
A Quantitative Systems Pharmacology Perspective on Cancer Immunology

1
Department of Basic Pharmaceutical Sciences, West Virginia University, Morgantown, WV 26506, USA
2
Department of Chemical Engineering and Mary Babb Randolph Cancer Center, West Virginia University, Morgantown, WV 26506, USA
3
Department of Microbiology, Immunology, and Cellular Biology, West Virginia University Morgantown, WV 26506, USA
*
Author to whom correspondence should be addressed.
Processes 2015, 3(2), 235-256; https://doi.org/10.3390/pr3020235
Submission received: 7 December 2014
/
Revised: 2 April 2015
/
Accepted: 13 April 2015
/
Published: 22 April 2015
(This article belongs to the Special Issue Modeling and Analysis of Signal Transduction Networks)
{kind=link}
{kind=link}
Abstract
:The return on investment within the pharmaceutical industry has exhibited an exponential decline over the last several decades. Contemporary analysis suggests that the rate-limiting step associated with the drug discovery and development process is our limited understanding of the disease pathophysiology in humans that is targeted by a drug. Similar to other industries, mechanistic modeling and simulation has been proposed as an enabling quantitative tool to help address this problem. Moreover, immunotherapies are transforming the clinical treatment of cure cancer and are becoming a major segment of the pharmaceutical research and development pipeline. As the clinical benefit of these immunotherapies seems to be limited to subset of the patient population, identifying the specific defect in the complex network of interactions associated with host immunity to a malignancy is a major challenge for expanding the clinical benefit. Understanding the interaction between malignant and immune cells is inherently a systems problem, where an engineering perspective may be helpful. The objective of this manuscript is to summarize this quantitative systems perspective, particularly with respect to developing immunotherapies for the treatment of cancer.
1. Introduction
Motivated by a desire to improve human health, the pharmaceutical industry leverages biological discoveries to develop drugs that aim to restore health at significant financial investment. Overall the costs associated with pharmaceutical research and development, as represented by the US share of the market, has been increasing exponentially. The current estimate to bring a new medical entity to market requires upwards of approximately a $1–2.5 billion investment in research and development [1,2]. To recoup these financial investments, pharmaceutical companies are provided with protection from competition for a limited time by patenting their inventions. However, the estimated return on investment by the pharmaceutical industry has been experiencing an exponential decline in the last several decades. This trend, sometimes referred to as the innovation gap, presents a challenge for economic sustainability of the current model for innovation within the pharmaceutical industry.
The cost of pharmaceutical development escalates as drugs progress further from bench to market. In particular, Phase II clinical trials have become a key pinch point in the research and development pipeline, as it combines both significant risk and cost. This phase is the first time the efficacy of the drug is tested in real patients within the target disease and therefore has the highest probability of failure. It is also one of the phases with the highest out-of-pocket cost for the developer and other stakeholders [3]. Clinical trials are predicated largely on positive pre-clinical studies using animal models of disease. Unfortunately, animal modeling, which presents lower financial and human health hazards, may not always recreate the specific molecular and cellular networks associated with the pathophysiology and adverse reactions in human subjects [4]. Therefore, to lessen the costs of development and risk to humans in clinical trials, it is important to use appropriate models of the disease to maximize efficacy, safety, and benefit to patients.
In the last 50 years, computer-aided modeling and simulation has transformed a variety of industries, including financial portfolio management and the aerospace industry. Modeling and simulation in the financial sector has enabled real-time evaluation of economic performance measures using a mathematical model of the particular business sector to predict future performance and to optimize financial return [5]. In the aerospace industry, modeling and simulation is used to design new airframes, which eliminates the need for multiple physical prototypes constructed at intermediate points during design and reduces the time from concept to production [6]. In both cases, mathematical modeling and simulation provide a quantitative framework to capture our conceptual understanding of the modeled process and interpret heterogeneous data acquired from the process. These two examples also represent extremes of our conceptual understanding. Financial markets are complex systems that are influenced by a variety of observed and unobserved factors. Assuming that the underlying structure of the market is not changing, future behavior can be predicted using empirical mathematical models that are constructed using historical data. At the opposite end of the spectrum, computer-aided design of airframes captures physical principles such as the conservation of mass, which implies two physical objects cannot occupy the same space, and the governing physics associated with the performance objectives of the airframe. Similar to these industries, mathematical modeling and simulation has been proposed as approach to improve our understanding of the biological mechanisms targeted by a particular therapy [7]. This could help predict the outcomes of human clinical trials and, thereby, help bridge the innovation gap between cell and animal models and human pathophysiology, while also providing a cost savings in development, as computationally “expensive” modeling is inherently more cost effective than additional physical and biological models of disease [8]. In a 2011 National Institutes of Health White Paper, recommendations for quantitative systems pharmacology using quantitative experimental studies and model-based computational analyses that also incorporate clinical “omics data” were given in hopes of addressing clinical Phase II study failures in drug development and physiological, chemical, and biological disconnects in preclinical research [9].
Given the oncology slice of pharmaceutical research and development and the recent shift towards immunotherapies for cancer, the objective of this review is to summarize how modeling and simulation has aided in the understanding of biological changes associated with oncogenesis as it relates to immunity. In subsequent paragraphs, we provide a brief overview of the cancer and immune systems, tumor somatic and clonal evolution properties, and how cancer cell heterogeneity complicates therapeutic aims. We will also discuss recent immunotherapeutic advancements and the computational models used to describe the interactions between cancer and the immune system.
2. Emerging View of Cancer as a System
Oncogenesis is attributed to the accumulation of genetic mutations that lead to uncontrolled cell growth and proliferation. These mutations alter function of the modified gene through overexpression of the corresponding protein or rearrangement of a gene to create an entirely new protein that has dysregulated activity [10]. Mutations in specific genes that can, in isolation, transform a normal cell into a malignant cell are called oncogenes. Cancer drug development over the past several decades has been focused on targeting these oncogene mutations by inhibiting the function of corresponding proteins using small molecule drugs [11]. Researchers have scrutinized the altered signaling pathways in malignant cells in hopes of finding the key protein conserved in oncogenesis and metastasis but that plays minimal role in normal cells [12]. However, these drugs are rarely as efficacious in the clinic, where de novo and emergent drug resistance is common [13].
The small molecule inhibitor segment of the pharmaceutical industry is also associated with a view of cancer as a disease driven by malignant alterations that are intrinsic to or driven by the cancer cell [14,15]. This view can be represented by the six hallmarks of cancer discussed by Hanahan and Weinberg 2000 [16]. The six hallmarks summarize how genetic alterations change how a malignant cell senses and responds to extracellular signals in ways detrimental to the host. Assuming cancer is driven by the autonomous actions of malignant cells, the in vitro study of a cell line can be an appropriate model for identifying new therapeutic leads. This idea underpins using a collection of cell lines as a way to screen drugs that inhibit cell proliferation of exhibit cytotoxic activity in a high-throughput manner, such as the NCI-60 [17,18,19,20].
Previously to recent breakthroughs in immunotherapy, small molecule inhibitors were standard of care for many non-resectable metastatic diseases. B-Raf, a Raf kinase member of the MAP Kinase/ERK signaling pathway and involved in cell growth and proliferation, is a commonly mutated gene in many human cancers, such as metastatic melanoma. Vemurafenib and Dabrafenib are two FDA-approved B-Raf inhibitors used in the clinic that targets cancer with the B-RAF V600E (valine at amino acid position 600 to glutamic acid) mutation. However, cancer cells without the V600E B-Raf mutation may proliferate more in response to the vemurafenib drug [21]. Additionally, most metastatic melanoma patients become chemoresistant to both of these B-Raf inhibitors within 6 to 7 months of treatment. Therefore, combination therapies, such as vemurafenib with MEK-inhibitors like FDA-approved trametinib, are preferred to overcome the resistance mechanism in advanced melanoma, and may extend progression-free survival in patients by an average of about 3 months [22]. However, resistance will eventually reoccur and the patient will fatally relapse.
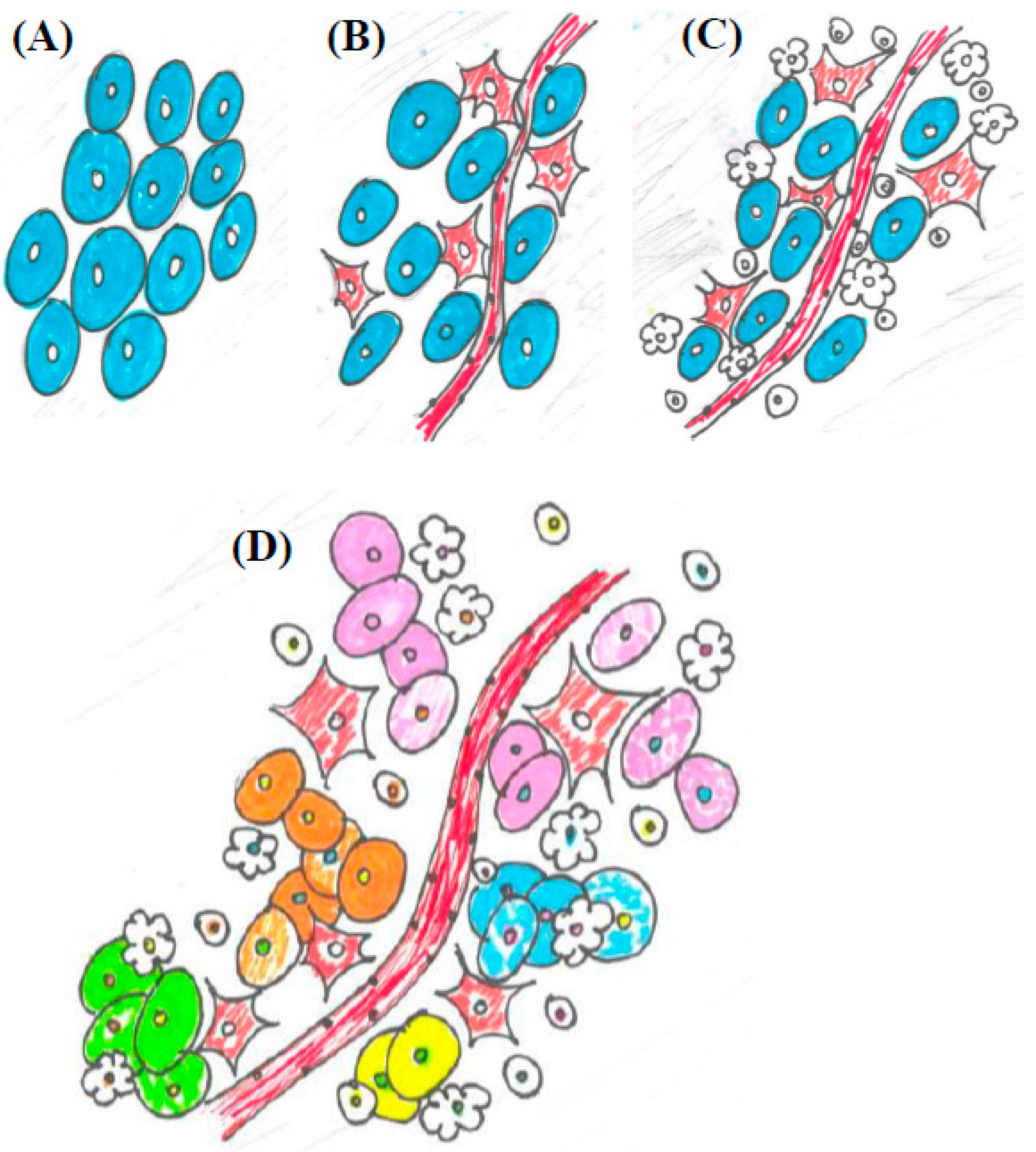
More recently, cancer research has expanded to include factors external to the malignant cell that contribute to oncogenesis. In 2011, Hanahan and Weinberg updated the hallmarks to include four new “emerging hallmarks and enabling characteristics”, which focus on changes associated with the malignancy that alter interactions among cells of the host [23]. The immune system was identified as having an influential role on tumor progression, and changes in metabolism and inflammation in the tumor microenvironment and throughout the body are known to have an effect on clinical outcomes, as well. This shift in perspective represents a malignancy as part of an integrated but dysfunctional system, rather than as an isolated mass of malignant clones. By incorporating the emerging hallmarks into the collective understanding of carcinogenesis, the ability of a malignant cell to manipulate its local environment and the immune system that it interacts with are being recognized as integral to tumor development and support the description of cancer as an evolutionary process (Figure 1). The ability of a malignant cell to maintain the tumor microenvironment hinges on dysfunctional intercellular communication [24,25].
A tumor is comprised of a variety of cell types, including a heterogeneous collection of malignant clones, various stromal cells that provide nutrients and facilitate remodeling of the extracellular matrix, and immune cells. Heterogeneity among malignant clones can exist within various morphologies or cellular phenotypes, producing cells originating from a similar origin, but may yet exhibit various structural, gene expression, signaling network, proliferative, metabolic, and metastatic differences (Figure 1). The existence of heterogeneity is a key element of evolutionary process. Somatic evolution is thought to change the dynamics of tissues [26,27], while evolutionary processes also maintains their own dynamics [28], both of which can influence development of heterogeneity within a particular cellular population. Resolving the issues surrounding multiple dynamics of biological systems and the influence of the immune and other cellular systems on tumors is technically difficult to replicate, particularly in in vitro models of cancer. The dynamics of cell communication can also influence the hallmarks of cancer of a potential malignancy and emerging hallmarks can alter the fitness, adaptive, or phenotypic landscapes [29,30,31]. Timing of response to signaling, especially that of the immune system, can mean the difference between malignancy proliferation and tumor eradication [32].
As part of the evolutionary process, recognizing the heterogeneity of the environment is crucial to understanding how alterations in cell signaling and immune response may promote tumors. Cancer cell heterogeneity within some tumors is linked to epigenetic differences in tumor cell genomes, perhaps from variances in cancer stem cells, while other examples of divergence from a single phenotype can be accounted for through clonal evolution, or even a combination of the two models of tumor propagation [33,34]. Therefore, we see that clonal evolution and heterogeneity are directly proportional to one another and will determine impact of immunological eradication or pharmaceutical treatment of the tumor. The remainder of this section will discuss models of somatic and clonal evolution and influence of heterogeneity of the tumor microenvironment on metastatic progression from a systemic perspective of cancer.
Figure 1.
An illustration of the conceptual progression of the tumor microenvironment from a reductionist view to cancer as a dynamic system. (A) A highly reductionist view. Tumors viewed as a homogenous population malignant clones; (B) Cancer cells integrated into the circulatory system, as represented by including blood vessels and fibroblasts, as seen in Hanahan and Weinberg 2000; (C) The tumor microenvironment becomes more complex. In addition to blood vessels and fibroblasts, immune cell types of various kinds are introduced into the system, as seen in Hanahan and Weinberg 2011; (D) The emerging view of a malignancy as a heterogeneous and dynamic system. Not only are blood vessels, fibroblasts, and immune cells in the tumor microenvironment, but there are distinct clonal populations of cancer cells intermingled with a variety of different immune cells and other cell types.
Figure 1.
An illustration of the conceptual progression of the tumor microenvironment from a reductionist view to cancer as a dynamic system. (A) A highly reductionist view. Tumors viewed as a homogenous population malignant clones; (B) Cancer cells integrated into the circulatory system, as represented by including blood vessels and fibroblasts, as seen in Hanahan and Weinberg 2000; (C) The tumor microenvironment becomes more complex. In addition to blood vessels and fibroblasts, immune cell types of various kinds are introduced into the system, as seen in Hanahan and Weinberg 2011; (D) The emerging view of a malignancy as a heterogeneous and dynamic system. Not only are blood vessels, fibroblasts, and immune cells in the tumor microenvironment, but there are distinct clonal populations of cancer cells intermingled with a variety of different immune cells and other cell types.

2.1. Somatic Evolution
Somatic evolution, as distinct from classical Darwinian evolution, is a driving force for carcinogenesis [35]. The majority of somatic mutations within the genome are neutral, but accumulations of even non-deleterious mutations can alter the fitness landscape of an organism via genetic drift [36,37]. Changes in the fitness landscape include alterations in cellular communication [38], such as upregulation or downregulation of receptors, intermediate players, or downstream products, alterations in pathway activity, or mutations in pathway protein partners. This can promote selection of advantageous clones [39]. More importantly, these malignant clones can change the fitness landscape in such a way that the local microenvironments become a favorable niche for malignant cells. A particular case of somatic evolution is clonal evolution, which considers how the environment influences the selection of pre-existing clones within a heterogeneous population and neglects the influence of mutagens to further diversify the clonal population. Somatic and clonal evolution may provide many benefits to a cell. Changes in the environment of the cell may force it down a particular path, providing a fitness advantage for growth and proliferation. While most of these particular mutations may provide benefit for survival of the host, these alterations can promote further genomic instability, leading to tumorigenesis.
Reductionist theories and experimentation usually examines one input and one output of a particular system, and therefore may be inadequate in describing cell processes with appropriate models, especially in relation to somatic and clonal evolution within a population. A cell is ever-sensing of its microenvironment, ready to respond to relevant extracellular signals present in its vicinity, whether the cues are coming from other cell types, other tissues, or from itself. These cues can also have a profound impact on the evolution of a cellular population, depending on duration, strength, and ability of the cell machinery to respond to the signal. This communication may come in the form of a soluble ligand or direct cell contact and provide positive or negative feedback to the cell.
As a heterogeneous environment, a variety of signals are occurring and being processed by the target cell at any time and are difficult to observe with traditional models. Various factors, driven by cellular communication, can affect the fitness of a particular population of tumor cells, such as availability of nutritional or metabolic resources, presence and strength of immune system influences, and ability of the cell to interact with the extracellular matrix. To combat this hurdle, development of a hybrid-discrete continuum (HDC) model to examine tumor morphology and metastatic potential was developed by Anderson et al. [40], which leverages the impact of multiple factors of the tumor microenvironment on somatic or clonal evolution. This mathematical model allows for “random” influence of particular set of variables, while retaining deterministic properties of known inputs with known outputs.
Related to factors surrounding use of the HDC computational model is the “clonal dominance theory” developed by Kerbel and colleagues. While clonal dominance was easily observed in vivo in mammary tumors, replication under ideal growth conditions was difficult. The group examined the growth of two subpopulations, non-metastatic SP1 and metastatic variant C1, of cells under both ideal and non-optimal growth conditions. Under optimal conditions, both subpopulations of cells grew at the same rate, while under the “stressed” conditions, emergence of the “dominant” metastatic subpopulation C1 became evident. In addition, transforming growth factor-beta (TGFβ) and extracellular matrix-driven cell-to-cell communication between the two subpopulations was indicated as being required for assistance of the dominant metastatic subpopulation to maximum capacity [41]. This study highlighted the importance of culture conditions and physiological dimensions, such as serum concentration and spheroid growth, for in vitro work to best replicate in vivo tumors, given the importance of cell-to-cell communication in tumor development and maintenance. However, researchers often overlook the effect of these conditions on clonal evolution, and therefore, how these conditions may affect whether a certain model is appropriate for judging efficacy of a treatment in in vitro or in vivo experiments.
Other features that impact clonal dominance in the evolution of cancer include consideration of compartment size. All tissues can be viewed as “compartments”. Clonal expansion readily occurs within these compartments and can be restricted by neighboring clones [42]. As shown by Michor et al. in a mathematical modeling study, clonal expansion within a small compartment is driven by genetic drift and is quickly dominated by a certain genetically unstable subpopulation. However, the expansion is readily limited by boundaries of the compartment and is therefore likely to be contained. Conversely, large compartments will contain a variety of subpopulations, some tumorigenic; however, the effect of clonal expansion is diluted unless both alleles of a particular gene are affected, or selective advantage favors the tumorigenic subpopulation [43]. Once a tumorigenic subpopulation has taken over the compartment, success of the neoplasm and resulting metastasis is a more probable scenario. This highlights the importance of cell-communication through cell-to-cell contact and local regulation of homeostasis in limiting genomic instability. Experimental cell models may reflect these effects, and therefore cellular confluency in vitro, or tumor transplant location in vivo may affect treatment outcomes.
Driver mutations and passenger mutations are another area of investigation in regards to computational models of the evolution of cancer. Driver mutations result in functional or morphological differences of cells, while passenger mutations are a neutral consequence of the evolutionary process [44]. In a meta-analysis study of lung and ovarian cancer, Youn and Simon found that metastasis as a result of cancer evolution was related to the age of the tumor and number of cell generations. The difference in metastatic potential as a function of generation or age of the tumor was related to the ability of the parental lineage to self-renew. For example, more generations were required for ovarian cancer to metastasize versus the lung cancer cell, which self-renews more gradually. The study was conducted by using passenger mutations, which do not confer a selective growth advantage in clonal expansion of the tumor, to estimate when the driver mutation, developed in the early stages of tumorigenesis and linked to causation of the tumor, had occurred and therefore, the approximate generational age of the tumor [45]. This study highlights the tumor progression differences in cell line or cancer types are largely attributed to clonal differences and how this may have an effect on treatment outcomes, not only in cell and animal model experiments, but also in the clinic.
2.2. Heterogeneity of the Tumor Microenvironment
We have examined the effects of clonal expansion in tumorigenesis and how compartment size and tissue dynamics may affect this process. Clonal expansion implies a somewhat homogenous subpopulation of cells dominates a certain microenvironment, compartment, or tissue. However, the heterogeneity of tumor microenvironments and tissues is well described [46]. Not only does the tumor stroma contain different cell types, such as immune cells, and structural support cells, but tumor cells themselves are part of a heterogeneous population. Different subpopulations of various clones exist, each expressing a variety of biomarkers and each clone with their own signaling dynamics, within the total tumor cell population.
Analysis of National Cancer Institute (NCI) and National Human Genome Research Institute (NHGRI) TCGA (the Cancer Genome Atlas) Project has revealed that sequencing of human tumors has provided a wealth of genomic information. The goal of this project was to determine common genetic alterations in various cancer types and subtypes, and use this information to improve clinical outcomes and tailor therapies to individuals. However, the impact of this data on improving clinical outcomes or identifying successful drug targets within corresponding patient groups remains limited. While the TCGA project was conceptually born out of the oncogene perspective, recognizing that tumors contain a variety of heterogeneous tumor clonal populations and prevalence within these clonal subsets of varying abundance could have a profound impact on the information gathered and possibly skew data interpretation [12]. Molecular-targeted therapies focusing on one set of genetic alterations would have little effect on a subpopulation expressing an entirely different set of alterations, as seen within the heterogeneous tumor. This may be one reason combination therapies have been so effective in treating certain types of cancer, as seen with the synergistic effect of p53 vaccines and chemotherapy in an evolutionary double bind study conducted by Anderson and colleagues [47].
As discussed previously, clonal evolution plays a critical role in the development of a tumor, but tumor heterogeneity may be a critical aspect for tumor persistence. Different tumor clonal populations may allow for evasion of the immune system. If tumor antigens produced vary from cell to cell within the tumor, a targeted immune system response will become diluted, allowing immunoescape of the tumor. This type of clonal selection provides for a phenomenon described as immunoselective pressure. By allowing for targeted killing of a specific tumor cell population expressing a given antigen, other tumor cell populations may be able to fill the tumor niche, especially cell populations that do not express an immunogenic antigen. Tumor cells sensitive to the inflammatory response, generalized apoptosis signals, or other factors in the microenvironment, may also be eliminated, while more resistant cells are allowed to proliferate. Additionally, cytokines may provide selective pressure to develop mutations that result in clones that are able to overcome these immune signals targeting the cells for destruction [48]. This phenomenon is called immunoediting. A deadly combination of elimination of immune-sensitive tumor cells, equilibrium of tumor cells that have survived elimination, and escape of resistant clones limits tumor eradication by the immune system [49].
Clonal heterogeneity can also provide survival advantages depending on the location of the clonal subset within the tumor microenvironment. Mutations that promote angiogenesis, for example, would provide a survival advantage in a tumor region not within immediate contact of the vasculature and perfusion of blood and nutrient resources. Alternatively, mutations that upregulate multidrug resistance transporters would provide a survival advantage in areas of the tumor that is most susceptible to contact with chemotherapeutic agents, such as in the periphery of the tumor that is most accessible to drug delivery. To explore this relationship between tumor heterogeneity and persistence, a computational study by Michor and colleagues examined the effect of immune system response and chemotherapeutic interventions in tumor escape [50]. They developed a mathematical model to describe original tumor cell and variant tumor cell fitness and number, mutation rate of the cancer cells, competition between variants, tumor cell elimination rate, interactions between tumor cells and cytotoxic T lymphocytes (CTLs), and the proliferation and decay of CTLs. They found that the more variants that exist within a tumor, the more likely the tumor will catastrophically or partially escape from immune system or chemotherapy attack. However, a certain level of homeostasis in variation must be maintained to support a functional genome for survival of the tumor.
In summary, a tumor is not only heterogeneous in its various clonal populations and expression of various tumor antigens, but is also heterogeneous in terms of weaknesses. By directing therapy at several weaknesses at once, instead of one at a time, or by targeting the interplay between tumor and other systems it interacts with, this lessens the probability of a certain clonal population gaining proliferative momentum in the tumor niche. Tumor heterogeneity is extremely important for the success of a tumor cell in the microenvironment and relies on the dynamics between the immune system, rates of genetic alterations, and sensitivity of variants to chemotherapeutic or immune system attack.
3. The Re-Emergence of Cancer Immunotherapy
The immune system has long been suspected as influential on cancer development. In the early 1890s, Dr. William Coley noticed a reduction in tumor size of patients with bacterial infections. He began injecting live bacteria into the tumors, and later developed a safer concoction, termed “Coley’s toxins”, with mixed results [51]. Over time, the field shifted away from cancer immunology research, as cytokines and immune cell types became well defined, yet cancer treatment with interleukins and other cytokines yielded variable results. Interleukin-2 showed efficacy in treating metastatic melanoma. However, its adverse side effects, inefficacy in large tumors, and high rates of toxicity such as allergic reactions and seizures discouraged widespread use in patients [52,53,54,55]. Interleukin-12 was also investigated as a cancer drug for its robust anti-angiogenic and immune cell-promoting activity. Although it had shown promise in preclinical trials, a patient death in a study designed to test safety diminished enthusiasm to pursue systemic delivery of IL-12 as a potential therapeutic [56,57].
Most of the failures of immune cell-promoting cytokines to eradicate tumors in patients could be attributed to inappropriate animal models and the lack of target specificity of these immunotherapeutic agents, as seen with the inability of cytokines to possess the sensitivity and specificity to function as appropriate biomarkers [58]. In an effort to gain specificity, antibodies, such as Rituximab, were developed and approved for the treatment of cancer [59]. This development of antibodies to target cancer has continued to this day, with the break-through of ipilimumab in metastatic melanoma [60]. Through this process of repeated success and failures, the view of cancer immunology has changed, and once considered a flame extinguished, has reignited from the embers.
3.1. The Cancer Immunology System and Systems Therapeutics
The view of cancer and the immune system as an integrated system is an important perspective to develop additional targets for cancer immunotherapy. Being cognizant of the interplay of systems is the first step to developing an appropriate model to test hypotheses. As described by Chen and colleagues, the “cancer-immunity cycle” involves an intricate process of cancer cell antigen recognition by the immune system, immune cell trafficking and tumor infiltration, and a localized immune response to eradicate the tumor [61]. At each step of the process, a variety of cytokines may be expressed to promote or inhibit the immune response. Checkpoints in this process exist to support a balance of effective activity to maintain tissue homeostasis and discourage fluctuations that could lead to the extremes of immunosenescence or autoimmunity. The following paragraphs, we will discuss oncolytic viruses, adoptive cell transfer, chimeric antigen receptors, and immune checkpoint modulators, in context of their role in the cancer-immunity cycle. Each is able to alter feedback mechanisms to promote the cancer-immunity cycle via various methods. The advantages and disadvantages of each of these therapies will also be discussed.
3.1.1. Oncolytic Viruses
Oncolytic viruses are used to target and kill cancer cells. There are two methods used to target the oncolytic viruses to kill only cancer cells, and they can be used in combination to promote efficacy of tumor-targeted killing. Transductional targeting of oncolytic viruses entails modifying the viral coat proteins of the virus to target malignant cells preferentially by inhibiting the entry of the virus into non-cancerous cells. Non-transductional targeting genetically alters the virus so that it may only replicate in the targeted cancer cell, whereby tumor-specific transcriptional promoters are used [62]. However, the host immune response to oncolytic viruses may vary between individuals, and mechanisms of resistance to and effective delivery of oncolytic viruses are poorly understood [63]. The variation among patients in immune response to oncolytic viruses provides a significant clinical barrier to their use and efficacy. However, oncolytic viruses may be used as an experimental tool to provide insight to mechanisms of cancer evolution and immune system response in virally-mediated cancers.
3.1.2. Adoptive Cell Transfer and Chimeric Antigen Receptors
A number of approaches have been proposed to jumpstart the cancer-immunity cycle and maintain its efficacy. Adoptive Cell Transfer (ACT) expands T cell immunity to a particular cancer antigen in the patient through isolation of T lymphocytes, population expansion, and reinfusion. Cells can be genetically modified to recognize certain antigens, to infiltrate tumors more readily, or to respond more robustly to cytokine cues [64]. One example of genetic modifications that can be integrated into T lymphocytes in ACT involves chimeric antigen receptors (CARs). These receptors are engineered to recognize a specific antigen, which allows this treatment method to become tailored to an individual type of cancer and the antigens it expresses [65]. Use of CARs in conjunction with ACT also overcomes a potential barrier with ACT and other immune modulation therapies, which assumes that a patient’s T lymphocytes or that of a donor will recognize the patient’s cancer cells and target them for killing. As shown by Chacon et al. [64], using tumor-infiltrating CD8+ lymphocytes in ACT can also have an impact on the rest of the tumor microenvironment, expanding a dynamically regulated and more competent T cell population for killing of the tumor.
ACT and CARs can be modeled mathematically in two ways: (1) through mathematical modeling of the system effects and response of directed ACT and CAR activity; and (2) by modeling molecular level CAR interactions. When designing CARs, it is important to remember the effect of this type of therapy on the host’s system. The plasmids used to express classical CARs use a signaling fragment, an extracellular spacer, a co-stimulating domain, and an antibody to direct a specific response against the tumor. However, using single-chain fragment variable (scFv) antibodies can cause activation of the immune system in an undesirable manner. Single variable heavy chain domains (VHH) have been shown to avoid immunogenicity. However, changing any component of the CAR can lead to poor interaction of the receptor with its target. To improve CAR targeting, molecular modeling can be used to predict interactions between targets and the CAR with various components, such as a VHH directed against a particular target [66]. At the system level, a recent study by James and colleagues modeled the target lysis achieved by alterations in chimeric T cell receptor (cTCR) expression density, target antigen density, and activation of the cTCR. They found that approximately 20,000 cTCRs per cell was an ideal density of expression of the surface of the T cell. Anything above this did not increase target lysis or increase sensitivity to the target antigen, and anything below this expression impaired target lysis activity, perhaps by causing antigen-induced T cell death [67].
3.1.3. Immune Checkpoint Modulators
Based on remarkable clinical success, the current immunotherapies that hold the most promise are immune checkpoint modulators. Immune checkpoints are considered natural negative feedback mechanisms that limit an adaptive immune response to minimize the risk of autoimmunity. Investigation of immune checkpoint modulators has been on the rise in academia, biotechnology group, and pharmaceutical company research [68]. It is also known that in late stage cancers, the immune system appears to be turned “off”. Part of this “immune switching” phenomenon can be credited to the tumor actively evading the immune response through its clonal evolution of certain cellular subpopulations [69]. As an example, metastatic melanoma is arguably one of the hardest cancers to treat and has very few treatment options in the clinic. As an immunogenic cancer, it is known the immune system plays a role in progression of the disease [70]. Therefore, the immune system was targeted as a potential solution to molecular inhibitor resistance in metastatic melanoma and other cancers. The particular proteins currently being examined for targeting include CTLA-4, cytotoxic T-lymphocyte antigen-4, and PD-1, programed death receptor-1, two regulatory molecules on the surface of T cells [71].
A neutralizing antibody against CTLA-4, called ipilimumab, improved overall and progression-free survival in metastatic melanoma [72] and was the first FDA-approved therapy to target cancer through an immune checkpoint blockade. While clinical benefit was observed in only a subset of patients, ipilimumab demonstrates an important proof-of-principle especially in metastatic melanoma. Anti-CTLA4 immunotherapy, such as found with ipilimumab, is able to target senescence of CD8+ cytotoxic and CD4+ T cell populations. It does this by blocking the effect of CTLA4, a T cell inhibitory molecule similar in function to costimulatory protein CD28, on the surface of the T lymphocyte that limits clonal expansion [73]. As this therapy globally increases T cell numbers, serious adverse side effects, such as autoimmunity, are directly linked to this function, by causing over-activation of the immune system. Ipilimumab is currently being investigated for potential treatment of other cancer types, while other immunotherapies targeting T cell response are also being tested for efficacy against melanoma and other cancer types to improve efficacy and safety [68].
Similar to the actions of CTLA-4, PD-1 is also an inhibitory protein expressed on the surface of chronically activated T cells, which show decreased TCR-mediated proliferative and cytokine-releasing ability. Its expression is increased, compared to peripheral blood cells, on tumor-infiltrating T-lymphocytes [74,75]. PD-1, along with its ligand that is also upregulated in the tumor microenvironment, PD-L1, is currently being investigated as a potential immunotherapy target in clinical trials. An anti-PD-L1 antibody was administered to patients with melanoma, colorectal, renal-cell, ovarian, pancreatic, gastric, breast, or non-small-cell lung cancers. Patients were treated for an average of 12 weeks and evaluated. A durable tumor regression response was recorded at 6%–17%, while 12%–41% of patients experienced prolonged disease stabilization over a period of 24 weeks [76]. Drugs targeting PD-1 or PD-L1 seem to have a slightly better safety profile compared with anti-CTLA4 and may prove to be more effective as combination therapies [32,77].
3.1.4. Summarizing the Rise of Immunotherapies
Overall, immune system modulators, ACT therapies, and oncolytic viruses all show promise clinically, but their considerable development and clinical costs, safety profiles, and limited efficacy in certain patient populations are a substantial obstacle to a broad clinical impact on patient response and management. However, these technologies and molecular targets may not only be useful as therapeutics to treat patients in the clinic, but can be used as preclinical tools to investigate and better define the interplay between cancer and immunology systems for further pharmaceutical development. To ensure these tools are used in a way that minimizes financial and patient risk in pharmaceutical development, it is necessary to use these tools in appropriate preclinical models for investigation and success of future cancer immunotherapies. In the next section, the use of appropriate system models that should be used with these potential preclinical and clinical tools for cancer treatment will be discussed.
3.2. Modeling the System for Pharmaceutical Aims
Knowing the particular causal suppression mechanisms at work in a cancer from observations of biological state is one of the most pervasive problems in the analysis of physiological systems. In engineering, this problem is called an identification problem, where causal relationships between system elements are inferred from a set of input cues and output responses [78]. In context of cancer, an input cue may be antibodies against tumor-specific epitopes and an output response may be tumor regression. Many approaches exist for the identification of simple-input-simple-output (SISO) systems―where a change in input causes a unique change in output.
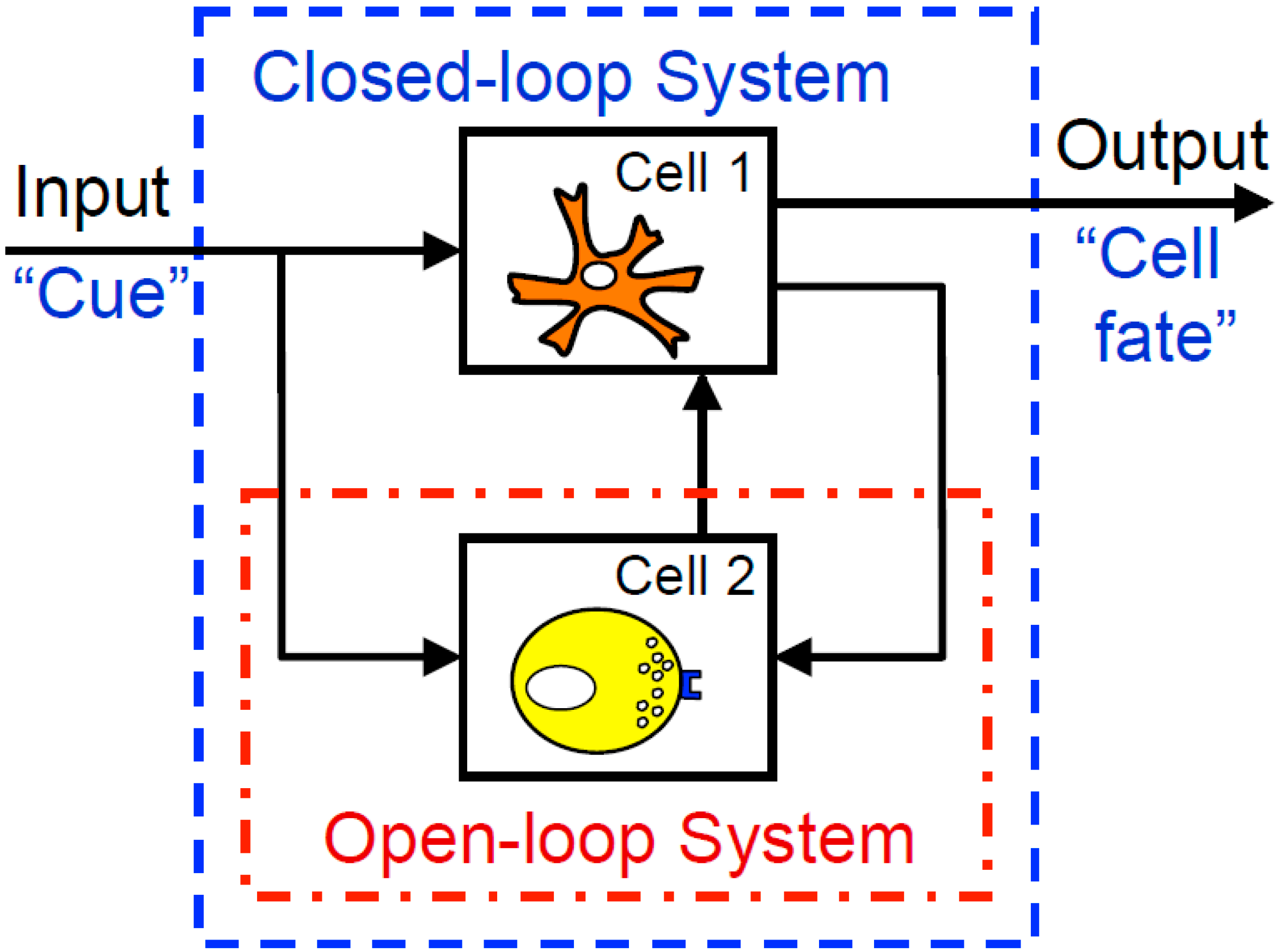
As a consequence of reductionist methods, there is a wealth of experimental data that characterize how isolated elements of physiological systems respond to inputs. However, approaches for identifying causal relationships among elements of more complex integrated closed-loop systems, like the immune system, are less well developed. Typically, a closed-loop system is defined as a multi-element system where the output (i.e., response) of one element provides the input (i.e., biochemical cue) to another element. A schematic diagram of a closed-loop system comprised of two cell types is shown in Figure 2. Closed-loop systems are particularly challenging as it is impossible to identify the relationships among cells of a system based upon overall input (e.g., tumor vaccines) and output (e.g., tumor regression) measurements. One of the reasons for this is that changes in the internal state of the system may alter the response of the system to a defined input, such that there is not a direct causal relationship between overall system input and output.
Figure 2.
Studying open-loop cellular systems, as indicated by the red open-dash box, involves directly examining at a target cell or a cell population, without regard to the other cells, inputs, or outputs that may be affecting the behavior of that particular cell. Closed-loop systems examine multiple components of the overall system, including cues going into the system, interactions within the cellular environment, and outputs resulting from the multiple dynamics of cellular signaling and communication.
Figure 2.
Studying open-loop cellular systems, as indicated by the red open-dash box, involves directly examining at a target cell or a cell population, without regard to the other cells, inputs, or outputs that may be affecting the behavior of that particular cell. Closed-loop systems examine multiple components of the overall system, including cues going into the system, interactions within the cellular environment, and outputs resulting from the multiple dynamics of cellular signaling and communication.

Historically, the causal mechanisms underlying the behavior of closed-loop systems in physiology have been identified via ingenious methods for isolating elements within the integrated system (i.e., “opening the loop”). A classic example of this is the discovery of insulin and its role in connecting food intake to substrate metabolism. As insulin is only produced by the endocrine pancreas, measuring plasma insulin provides a direct measure of the organ-level communication between food intake and substrate metabolism in the peripheral tissues. The pancreas can then be approximated as a SISO system where the glucose concentration in the portal vein is the input and insulin release into the plasma is the output, as depicted in the Minimal Model for the regulation of blood glucose [79]. Measuring changes in insulin in the blood in response to changes in plasma glucose provide the basis for partitioning alterations in system response (e.g., diabetes) into deficiencies in insulin production (i.e., type 1 diabetes) and insulin action (i.e., type 2 diabetes). Treatment for diabetes is tailored to the deficiency in component function that exists in the patient. In diabetes, “opening the loop” means identifying organ-level cross-talk using blood measurements. In contrast, the cell-level cross-talk between tumor and immune cells occurs locally within the tumor microenvironment and may not reach a titer sufficient enough to detect using blood measurements.
3.2.1. Methods to Quantify and Characterize Cross-Talk: Developing Appropriate Models
Because biological systems are dynamic and require inputs from a variety of sources to maintain homeostasis, pathway cross-talk is essential for regulation of equilibrium. Biologists have often depended on methods to quantify this cross-talk that introduce scientific bias. Immunohistology, for example, requires the researcher to know what they are probing for. The same concern exists in Western blotting, classical reverse transcriptase-polymerase chain reaction (RT-PCR), ELISAs, and flow cytometry, which require labeling or probing for a specific molecular component. Because levels of gene expression and proteins are dynamic in that they change with respect to both time and response to a stimulus, it is difficult to say with certainty that a particular stimulus is direct cause of protein expression or whether cross-talk between molecular pathways is involved and causing an upregulation or downregulation of a particular gene and its translation into protein gene product. This requires the researcher to have extensive knowledge of “known” pathways and relies on previous data to support the hypothesis and conclusions.
Discovery-based research, using high-throughput methods to monitor expression level changes in a wide variety of targets, was one way to combat this “known pathway” requirement in cancer biomarker research. Discovery-based research, because it looks at a variety of targets, which may or may not be known to have pathway association, may be thought of as a way to combat methodological bias. However, in his Nature opinion paper regarding biomarker research, Ransohoff argued target-driven discovery-based research methods still portray a bias component, as “cancer group” samples may be handled in a particular way or undergo particular procedures during collection and storage, whereas “normal tissue” would not. In sensitive assays, which themselves may have these molecular pathway biases, this can make data interpretation, statistical analyses, and conclusions difficult and a technical bias may be inadvertently contributed to the protocol. Ransohoff also discusses “fitting” of the model during biomarker research and how this can become a bias in data analysis and conclusions [80]. We expect certain associations of molecules or patterns in signaling, and the researcher then tries to fit these patterns to a certain disease. However, as protein and mRNA expression levels are highly variable and individual samples or subjects can have divergent baselines, fitting the data to a certain model of gene or protein expression may not be a dependable method of determining disease status conclusions.
One of the great challenges of biochemical research is that the biological activity of many gene products are unknown, and therefore, it is unclear as to how they may influence cellular response mechanisms. Proteomics workflow methods may remove some of the bias of probing for known protein products. Studies conducted by our laboratory on the secretomes of a “normal” breast epithelial line compared to the breast cancer lines BT474 and SKBR3 provided a wealth of information on protein products that may be selectively secreted by breast cancers. We performed this workflow by isolating and enriching the secretomes from the cell lines, running the samples on a 2DE-gel and using MALDI-TOF analysis to identify proteins secreted [81]. To gain insight into common mechanisms for altering local intercellular communication in breast cancer, we used computational tools to identify common pathway alterations an as a way to counter uncertainty due to the inherent variability among samples and limits to sensitivity of this experimental approach.
An alternative approach to identify alterations in intracellular communication is to develop phenotypic screening assays. While the contemporary focus has largely been on target-based drug discovery, phenotypic screening produced greater than 60% of first-in-class small molecule drugs approved by the FDA between 1999 and 2008 [82]. Phenotypic screening assays are one method of discover-based research that does not rely on target-based screening, as do many discovery-based research methods, and is making a return to the field of pharmaceutical research [83]. Phenotypic screening assays rely on observing the effects of a particular stimulus on a given outcome, rather than the players that may cause that outcome. It allows for some uncertainty on what particular pathways are involved to effect disease status and can give insight to unknown molecular interactions and molecules acting via previously undiscovered mechanisms. To illustrate the approach, we developed a phenotypic screening assay to identify biochemical cues responsible for local immunosuppression in vitro, validated these mechanisms in human tumor biopsy specimens, and correlated these putative immunosuppressive mechanisms with clinical outcomes [84,85]. The phenotypic screening assay also incorporated a mathematical model that provides a quantitative prediction of a T cell response to Interleukin-12 in terms of cytokine production and cell fate. As multiple factors were observed in the phenotypic screening assay, the mathematical model provided a quantitative context to determine whether the observed factors were sufficient to explain the observed changes in T cell response or whether there were additional behaviors that were unexplained.
3.2.2. Computer Simulation Provides a Translational Bridge Across Model Systems
Even after defining signaling pathway components, it is still difficult for scientists to predict the overall behavior of the system. Animal models are most commonly used to predict the human response to a particular therapy and to replicate human systems. However, success of therapeutic interventions seems to be more easily attained in animals and brings to question the translational viability of animal models of disease [86]. As discussed previously, methodological bias may play a role in these discrepancies. Moreover, humans are known to have fundamental differences in biology, regardless of whether molecular components are conserved, and animal models are usually made of clones of a particular population. Therefore, this does not account for the heterogeneity of the human population, which will attribute different clinical responses. Additionally, it is unclear how conserved cellular signaling networks across different model systems. The heterogeneity of tumor cell populations and individual metabolic and disease state differences may further cloud this issue, even when “normal” human cellular networks are well-defined.
Novel drug targets are often difficult to predict, due to the interplay of multiple genes and systems and the differences between human and animal models. To overcome these barriers to disease modeling, computer simulation using previous molecular interaction and human clinical data may be able to elucidate these signaling network differences [9,87]. Quantitative systems pharmacology is now being used to analyze drug interactions and provide insight to adverse effects, and has been suggested as a method of selecting better drug candidates for development [9]. Two recent studies highlight the different functions computer modeling may perform in aiding in drug development.
In a study examining prostate cancer malignancies, human and mouse model data was analyzed to determine master regulators of prostate cancer using the Algorithm for the Reconstruction of Accurate Cellular Networks (ARACNe), which uses microarray data to predict direct molecular interactions [88,89]. A Master Regulator Inference algorithm (MARINa) was then used to determine gene regulators of prostate cancer. Co-expression of FOXM1 and CENPF was determined to promote prostate cancer malignancy by synergistically acting via the PI3K and MAPK pathways. This suggests a complicated feedback system that may be therapeutically difficult to target with current treatments [88]. Using this analysis could serve as a way to determine gene interactions in tumor development and to help to better classify tumors based on their phenotypes and genotypes.
Knowing gene regulators of malignancy could also be helpful in finding new targets for therapy and determining adverse effects before they are observed in the clinic. In a study regarding drug-induced peripheral neuropathy, data from DrugBank and Therapeutic Target Database was used to create pharmacological networks of peripheral neuropathy-inducing drugs, their known targets, and the diseases for which they are used to treat. The Database for Annotation, Visualization, and Integrated Discovery (DAVID) was then used to identify connections between drug targets and cellular pathways. Regulation of two genes, MYC and PAF15, were found to be correlated with a higher incidence of drug-induced peripheral neuropathy and suggested drugs that may produce this adverse effect, as well as targets for future studies to alleviate neuropathy symptoms in the clinic [90].
4. Conclusions
One of the most costly decisions a pharmaceutical company can make in regards to a cancer immunotherapy is selecting the patient population to conduct the clinical trial. Success of the drug is often determined by which patient populations the study is conducted in. If an inappropriate cancer phenotype is selected for the study, adverse events or inefficacy can stop the study and discourage the drug from continuing in the development pipeline. Troubleshooting and optimizing the drug could prove more costly than developing an entirely different drug. Most cell lines do not contain heterogeneous populations of cancer cells, let alone T cells and other stromal cells, which can lead to misleading results in preclinical trials for determining optimal cancer phenotypes.
Given validation of these computational methods, and by combining methods to investigate both drug response and molecular signaling, we can begin to properly examine cellular networks to infer on whether our biological experimental methods correctly forecast patient outcomes and portray an accurate representation of biological activity of a certain therapy or whether that therapy is appropriate for use in a certain patient population. Using computational tools to model relevant signaling networks and cancer immunity may also remove some of the bias associated with sample processing and inappropriate biological method selection. To improve the translational value of these approaches, they should account for clonal evolution and heterogeneity of the system. Combining system-targeting drugs with system modeling and phenotypic assays is in line with NIH recommendations for more quantitative and systems pharmacology research, and could prove to be our best resource in engaging host immunity in the fight against cancer. Given our currently known limitations and the recent developments in cancer immunotherapy research, the pharmaceutical industry can reinvigorate their model for innovation with the help of quantitative and systems pharmacology techniques and focusing on a systems perspective with respect to cancer.
Acknowledgments
This work was supported by grants from the National Science Foundation (NSF) CAREER 1053490 and the National Cancer Institute (NCI) R15CA123123. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NSF, the NCI, or the National Institutes of Health.
Author Contributions
C.B.H. and D.J.K. wrote, reviewed, and edited the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pharmaceutical Research and Manufacturers of America (PhRMA). 2013 Biopharmaceutical Research Industry Profile; PhRMA: Washington, DC, USA, 2013. [Google Scholar]
- Mullin, R. Tufts study finds big rise in cost of drug development. Chemical & Engineering News, 20 November 2014. [Google Scholar]
- Chen, C.; Beckman, R.A. Maximizing return on socioeconomic investment in phase ii proof-of-concept trials. Clin. Cancer Res. 2014, 20, 1730–1734. [Google Scholar] [CrossRef] [PubMed]
- Wartha, K.; Herting, F.; Hasmann, M. Fit-for purpose use of mouse models to improve predictivity of cancer therapeutics evaluation. Pharmacol. Ther. 2014, 142, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Dailami, M.; Lipkovich, I.; Dyck, V.J. Infrisk: A Computer Simulation Approach to Risk Management in Infrastructure Project Finance Transactions; Economic Development Institute of the World Bank: Washington, DC, USA, 1999. [Google Scholar]
- Mavris, D.; Bandte, O.; DeLaurentis, D.A. Robust design simulation: A probabilistic approach to multidisciplinary design. J. Aircr. 1999, 36, 298–307. [Google Scholar] [CrossRef]
- Lauffenburger, D.; Giacomini, K. Systems biology and systems pharmacology. Bridge Converg. Eng. Life Sci. 2013, 43, 26–33. [Google Scholar]
- Ananthakrishnan, R.; Gona, P. Pharmacological modeling and biostatistical analysis of a new drug. Open Access J. Clin. Trials 2010, 2, 59–82. [Google Scholar] [CrossRef]
- Sorger, P. Quantitative and Systems Pharmacology in the Post-Genomic Era: New Approaches to Discovering Drugs and Understanding Therapeutic Mechanisms; National Institutes of Health: Bethesda, MD, USA, 2011. [Google Scholar]
- Radulescu, R.T. Oncoprotein metastasis: An expanded topography. Romanian J. Morphol. Embryol. 2013, 54, 237–239. [Google Scholar]
- Arnedos, M.; Soria, J.C.; Andre, F.; Tursz, T. Personalized treatments of cancer patients: A reality in daily practice, a costly dream or a shared vision of the future from the oncology community? Cancer Treat. Rev. 2014, 40, 1192–1198. [Google Scholar] [CrossRef] [PubMed]
- Li, S.C.; Tachiki, L.M.; Kabeer, M.H.; Dethlefs, B.A.; Anthony, M.J.; Loudon, W.G. Cancer genomic research at the crossroads: Realizing the changing genetic landscape as intratumoral spatial and temporal heterogeneity becomes a confounding factor. Cancer Cell Int. 2014, 14, 115. [Google Scholar] [CrossRef] [PubMed]
- Ramos, P.; Bentires-Alj, M. Mechanism-based cancer therapy: Resistance to therapy, therapy for resistance. Oncogene 2014. [Google Scholar] [CrossRef]
- Weinstein, I.B.; Joe, A.K. Mechanisms of disease: Oncogene addiction―A rationale for molecular targeting in cancer therapy. Nat. Clin. Pract. Oncol. 2006, 3, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.M.; Hicklin, D.J. Resistance to targeted therapies: Refining anticancer therapy in the era of molecular oncology. Clin. Cancer Res. 2009, 15, 7471–7478. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, R.H. The nci60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A.; et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Paull, K.D.; Shoemaker, R.H.; Hodes, L.; Monks, A.; Scudiero, D.A.; Rubinstein, L.; Plowman, J.; Boyd, M.R. Display and analysis of patterns of differential activity of drugs against human tumor cell lines: Development of mean graph and compare algorithm. J. Natl. Cancer Inst. 1989, 81, 1088–1092. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.; Cheng, K.W.; Rigas, B. Preclinical predictors of anticancer drug efficacy: Critical assessment with emphasis on whether nanomolar potency should be required of candidate agents. J. Pharmacol. Exp. Ther. 2012, 341, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with braf v600e mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined braf and mek inhibition in melanoma with braf v600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Haviv, I.; Campbell, I.G. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009, 25, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Tlsty, T.D.; Coussens, L.M. Tumor stroma and regulation of cancer development. Annu. Rev. Pathol. 2006, 1, 119–150. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Vincent, T.L. An evolutionary model of carcinogenesis. Cancer Res. 2003, 63, 6212–6220. [Google Scholar] [PubMed]
- Michor, F.; Iwasa, Y.; Nowak, M.A. Dynamics of cancer progression. Nat. Rev. Cancer 2004, 4, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.A.; Michor, F.; Iwasa, Y. The linear process of somatic evolution. Proc. Natl. Acad. Sci. USA 2003, 100, 14966–14969. [Google Scholar] [CrossRef] [PubMed]
- Klinke, D.J., II. An evolutionary perspective on anti-tumor immunity. Front. Oncol. 2012, 2, 202. [Google Scholar] [PubMed]
- Mahadevan, N.R.; Zanetti, M. Tumor stress inside out: Cell-extrinsic effects of the unfolded protein response in tumor cells modulate the immunological landscape of the tumor microenvironment. J. Immunol. 2011, 187, 4403–4409. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Rech, A.J.; Vonderheide, R.H. Dynamic interplay of oncogenes and t cells induces pd-l1 in the tumor microenvironment. Cancer Discov. 2013, 3, 1330–1332. [Google Scholar] [CrossRef] [PubMed]
- LaBarge, M.A. The difficulty of targeting cancer stem cell niches. Clin. Cancer Res. 2010, 16, 3121–3129. [Google Scholar] [CrossRef] [PubMed]
- Shackleton, M.; Quintana, E.; Fearon, E.R.; Morrison, S.J. Heterogeneity in cancer: Cancer stem cells versus clonal evolution. Cell 2009, 138, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Merlo, L.M.; Pepper, J.W.; Reid, B.J.; Maley, C.C. Cancer as an evolutionary and ecological process. Nat. Rev. Cancer 2006, 6, 924–935. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G. Two genetic hits (more or less) to cancer. Nat. Rev. Cancer 2001, 1, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Gillies, R.J. A microenvironmental model of carcinogenesis. Nat. Rev. Cancer 2008, 8, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Casas-Selves, M.; Degregori, J. How cancer shapes evolution, and how evolution shapes cancer. Evolution (N.Y.) 2011, 4, 624–634. [Google Scholar]
- Anderson, A.R.; Weaver, A.M.; Cummings, P.T.; Quaranta, V. Tumor morphology and phenotypic evolution driven by selective pressure from the microenvironment. Cell 2006, 127, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Rak, J.W.; Kerbel, R.S. Growth advantage (“clonal dominance”) of metastatically competent tumor cell variants expressed under selective two- or three-dimensional tissue culture conditions. In Vitro Cell Dev. Biol. Anim. 1993, 29A, 742–748. [Google Scholar] [CrossRef]
- Mintz, B. Gene control of mammalian pigmentary differentiation. I. Clonal origin of melanocytes. Proc. Natl. Acad. Sci. USA 1967, 58, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Michor, F.; Frank, S.A.; May, R.M.; Iwasa, Y.; Nowak, M.A. Somatic selection for and against cancer. J. Theor. Biol. 2003, 225, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Cunningham, J.J.; Brown, J.S. Evolutionary triage governs fitness in driver and passenger mutations and suggests targeting never mutations. Nat. Commun. 2014, 5, 5499. [Google Scholar] [CrossRef] [PubMed]
- Youn, A.; Simon, R. Using passenger mutations to estimate the timing of driver mutations and identify mutator alterations. BMC Bioinform. 2013, 14, 363. [Google Scholar] [CrossRef]
- Peterson, E.A.; Chavan, S.S.; Bauer, M.A.; Heuck, C.J.; Johann, D.J. Revealing the inherent heterogeneity of human malignancies by variant consensus strategies coupled with cancer clonal analysis. BMC Bioinform. 2014, 15, S9. [Google Scholar] [CrossRef]
- Basanta, D.; Gatenby, R.A.; Anderson, A.R. Exploiting evolution to treat drug resistance: Combination therapy and the double bind. Mol. Pharm. 2012, 9, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Mumm, J.B.; Oft, M. Cytokine-based transformation of immune surveillance into tumor-promoting inflammation. Oncogene 2008, 27, 5913–5919. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef] [PubMed]
- Iwami, S.; Haeno, H.; Michor, F. A race between tumor immunoescape and genome maintenance selects for optimum levels of (epi)genetic instability. PLoS Comput. Biol. 2012, 8, e1002370. [Google Scholar] [CrossRef] [PubMed]
- Zacharski, L.R.; Sukhatme, V.P. Coley’s toxin revisited: Immunotherapy or plasminogen activator therapy of cancer? J. Thromb. Haemost. 2005, 3, 424–427. [Google Scholar] [CrossRef] [PubMed]
- Heywood, G.R.; Rosenberg, S.A.; Weber, J.S. Hypersensitivity reactions to chemotherapy agents in patients receiving chemoimmunotherapy with high-dose interleukin 2. J. Natl. Cancer Inst. 1995, 87, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Karp, B.I.; Yang, J.C.; Khorsand, M.; Wood, R.; Merigan, T.C. Multiple cerebral lesions complicating therapy with interleukin-2. Neurology 1996, 47, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Alexandrescu, D.T.; Maddukuri, P.; Wiernik, P.H.; Dutcher, J.P. Thrombotic thrombocytopenic purpura/hemolytic uremic syndrome associated with high-dose interleukin-2 for the treatment of metastatic melanoma. J. Immunother. 2005, 28, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Ramirez, D.; Ales-Martinez, M.; Ferrandiz, L. Fast-growing in-transit melanoma metastasis after intratumoral interleukin-2. Cancer Immunol. Immunother. 2014, 63. [Google Scholar] [CrossRef]
- Zagozdzon, R.; Golab, J. Immunomodulation by anticancer chemotherapy: More is not always better (review). Int. J. Oncol. 2001, 18, 417–424. [Google Scholar] [PubMed]
- Zagozdzon, R.; Golab, J.; Mucha, K.; Foroncewicz, B.; Jakobisiak, M. Potentiation of antitumor effects of il-12 in combination with paclitaxel in murine melanoma model in vivo. Int. J. Mol. Med. 1999, 4, 645–648. [Google Scholar] [PubMed]
- Schetter, A.J.; Heegaard, N.H.; Harris, C.C. Inflammation and cancer: Interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis 2010, 31, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Grillo-Lopez, A.J.; White, C.A.; Dallaire, B.K.; Varns, C.L.; Shen, C.D.; Wei, A.; Leonard, J.E.; McClure, A.; Weaver, R.; Cairelli, S.; et al. Rituximab: The first monoclonal antibody approved for the treatment of lymphoma. Curr. Pharm. Biotechnol. 2000, 1, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Hodi, F.S.; Weber, J.S.; Allison, J.P.; Urba, W.J.; Robert, C.; O’Day, S.J.; Hoos, A.; Humphrey, R.; Berman, D.M.; et al. Development of ipilimumab: A novel immunotherapeutic approach for the treatment of advanced melanoma. Ann. N.Y. Acad. Sci. 2013, 1291, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Verheije, M.H.; Rottier, P.J. Retargeting of viruses to generate oncolytic agents. Adv. Virol. 2012, 2012, 798526. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.H.; Lemoine, N.R.; Wang, Y. Oncolytic viruses for cancer therapy: Overcoming the obstacles. Viruses 2010, 2, 78–106. [Google Scholar] [CrossRef] [PubMed]
- Chacon, J.; Sarnaik, A.; Chen, J.; Creasy, C.; Kale, C.; Robinson, J.; Weber, J.; Hwu, P.; Pilon-Thomas, S.; Radvanyi, L.G. Manipulating the tumor microenvironment ex vivo for enhanced expansion of tumor-infiltrating lymphocytes for adoptive cell therapy. Clin. Cancer Res. 2014, 21, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Kalos, M.; June, C.H. Adoptive T cell transfer for cancer immunotherapy in the era of synthetic biology. Immunity 2013, 39, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Pirooznia, N.; Hasannia, S.; Taghdir, M.; Rahbarizadeh, F.; Eskandani, M. The construction of chimeric t-cell receptor with spacer base of modeling study of vhh and muc1 interaction. J. Biomed. Biotechnol. 2011, 2011, 578128. [Google Scholar] [CrossRef] [PubMed]
- James, S.E.; Greenberg, P.D.; Jensen, M.C.; Lin, Y.; Wang, J.; Budde, L.E.; Till, B.G.; Raubitschek, A.A.; Forman, S.J.; Press, O.W. Mathematical modeling of chimeric tcr triggering predicts the magnitude of target lysis and its impairment by tcr downmodulation. J. Immunol. 2010, 184, 4284–4294. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, G.; Derhovanessian, E.; Larbi, A. Immunosenescence and cancer. Crit. Rev. Oncol. Hematol. 2010, 75, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Bombelli, F.B.; Webster, C.A.; Moncrieff, M.; Sherwood, V. The scope of nanoparticle therapies for future metastatic melanoma treatment. Lancet Oncol. 2014, 15, e22–e32. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hodi, F.S.; Robert, C. Ctla-4 and pd-1/pd-l1 blockade: New immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin. Cancer Res. 2013, 19, 5300–5309. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.L. Combination checkpoint blockade—Taking melanoma immunotherapy to the next level. N. Engl. J. Med. 2013, 369, 187–189. [Google Scholar] [CrossRef] [PubMed]
- Mockler, M.B.; Conroy, M.J.; Lysaght, J. Targeting T cell immunometabolism for cancer immunotherapy; understanding the impact of the tumor microenvironment. Front. Oncol. 2014, 4, 107. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific cd8 t cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-pd-l1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Irving, B.A.; Hodi, F.S. Molecular pathways: Next-generation immunotherapy—Inhibiting programmed death-ligand 1 and programmed death-1. Clin. Cancer Res. 2012, 18, 6580–6587. [Google Scholar] [CrossRef] [PubMed]
- Khoo, M.C.K. Identification of physiological control systems. In Physiological Control Systems; John Wiley & Sons, Inc.: New York, NY, USA, 1999; pp. 159–202. [Google Scholar]
- Bergman, R.N. Toward physiological understanding of glucose tolerance: Minimal-model approach. Diabetes 1989, 38, 1512–1527. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, D.F. Bias as a threat to the validity of cancer molecular-marker research. Nat. Rev. Cancer 2005, 5, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Klinke, D.J., 2nd; Kulkarni, Y.M.; Wu, Y.; Byrne-Hoffman, C. Inferring alterations in cell-to-cell communication in her2+ breast cancer using secretome profiling of three cell models. Biotechnol. Bioeng. 2014, 111, 1853–1863. [Google Scholar] [CrossRef] [PubMed]
- Swinney, D.C.; Anthony, J. How were new medicines discovered? Nat. Rev. Drug Discov. 2011, 10, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Kotz, J. Phenotypic screening, take two. SciBX 2012, 5. [Google Scholar] [CrossRef]
- Kulkarni, Y.M.; Chambers, E.; McGray, A.J.; Ware, J.S.; Bramson, J.L.; Klinke, D.J., II. A quantitative systems approach to identify paracrine mechanisms that locally suppress immune response to interleukin-12 in the b16 melanoma model. Integr. Biol. (Camb.) 2012, 4, 925–936. [Google Scholar] [CrossRef]
- Klinke, D.J., II. Induction of wnt-inducible signaling protein-1 correlates with invasive breast cancer oncogenesis and reduced type 1 cell-mediated cytotoxic immunity: A retrospective study. PLoS Comput. Biol. 2014, 10, e1003409. [Google Scholar] [CrossRef] [PubMed]
- Wen, F.T.; Thisted, R.A.; Rowley, D.A.; Schreiber, H. A systematic analysis of experimental immunotherapies on tumors differing in size and duration of growth. Oncoimmunology 2012, 1, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Coumans, J.V.; Gau, D.; Poljak, A.; Wasinger, V.; Roy, P.; Moens, P.D. Profilin-1 overexpression in mda-mb-231 breast cancer cells is associated with alterations in proteomics biomarkers of cell proliferation, survival, and motility as revealed by global proteomics analyses. Omics 2014, 18, 778–791. [Google Scholar] [CrossRef] [PubMed]
- Aytes, A.; Mitrofanova, A.; Lefebvre, C.; Alvarez, M.J.; Castillo-Martin, M.; Zheng, T.; Eastham, J.A.; Gopalan, A.; Pienta, K.J.; Shen, M.M.; et al. Cross-species regulatory network analysis identifies a synergistic interaction between foxm1 and cenpf that drives prostate cancer malignancy. Cancer Cell 2014, 25, 638–651. [Google Scholar] [CrossRef] [PubMed]
- Margolin, A.A.; Nemenman, I.; Basso, K.; Wiggins, C.; Stolovitzky, G.; Dalla Favera, R.; Califano, A. Aracne: An algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinform. 2006, 7, S7. [Google Scholar] [CrossRef]
- Hur, J.; Guo, A.Y.; Loh, W.Y.; Feldman, E.L.; Bai, J.P. Integrated systems pharmacology analysis of clinical drug-induced peripheral neuropathy. CPT Pharmacomet. Syst. Pharmacol. 2014, 3, e114. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Byrne-Hoffman, C.; II, D.J.K. A Quantitative Systems Pharmacology Perspective on Cancer Immunology. Processes 2015, 3, 235-256. https://doi.org/10.3390/pr3020235
AMA Style
Byrne-Hoffman C, II DJK. A Quantitative Systems Pharmacology Perspective on Cancer Immunology. Processes. 2015; 3(2):235-256. https://doi.org/10.3390/pr3020235
Chicago/Turabian StyleByrne-Hoffman, Christina, and David J. Klinke II. 2015. "A Quantitative Systems Pharmacology Perspective on Cancer Immunology" Processes 3, no. 2: 235-256. https://doi.org/10.3390/pr3020235