
Congenital Cystic Adenomatoid Malformation (CCAM) Type II: A Rare Case of Sudden Infant Death

 , ,
, ,  and
and 
Abstract
:1. Introduction
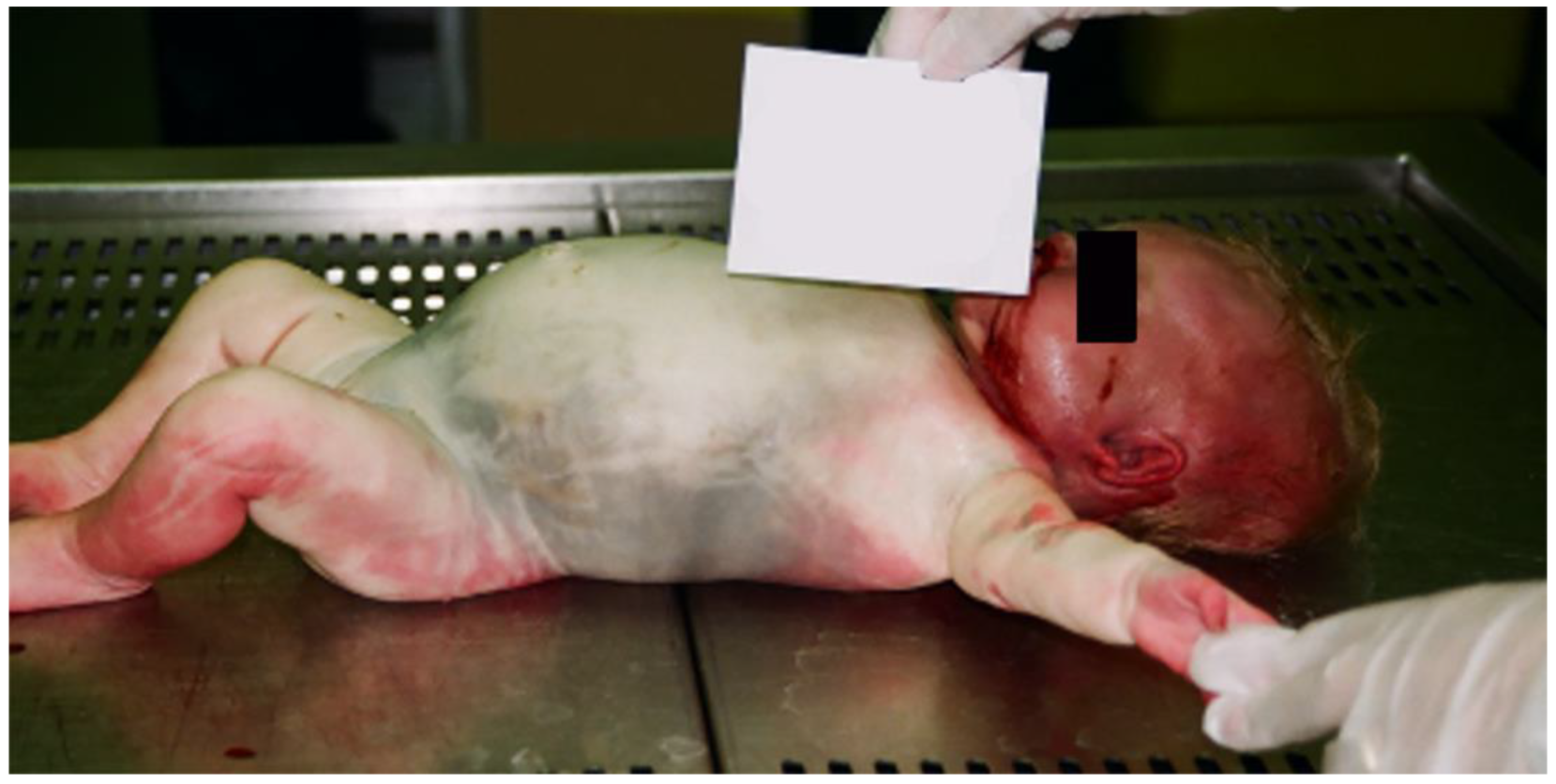
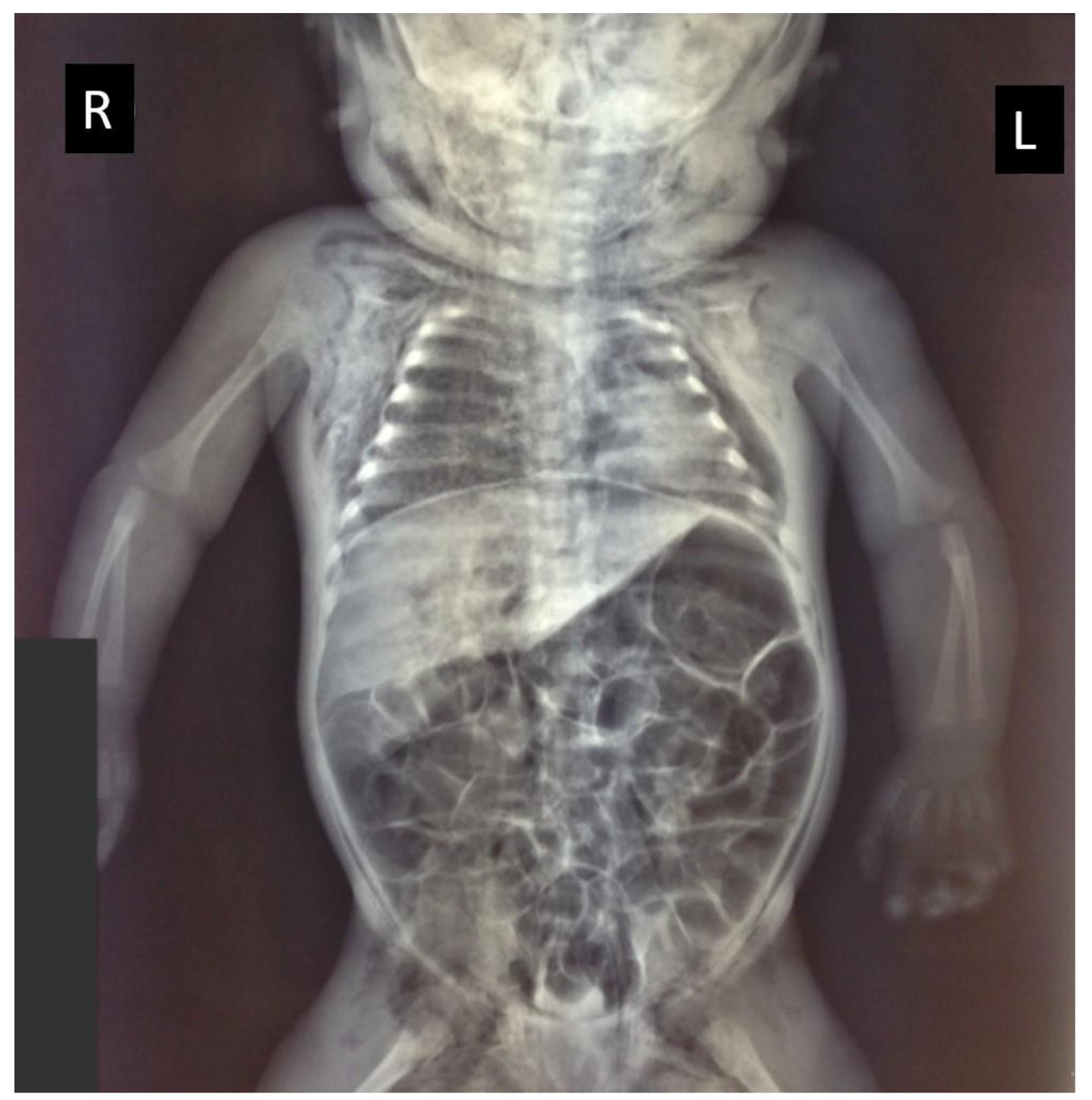
2. Case Presentation
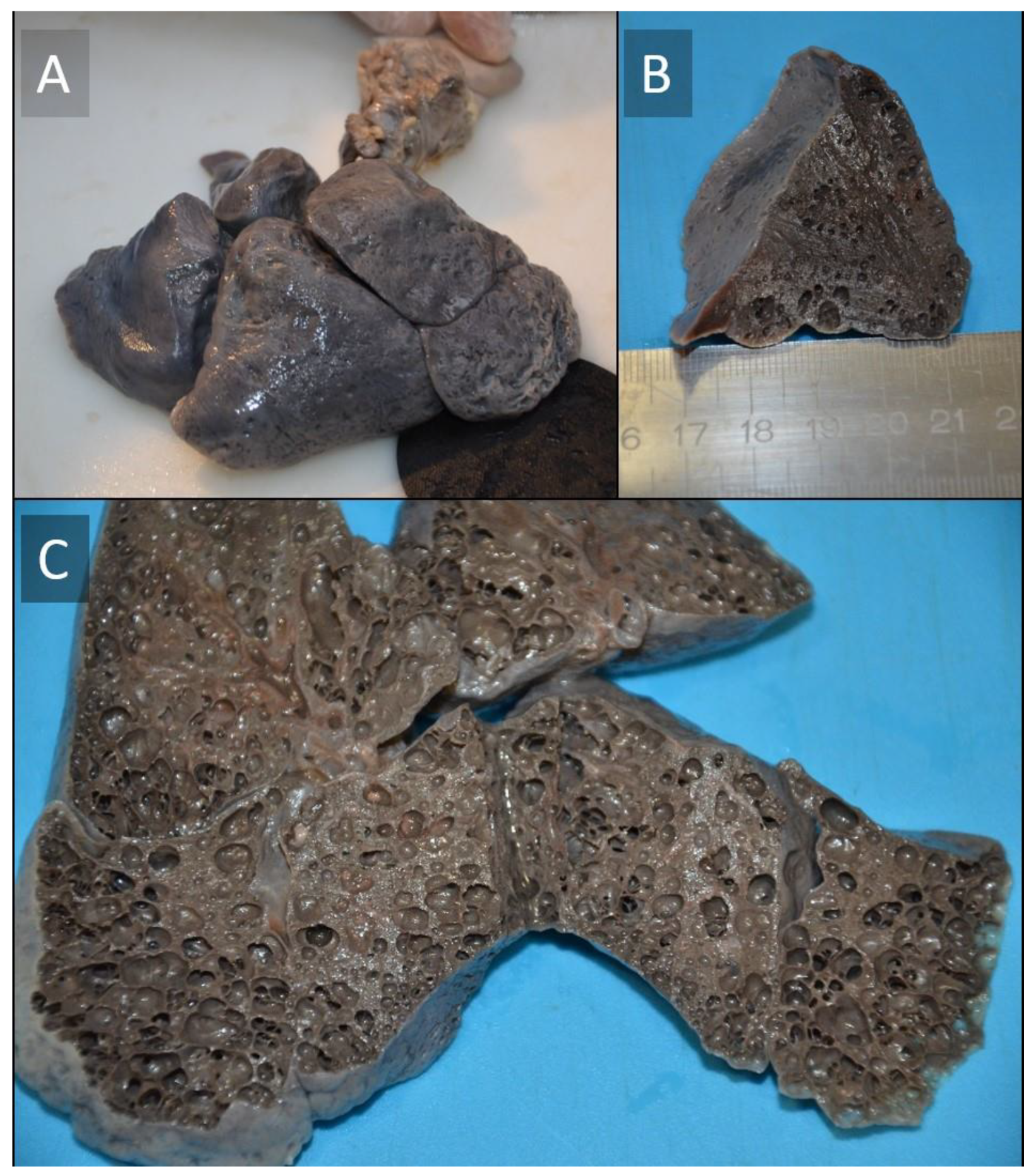
Histopathological Investigation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sirithangkul, S.; Chuengchitraks, S.; Staworn, D.; Laohapand, C.; Silarat, T. Late Manifestation of Congenital Cystic Adenomatoid Malformation with Lung Abscess: A Case Report. J. Med. Assoc. Thail. 2010, 93 (Suppl. 6), 223–227. [Google Scholar]
- Strumillo, B.; Jóźwiak, A.; Palka, A.; Szaflik, K.; Piaseczna-Piotrowska, A. Congenital Cystic Adenomatoid Malformation-Diagnostic and Therapeutic Procedure: 8-Year Experience of One Medical Centre. Kardiochirurgia i Torakochirurgia Polska 2018, 15, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Viana, R.; Carvalho, L.; Santos, C. Congenital Pulmonary Airway Malformation: A Rare Diagnosis in Adulthood. Respirol. Case Rep. 2022, 10, e0895. [Google Scholar] [CrossRef] [PubMed]
- Koga, H.; Ochi, T.; Hirayama, S.; Watanabe, Y.; Ueno, H.; Imashimizu, K.; Suzuki, K.; Kuwatsuru, R.; Nishimura, K.; Lane, G.J.; et al. Congenital Pulmonary Airway Malformation in Children: Advantages of an Additional Trocar in the Lower Thorax for Pulmonary Lobectomy. Front. Pediatr. 2021, 9, 722428. [Google Scholar] [CrossRef] [PubMed]
- Stocker, J.T.; Madewell, J.E.; Drake, R.M. Congenital Cystic Adenomatoid Malformation of the Lung. Classification and Morphologic Spectrum. Hum. Pathol. 1977, 8, 155–171. [Google Scholar] [CrossRef]
- Widiyanto, M.K.; Ismail, D.; Hayuningrat, P.K.; Muhammad, F. Congenital Cystic Adenomatoid Malformation Type i in a Newborn with Sepsis: A Case Report. Malays. J. Med. Health Sci. 2021, 17, 179–181. [Google Scholar]
- Kotecha, S.; Barbato, A.; Bush, A.; Claus, F.; Davenport, M.; Delacourt, C.; Deprest, J.; Eber, E.; Frenckner, B.; Greenough, A.; et al. Antenatal and Postnatal Management of Congenital Cystic Adenomatoid Malformation. Paediatr. Respir. Rev. 2012, 13, 162–171. [Google Scholar] [CrossRef]
- Stocker, J.T. Cystic Lung Disease in Infants and Children. Fetal. Pediatr. Pathol. 2009, 28, 155–184. [Google Scholar] [CrossRef]
- Argeitis, J.; Botsis, D.; Kairi-Vassilatou, E.; Hasiakos, D.; Papakonstantinou, K.; Kondi-Pafiti, A. Congenital Cystic Adenomatoid Lung Malformation: Report of Two Cases and Literature Review. Clin. Exp. Obstet. Gynecol. 2008, 35, 76–80. [Google Scholar]
- Yamashita, A.; Hidaka, N.; Yamamoto, R.; Nakayama, S.; Sasahara, J.; Ishii, K.; Mitsuda, N. In Utero Resolution of Microcystic Congenital Cystic Adenomatoid Malformation after Prenatal Betamethasone Therapy: A Report of Three Cases and a Literature Review. J. Clin. Ultrasound 2015, 43, 451–457. [Google Scholar] [CrossRef]
- Miller, J.A.; Corteville, J.E.; Langer, J.C. Congenital Cystic Adenomatoid Malformation in the Fetus: Natural History and Predictors of Outcome. J. Pediatr. Surg. 1996, 31, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Kersten, C.M.; Hermelijn, S.M.; Mullassery, D.; Muthialu, N.; Cobanoglu, N.; Gartner, S.; Bagolan, P.; Mesas Burgos, C.; Sgrò, A.; Heyman, S.; et al. The Management of Asymptomatic Congenital Pulmonary Airway Malformation: Results of a European Delphi Survey. Children 2022, 9, 1153. [Google Scholar] [CrossRef] [PubMed]
- Mardy, C.; Blumenfeld, Y.J.; Arunamata, A.A.; Girsen, A.I.; Sylvester, K.G.; Halabi, S.; Rubesova, E.; Hintz, S.R.; Tacy, T.A.; Maskatia, S.A. In Fetuses with Congenital Lung Masses, Decreased Ventricular and Atrioventricular Valve Dimensions Are Associated with Lesion Size and Clinical Outcome. Prenat. Diagn. 2020, 40, 206–215. [Google Scholar] [CrossRef]
- Pomara, C.; Fineschi, V. Forensic and Clinical Forensic Autopsy. An Atlas and Handbook—2nd Edition; Pomara, C., Fineschi, V., Eds.; CRC Press: Boca Raton, FL, USA, 2020; ISBN 9780367330712. [Google Scholar]
- Mackenzie, T.C.; Guttenberg, M.E.; Nisenbaum, H.L.; Johnson, M.P.; Adzick, N.S. A Fetal Lung Lesion Consisting of Bronchogenic Cyst, Bronchopulmonary Sequestration, and Congenital Cystic Adenomatoid Malformation: The Missing Link? Fetal Diagn. Ther. 2001, 16, 193–195. [Google Scholar] [CrossRef] [PubMed]
- Seear, M.; Townsend, J.; Hoepker, A.; Jamieson, D.; McFadden, D.; Daigneault, P.; Glomb, W. A Review of Congenital Lung Malformations with a Simplified Classification System for Clinical and Research Use. Pediatr. Surg. Int. 2017, 33, 657–664. [Google Scholar] [CrossRef]
- Müller, A.M. Pathology of Pediatric Lung Diseases and Malformations|Pathologie von Pädiatrischen Lungenerkrankungen Und Fehlbildungen. Pneumologe 2015, 12, 52–58. [Google Scholar] [CrossRef]
- Annam, V.; Korishetty, S.I.; Yelikar, B.R.; Hippargi, S.B.; Shivalingappa, D.B. Bilateral Congenital Cystic Adenomatoid Malformation, Stocker Type III with Associated Findings and Review of Literature. Indian J. Pathol. Microbiol. 2010, 53, 331–333. [Google Scholar] [CrossRef]
- Kurland, G.; Deterding, R.R.; Hagood, J.S.; Young, L.R.; Brody, A.S.; Castile, R.G.; Dell, S.; Fan, L.L.; Hamvas, A.; Hilman, B.C.; et al. An Official American Thoracic Society Clinical Practice Guideline: Classification, Evaluation, and Management of Childhood Interstitial Lung Disease in Infancy. Am. J. Respir. Crit. Care Med. 2013, 188, 376–394. [Google Scholar] [CrossRef] [Green Version]
- Deutsch, G.H.; Young, L.R.; Deterding, R.R.; Fan, L.L.; Dell, S.D.; Bean, J.A.; Brody, A.S.; Nogee, L.M.; Trapnell, B.C.; Langston, C.; et al. Diffuse Lung Disease in Young Children: Application of a Novel Classification Scheme. Am. J. Respir. Crit. Care Med. 2007, 176, 1120–1128. [Google Scholar] [CrossRef] [Green Version]
- Nogee, L.M. Interstitial Lung Disease in Newborns. Semin. Fetal Neonatal Med. 2017, 22, 227–233. [Google Scholar] [CrossRef]
- Khan, H.; Kurup, M.; Saikia, S.; Desai, A.; Mathew, M.; Sheikh, A.; Goonasekera, C.D.A. Morbidity after Thoracoscopic Resection of Congenital Pulmonary Airway Malformations (CPAM): Single Center Experience over a Decade. Pediatr. Surg. Int. 2021, 37, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Hermelijn, S.M.; Mackenbach, M.J.; van Horik, C.; Ciet, P.; Wolf, J.L.; von der Thüsen, J.H.; Wijnen, R.M.H.; Tiddens, H.A.W.M.; Schnater, J.M. Quantitative CT Imaging Analysis to Predict Pathology Features in Patients with a Congenital Pulmonary Airway Malformation. J. Pediatr. Surg. 2022, 57, 1567–1572. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, H.; Hafiz, A.; Rabiu, B.; Abdullahi, U.; Ghidazuka, Y.; Aliyu, I. Spontaneous Resolution of a Congenital Multicystic Lung Lesion in a Newborn. Med. J. Dr. D.Y. Patil Vidyapeeth 2019, 12, 352–355. [Google Scholar] [CrossRef]
- Garg, S.; Singh, R.S.; Singh, H. Congenital Cystic Adenomatoid Malformation of the Lung in Adults: Report of Two Cases and Review of the Literature. Indian J. Thorac. Cardiovasc. Surg. 2018, 34, 488–490. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Pannu, M.S.; Lata, N.; Dhillon, S.P.S.; Singh, N.; Sareen, A. Rare Presentation of Congenital Cystic Adenomatoid Malformation of the Lung. J. Nepal Paediatr. Soc. 2016, 36, 91–93. [Google Scholar] [CrossRef]
- Badour, M.; Hussain, B.; Hammed, A.; Sawssan, A.; Saeed, F. A Rare Case of Congenital Cystic Adenomatoid Malformation: Mimics Pneumonia Manifestations. Ann. Med. Surg. 2021, 66, 102433. [Google Scholar] [CrossRef]
- Hermelijn, S.M.; Elders, B.B.L.J.; Ciet, P.; Wijnen, R.M.H.; Tiddens, H.A.W.M.; Schnater, J.M. A Clinical Guideline for Structured Assessment of CT-Imaging in Congenital Lung Abnormalities. Paediatr. Respir. Rev. 2021, 37, 80–88. [Google Scholar] [CrossRef]
- Zhang, Z.-J.; Huang, M.-X. Children with Congenital Cystic Adenomatoid Malformation of the Lung CT Diagnosis. Int. J. Clin. Exp. Med. 2015, 8, 4415–4419. [Google Scholar]
- Sessa, F.; Esposito, M.; Messina, G.; di Mizio, G.; di Nunno, N.; Salerno, M. Sudden Death in Adults: A Practical Flow Chart for Pathologist Guidance. Healthcare 2021, 9, 870. [Google Scholar] [CrossRef]
- di Nunno, N.; Patanè, F.G.; Amico, F.; Asmundo, A.; Pomara, C. The Role of a Good Quality Autopsy in Pediatric Malpractice Claim: A Case Report of an Unexpected Death in an Undiagnosed Thymoma. Front. Pediatr. 2020, 8, 8. [Google Scholar] [CrossRef]
- Schiavone, S.; Mhillaj, E.; Neri, M.; Morgese, M.G.; Tucci, P.; Bove, M.; Valentino, M.; di Giovanni, G.; Pomara, C.; Turillazzi, E.; et al. Early Loss of Blood-Brain Barrier Integrity Precedes NOX2 Elevation in the Prefrontal Cortex of an Animal Model of Psychosis. Mol. Neurobiol. 2017, 54, 2031–2044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neri, M.; Othman, S.M.; Cantatore, S.; de Carlo, D.; Pomara, C.; Riezzo, I.; Turillazzi, E.; Fineschi, V. Sudden Infant Death in an 8-Month-Old Baby with Dengue Virus Infection: Searching for Virus in Postmortem Tissues by Immunohistochemistry and Western Blotting. Pediatr. Infect. Dis. J. 2012, 31, 878–880. [Google Scholar] [CrossRef] [PubMed]
- Bertozzi, G.; Maglietta, F.; Baldari, B.; Besi, L.; Torsello, A.; di Gioia, C.R.T.; Sessa, F.; Aromatario, M.; Cipolloni, L. Mistrial or Misdiagnosis: The Importance of Autopsy and Histopathological Examination in Cases of Sudden Infant Bronchiolitis-Related Death. Front. Pediatr. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Santacroce, R.; Santoro, R.; Sessa, F.; Iannaccaro, P.; Sarno, M.; Longo, V.; Gallone, A.; Vecchione, G.; Muleo, G.; Margaglione, M. Screening of Mutations of Hemophilia A in 40 Italian Patients: A Novel G-to-A Mutation in Intron 10 of the F8 Gene as a Putative Cause of Mild Hemophilia a in Southern Italy. Blood Coagul. Fibrinolysis 2008, 19, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Sintim-Damoa, A.; Cohen, H.L. Fetal Imaging of Congenital Lung Lesions with Postnatal Correlation. Pediatr. Radiol. 2022, 52, 1921–1934. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Cheng, K.; He, M.; Wang, Y.; Shen, P.; He, K.; Xu, C.; Zhang, B.; Liu, Z. Diagnostic Value of Congenital Pulmonary Airway Malformation Volume Ratio for Fetal Hydrops Due to Congenital Lung Malformations: A Systematic Review and Meta-Analysis. Orphanet J. Rare Dis. 2022, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-Y.; Chang, Y.-J.; Chen, L.-J.; Lee, C.-H.; Chen, H.-N.; Chen, J.-Y.; Chen, M.; Hsiao, C.-C. Survival of Hydrops Fetalis with and without Fetal Intervention. Children 2022, 9, 530. [Google Scholar] [CrossRef]
- Jeong, B.-D.; An, S.-A.; Lee, M.-Y.; Won, H.-S.; Han, M.; Yoon, H.; Lee, J.-H.; Cho, Y.-J. Comparison of the Prognostic Factors of Fetuses with Congenital Pulmonary Airway Malformations According to Type. J. Ultrasound Med. 2020, 39, 2243–2252. [Google Scholar] [CrossRef]
- Mon, R.A.; Johnson, K.N.; Ladino-Torres, M.; Heider, A.; Mychaliska, G.B.; Treadwell, M.C.; Kunisaki, S.M. Diagnostic Accuracy of Imaging Studies in Congenital Lung Malformations. Arch. Dis. Child. Fetal Neonatal Ed. 2018, 104, F372-2018. [Google Scholar] [CrossRef]
- Chaouachi, S.; ben Hamida, E.; ben Fraj, N.; Blibèche, S.; Marrakchi, Z. Congenital Cystic Adenomatoid Malformation of the Lung: Two Cases Report. Tunis. Med. 2011, 89, 55–58. [Google Scholar]
- Shupe, M.P.; Kwon, H.P.; Morris, M.J. Spontaneous Pneumothorax in a Teenager with Prior Congenital Pulmonary Airway Malformation. Respir. Med. Case Rep. 2014, 11, 18–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barikbin, P.; Roehr, C.C.; Wilitzki, S.; Kalache, K.; Degenhardt, P.; Bührer, C.; Schmalisch, G. Postnatal Lung Function in Congenital Cystic Adenomatoid Malformation of the Lung. Ann. Thorac. Surg. 2015, 99, 1164–1169. [Google Scholar] [CrossRef] [PubMed]
- Isabellea, V.; Wildhaber Barbara, E.; Ueli, M.; Nicolas, R.; Daniel, T.; Dietmar, C.; Juerg, B.; Constance, B.-A.; Isabelle, R.-M. A Swiss Database and Biobank to Better Understand and Manage Congenital Lung Anomalies. Swiss Med. Wkly. 2019, 149, w20081. [Google Scholar] [CrossRef] [Green Version]
- Davenport, M.; Warne, S.A.; Cacciaguerra, S.; Patel, S.; Greenough, A.; Nicolaides, K. Current Outcome of Antenally Diagnosed Cystic Lung Disease. J. Pediatr. Surg. 2004, 39, 549–556. [Google Scholar] [CrossRef]
- Stanton, M.; Davenport, M. Management of Congenital Lung Lesions. Early Hum. Dev. 2006, 82, 289–295. [Google Scholar] [CrossRef]
- Tsai, A.Y.; Liechty, K.W.; Hedrick, H.L.; Bebbington, M.; Wilson, R.D.; Johnson, M.P.; Howell, L.J.; Flake, A.W.; Adzick, N.S. Outcomes after Postnatal Resection of Prenatally Diagnosed Asymptomatic Cystic Lung Lesions. J. Pediatr. Surg. 2008, 43, 513–517. [Google Scholar] [CrossRef]
- Jaffé, A.; Chitty, L.S. Congenital Cystic Adenomatoid Malformations May Not Require Surgical Intervention. Arch. Dis. Child. Fetal Neonatal Ed. 2006, 91, F464. [Google Scholar] [CrossRef]
- Chetcuti, P.A.J.; Crabbe, D.C.G. CAM Lungs: The Conservative Approach. Arch. Dis. Child Fetal Neonatal Ed. 2006, 91, F463. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Developmental diseases | Genetic mutations |
|---|---|
|
|
| Growth disorders | Undefined etiology |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salerno, M.; Sessa, F.; Cocimano, G.; Roccuzzo, S.; Esposito, M.; Pomara, C. Congenital Cystic Adenomatoid Malformation (CCAM) Type II: A Rare Case of Sudden Infant Death. Children 2022, 9, 1830. https://doi.org/10.3390/children9121830
Salerno M, Sessa F, Cocimano G, Roccuzzo S, Esposito M, Pomara C. Congenital Cystic Adenomatoid Malformation (CCAM) Type II: A Rare Case of Sudden Infant Death. Children. 2022; 9(12):1830. https://doi.org/10.3390/children9121830
Chicago/Turabian StyleSalerno, Monica, Francesco Sessa, Giuseppe Cocimano, Salvatore Roccuzzo, Massimiliano Esposito, and Cristoforo Pomara. 2022. "Congenital Cystic Adenomatoid Malformation (CCAM) Type II: A Rare Case of Sudden Infant Death" Children 9, no. 12: 1830. https://doi.org/10.3390/children9121830