Scale-up of the Reversible Addition-Fragmentation Chain Transfer (RAFT) Polymerization Using Continuous Flow Processing

Abstract
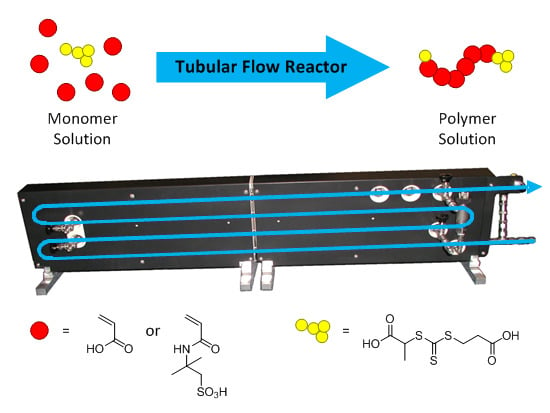
:
1. Introduction
2. Experimental Section
2.1. Materials and Analysis
2.2. RAFT Polymer Synthesis in Batch Using a Microwave Reactor—Scale: Up to 20 mL
2.3. RAFT Polymer Synthesis in Batch Using an Oil Bath—Scale: Up to 500 mL

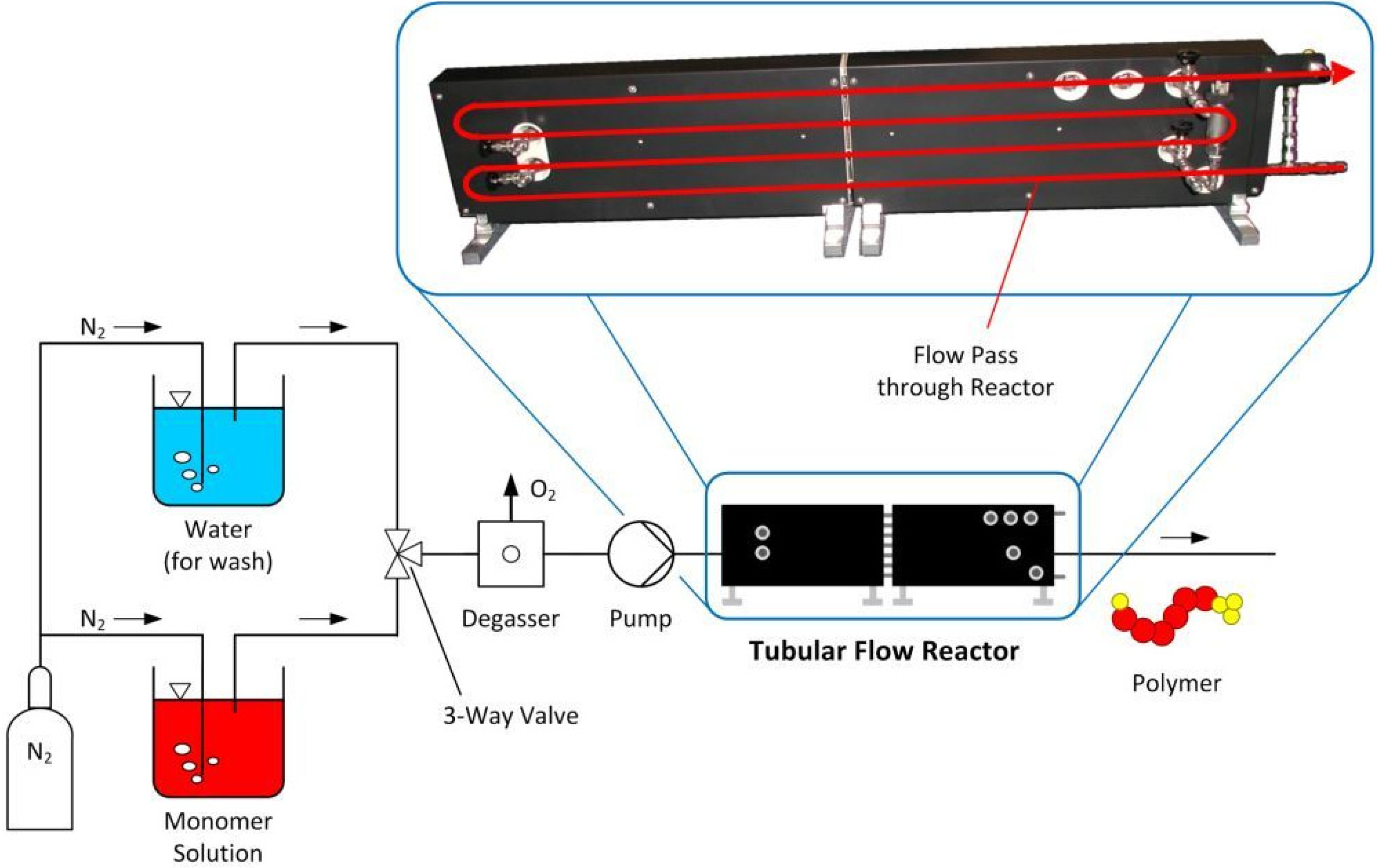
2.4. RAFT Polymer Synthesis in Continuous Flow—Scale: 500 mL

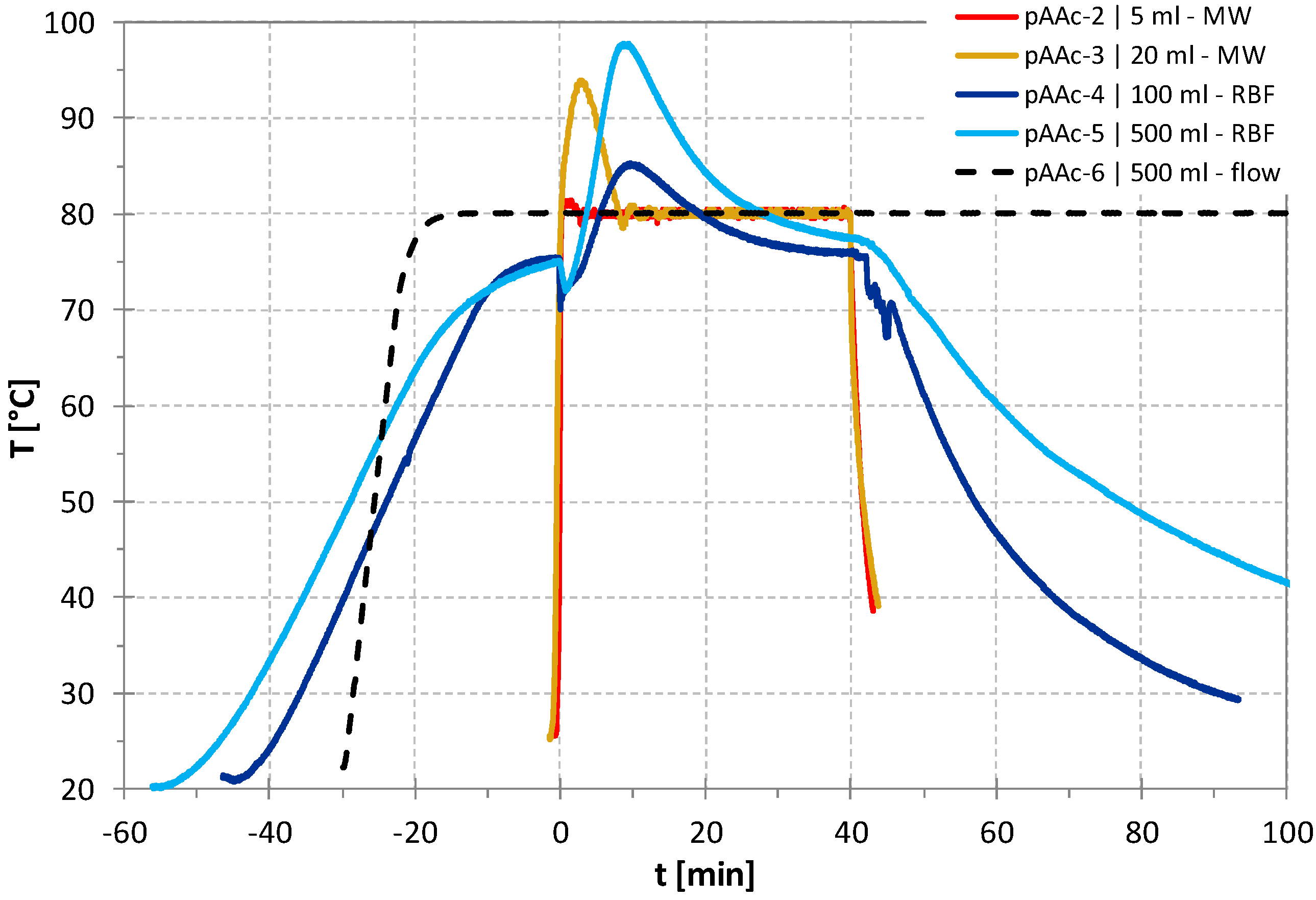
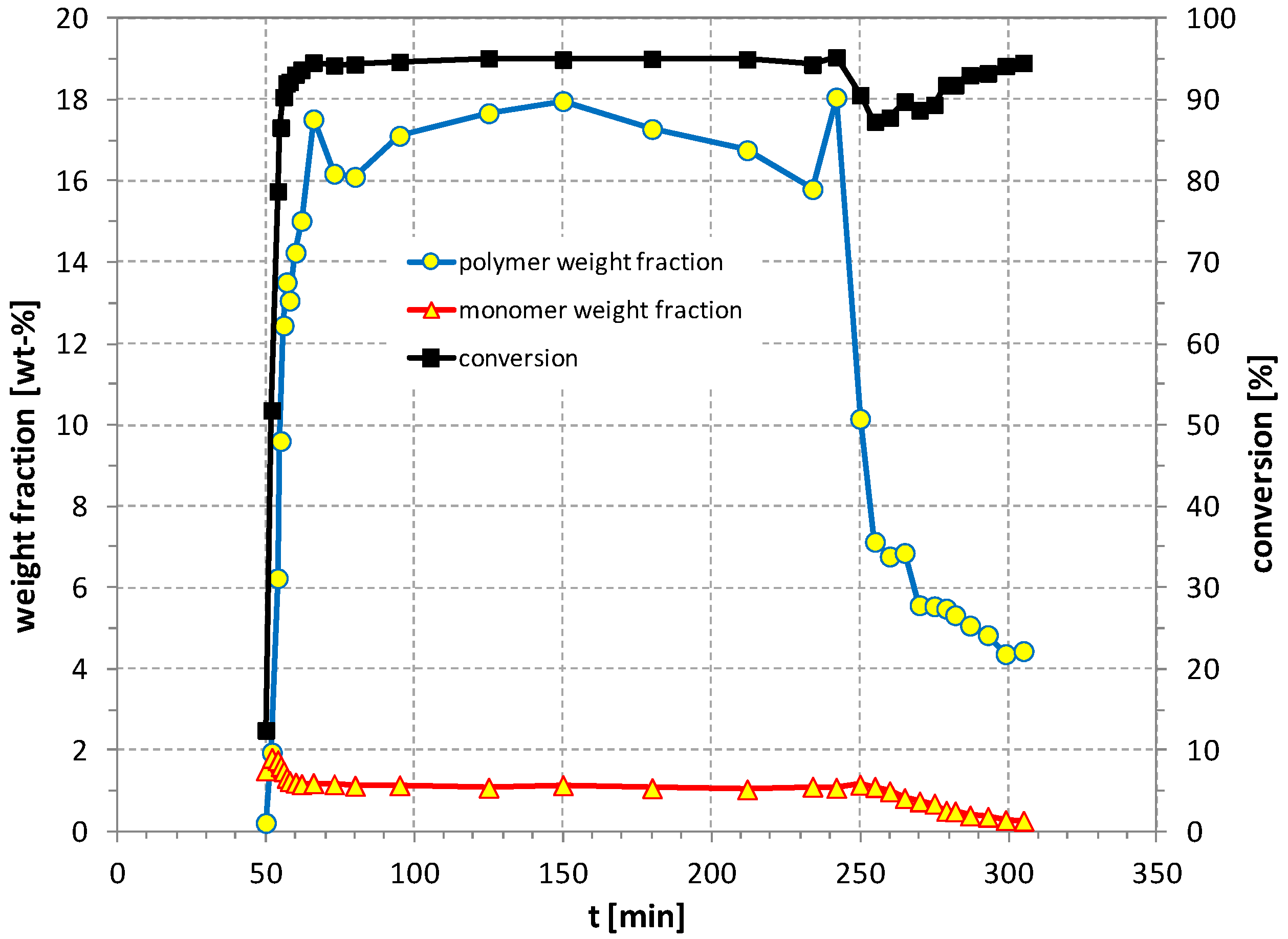
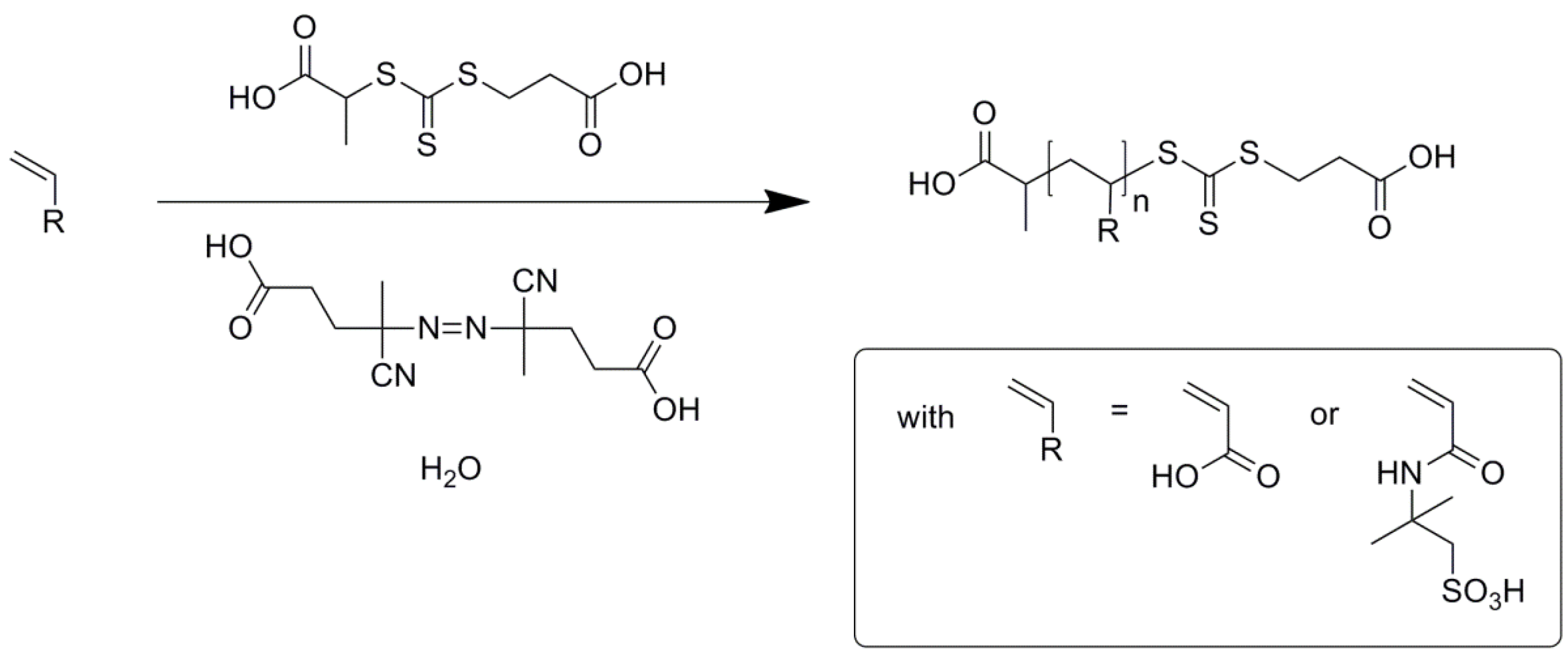
3. Results and Discussion


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Process | Scale (mL) | Monomer wt-% c | T (°C) | t (min) | Conversion (%) | Mn (g/mol) | Ð |
|---|---|---|---|---|---|---|---|---|
| pAAc-1 | batch a | 3 | 17.7 | 80 | 30 | 95.4 | 22,700 f | 1.35 f |
| pAAc-2 | batch a | 5 | 17.7 | 80 | 40 | 95.5 | 20,200 | 1.37 |
| pAAc-3 | batch a | 20 | 17.7 | 80 | 40 | 96.7 | 27,000 | 1.45 |
| pAAc-4 | batch b | 100 | 17.7 | 80 | 40 d | 94.2 d | 24,900 | 1.36 |
| pAAc-5 | batch b | 500 | 17.7 | 80 | 40 e | 97.4 e | 21,600 | 1.45 |
| pAAc-6 | cont. | 500 | 17.7 | 80 | 40 | 94.7 | 23,200 | 1.53 |
| pAMPS-1 | batch a | 5 | 30.0 | 80 | 30 | 96.3 | 32,200 f | 1.44 f |
| pAMPS-2 | cont. | 500 | 30.0 | 80 | 40 | 92.6 | 31,500 | 1.50 |


4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Chiefari, J.; Chong, Y.K.; Ercole, F.; Krstina, J.; Jeffery, J.; Le, T.P.T.; Mayadunne, R.T.A.; Meijs, G.F.; Moad, C.L.; Moad, G.; et al. Living free-radical polymerization by reversible addition—Fragmentation chain transfer: The RAFT process. Macromolecules 1998, 31, 5559–5562. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living radical polymerization by the RAFT process. Aust. J. Chem. 2005, 58, 379–410. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living radical polymerization by the RAFT process—A first update. Aust. J. Chem. 2006, 59, 669–692. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living radical polymerization by the RAFT process—A second update. Aust. J. Chem. 2009, 62, 1402–1472. [Google Scholar] [CrossRef]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living radical polymerization by the RAFT process—A third update. Aust. J. Chem. 2012, 65, 985–1076. [Google Scholar] [CrossRef]
- Barner-Kowollik, C. Handbook of RAFT Polymerization; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Hessel, V.; Renken, A.; Schouten, J.C.; Yoshida, J. Micro Process Engineering: A Comprehensive Handbook, 3 Volume Set; Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]
- Hessel, V.; Hardt, S.; Löwe, H.; Müller, A.; Kolb, G. Chemical Micro Process Engineering, 2 Volume Set; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Ehrfeld, W.; Hessel, V.; Löwe, H. Microreactors: New Technology for Modern Chemistry; Wiley-VCH Verlag GmbH: Weinheim ,Germany, 2000. [Google Scholar]
- Wirth, T. Microreactors in Organic Synthesis and Catalysis; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Deal, M. Continuous Flow Chemistry in Medicinal Chemistry. In RSC Drug Discovery; Farrant, E., Ed.; Royal Society of Chemistry: Cambridge, UK, 2012; Chapter 5; pp. 90–125. [Google Scholar]
- Fukuyama, T.; Ryu, I. Radical Chemistry by Using Flow Microreactor Technology. In Encyclopedia of Radicals in Chemistry, Biology and Materials; Chatgilialoglu, C., Studer, A., Eds.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2012; pp. 1243–1258. [Google Scholar]
- Baxendale, I.R.; Hornung, C.; Ley, S.V.; de Mata Muñoz Molina, J.; Wikström, A. Flow microwave technology and microreactors in synthesis. Aust. J. Chem. 2013, 66, 131–144. [Google Scholar] [CrossRef]
- Mawatari, K.; Kazoe, Y.; Aota, A.; Tsukahara, T.; Sato, K.; Kitamori, T. Microflow systems for chemical synthesis and analysis: Approaches to full integration of chemical process. J. Flow Chem. 2012, 1, 3–12. [Google Scholar] [CrossRef]
- Wiles, C.; Watts, P. Continuous flow reactors: A perspective. Green Chem. 2012, 14, 38–54. [Google Scholar]
- Hartman, R.L.; McMullen, J.P.; Jensen, K.F. Deciding whether to go with the flow: Evaluating the merits of flow reactors for synthesis. Angew. Chem. Int. Ed. 2011, 50, 7502–7519. [Google Scholar] [CrossRef]
- Oelgemöller, M.; Shvydkiv, O. Recent advances in microflow photochemistry. Molecules 2011, 16, 7522–7550. [Google Scholar] [CrossRef]
- Valera, F.E.; Quaranta, M.; Moran, A.; Blacker, J.; Armstrong, A.; Cabral, J.T.; Blackmond, D.G. The flow’s the thing...or is it? Assessing the merits of homogeneous reactions in flask and flow. Angew. Chem. Int. Ed. 2010, 49, 2478–2485. [Google Scholar]
- Ley, S.V.; Baxendale, I.R. The changing face of organic synthesis. CHIMIA Int. J. Chem. 2008, 62, 162–168. [Google Scholar] [CrossRef]
- Hessel, V.; Löb, P.; Löwe, H. Industrial and Real-life Applications of Micro-reactor Process Engineering for Fine and Functional Chemistry. In New Development and Application in Chemical Reaction Engineering; Proceedings of the 4th Asia-Pacific Chemical Reaction Engineering Symposium (APCRE ’05), Gyeongju, Korea, 12–15 June 2005; Elsevier: Amsterdam, The Netherlands, 2006; Volume 159, pp. 35–46. [Google Scholar]
- Seyler, H.; Haid, S.; Kwon, T.-H.; Jones, D.J.; Bäuerle, P.; Holmes, A.B.; Wong, W.W.H. Continuous flow synthesis of organic electronic materials—Case studies in methodology translation and scale-up. Aust. J. Chem. 2013, 66, 151–156. [Google Scholar] [CrossRef]
- Browne, D.L.; Wright, S.; Deadman, B.J.; Dunnage, S.; Baxendale, I.R.; Turner, R.M.; Ley, S.V. Continuous flow reaction monitoring using an on-line miniature mass spectrometer Rapid Commun. Mass Spectrom. 2012, 26, 1999–2010. [Google Scholar]
- Hornung, C.H.; Hallmark, B.; Baumann, M.; Baxendale, I.R.; Ley, S.V.; Hester, P.; Clayton, P.; Mackley, M.R. Multiple microcapillary reactor for organic synthesis. Ind. Eng. Chem. Res. 2010, 49, 4576–4582. [Google Scholar]
- Hornung, C.H.; Mackley, M.R.; Baxendale, I.R.; Ley, S.V. A microcapillary flow disc reactor for organic synthesis. Org. Process Res. Dev. 2007, 11, 399–405. [Google Scholar] [CrossRef]
- Van Rens, L.; van Dijk, H.; Mulder, J.; Nieuwland, P. Using a web application to conduct and investigate syntheses of methyl orange remotely. J. Chem. Educ. 2013, 90, 574–577. [Google Scholar] [CrossRef]
- McMullen, J.P.; Jensen, K.F. Rapid determination of reaction kinetics with an automated microfluidic system. Org. Process Res. Dev. 2011, 15, 398–407. [Google Scholar] [CrossRef]
- Serra, C.A.; Chang, Z. Microfluidic-assisted synthesis of polymer particles. Chem. Eng. Technol. 2008, 31, 1099–1115. [Google Scholar] [CrossRef]
- Iwasaki, T.; Yoshida, J.-i. Free radical polymerization in microreactors. Significant improvement in molecular weight distribution control. Macromolecules 2005, 38, 1159–1163. [Google Scholar] [CrossRef]
- Iwasaki, T.; Kawano, N.; Yoshida, J. Radical polymerization using microflow system: Numbering-up of microreactors and continuous operation. Org. Process Res. Dev. 2006, 10, 1126–1131. [Google Scholar] [CrossRef]
- Rosenfeld, C.; Serra, C.; Brochon, C.; Hadziioannou, G. High-temperature nitroxide-mediated radical polymerization in a continuous microtube reactor: Towards a better control of the polymerization reaction. Chem. Eng. Sci. 2007, 62, 5245–5250. [Google Scholar] [CrossRef]
- Wilms, D.; Klos, J.; Frey, H. Microstructured reactors for polymer synthesis: A renaissance of continuous flow processes for tailor-made macromolecules? Macromol. Chem. Phys. 2008, 209, 343–356. [Google Scholar] [CrossRef]
- Schork, F.J.; Guo, J. Continuous miniemulsion polymerization. Macromol. React. Eng. 2008, 2, 287–303. [Google Scholar] [CrossRef]
- Kessler, D.; Löwe, H.; Theato, P. Synthesis of defined poly(silsesquioxane)s: Fast polycondensation of trialkoxysilanes in a continuous-flow microreactor. Macromol. Chem. Phys. 2009, 210, 807–813. [Google Scholar] [CrossRef]
- Enright, T.E.; Cunningham, M.F.; Keoshkerian, B. Nitroxide-mediated bulk and miniemulsion polymerization in a continuous tubular reactor: Synthesis of homo-, di- and triblock copolymers. Macromol. React. Eng. 2010, 4, 186–196. [Google Scholar] [CrossRef]
- Nagaki, A.; Takahashi, Y.; Akahori, K.; Yoshida, J.-i. Living anionic polymerization of tert-butyl acrylate in a flow microreactor system and its applications to the synthesis of block copolymers. Macromol. React. Eng. 2012, 6, 467–472. [Google Scholar] [CrossRef]
- Vandenbergh, J.; de Moraes Ogawa, T.; Junkers, T. Precision synthesis of acrylate multiblock copolymers from consecutive microreactor RAFT polymerizations. J. Polym. Sci. A 2013, 51, 2366–2374. [Google Scholar] [CrossRef]
- Hornung, C.H.; Guerrero-Sanchez, C.; Brasholz, M.; Saubern, S.; Chiefari, J.; Moad, G.; Rizzardo, E.; Thang, S.H. Controlled RAFT polymerization in a continuous flow microreactor. Org. Process Res. Dev. 2011, 15, 593–601. [Google Scholar] [CrossRef]
- Hornung, C.H.; Postma, A.; Saubern, S.; Chiefari, J. A continuous flow process for the radical induced end group removal of RAFT polymers. Macromol. React. Eng. 2012, 6, 246–251. [Google Scholar] [CrossRef]
- Hornung, C.H.; Nguyen, X.; Dumsday, G.; Saubern, S. Integrated continuous processing and flow characterization of RAFT polymerization in tubular flow reactors. Macromol. React. Eng. 2012, 6, 458–466. [Google Scholar] [CrossRef]
- Hornung, C.H.; Nguyen, X.; Kyi, S.; Chiefari, J.; Saubern, S. Synthesis of RAFT block copolymers in a multi-stage continuous flow process inside a tubular reactor. Aust. J. Chem. 2013, 66, 192–198. [Google Scholar] [CrossRef]
- Chiefari, J.; Hornung, C.H.; Saubern, S. Continuous Flow Polymerisation Process. WO2012037596, 22 September 2010. [Google Scholar]
- Chiefari, J.; Hornung, C.H.; Postma, A.; Saubern, S. RAFT Polymers. WO2013086585, 14 December 2011. [Google Scholar]
- Wang, R.; McCormick, C.L.; Lowe, A.B. Synthesis and evaluation of new dicarboxylic acid functional trithiocarbonates: RAFT synthesis of telechelic poly(n-butyl Acrylate)s. Macromolecules 2011, 38, 9518–9525. [Google Scholar]
- Biotage. Available online: http://www.biotage.com/ (accessed on 7 November 2013).
- Cambridge Reactor Design Ltd. Available online: http://www.cambridgereactordesign.com/ (accessed on 7 November 2013).
- Saldivar-Guerra, E.; Vivaldo-Lima, E. Handbook of Polymer Synthesis, Characterisation and Processing; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Micic, N.; Young, A.; Rosselgong, J.; Hornung, C.H. Scale-up of the Reversible Addition-Fragmentation Chain Transfer (RAFT) Polymerization Using Continuous Flow Processing. Processes 2014, 2, 58-70. https://doi.org/10.3390/pr2010058
Micic N, Young A, Rosselgong J, Hornung CH. Scale-up of the Reversible Addition-Fragmentation Chain Transfer (RAFT) Polymerization Using Continuous Flow Processing. Processes. 2014; 2(1):58-70. https://doi.org/10.3390/pr2010058
Chicago/Turabian StyleMicic, Nenad, Alan Young, Julien Rosselgong, and Christian H. Hornung. 2014. "Scale-up of the Reversible Addition-Fragmentation Chain Transfer (RAFT) Polymerization Using Continuous Flow Processing" Processes 2, no. 1: 58-70. https://doi.org/10.3390/pr2010058