
EndoNet: A Model for the Automatic Calculation of H-Score on Histological Slides
 , , ,
, , ,  , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Methods
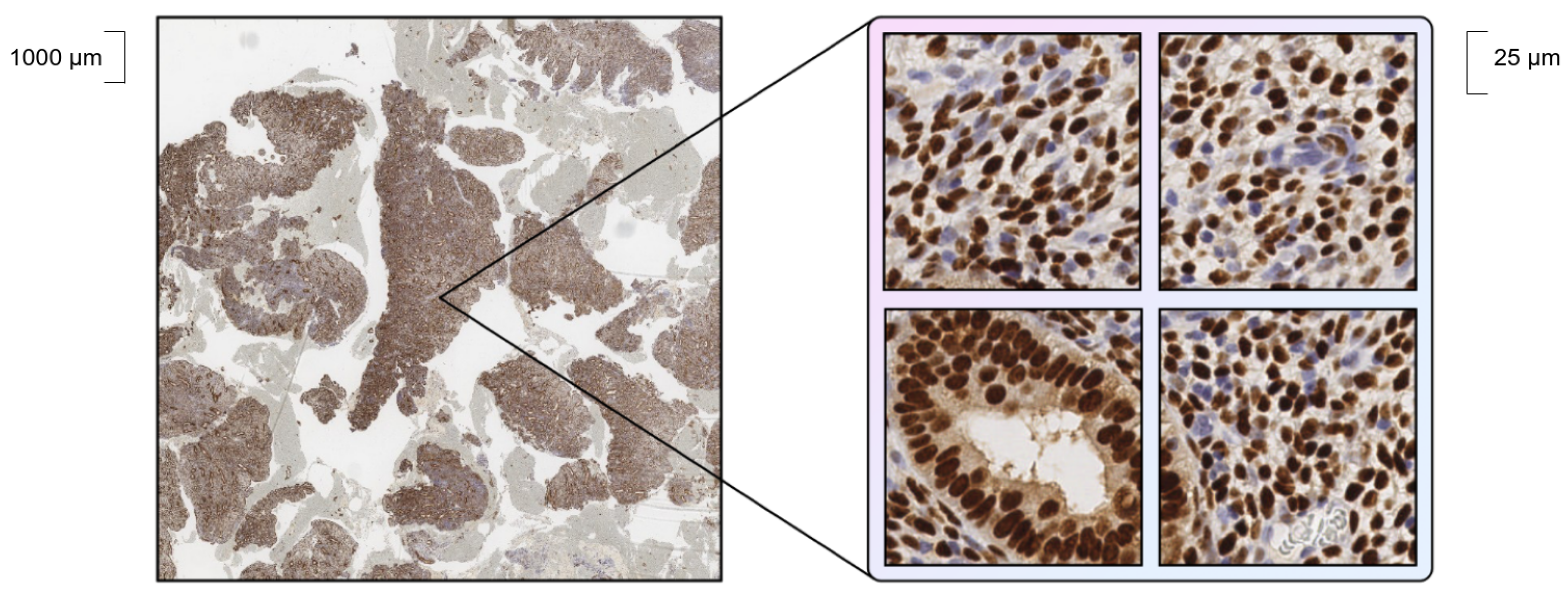
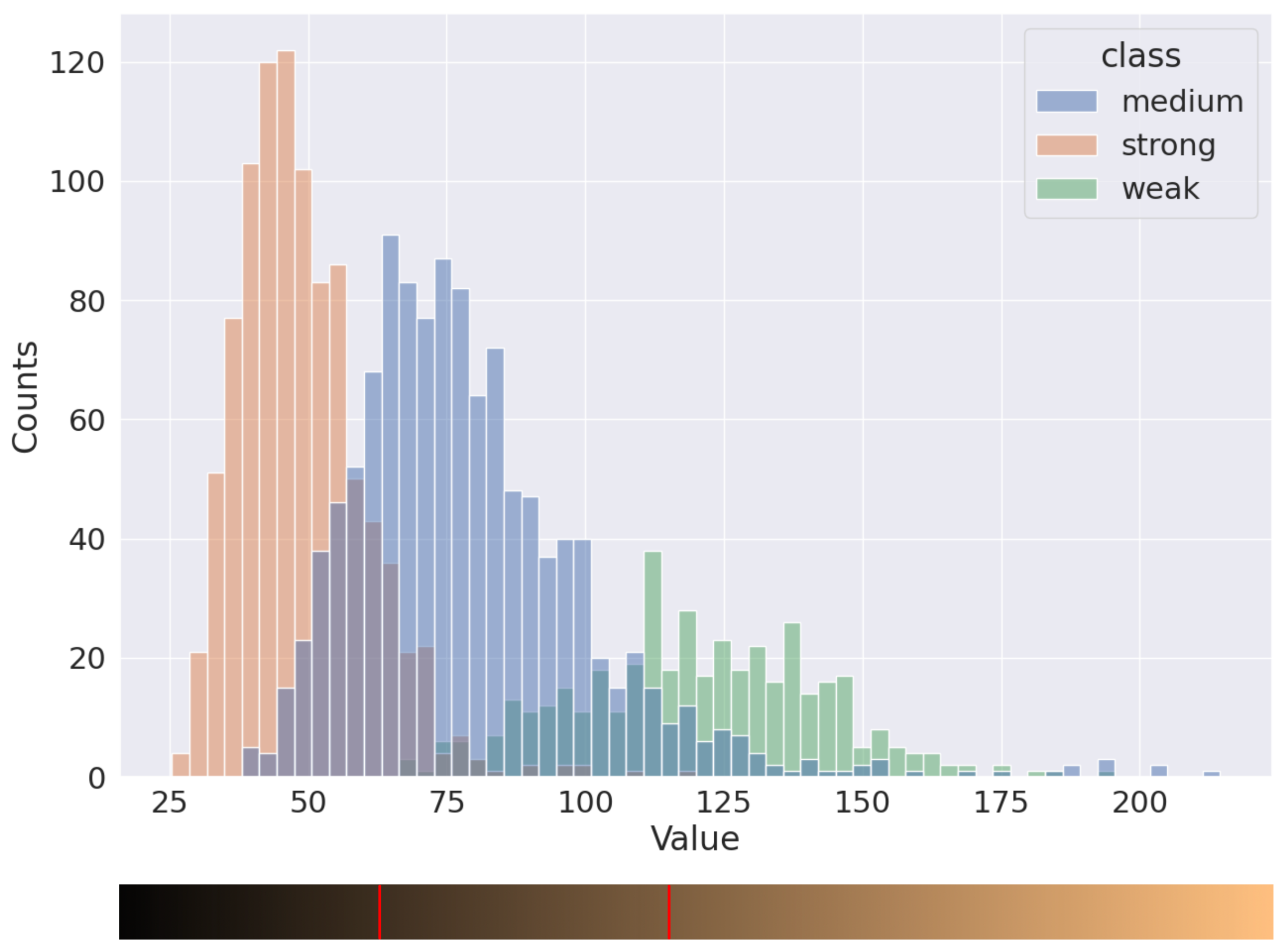
2.1. Data
- = point locations;
- s = scale parameter, which equals the mean square nuclei radius.
2.2. General Architecture
2.3. Detection Model Architecture
2.4. Training of Detection Model
- y = true label;
- = predicted label;
- = the threshold where the Huber loss function transitions from quadratic to linear.
2.5. Pre-Training
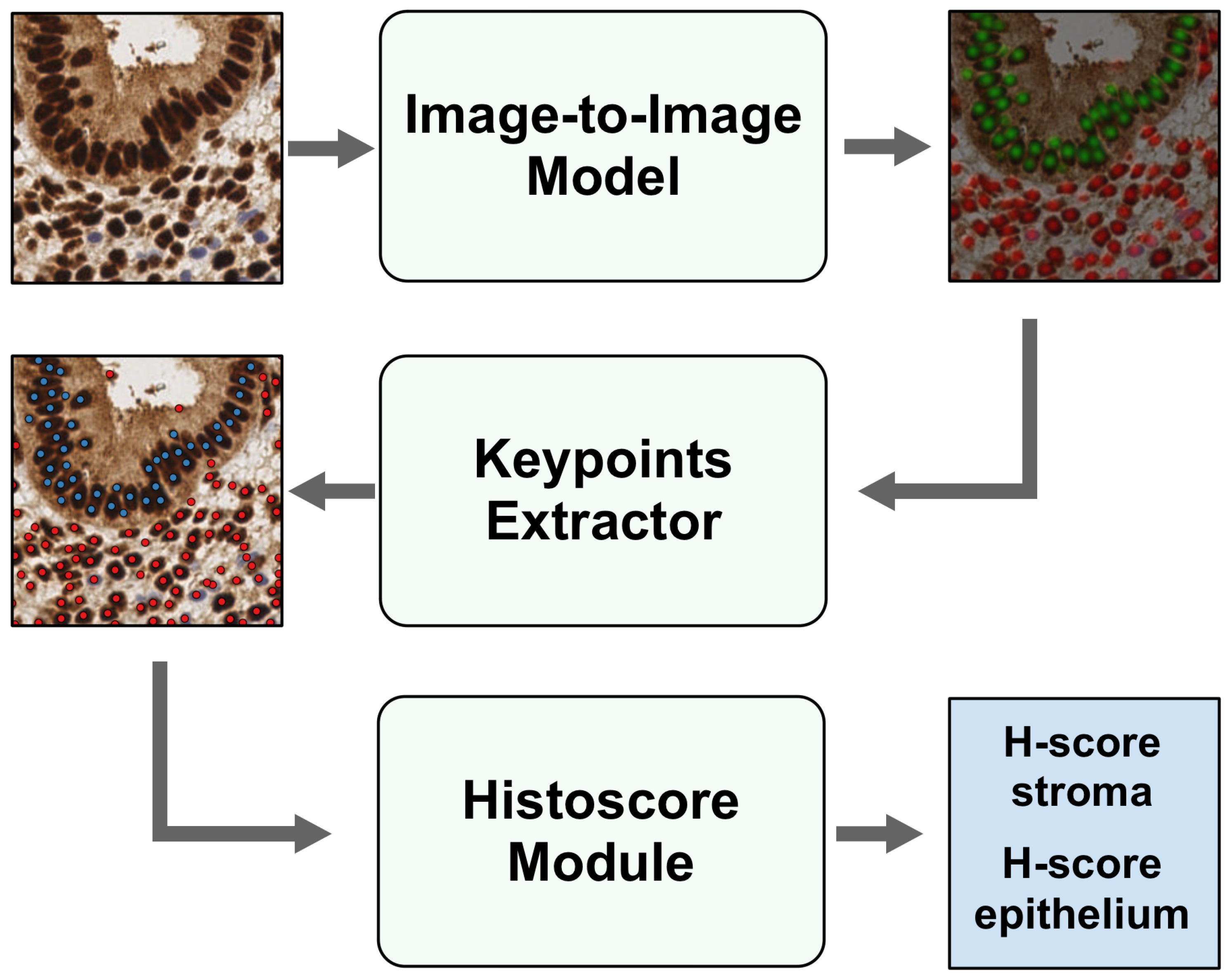
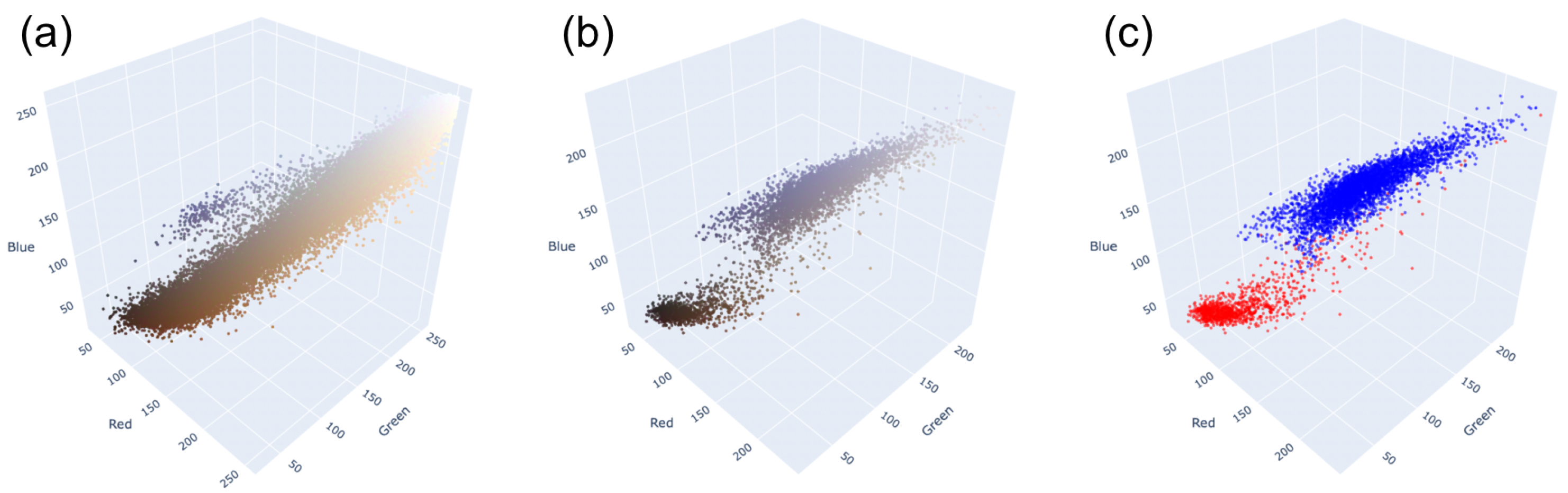
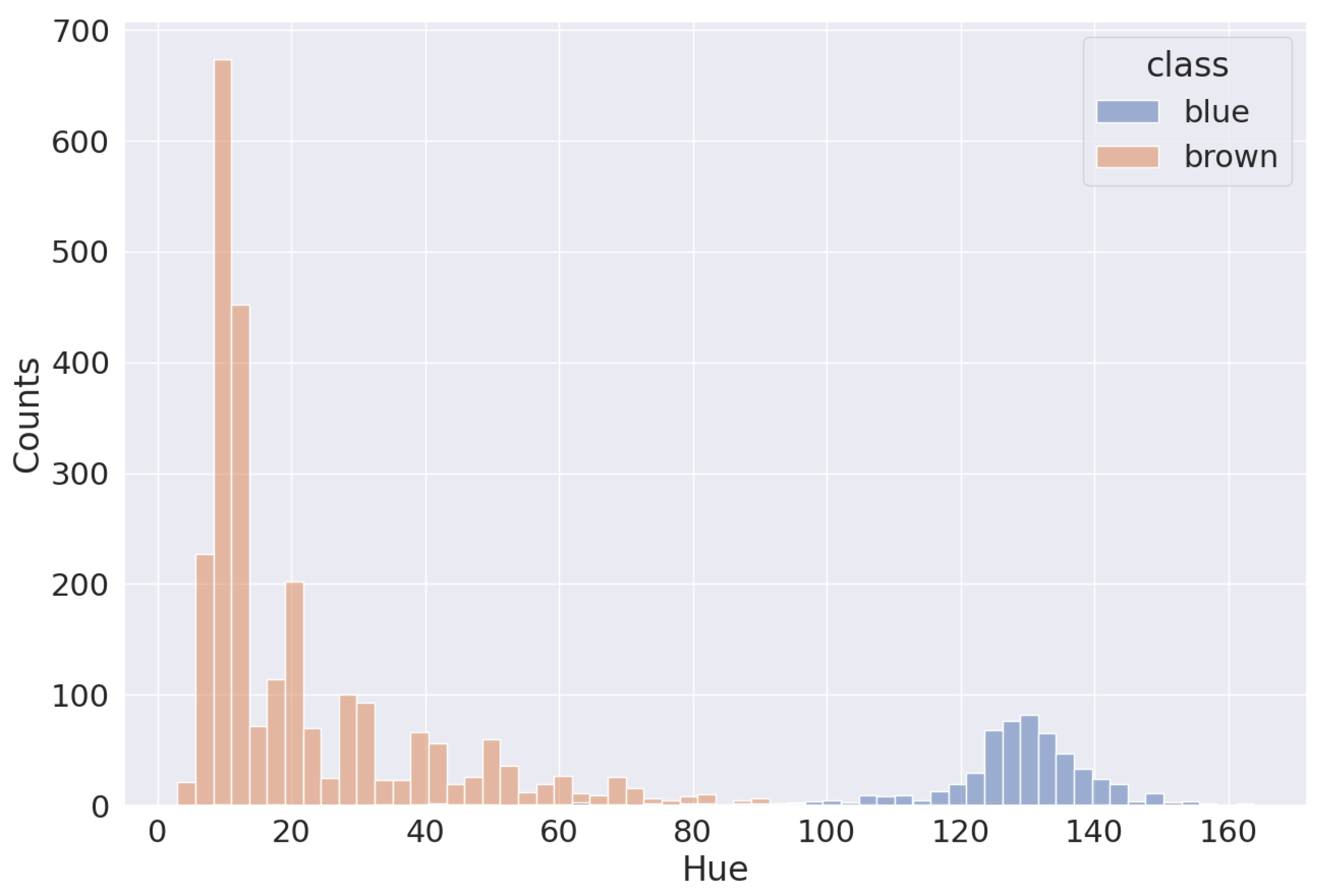
2.6. H-Score Module
2.7. Statistical Testing
3. Results
3.1. Pre-Training Results
3.2. Training
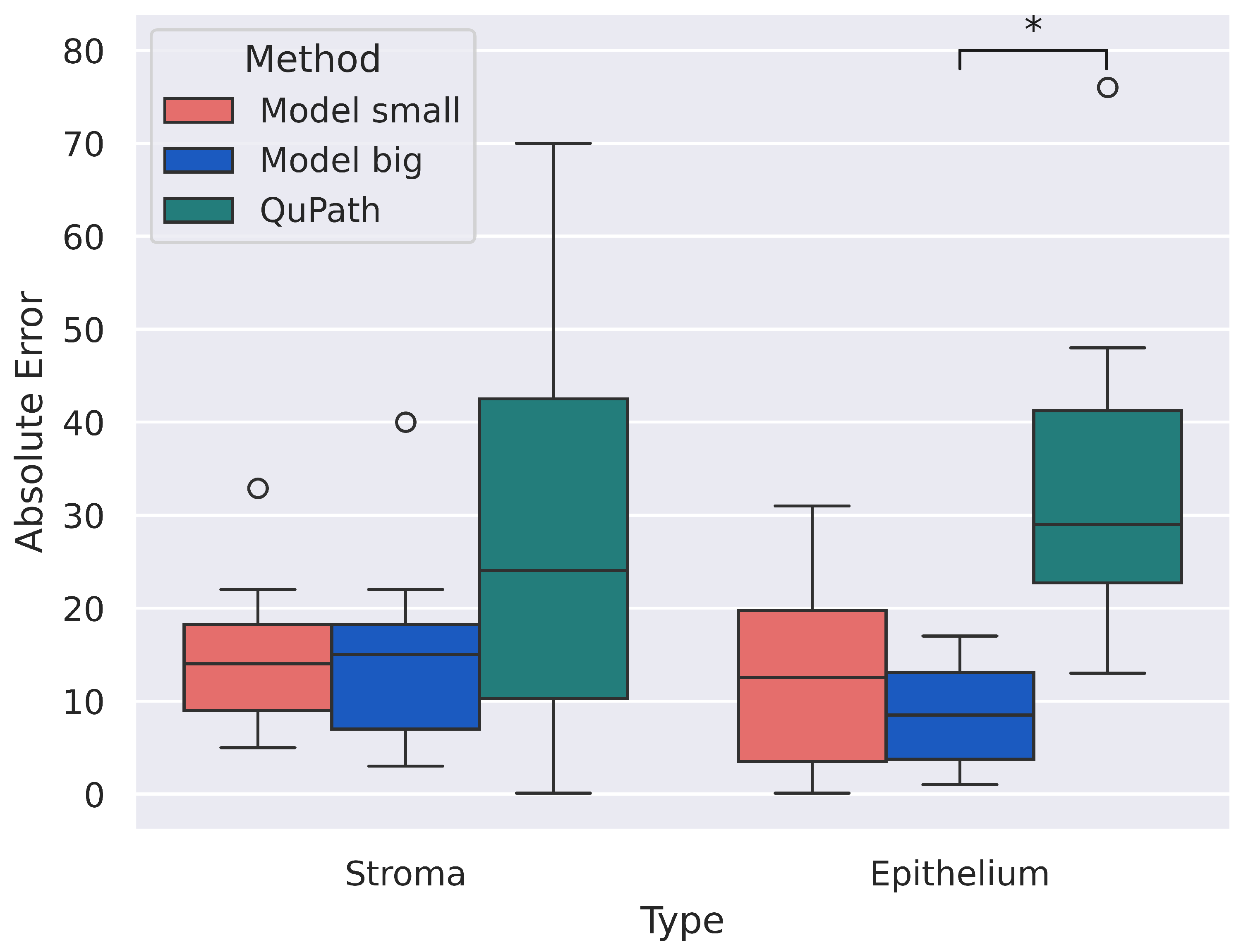
3.3. H-Score
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rizzardi, A.E.; Johnson, A.T.; Vogel, R.I.; Pambuccian, S.E.; Henriksen, J.; Skubitz, A.P.; Metzger, G.J.; Schmechel, S.C. Quantitative comparison of immunohistochemical staining measured by digital image analysis versus pathologist visual scoring. Diagn. Pathol. 2012, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Srinidhi, C.L.; Ciga, O.; Martel, A.L. Deep neural network models for computational histopathology: A survey. Med Image Anal. 2021, 67, 101813. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, O.; Kanavati, F.; Kato, K.; Rambeau, M.; Arihiro, K.; Tsuneki, M. Deep Learning Models for Histopathological Classification of Gastric and Colonic Epithelial Tumours. Sci. Rep. 2020, 10, 1504. [Google Scholar] [CrossRef] [PubMed]
- Budwit-Novotny, D.A.; McCarty, K.S.; Cox, E.B.; Soper, J.T.; Mutch, D.G.; Creasman, W.T.; Flowers, J.L.; McCarty, K.S., Jr. Immunohistochemical analyses of estrogen receptor in endometrial adenocarcinoma using a monoclonal antibody. Cancer Res. 1986, 46, 5419–5425. [Google Scholar] [PubMed]
- van Netten, J.P.; Thornton, I.G.; Carlyle, S.J.; Brigden, M.L.; Coy, P.; Goodchild, N.L.; Gallagher, S.; George, E.J. Multiple microsample analysis of intratumor estrogen receptor distribution in breast cancers by a combined biochemical/immunohistochemical method. Eur. J. Cancer Clin. Oncol. 1987, 23, 1337–1342. [Google Scholar] [CrossRef] [PubMed]
- Babu, R.; Balaji, D.; Reddy, G.; Paramaswamy, B.; Ramasundaram, M.; Agarwal, P.; Joseph, L.; D’Cruze, L.; Sundaram, S. Androgen receptor expression in hypospadias. J. Indian Assoc. Pediatr. Surg. 2020, 25, 6. [Google Scholar] [CrossRef]
- Pierceall, W.E.; Wolfe, M.; Suschak, J.; Chang, H.; Chen, Y.; Sprott, K.M.; Kutok, J.L.; Quan, S.; Weaver, D.T.; Ward, B.E. Strategies for H-score Normalization of Preanalytical Technical Variables with Potential Utility to Immunohistochemical-Based Biomarker Quantitation in Therapeutic Reponse Diagnostics. Anal. Cell. Pathol. 2011, 34, 159–168. [Google Scholar] [CrossRef]
- Sharada, P.; Swaminathan, U.; Nagamalini, B.; Vinod Kumar, K.; Ashwini, B. Histoscore and Discontinuity Score - A Novel Scoring System to Evaluate Immunohistochemical Expression of COX-2 and Type IV Collagen in Oral Potentially Malignant Disorders and Oral Squamous Cell Carcinoma. J. Orofac. Sci. 2021, 13, 96–104. [Google Scholar] [CrossRef]
- Ram, S.; Vizcarra, P.; Whalen, P.; Deng, S.; Painter, C.L.; Jackson-Fisher, A.; Pirie-Shepherd, S.; Xia, X.; Powell, E.L. Pixelwise H-score: A novel digital image analysis-based metric to quantify membrane biomarker expression from immunohistochemistry images. PLoS ONE 2021, 16, e0245638. [Google Scholar] [CrossRef]
- Pantanowitz, L.; Sharma, A.; Carter, A.B.; Kurc, T.; Sussman, A.; Saltz, J. Twenty Years of Digital Pathology: An Overview of the Road Travelled, What is on the Horizon, and the Emergence of Vendor-Neutral Archives. J. Pathol. Inform. 2018, 9, 40. [Google Scholar] [CrossRef]
- Gurcan, M.; Boucheron, L.; Can, A.; Madabhushi, A.; Rajpoot, N.; Yener, B. Histopathological Image Analysis: A Review. IEEE Rev. Biomed. Eng. 2009, 2, 147–171. [Google Scholar] [CrossRef]
- Madabhushi, A.; Lee, G. Image analysis and machine learning in digital pathology: Challenges and opportunities. Med Image Anal. 2016, 33, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Veta, M.; Pluim, J.P.W.; van Diest, P.J.; Viergever, M.A. Breast Cancer Histopathology Image Analysis: A Review. IEEE Trans. Biomed. Eng. 2014, 61, 1400–1411. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chen, H.; Li, X.; Xu, N.; Hu, Z.; Xue, D.; Qi, S.; Ma, H.; Zhang, L.; Sun, H. A review for cervical histopathology image analysis using machine vision approaches. Artif. Intell. Rev. 2020, 53, 4821–4862. [Google Scholar] [CrossRef]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. ImageNet Classification with Deep Convolutional Neural Networks. In Advances in Neural Information Processing Systems; Pereira, F., Burges, C., Bottou, L., Weinberger, K., Eds.; Curran Associates, Inc.: San Diego, CA, USA, 2012; Volume 25. [Google Scholar]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Delving Deep into Rectifiers: Surpassing Human-Level Performance on ImageNet Classification. In Proceedings of the IEEE International Conference on Computer Vision (ICCV), Santiago, Chile, 11–18 December 2015. [Google Scholar]
- Dosovitskiy, A.; Beyer, L.; Kolesnikov, A.; Weissenborn, D.; Zhai, X.; Unterthiner, T.; Dehghani, M.; Minderer, M.; Heigold, G.; Gelly, S.; et al. An Image is Worth 16x16 Words: Transformers for Image Recognition at Scale. arXiv 2020, arXiv:2010.11929. [Google Scholar]
- Sun, Y.; Huang, X.; Zhou, H.; Zhang, Q. SRPN: Similarity-based region proposal networks for nuclei and cells detection in histology images. Med Image Anal. 2021, 72, 102142. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep Residual Learning for Image Recognition. arXiv 2015, arXiv:1512.03385. [Google Scholar]
- Avranas, A.; Kountouris, M. Coded ResNeXt: A network for designing disentangled information paths. arXiv 2022, arXiv:2202.05343. [Google Scholar]
- Ren, S.; He, K.; Girshick, R.; Sun, J. Faster R-CNN: Towards Real-Time Object Detection with Region Proposal Networks. arXiv 2015, arXiv:1506.01497. [Google Scholar] [CrossRef]
- Naumov, A.; Ushakov, E.; Ivanov, A.; Midiber, K.; Khovanskaya, T.; Konyukova, A.; Vishnyakova, P.; Nora, S.; Mikhaleva, L.; Fatkhudinov, T.; et al. EndoNuke: Nuclei Detection Dataset for Estrogen and Progesterone Stained IHC Endometrium Scans. Data 2022, 7, 75. [Google Scholar] [CrossRef]
- Ronchi, M.R.; Perona, P. Benchmarking and Error Diagnosis in Multi-instance Pose Estimation. In Proceedings of the 2017 IEEE International Conference on Computer Vision (ICCV), Venice, Italy, 22–29 October 2017. [Google Scholar] [CrossRef]
- Kuhn, H.W. The Hungarian method for the assignment problem. Nav. Res. Logist. Q. 1955, 2, 83–97. [Google Scholar] [CrossRef]
- Cohen, J. A Coefficient of Agreement for Nominal Scales. Educ. Psychol. Meas. 1960, 20, 37–46. [Google Scholar] [CrossRef]
- Solovyev, R.; Wang, W.; Gabruseva, T. Weighted boxes fusion: Ensembling boxes from different object detection models. Image Vis. Comput. 2021, 107, 104117. [Google Scholar] [CrossRef]
- Lin, T.Y.; Goyal, P.; Girshick, R.; He, K.; Dollár, P. Focal Loss for Dense Object Detection. arXiv 2017, arXiv:1708.02002. [Google Scholar]
- Chen, T.; Kornblith, S.; Norouzi, M.; Hinton, G. A Simple Framework for Contrastive Learning of Visual Representations. arXiv 2020, arXiv:2002.05709. [Google Scholar]
- Ushakov, E.; Naumov, A.; Fomberg, V. EndoNet: Code and Weights. Available online: https://github.com/ispras/endonet (accessed on 26 November 2023).
- Sun, H.; Zeng, X.; Xu, T.; Peng, G.; Ma, Y. Computer-Aided Diagnosis in Histopathological Images of the Endometrium Using a Convolutional Neural Network and Attention Mechanisms. IEEE J. Biomed. Health Inform. 2020, 24, 1664–1676. [Google Scholar] [CrossRef]
- Lahiani, A.; Gildenblat, J.; Klaman, I.; Navab, N.; Klaiman, E. Generalising multistain immunohistochemistry tissue segmentation using end-to-end colour deconvolution deep neural networks. IET Image Process. 2019, 13, 1066–1073. [Google Scholar] [CrossRef]
- Sharma, H.; Zerbe, N.; Klempert, I.; Hellwich, O.; Hufnagl, P. Deep convolutional neural networks for automatic classification of gastric carcinoma using whole slide images in digital histopathology. Comput. Med Imaging Graph. 2017, 61, 2–13. [Google Scholar] [CrossRef]
- Chen, T.; Chefd’hotel, C. Deep Learning Based Automatic Immune Cell Detection for Immunohistochemistry Images. In Machine Learning in Medical Imaging; Springer International Publishing: Berlin/Heidelberg, Germany, 2014; pp. 17–24. [Google Scholar] [CrossRef]
- Krajewska, M.; Smith, L.H.; Rong, J.; Huang, X.; Hyer, M.L.; Zeps, N.; Iacopetta, B.; Linke, S.P.; Olson, A.H.; Reed, J.C.; et al. Image Analysis Algorithms for Immunohistochemical Assessment of Cell Death Events and Fibrosis in Tissue Sections. J. Histochem. Cytochem. 2009, 57, 649–663. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SimCLR Pre-Trained | ImageNET | Confidence Interval—Lower Bound | Confidence Interval—Upper Bound | |
|---|---|---|---|---|
| Stroma AP | 0.8577 | 0.8544 | −0.00666 | 0.01840 |
| Epithelium AP | 0.7576 | 0.7256 | 0.00461 | 0.07010 |
| mAP | 0.8077 | 0.7900 | −0.00024 | 0.04115 |
| EndoNuke | PathLab | Combined | |
|---|---|---|---|
| Stroma AP | 0.85 | 0.83 | 0.85 |
| Epithelium AP | 0.69 | 0.84 | 0.69 |
| mAP | 0.77 | 0.84 | 0.77 |
| Annonator | Slide | Left | Right |
|---|---|---|---|
| 1st | 1 | 80 | 120 |
| 2 | 80 | 125 | |
| 3 | 80 | 120 | |
| 4 | 80 | 125 | |
| 2nd | 4 | 80 | 135 |
| 5 | 80 | 120 | |
| 6 | 75 | 130 | |
| 7 | 80 | 130 |
| Annotator | Slide | Stroma | Epithelium | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Man. | Model Small | Model Big | QP | Man. | Model Small | Model Big | QP | ||
| 1st | 1 | 137 | 120 | 122 | 205 | 164 | 145 | 161 | 203 |
| 2 | 149 | 165 | 164 | 219 | 180 | 182 | 181 | 193 | |
| 3 | 138 | 128 | 116 | 138 | 144 | 144 | 128 | 112 | |
| 4 | 183 | 178 | 179 | 201 | 137 | 159 | 141 | 161 | |
| 2nd | 4 | 187 | 181 | 184 | 201 | 150 | 167 | 159 | 176 |
| 5 | 131 | 109 | 114 | 100 | 150 | 142 | 138 | 169 | |
| 6 | 198 | 165 | 158 | 164 | 57 | 88 | 65 | 9 | |
| 7 | 180 | 168 | 188 | 182 | 202 | 198 | 219 | 278 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ushakov, E.; Naumov, A.; Fomberg, V.; Vishnyakova, P.; Asaturova, A.; Badlaeva, A.; Tregubova, A.; Karpulevich, E.; Sukhikh, G.; Fatkhudinov, T. EndoNet: A Model for the Automatic Calculation of H-Score on Histological Slides. Informatics 2023, 10, 90. https://doi.org/10.3390/informatics10040090
Ushakov E, Naumov A, Fomberg V, Vishnyakova P, Asaturova A, Badlaeva A, Tregubova A, Karpulevich E, Sukhikh G, Fatkhudinov T. EndoNet: A Model for the Automatic Calculation of H-Score on Histological Slides. Informatics. 2023; 10(4):90. https://doi.org/10.3390/informatics10040090
Chicago/Turabian StyleUshakov, Egor, Anton Naumov, Vladislav Fomberg, Polina Vishnyakova, Aleksandra Asaturova, Alina Badlaeva, Anna Tregubova, Evgeny Karpulevich, Gennady Sukhikh, and Timur Fatkhudinov. 2023. "EndoNet: A Model for the Automatic Calculation of H-Score on Histological Slides" Informatics 10, no. 4: 90. https://doi.org/10.3390/informatics10040090