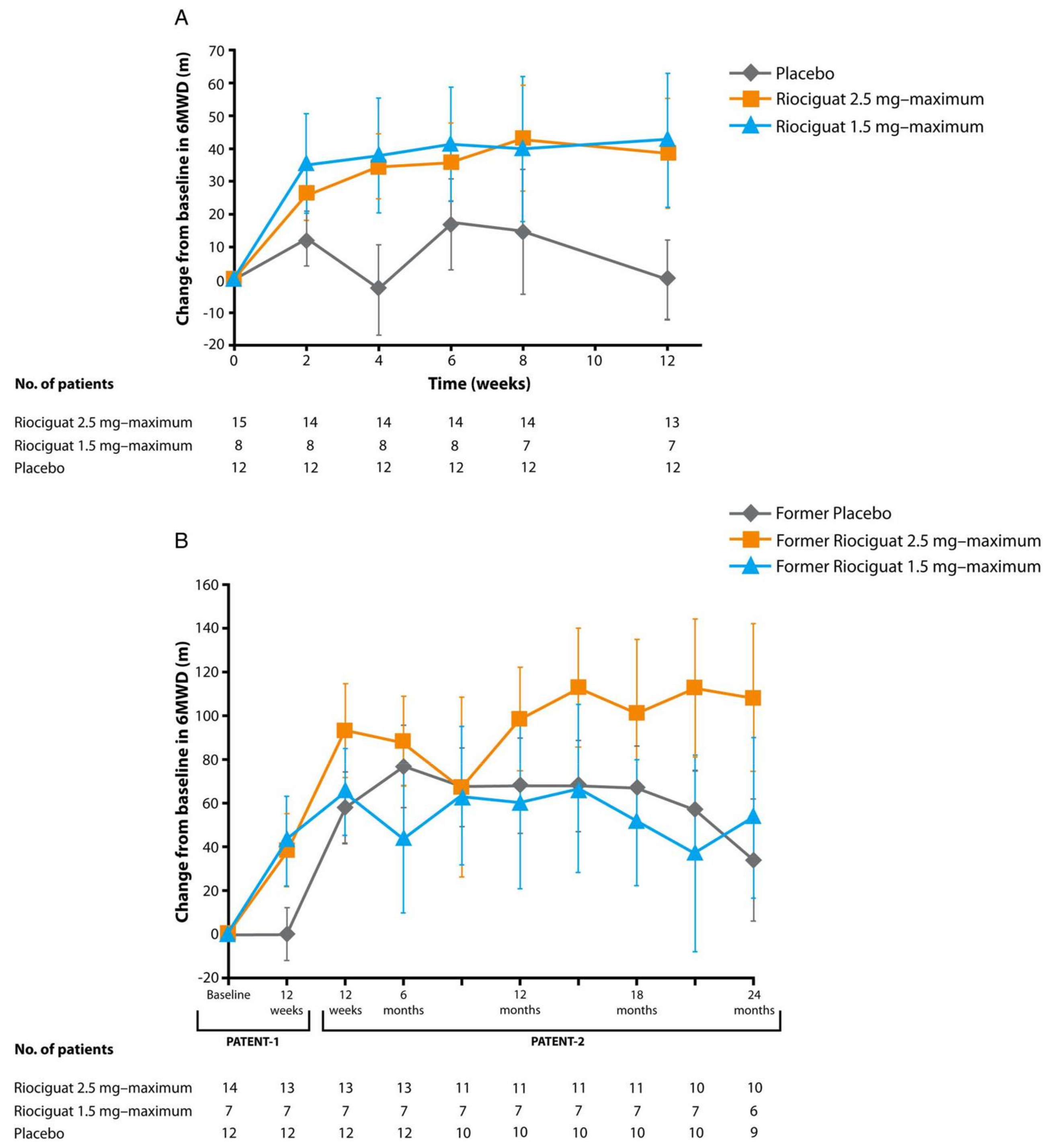
- Spreemann, T.; Bertram, H.; Happel, C.M.; Kozlik-Feldmann, R.; Hansmann, G. First-in-child use of the oral soluble guanylate cyclase stimulator riociguat in pulmonary arterial hypertension. Pulm. Circ. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Barbera, J.A.; Frost, A.E.; Ghofrani, H.-A.; Hoeper, M.M.; McLaughlin, V.V.; Peacock, A.J.; Simonneau, G.; Vachiery, J.-L.; Grünig, E.; et al. Initial Use of Ambrisentan plus Tadalafil in Pulmonary Arterial Hypertension. N. Engl. J. Med. 2015, 373, 834–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitbon, O.; Sattler, C.; Bertoletti, L.; Savale, L.; Cottin, V.; Jaïs, X.; De Groote, P.; Chaouat, A.; Chabannes, C.; Bergot, E.; et al. Initial dual oral combination therapy in pulmonary arterial hypertension. Eur. Respir. J. 2016, 47, 1727–1736. [Google Scholar] [CrossRef] [PubMed]
- Chiu, J.S.; Zuckerman, W.A.; Turner, M.E.; Richmond, M.E.; Kerstein, D.; Krishnan, U.; Torres, A.; Vincent, J.A.; Rosenzweig, E.B. Balloon atrial septostomy in pulmonary arterial hypertension: Effect on survival and associated outcomes. J. Heart Lung Transplant. 2015, 34, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, J.; Gaspar, J.; Pulido, T.; Bautista, E.; Martínez-Guerra, M.L.; Zeballos, M.; Palomar, A.; Gómez, A. Graded balloon dilation atrial septostomy in severe primary pulmonary hypertension. A therapeutic alternative for patients nonresponsive to vasodilator treatment. J. Am. Coll. Cardiol. 1998, 32, 297–304. [Google Scholar] [CrossRef]
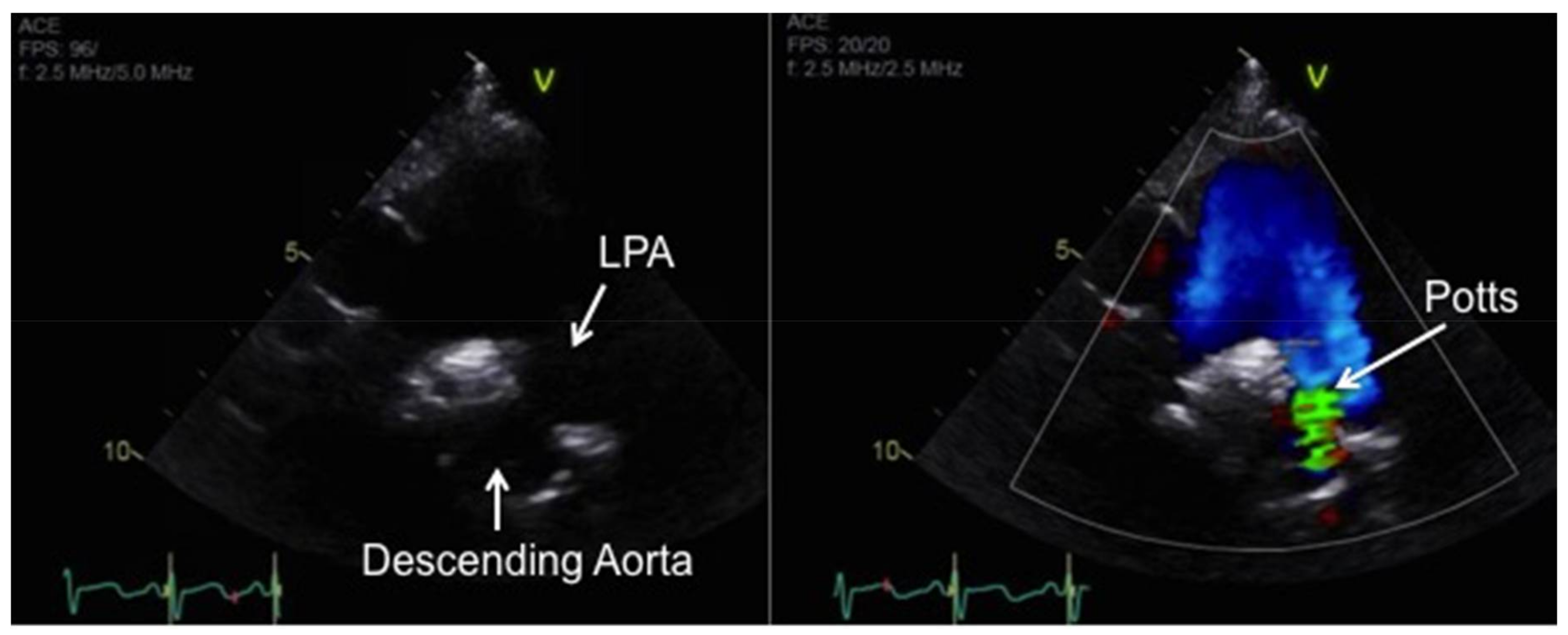
- Baruteau, A.E.; Serraf, A.; Levy, M.; Petit, J.; Bonnet, D.; Jais, X.; Vouhé, P.; Simonneau, G.; Belli, E.; Humbert, M. Potts shunt in children with idiopathic pulmonary arterial hypertension: Long-term results. Ann. Thorac. Surg. 2012, 94, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Blanc, J.; Vouhe, P.; Bonnet, D. Potts shunt in patients with pulmonary hypertension. N. Engl. J. Med. 2004, 350, 623. [Google Scholar] [CrossRef] [PubMed]
- Grady, R.M.; Eghtesady, P. Potts Shunt and Pediatric Pulmonary Hypertension: What We Have Learned. Ann. Thorac. Surg. 2015, 101, 1539–1543. [Google Scholar] [CrossRef] [PubMed]
- Aurora, P.; Boucek, M.M.; Christie, J.; Boucek, M.M.; Aurora, P.; Taylor, D.O.; Dobbels, F.; Rahmel, A.O.; Keck, B.M.; Hertz, M.I.; et al. Registry of the International Society for Heart and Lung Transplantation: Tenth official pediatric lung and heart/lung transplantation report—2007. J. Heart Lung Transplant. 2007, 26, 1223–1228. [Google Scholar] [CrossRef] [PubMed]
- Mallory, G.B.; Spray, T.L. Paediatric lung transplantation. Eur. Resp. J. 2004, 24, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, Y.; Thacker, J.; Santos, R.; Nguyen, D.; Bhama, J.; Bermudez, C.; Kormos, R.; Johnson, B.; Crespo, M.; Pilewski, J.; et al. Long-term outcome of lung and heart-lung transplantation for idiopathic pulmonary arterial hypertension. Ann. Thorac. Surg. 2008, 86, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.; Maxwell, B.; Boulate, D.; Haddad, F.; Ha, R.; Afshar, K.; Weill, D.; Dhillon, G.S. Heart-lung vs. double-lung transplantation for idiopathic pulmonary arterial hypertension. Clin. Transplant. 2015, 29, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Kirkby, S.; Hayes, D., Jr. Pediatric lung transplantation: Indications and outcomes. J. Thorac. Dis. 2014, 6, 1024–1031. [Google Scholar] [PubMed]
- Benden, C.; Edwards, L.B.; Kucheryavaya, A.Y.; Kucheryavaya, A.Y.; Benden, C.; Christie, J.D.; Dobbels, F.; Lund, L.H.; Rahmel, A.O.; Yusen, R.D. The Registry of the International Society for Heart and Lung Transplantation: Sixteenth Official Pediatric Lung and Heart-Lung Transplantation Report—2013; Focus theme: Age. J. Heart Lung Transplant. 2013, 32, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.; Heinle, J.S.; Samayoa, A.X.; Adachi, I.; Schecter, M.G.; Mallory, G.B.; Morales, D.L. Is lung transplantation survival better in infants? Analysis of over 80 infants. J. Heart Lung Transplant. 2013, 32, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Olsson, K.M.; Delcroix, M.; Ghofrani, H.A.; Tiede, H.; Huscher, D.; Speich, R.; Grünig, E.; Staehler, G.; Rosenkranz, S.; Halank, M.; et al. Anticoagulation and survival in pulmonary arterial hypertension: Results from the Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA). Circulation 2014, 129, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Barst, R.J.; Abenhaim, L. Fatal pulmonary arterial hypertension associated with phenylpropanolamine exposure. Heart 2004, 90, e42. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frank, B.S.; Ivy, D.D. Diagnosis, Evaluation and Treatment of Pulmonary Arterial Hypertension in Children. Children 2018, 5, 44. https://doi.org/10.3390/children5040044
Frank BS, Ivy DD. Diagnosis, Evaluation and Treatment of Pulmonary Arterial Hypertension in Children. Children. 2018; 5(4):44. https://doi.org/10.3390/children5040044
Chicago/Turabian StyleFrank, Benjamin S., and D. Dunbar Ivy. 2018. "Diagnosis, Evaluation and Treatment of Pulmonary Arterial Hypertension in Children" Children 5, no. 4: 44. https://doi.org/10.3390/children5040044
APA StyleFrank, B. S., & Ivy, D. D. (2018). Diagnosis, Evaluation and Treatment of Pulmonary Arterial Hypertension in Children. Children, 5(4), 44. https://doi.org/10.3390/children5040044