Melt Stable Functionalized Organosolv and Kraft Lignin Thermoplastic


1
Chemistry of Interfaces, Division of Chemical Engineering, Department of Civil, Environmental and Natural Resources Engineering, Luleå University of Technology, 97187 Luleå, Sweden
2
Biochemical Process Engineering, Division of Chemical Engineering, Department of Civil, Environmental and Natural Resources Engineering, Luleå University of Technology, 97187 Luleå, Sweden
*
Author to whom correspondence should be addressed.
Processes 2020, 8(9), 1108; https://doi.org/10.3390/pr8091108
Submission received: 23 July 2020
/
Revised: 28 August 2020
/
Accepted: 3 September 2020
/
Published: 5 September 2020
(This article belongs to the Special Issue Biochemical and Thermochemical Conversion Processes of Lignicellulosic Biomass Fractionated Streams)
Abstract
:A shift towards an economically viable biomass biorefinery concept requires the use of all biomass fractions (cellulose, hemicellulose, and lignin) for the production of high added-value products. As lignin is often underutilized, the establishment of lignin valorization routes is highly important. In-house produced organosolv as well as commercial Kraft lignin were used in this study. The aim of the current work was to make a comparative study of thermoplastic biomaterials from two different types of lignins. Native lignins were alkylate with two different alkyl iodides to produce ether-functionalized lignins. Successful etherification was verified by FT-IR spectroscopy, changes in the molecular weight of lignin, as well as 13C and 1H Nuclear Magnetic Resonance (NMR). The thermal stability of etherified lignin samples was considerably improved with the T2% of organosolv to increase from 143 °C to up to 213 °C and of Kraft lignin from 133 °C to up to 168 °C, and glass transition temperature was observed. The present study shows that etherification of both organosolv and Kraft lignin with alkyl halides can produce lignin thermoplastic biomaterials with low glass transition temperature. The length of the alkyl chain affects thermal stability as well as other thermal properties.
Keywords:
lignin; organosolv; Kraft lignin; etherification; lignin functionalization; thermoplastics1. Introduction
Lignin is an aromatic heteropolymer, the second most abundant biopolymer in the world after cellulose. It constitutes 15–35% w/w of a plant’s cell wall and plants are estimated to generate 0.5–3.6 billion tons of lignin annually [1,2]. The global annual production of lignin is estimated at approximately 100 million tons, of which only 2% is used commercially (primarily in dispersants, adhesives, and surfactants); whereas the rest is burned as low-value fuel [3,4,5]. The main sources of lignin are technical lignin from the pulp industry, namely Kraft and ligno-sulphonate. Organosolv lignin (accounting for approximately 2% of the total lignin) has started to gain attention owing to advances in biomass biorefinery technology, its high purity, and a chemical structure close to that of natural lignin [3,6]. The ongoing worldwide construction of second-generation cellulosic ethanol plants is expected to further increase the availability of lignin [7]. Kraft lignin is produced through the sulphate cooking process, in which fibers are treated at temperatures of 165–175 °C for 1–2 h in the presence of sodium hydroxide and sodium sulphite [5]. Ligno-sulphonates are a by-product of sulphite cooking in which the fibers are treated by HSO3− and SO32− ions and the digestion is typically operated at 120–180 °C for 1–5 h [1,5].
On the contrary, organosolv lignin is produced through organosolv pulping, in which the biomass is treated in aqueous-organic solvent mixtures with or without the addition of an inorganic catalyst [8,9]. Ligno-sulfonates and Kraft lignin are sulfur-containing lignin, with the sulfur to vary in the range 1–3% for Kraft and 3.5–8% for lignosulfonates and they consist of a by-product of chemical pulping process from the paper industry [3]. On the contrary, organosolv lignin is practically sulfur-free with a very low ash content [10]. Another major difference between the first two lignins and organosolv is that organosolv retains a considerable fraction of the β-O-4 linkages, whereas in lignin from chemical pulping, the β-O-4 linkages represent fewer than 10% of the connections and promote the formation of carbon–carbon bonds [5,11]. Due to the high lignin content of lignocellulosic biomass, its valorization is crucial for the financial viability of biorefineries. However, existing technologies for lignin valorization are still not as advanced as those applied to carbohydrate fractions [1]. New technologies for the conversion of lignin into high added-value products represent an important cornerstone towards establishing economically viable biomass biorefinery processes that can reduce our dependency on fossil feedstock.
Thermoplastic-based polymers are widely used in various applications due to their flexible nature. The development of polymer technology has seen a continuous demand for eco-friendly, biodegradable thermoplastic materials. Thermoplasticity is defined as the use of heating to weaken intermolecular forces that connect polymer chains, without altering the chemical structure of the polymer. Thus, thermoplastic materials can be melted and shaped multiple times into desired structures without eliciting any chemical changes. In this regard, lignin is a very promising raw material as it is readily available, sustainable, and can be functionalized to achieve the desired physical properties.
Lignin, however, presents also some challenges as it is brittle, exhibits poor mechanical properties, has a relative high glass transition temperature (for Kraft lignin 124–174 °C, organosolv lignin 91–97 °C [12]), and its thermal processing at elevated temperatures is often impaired by radical-induced self-condensation, which limits its application to thermosets [13,14]. Self-polymerization of lignin during heating is often manifested by an increase in molecular weight, changes in chemical behavior (e.g., loss of solubility in common organic solvents such as tetrahydrofuran—THF) or ‘loss’ of Tg after repeated heating and cooling cycles [15]. The elevated hydroxyl content of lignin could induce swelling due to water adsorption, causing expansions in the composite [16] and, in turn, poor dimensional stability. However, the high reactivity of hydroxyl groups allows also lignin functionalization, which can significantly improve Tg, thermal stability, and compatibility with polymers [13,14].
Various efforts have been made to achieve functionalized lignins with low Tg and improved thermal properties for thermoplastic application [13,17]. Lignin esterification is one of the oldest methods to decrease Tg, whereas acetylation augments the solubility of lignin for molecular weight and structural analysis. Lewis and Brauns were the first to describe the solubility and thermal properties of lignin-based esters [18]. Later, Glasser and Jain reported that Tg of lignin esters decreased linearly with increasing length of the acyl group [19]. In most cases, lignin esterification was achieved using carboxylic acids and particularly acid chlorides or acid anhydrides. Bi-functional reagents, such as dicarboxylic aliphatic (sebacoyl) or aromatic (terephthaloyl) acid chlorides, are also promising in the production of lignin-based polyester network materials [20,21]. Another such reagent is dimer acid [22], which can be produced during the processing of soybean, tall, and cottonseed oil by a Diels–Alder cycloaddition reaction. Dimer acid-based lignin polyesters are fully renewable materials for polymer applications. Recently, polybutadiene-based lignin polyester thermoplastic with network structure and very low Tg was reported [2].
Graft modification is another efficient way to produce integrated lignin-polymer grafted materials with improved thermal processing and stability. Both “grafting onto” and “grafting from” modification methods have been reported. Korich et al. were the first to introduce boronic-acids based reagents for “grafting onto” modification of lignin, resulting in promising thermal properties and Tg in the range of −30 °C to −50 °C [23]. A similar lignin-based copolymer with low Tg was obtained by using azide–alkyne Huisgen cycloaddition “click chemistry” tools [24]. In comparison, the “grafting from” modification method allowed de Oliveira and Glasser to chemically modify hydroxypropylated lignin with ring-opening polymerization of ε-caprolactone [25] and subsequently lower Tg. Another widely used “grafting from” method is atom transfer radical polymerization (ATRP) [26]. ATRP was used by Hilburg et al. to prepare lignin-based thermoplastic with improved mechanical properties, and by Kim and Kadla to obtain lignin-based thermoresponsive thermoplastic [27,28].
Two other prominent etherification methods for obtaining lignin-based thermoplastic include methylation and hydroxyalkylation [29,30]. However, no systematic studies of etherification reactions with higher alkyl chain congeners have been reported [13,31]. Furthermore, comparative studies regarding hydrophobic lignin-based thermoplastic from different types of lignins are also scarcely documented. Thus, in this work we have chosen our in house organosolv lignin as well as commercial Kraft lignin for comparative studies. Both lignins were alkylated by etherification reaction with two different alkyl iodides to produce hydrophobic thermoplastic. All lignin based hydrophobic thermoplastics were characterized and comparative studies has been discussed in this work.
2. Materials and Methods
2.1. Materials and Chemicals
Kraft lignin (product no. 370959; Sigma-Aldrich, St. Louis, MO, USA) was used without any further treatment. Organosolv lignin was produced from a hybrid organosolv—steam explosion reactor as described previously [32]. Specifically, birch chips from mills in northern Sweden were milled to <1 mm particle size and used as raw material for organosolv treatment at 200 °C for 30 min with 30% (v/v) ethanol. The resulting pretreated solids were separated from the liquor by vacuum filtration and the liquor was collected for lignin isolation. The ethanol was removed under vacuum in a rotary evaporator and, finally, lignin was separated from the liquor by centrifugation at 29,416× g for 15 min at 4 °C. The lignin was air-dried until further use. Approximately 12 g of lignin were recovered per 100 g treated birch chips (dry basis). Contaminants in lignin were analyzed as previously described [32]; they amounted to 2.73% w/w hemicellulose and 0.12% w/w ash, but no cellulose was detected.
Anhydrous potassium carbonate was purchased from Sigma-Aldrich and dried overnight in the oven at 130 °C before every reaction. Dimethylformamide (DMF) was dried through a 4-Å molecular sieve. 1-iododecane and 1-iodohexadecane were purchased from Sigma-Aldrich and used without further purification.
2.2. Etherification Reaction
Prior to etherification, ~200 mg of lignin was weighted and heated in the oven (up to 60 °C) for 1 h, followed by drying under vacuum in a two-neck round-bottom flask for 2 h. Then, 5 mL of dry DMF was added under nitrogen atmosphere and the mixture was stirred at room temperature until a homogeneous solution was formed. Subsequently, 4 mol equivalents of anhydrous K2CO3 were added and the mixture was heated up to 65 °C under nitrogen atmosphere. Next, 1.2 mol equivalents of alkyl iodide were added dropwise with continuous stirring and heating, after which the mixture was heated at 60–65 °C for 16 h under an inert atmosphere. Upon completion, the reaction was quenched by the addition of water (50 mL). The reaction mass was stirred and a solid precipitate was formed. The solid mass was washed several times with water, dried under vacuum, and mixed with diethyl ether to dissolve the alkylated hydrophobic lignin. The ether-soluble part was separated and, upon evaporation, a solid mass of 250–260 mg was recovered. The sample was kept under vacuum for 48 h before further characterization.
2.3. Nuclear Magnetic Resonance (NMR) Characterization
A Bruker Ascend Aeon WB 400 (Bruker BioSpin AG, Fällanden, Switzerland) NMR spectrometer was used with a working frequency of 400.22 MHz for 1H, 100.65 MHz for 13C, and 162.02 MHz for 31P with a 10-mm Z-gradient Broadband Observe (BBO) probe. Bruker Topspin 3.5pl2 software was used for data processing. All spectra were recorded at 25 °C. Chemical shifts were expressed in ppm (δ) downfield from tetramethylsilane (TMS), using the solvent as internal standard (CDCl3, δ = 7.26). 31P NMR spectra were acquired at an inverse gated pulse sequence with 90° pulse and Waltz 16 decoupling technique. A total of 72 scans were acquired with a 10-s recycle delay. 1H NMR spectra were analyzed with the Bruker standard pulse program based on a total of 32 scans, while 13C NMR spectra included a total of 1024 scans.
2.3.1. Quantitative 31P NMR
31P NMR analysis was performed according to a previously published method [33], modified so as to be analyzed in 10 mm NMR probe. Approximately 120 mg of lignin was dissolved in 1.6 mL anhydrous CDCl3/pyridine (1:1.6 v/v) solution. A 0.1 M standard solution containing cholesterol as internal standard was prepared in anhydrous CDCl3/pyridine solution and 5 mg/mL Cr(III) acetylacetonate was added as relaxation reagent. Then, 400 µL of this solution was added to the above prepared lignin solution. The mixture was stirred vigorously and 400 µL of phosphitylating reagent II (2-chloro-4,4,5,5-tetramethyl-1,2,3-dioxaphospholane) was added. The reaction mixture was stirred for 2 h at room temperature and then transferred to a 10-mm NMR tube for 31P NMR analysis. 31P NMR measurements were performed to determine hydroxyl and carboxyl group content in organosolv as well as in commercial Kraft lignin. 31P NMR results were expressed in mmol g−1 and were used to calculate the reactants’ molar ratio for the etherification reaction.
2.3.2. 1H and 13C NMR
Approximately 200 mg of ether-functionalized lignin was dissolved in 3 mL CDCl3 and then transferred to a 10-mm NMR tube. NMR spectra were acquired at 25 °C with sample spinning frequency of 20 Hz. All ether-functionalized lignin samples were referenced with solvent signal at 7.26 ppm (CDCl3).
2.4. Fourier Transform Infrared (FT-IR) Measurements
FT-IR spectra were recorded on a Bruker IFS 80v vacuum FT-IR spectrometer equipped with a deuterated triglycine sulphate detector. Samples were prepared with KBr as disks. All spectra were recorded at room temperature (~22 °C) using the double-side forward-backward acquisition mode under vacuum. A total of 128 scans were co-added and signal-averaged at an optical resolution of 4 cm−1.
2.5. Thermogravimetric Analysis (TGA)
TGA of lignin samples was performed on a PerkinElmer 8000 TGA instrument (Waltham, MA, USA) at 30–800 °C with a heating ramp of 10 °C/min under nitrogen atmosphere. Approximately 1–2 mg of lignin sample was used for each experiment and placed on PerkinElmer ceramic pans. All lignin samples were dried for 48 h under vacuum before TGA analysis to remove moisture from the samples.
2.6. Differential Scanning Calorimetry (DSC) Analysis
DSC of lignin samples was determined on a PerkinElmer DSC 6000 single-furnace instrument (Waltham, MA, USA) between −80 °C and 100 °C using an intracooler with a heating and cooling ramp of 5 °C/min under nitrogen atmosphere. Approximately 1–2 mg of lignin sample was placed in aluminum pans and sealed manually. At the beginning of the analysis, the sample was heated to 100 °C to form a thin layer inside the aluminum pan and then cooled gradually to −80 °C.
2.7. Gel Permeation Chromatography (GPC)
To determine the molecular weight of lignin samples, GPC was performed on a PerkinElmer Flexar HPLC apparatus equipped with a UV detector (PerkinElmer, Waltham, MA, USA) and a Waters (Milford, MA, USA) Styragel HR 4E column at 40 °C. THF was used as mobile phase at a flow rate of 0.6 mL/min. The detector was set at 280 nm. Prior to analysis, lignin was acetobrominated as previously described [34]. The calibration curve was acquired with polystyrene standards (Sigma-Aldrich, St. Louis, MO, USA).
3. Results and Discussion
3.1. Selection of Appropriate Functionalization Conditions
Etherification is one of the most well-known functionalization methods for generating lignin-based thermoplastic. Lignin ether linkages are more thermally stable and chemically resistant than lignin ester linkages, making them more suitable for further thermoplastic applications. Our initial objective was to prepare hydrophobic lignin thermoplastic by functionalization of organosolv lignin under mild reaction conditions. The most commonly used etherification procedure involves the reaction of an alkyl halide with lignin in the presence of a base. Lignin contains both aliphatic and phenolic hydroxyl groups that are susceptible to etherification with the alkyl halide. However, due to the high pKa value of the aliphatic hydroxyl group, etherification occurs exclusively at the phenolic hydroxyl position under normal reaction conditions. We hypothesized that the use of strong base (e.g., NaOH or KOH) at an elevated temperature could result in partial decomposition of native lignin. In addition to this, a strong base can promote further competitive side hydrolysis reaction of alkyl halide with strongly nucleophilic hydroxide ions (-OH). In this regard, K2CO3 is the best choice of base when aiming at milder reaction condition and it has been widely used for the deprotonation of phenolic compounds. However, as K2CO3 is highly soluble in water, it may also generate hydroxide ions under these conditions and hydrolyze alkyl halides. To avoid this side reaction, etherification was carried out under a nitrogen atmosphere with the use of anhydrous K2CO3 and DMF. DMF is the most compatible solvent for the partial dissolution of K2CO3 particles, and allows for good solubility of lignin and most alkyl halides.
Previously Ren et al. carried out the alkylation of alkaline lignin with n-dodecyl bromide for the preparation of dodecylated lignin under reflux conditions in water for 2.5 days [35], which further suggest the slower reactivity of alkyl bromide. Thus, the selection of alkyl halides was narrowed to alkyl iodides, as these are more reactive compared to other alkyl halide congeners. Most alkyl iodides undergo reaction with phenolic compounds at room temperature, but long-chain alkyl iodides require elevated temperatures (60–65 °C) perhaps due to size flexibility or steric hindrance. We selected two long-chain alkyl iodides, 1-iodohexadecane and 1-iododecane, to evaluate the effect of alkyl chain length on the properties of functionalized lignin. Finally, to examine how the source of lignin affected the etherification process, we compared in-house organosolv birch lignin with commercially available Kraft lignin.
31P NMR analysis provided an estimate of hydroxyl as well as carboxyl group content. As summarized in Table 1, total phenol and carboxyl content amounted to 2.50 and 0.19 mmol g−1 in organosolv lignin and 3.92 and 0.35 mmol g−1 in Kraft lignin. In contrast, the aliphatic hydroxyl group content was higher in organosolv lignin (3.00 mmol g−1) than in Kraft lignin (2.18 mmol g−1). Etherification of lignin was carried out on the basis of total phenol as well as carboxyl group content.
3.2. Characterization of Lignins
Lignin is one of the most complex biopolymers as it contains various kinds of bonds and functional groups. Its complex nature often results in overlapped infrared spectra, which make it difficult to assign the correct identities. In spite of band overlapping in the fingerprint region, FT-IR spectra of native organosolv lignin and its ether functionalized forms (Figure 1), as well as the corresponding Kraft lignins (Figure 2), allowed bands to be assigned according to previously published data (Table 2) [36,37,38,39,40,41,42,43,44].
The band around 3438 cm−1 in organosolv (OS) lignin and 3427 cm−1 in Kraft (K) lignin corresponded to the O-H stretching frequency in aliphatic or phenolic hydroxyl groups. This band decreased significantly after functionalization in both OS and K lignin due to formation of an ether linkage with the aromatic hydroxyl group. The bands in the 2924 cm−1 to 2938 cm−1 and 2842 cm−1 to 2854 cm−1 regions corresponded to C-H stretching in methyl and methylene groups, respectively. A sharp increase in the C-H stretching frequency after functionalization confirmed the presence of an alkyl chain, demonstrating the successful etherification of lignin. The carbonyl/carboxyl region between 1711 cm−1 and 1730 cm−1 exhibited a different pattern in OS and K lignin before and after etherification. In the case of OS lignin, the band around 1711 cm−1 demonstrated the presence of aryl saturated carboxylic acids, accompanied by a shift to ~1725 cm−1 that suggested esterification at the carboxylic acid groups. In the case of K lignin, a broad aromatic conjugated carboxylate band around 1661 cm−1 was observed. After etherification, the carboxylate band shifted to 1725 cm−1, perhaps due to side esterification of the carboxylate group with alkyl iodide.
Aromatic skeletal vibration at 1590 cm−1 to 1598 cm−1, as well as aromatic ring vibration at 1506 cm−1 to 1514 cm−1 and 1419 cm−1 to 1427 cm−1 were common for all types of lignin. Band intensity varied between samples depending on texture. Asymmetric deformations of C-H bonds in methoxy and methylene groups appeared at 1452 cm−1 to 1466 cm−1, but displayed no change in intensity and position for both lignin types before and after etherification. Common weak bands at around 1367 cm−1 to 1378 cm−1 were found in all lignin samples; they originated from aliphatic C-H symmetric deformation in methyl groups and O-H deformation in phenolic groups. Interestingly, the band around 1328 cm−1 corresponding to the syringyl ring breathing vibration was present only in organosolv samples (Figure 1). This is probably due to the fact that Kraft lignin originated from spruce and the syringyl unit is absent from this source. The bands around 1264 cm−1 to 1269 cm−1 were assigned to the guaicyl ring breathing vibration and were present in all lignin samples; however, their intensity was higher in K lignin, confirming its provenance from spruce, which contains mainly guaicyl units.
Aromatic C-H in-plane deformation was observed at 1122 cm−1 to 1127 cm−1 in all lignin samples with different intensities. On the one hand, the small hump of C-O stretching from secondary alcohols and aliphatic ethers at 1082 cm−1 was found in all Kraft lignin samples. On the other hand, aromatic C-H in-plane deformations in G units along with C-O deformations in primary alcohols at 1034 cm−1 were found in all lignin samples. The same was true for C-H out-of-plane deformations corresponding to a weak band around 835 cm−1 to 855 cm−1. Overall, no significant changes were observed in the region from 835 cm−1 to 1750 cm−1, demonstrating that the core structure of lignin was maintained after mild etherification reaction.
NMR analysis based on 1H and 13C NMR spectra is a powerful tool for characterizing lignin functionalization. After etherification, lignin samples become highly hydrophobic in nature and thus highly soluble in low-polarity solvents, such as chloroform, diethylether. In this case CDCl3 was used as NMR solvent due to high solubility of etherified lignin samples. In 1H NMR spectra, the broad triplet signal around 0.87 ppm was assigned to methyl protons of C10–C16 alkyl chain units (Figure 3). The strong broad peaks around 1.2 ppm were assigned to all methylene protons (except α, β methylene protons) of C10–C16 alkyl chain units. The broad signal around 1.5–1.8 ppm was attributed to β methylene protons of C10–C16, whereas that of α methylene protons appeared around 3.8 ppm as a broad signal together with lignin methoxy groups. Aromatic and vinylic protons of lignin appeared around 6.1–7.0 ppm, although signal intensity was very low compared to aliphatic signals of C10–C16 chain units.
In 13C NMR spectra, the methyl carbon of C10–C16 chain units appeared around 14.4 ppm (Figure 4). Signals from methylene carbons (except α, β methylene carbons) of C10–C16 alkyl chains appeared between 22.9–30 ppm. The signals between 32.1–32.2 ppm were assigned to the β carbon of C10–C16 aliphatic chain units, whereas that of the α carbon was found around 69 ppm. The methoxy carbons of lignin appeared around 56.6 ppm. Aromatic carbons of lignin showed a very low signal between 102–156 ppm compared to C10–C16 aliphatic carbons. NMR signals of etherified lignin samples reflected previously reported O-alkyl phenolic compounds and thus further confirmed the successful etherification of lignin [45,46], as suggested also by high solubility in low-polarity solvents, such as chloroform and diethylether.
Molecular weights of lignin samples before and after etherification were compared and are reported in Table 3. Kraft lignin displayed generally higher molecular weight compared to organosolv lignin and was accompanied by a high polydispersity index. In all cases, etherification increased the molecular weight as a result of the newly attached alkyl chains, further attesting to successful etherification of both lignin types. The increase in lignin molecular weight has been previously used to verify the success of esterification reactions [14,19,47]. Finally, etherification was seen to lower the polydispersity index, as observed for the esterification of lignin [19,47].
3.3. Thermal Characterization of Functionalized Lignins
Thermal stability is one of the main parameters for thermoplastic applications, which can be assessed by measuring TGA analysis. There are very few reports available on systematic thermal analysis data of alkylated lignins with higher alkyl chains. Previously Chen et al. reported on C12 alkylated lignin for PP blends and observed 10% weight loss at 200 °C [48]. Ramp TGA is a widespread method for the initial thermal stability screening of any compound. Short-term thermal stability based on Twt% from ramped TGA data represents the fastest way to measure thermal stability of any materials [49]. Here, short-term thermal stability of T0.5%, T1%, and T2% corresponded to the temperature at which a weight loss of 0.5%, 1%, and 2% was observed in lignin samples (Table 4). Thermal stability of all lignin samples was analyzed by TGA with a heating ramp of 10 °C/min up to 800 °C under nitrogen atmosphere. In this study, two sets of lignin samples were analyzed by thermogravimetric method.
In the first case, organosolv lignin and its alkylated thermoplastic lignins were analyzed. In our case degradation of native organosolv lignin was observed relatively easily, even at <80 °C, possibly due to internal radical coupling reactions [50]. Initial 2% weight loss of native organosolv lignin were observed around 143 °C. From the TGA plot (Figure 5), it is evident that thermal stability increased significantly after ether functionalization and was highest in case of organosolv lignin etherified with C10. T0.5% of OS-C10 was observed around 174 °C; whereas OS-C16 reached 135 °C. The highest T1% was observed at 193 °C for OS-C10; whereas T1% for OS-C16 was 140 °C. Similarly, T2% of OS-C10 was around 213 °C, which is substantially more than the values for OS-C16, which was 148 °C.
On the other hand, the degradation of native Kraft lignin was also relatively facile like organosolv lignin and started even <80 °C. However, a 2% weight loss in native Kraft lignin was observed around 133 °C. Interestingly, etherified Kraft lignin also displayed good thermal stability (Figure 6), with the K-C16 sample exhibiting 0.5% decomposition at 148 °C, whereas K-C10 showed very poor thermal stability T0.5% at 75 °C. Furthermore, T1% of K-C16 and K-C10 were found to be 156 °C and 88 °C. Although 2% degradation of K-C16 was comparatively high 168 °C but K-C10 showed lower thermal stability 104 °C than its native Kraft lignin 133 °C. Finally, non-functionalized lignins (especially the Kraft one) produced a higher amount of char at 800 °C compared to etherified lignins. This is probably due to formation of highly fused polycyclic aromatic hydrocarbon compounds via coupling of free phenolic hydroxyl groups.
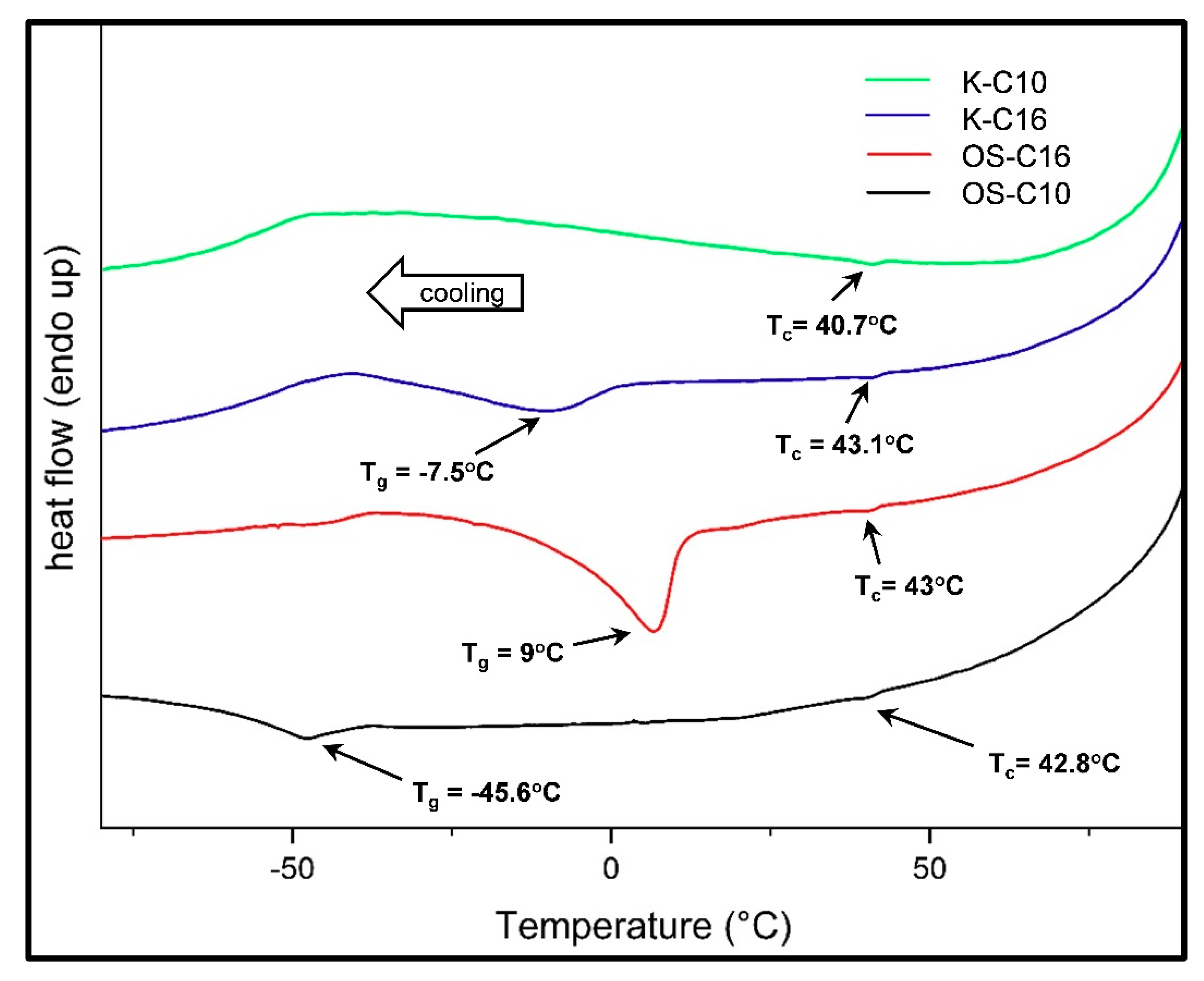
Tg and melting temperature (Tm) are crucial for choosing the most appropriate candidate thermoplastic application and can be measured by DSC analysis. Tg of non-functionalized organosolv lignin was found to be very broad and centered at 117.1 °C and for commercial Kraft lignin is reported at 153 °C [51], although both lignin starts to decompose also at lower temperature. This finding indicated that non-functionalized lignin did not exhibit true Tg and could not be used in thermoplastic applications, as it would decompose during the thermoforming process. On the contrary, ether functionalization of organosolv lignin led to a sharp Tg peak and the detection of Tm (Figure 7). Importantly, controlling the alkyl chain length during etherification allowed the control of Tg, opening the opportunity for fine-tuning the thermal properties of the generated thermoplastics in accordance with the required application. During DSC analysis, samples were initially heated to 100 °C to form a thin film and then gradually cooled to −80 °C prior to starting up the heating/cooling cycles. During the cooling part, a small dip around 40–43 °C was observed for almost in all samples and it was ascribed to the endothermic peak of crystallization (Tc). As all lignin samples were highly amorphous, the endothermic peak of Tc was small and was observed only during the cooling cycle. This small endothermic peak of crystallization occurred possibly due to the introduction of alkyl chain into the lignin structure, which induces alkyl stacking in lignin thermoplastic [52,53,54,55]. Another endothermic peak, corresponding to Tg, was observed when lignins were cooled further to a lower temperature. OS-C10 presented the lowest Tg (−45.6 °C), while OS-C16 showed a sharp endothermic peak around 9 °C, further confirming how variations in alkyl chain length could serve to fine-tune the thermoplastic properties. Finally, the Kraft lignin K-C16 sample exhibited glass transition around −7.5 °C (Figure 7), whereas K-C10 failed to show any Tg throughout the heating or cooling cycle. This was probably due to its broad molecular weight distribution with high polydispersity index (3.38) compared to other lignin samples.
Based on the above, it was shown that lignin can serve as raw material for the production of thermoplastic. Utilization of all the biomass fractions towards the production of bio-based products is important for an economically viable biomass biorefinery. In this context, organosolv fractionation serves as an excellent option to deliver fractions of cellulose, hemicellulose and lignin that can be used at different applications. Recently, in a techno-economic analysis of a biomass biorefinery based on organosolv fractionation of birch and spruce biomass it was shown that the process can be economically viable for the production of ethanol from cellulose, when hemicellulose and lignin are also products [56]. As it was discussed previously, lignin is often underutilized and there is a need to further develop a process for its conversion to high added-value products that aim to provide additional profit to the process. In this context, the production of thermoplastic materials from lignin can serve as a promising alternative towards establishing economically viable biorefinery processes.
4. Conclusions
Valorization of lignin towards high added-value products is a requirement for establishing an economically viable biorefinery paradigm. Most traditional processes for the valorization of lignocellulosic biomass result in the production of lignin contaminated with sugars, ash or microbial cells. Organosolv fractionation can produce elevated yields of highly pure and practically ash-free lignin with minimal sugar contaminations. Here, we demonstrated the comparative functionalization characteristics of organosolv-isolated lignin as well as Kraft lignin. Organosolv lignin can also serve as a promising material towards production of green thermoplastics with tunable characteristics compared to commercial lignin.
Author Contributions
Conceptualization, S.B., L.M., U.R. and P.C.; methodology, S.B. and L.M.; investigation, S.B. and L.M.; resources, U.R. and P.C.; writing—original draft preparation, S.B. and L.M.; writing—review and editing, U.R. and P.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors would like to thank Sveaskog, Sweden, for providing birch chips. L.M., U.R., and P.C. would like to thank Bio4Energy, a strategic research environment appointed by the Swedish government, for supporting this work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Azadi, P.; Inderwildi, O.R.; Farnood, R.R.; King, D.A. Liquid fuels, hydrogen and chemicals from lignin: A critical review. Renew. Sustain. Energy Rev. 2013, 21, 506–523. [Google Scholar] [CrossRef]
- Saito, T.; Perkins, J.H.; Jackson, D.; Trammel, N.E.; Hunt, M.A.; Naskar, A.K. Development of lignin-based polyurethane thermoplastics. RSC Adv. 2013, 3, 21832. [Google Scholar] [CrossRef]
- Bajwa, D.; Pourhashem, G.; Ullah, A.; Bajwa, S. A concise review of current lignin production, applications, products and their environmental impact. Ind. Crop. Prod. 2019, 139, 111526. [Google Scholar] [CrossRef]
- Hrůzová, K.; Matsakas, L.; Sand, A.; Rova, U.; Christakopoulos, P. Organosolv lignin hydrophobic micro- and nanoparticles as a low-carbon footprint biodegradable flotation collector in mineral flotation. Bioresour. Technol. 2020, 306, 123235. [Google Scholar] [CrossRef]
- Gillet, S.; Aguedo, M.; Petitjean, L.; Morais, A.R.C.; Lopes, A.M.D.C.; Lukasik, R.M.; Anastas, P.T. Lignin transformations for high value applications: Towards targeted modifications using green chemistry. Green Chem. 2017, 19, 4200–4233. [Google Scholar] [CrossRef]
- Mu, L.; Wu, J.; Matsakas, L.; Chen, M.; Rova, U.; Christakopoulos, P.; Zhu, J.; Shi, Y. Two important factors of selecting lignin as efficient lubricating additives in poly (ethylene glycol): Hydrogen bond and molecular weight. Int. J. Boil. Macromol. 2019, 129, 564–570. [Google Scholar] [CrossRef]
- Beckham, G.T.; Johnson, C.W.; Karp, E.M.; Salvachúa, D.; Vardon, D.R. Opportunities and challenges in biological lignin valorization. Curr. Opin. Biotechnol. 2016, 42, 40–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsakas, L.; Karnaouri, A.; Cwirzen, A.; Rova, U.; Christakopoulos, P. Formation of Lignin Nanoparticles by Combining Organosolv Pretreatment of Birch Biomass and Homogenization Processes. Molecules 2018, 23, 1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoresen, P.P.; Matsakas, L.; Rova, U.; Christakopoulos, P. Recent advances in organosolv fractionation: Towards biomass fractionation technology of the future. Bioresour. Technol. 2020, 306, 123189. [Google Scholar] [CrossRef] [PubMed]
- Kalogiannis, K.G.; Matsakas, L.; Aspden, J.; Lappas, A.; Rova, U.; Christakopoulos, P. Acid Assisted Organosolv Delignification of Beechwood and Pulp Conversion towards High Concentrated Cellulosic Ethanol via High Gravity Enzymatic Hydrolysis and Fermentation. Molecules 2018, 23, 1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinaldi, R.; Jastrzebski, R.; Clough, M.T.; Ralph, J.; Kennema, M.; Bruijnincx, P.C.; Weckhuysen, B.M. Paving the Way for Lignin Valorisation: Recent Advances in Bioengineering, Biorefining and Catalysis. Angew. Chem. Int. Ed. 2016, 55, 8164–8215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glasser, W.G. Classification of Lignin According to Chemical and Molecular Structure; American Chemical Society (ACS): Washington, DC, USA, 1999; Volume 742, pp. 216–238. [Google Scholar]
- Wang, C.; Kelley, S.S.; Venditti, R. Lignin-Based Thermoplastic Materials. ChemSusChem 2016, 9, 770–783. [Google Scholar] [CrossRef] [PubMed]
- Gordobil, O.; Robles, E.; Egüés, I.; Labidi, J. Lignin-ester derivatives as novel thermoplastic materials. RSC Adv. 2016, 6, 86909–86917. [Google Scholar] [CrossRef]
- Sen, S.; Patil, S.; Argyropoulos, D.S. Thermal properties of lignin in copolymers, blends, and composites: A review. Green Chem. 2015, 17, 4862–4887. [Google Scholar] [CrossRef]
- Ashori, A.; Sheshmani, S. Hybrid composites made from recycled materials: Moisture absorption and thickness swelling behavior. Bioresour. Technol. 2010, 101, 4717–4720. [Google Scholar] [CrossRef]
- Laurichesse, S.; Averous, L. Chemical modification of lignins: Towards biobased polymers. Prog. Polym. Sci. 2014, 39, 1266–1290. [Google Scholar] [CrossRef]
- Lewis, H.F.; Brauns, F.E. Esters of Lignin Material. U.S. Patent Application No. 2,429,102, 14 October 1944. [Google Scholar]
- Glasser, W.G.; Jain, R.K. Lignin derivatives. I. Alkanoates. Holzforschung 1993, 47, 225–233. [Google Scholar] [CrossRef]
- Guo, Z.-X.; Gandini, A.; Pla, F. Polyesters from lignin. 1. The reaction of Kraft lignin with dicarboxylic acid chlorides. Polym. Int. 1992, 27, 17–22. [Google Scholar] [CrossRef]
- Guo, Z.-X.; Gandini, A. Polyesters from lignin—2. The copolyesterification of Kraft lignin and polyethylene glycols with dicarboxylic acid chlorides. Eur. Polym. J. 1991, 27, 1177–1180. [Google Scholar] [CrossRef]
- Fang, R.; Cheng, X.-S.; Lin, W.-S. Preparation and application of dimer acid/lignin graft copolymer. BioResources 2011, 6, 2874–2884. [Google Scholar]
- Korich, A.L.; Fleming, A.B.; Walker, A.R.; Wang, J.; Tang, C.; Iovine, P.M. Chemical modification of organosolv lignin using boronic acid-containing reagents. Polymer 2012, 53, 87–93. [Google Scholar] [CrossRef]
- Han, Y.; Yuan, L.; Li, G.; Huang, L.; Qin, T.; Chu, F.; Tang, C. Renewable polymers from lignin via copper-free thermal click chemistry. Polymer 2016, 83, 92–100. [Google Scholar] [CrossRef]
- De Oliveira, W.; Glasser, W.G. Multiphase materials with lignin. 11. Starlike copolymers with caprolactone. Macromolecules 1994, 27, 5–11. [Google Scholar] [CrossRef]
- Chung, H.; Al-Khouja, A.; Washburn, N.R. Lignin-Based Graft CopolymersviaATRP and Click Chemistry. In Green Polymer Chemistry: Biocatalysis and Materials II; American Chemical Society (ACS): Washington, DC, USA, 2013; Volume 1144, pp. 373–391. [Google Scholar]
- Kim, Y.S.; Kadla, J.F. Preparation of a Thermoresponsive Lignin-Based Biomaterial through Atom Transfer Radical Polymerization. Biomacromolecules 2010, 11, 981–988. [Google Scholar] [CrossRef]
- Hilburg, S.L.; Elder, A.N.; Chung, H.; Ferebee, R.; Bockstaller, M.; Washburn, N.R. A universal route towards thermoplastic lignin composites with improved mechanical properties. Polymer 2014, 55, 995–1003. [Google Scholar] [CrossRef]
- Sadeghifar, H.; Cui, C.; Argyropoulos, D.S. Toward Thermoplastic Lignin Polymers. Part 1. Selective Masking of Phenolic Hydroxyl Groups in Kraft Lignins via Methylation and Oxypropylation Chemistries. Ind. Eng. Chem. Res. 2012, 51, 16713–16720. [Google Scholar] [CrossRef]
- Glasser, W.G.; Barnett, C.A.; Rials, T.G.; Saraf, V.P. Engineering plastics from lignin II. Characterization of hydroxyalkyl lignin derivatives. J. Appl. Polym. Sci. 1984, 29, 1815–1830. [Google Scholar] [CrossRef]
- Kazzaz, A.E.; Feizi, Z.H.; Fatehi, P. Grafting strategies for hydroxy groups of lignin for producing materials. Green Chem. 2019, 21, 5714–5752. [Google Scholar] [CrossRef] [Green Version]
- Matsakas, L.; Nitsos, C.; Raghavendran, V.; Yakimenko, O.; Persson, G.; Olsson, E.; Rova, U.; Olsson, L.; Christakopoulos, P. A novel hybrid organosolv: Steam explosion method for the efficient fractionation and pretreatment of birch biomass. Biotechnol. Biofuels 2018, 11, 160. [Google Scholar] [CrossRef]
- Meng, X.; Crestini, C.; Ben, H.; Hao, N.; Pu, Y.; Ragauskas, A.J.; Argyropoulos, D.S. Determination of hydroxyl groups in biorefinery resources via quantitative 31P NMR spectroscopy. Nat. Protoc. 2019, 14, 2627–2647. [Google Scholar] [CrossRef] [PubMed]
- Lange, H.; Rulli, F.; Crestini, C. Gel Permeation Chromatography in Determining Molecular Weights of Lignins: Critical Aspects Revisited for Improved Utility in the Development of Novel Materials. ACS Sustain. Chem. Eng. 2016, 4, 5167–5180. [Google Scholar] [CrossRef]
- Ren, W.; Pan, X.; Wang, G.; Cheng, W.; Liu, Y. Dodecylated lignin-g-PLA for effective toughening of PLA. Green Chem. 2016, 18, 5008–5014. [Google Scholar] [CrossRef]
- Hergert, H.L. Infrared Spectra of Lignin and Related Compounds. II. Conifer Lignin and Model Compounds 1,2. J. Org. Chem. 1960, 25, 405–413. [Google Scholar] [CrossRef]
- Faix, O. Classification of Lignins from Different Botanical Origins by FT-IR Spectroscopy. Holzforschung 1991, 45, 21–28. [Google Scholar] [CrossRef]
- Boeriu, C.G.; Bravo, D.; Gosselink, R.J.; Van Dam, J.E. Characterisation of structure-dependent functional properties of lignin with infrared spectroscopy. Ind. Crop. Prod. 2004, 20, 205–218. [Google Scholar] [CrossRef]
- Derkacheva, O.Y.; Sukhov, D. Investigation of Lignins by FTIR Spectroscopy. Macromol. Symp. 2008, 265, 61–68. [Google Scholar] [CrossRef]
- Gabov, K.; Gosselink, R.J.A.; Smeds, A.I.; Fardim, P. Characterization of Lignin Extracted from Birch Wood by a Modified Hydrotropic Process. J. Agric. Food Chem. 2014, 62, 10759–10767. [Google Scholar] [CrossRef]
- Zhang, L.; Yan, L.; Wang, Z.; Laskar, D.D.; Swita, M.S.; Cort, J.R.; Yang, B. Characterization of lignin derived from water-only and dilute acid flowthrough pretreatment of poplar wood at elevated temperatures. Biotechnol. Biofuels 2015, 8, 203. [Google Scholar] [CrossRef] [Green Version]
- Todorciuc, T.; Cǎpraru, A.-M.; Kratochvílová, I.; Popa, V.I. Characterization of non-wood lignin and its hydoxymethylated derivatives by spectroscopy and self-assembling investigations. Cellul. Chem. Technol. 2009, 43, 399–408. [Google Scholar]
- Sammons, R.J.; Harper, D.P.; Labbé, N.; Bozell, J.J.; Elder, T.; Rials, T.G. Characterization of Organosolv Lignins using Thermal and FT-IR Spectroscopic Analysis. BioResources 2013, 8, 2752–2767. [Google Scholar] [CrossRef]
- Hergert, H.L. Infrared spectra. In Lignins: Occurrence, Formation, Structures and Reactions; Sarkanen, K.V., Hergert, H.L., Eds.; John Wiley & Sons, Inc.: New York, NY, USA, 1971; pp. 267–297. [Google Scholar]
- Swapna, K.; Murthy, S.N.; Jyothi, M.T.; Nageswar, Y.V.D. Recyclable heterogeneous copper oxide on alumina catalyzed coupling of phenols and alcohols with aryl halides under ligand-free conditions. Org. Biomol. Chem. 2011, 9, 5978–5988. [Google Scholar] [CrossRef] [PubMed]
- Jammi, S.; Sakthivel, S.; Rout, L.; Mukherjee, T.; Mandal, S.; Mitra, R.; Saha, P.; Punniyamurthy, T. ChemInform Abstract: CuO Nanoparticles Catalyzed C-N, C-O, and C-S Cross-Coupling Reactions: Scope and Mechanism. ChemInform 2009, 40, 1971–1976. [Google Scholar] [CrossRef]
- Hult, E.-L.; Koivu, K.; Asikkala, J.; Ropponen, J.; Wrigstedt, P.; Sipilä, J.; Poppius-Levlin, K. Esterified lignin coating as water vapor and oxygen barrier for fiber-based packaging. Holzforschung 2013, 67, 899–905. [Google Scholar] [CrossRef]
- Chen, F.; Dai, H.; Dong, X.; Yang, J.; Zhong, M. Physical properties of lignin-based polypropylene blends. Polym. Compos. 2011, 32, 1019–1025. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Shah, F.U. Thermal stability of choline based amino acid ionic liquids. J. Mol. Liq. 2018, 266, 597–602. [Google Scholar] [CrossRef]
- Durbeej, B.; Eriksson, L. A Density Functional Theory Study of Coniferyl Alcohol Intermonomeric Cross Linkages in Lignin—Three-Dimensional Structures, Stabilities and the Thermodynamic Control Hypothesis. Holzforschung 2003, 57, 150–164. [Google Scholar] [CrossRef]
- An, Y.-X.; Li, N.; Wu, H.; Lou, W.-Y.; Zong, M.-H. Changes in the Structure and the Thermal Properties of Kraft Lignin during Its Dissolution in Cholinium Ionic Liquids. ACS Sustain. Chem. Eng. 2015, 3, 2951–2958. [Google Scholar] [CrossRef]
- Cao, Z.; Galuska, L.; Qian, Z.; Zhang, S.; Huang, L.; Prine, N.; Li, T.; He, Y.; Hong, K.; Gu, X. The effect of side-chain branch position on the thermal properties of poly(3-alkylthiophenes). Polym. Chem. 2020, 11, 517–526. [Google Scholar] [CrossRef]
- Prasad, S.; Jiang, Z.; Sinha, S.K.; Dhinojwala, A. Partial Crystallinity in Alkyl Side Chain Polymers Dictates Surface Freezing. Phys. Rev. Lett. 2008, 101, 065505. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.T. Crystallinity in isotactic polyolefins with unbranched side chains. Makromol. Chem. 1964, 71, 1–32. [Google Scholar] [CrossRef]
- Cowie, J.M.G.; Reid, V.M.C.; McEwen, I.J. Effect of side chain length on the glass transition of copolymers from styrene with n -alkyl citraconimides and with n -alkyl itaconimides. Br. Polym. J. 1990, 23, 353–357. [Google Scholar] [CrossRef]
- Mesfun, S.; Matsakas, L.; Rova, U.; Christakopoulos, P. Technoeconomic Assessment of Hybrid Organosolv–Steam Explosion Pretreatment of Woody Biomass. Energies 2019, 12, 4206. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
FT-IR spectra of nonfunctionalized organosolv (OS) lignin etherified with C10 (OS-C10) and C16 (OS-C16) alkyl chains.
Figure 1.
FT-IR spectra of nonfunctionalized organosolv (OS) lignin etherified with C10 (OS-C10) and C16 (OS-C16) alkyl chains.
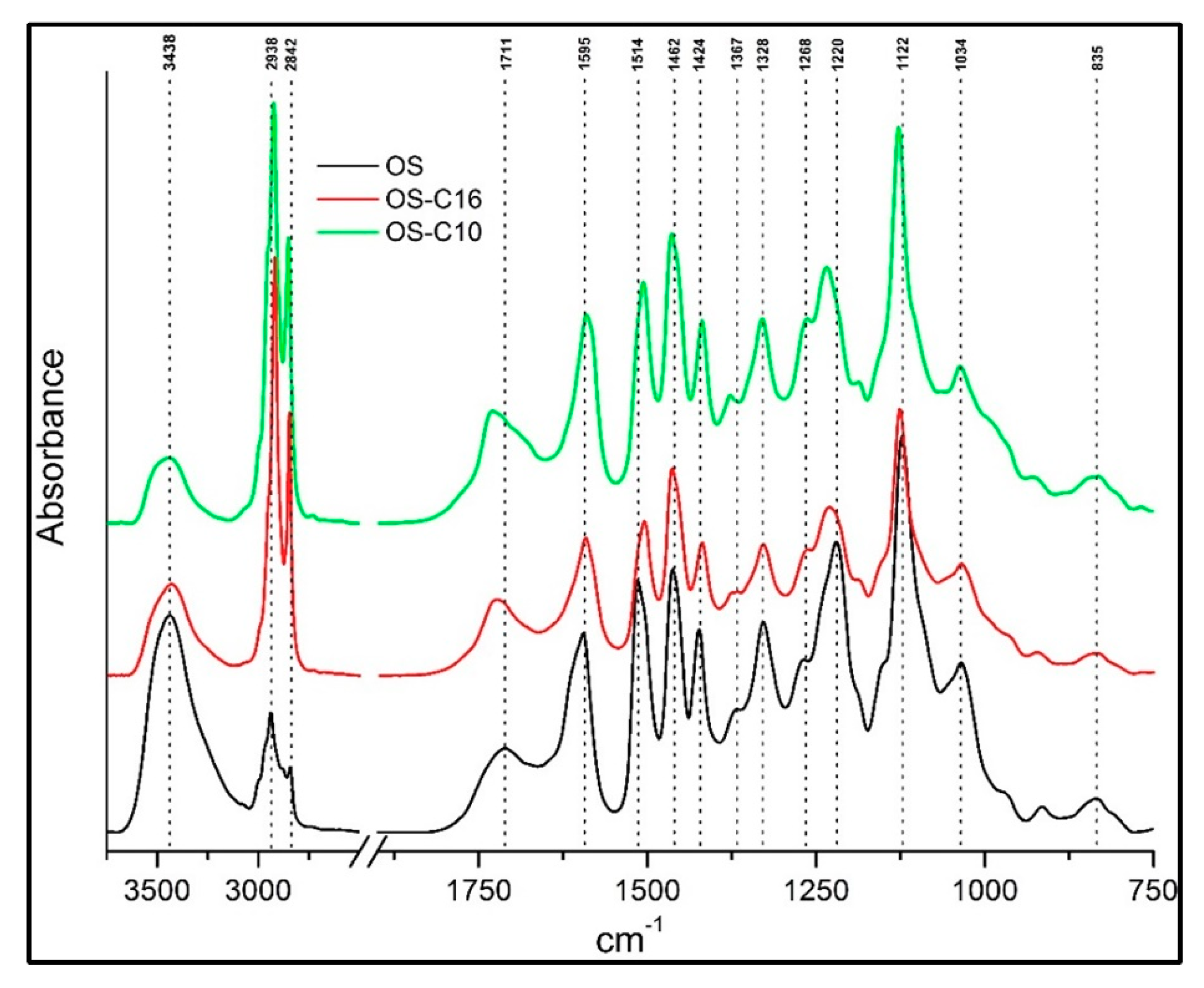
Figure 2.
FT-IR spectra of nonfunctionalized Kraft (K) lignin etherified with C10 (K-C10) and C16 alkyl chains.
Figure 2.
FT-IR spectra of nonfunctionalized Kraft (K) lignin etherified with C10 (K-C10) and C16 alkyl chains.
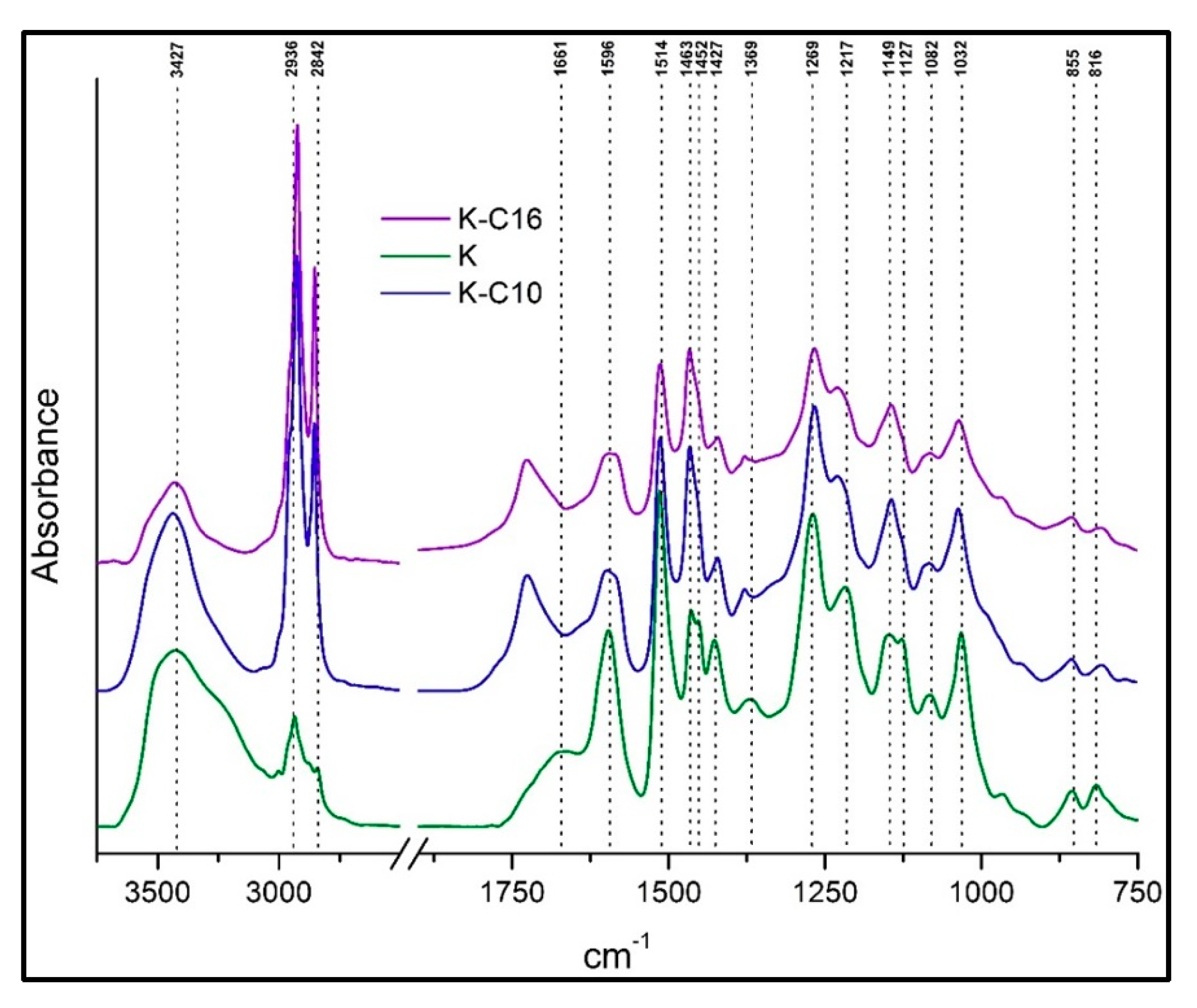
Figure 3.
1H NMR spectra of functionalized organosolv (OS) and Kraft (K) lignins.
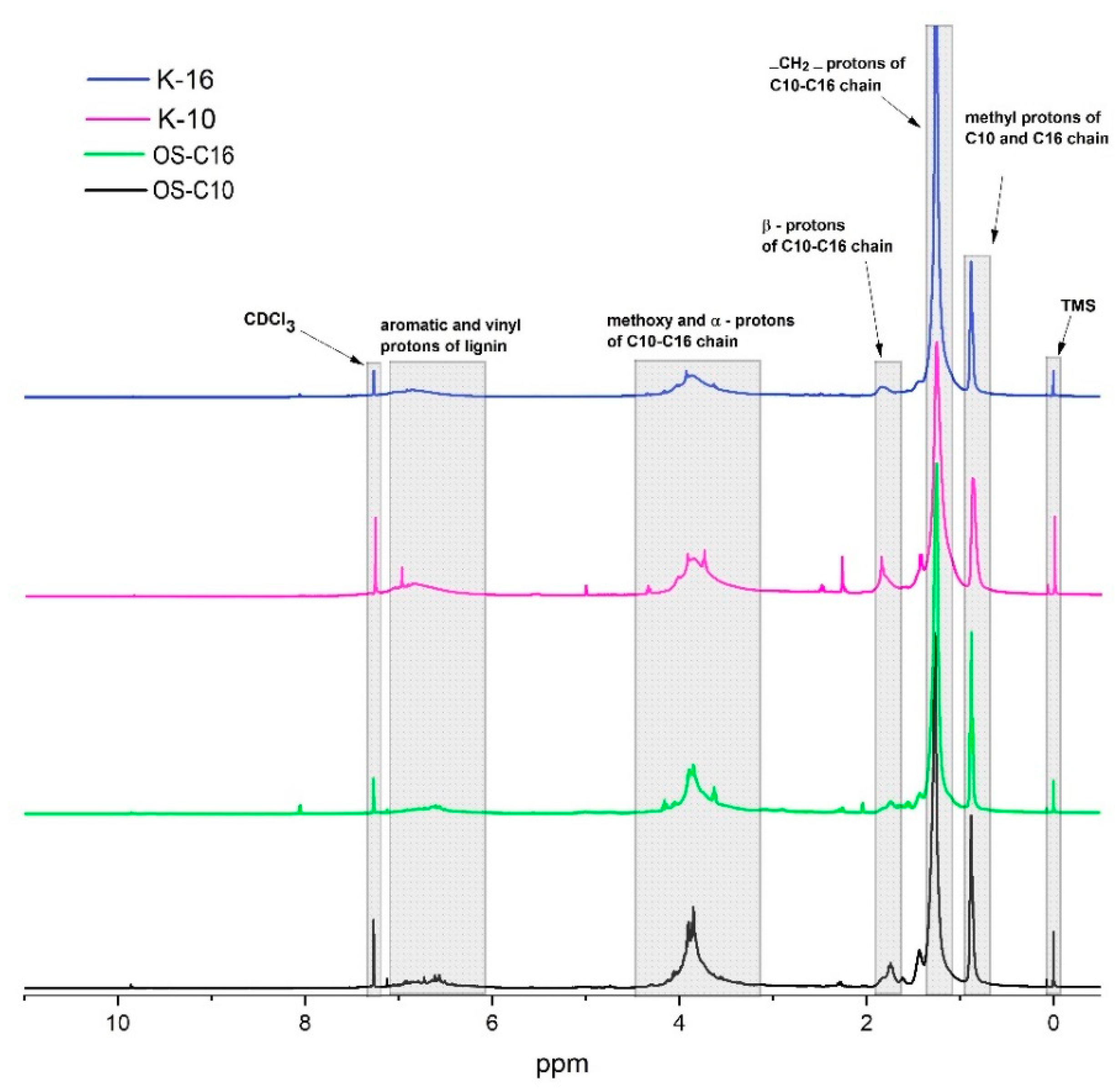
Figure 4.
13C NMR spectra of functionalized organosolv (OS) and Kraft (K) lignins.

Figure 5.
Thermal decomposition graphs obtained under nitrogen atmosphere of non-functionalized and etherified organosolv lignin.
Figure 5.
Thermal decomposition graphs obtained under nitrogen atmosphere of non-functionalized and etherified organosolv lignin.
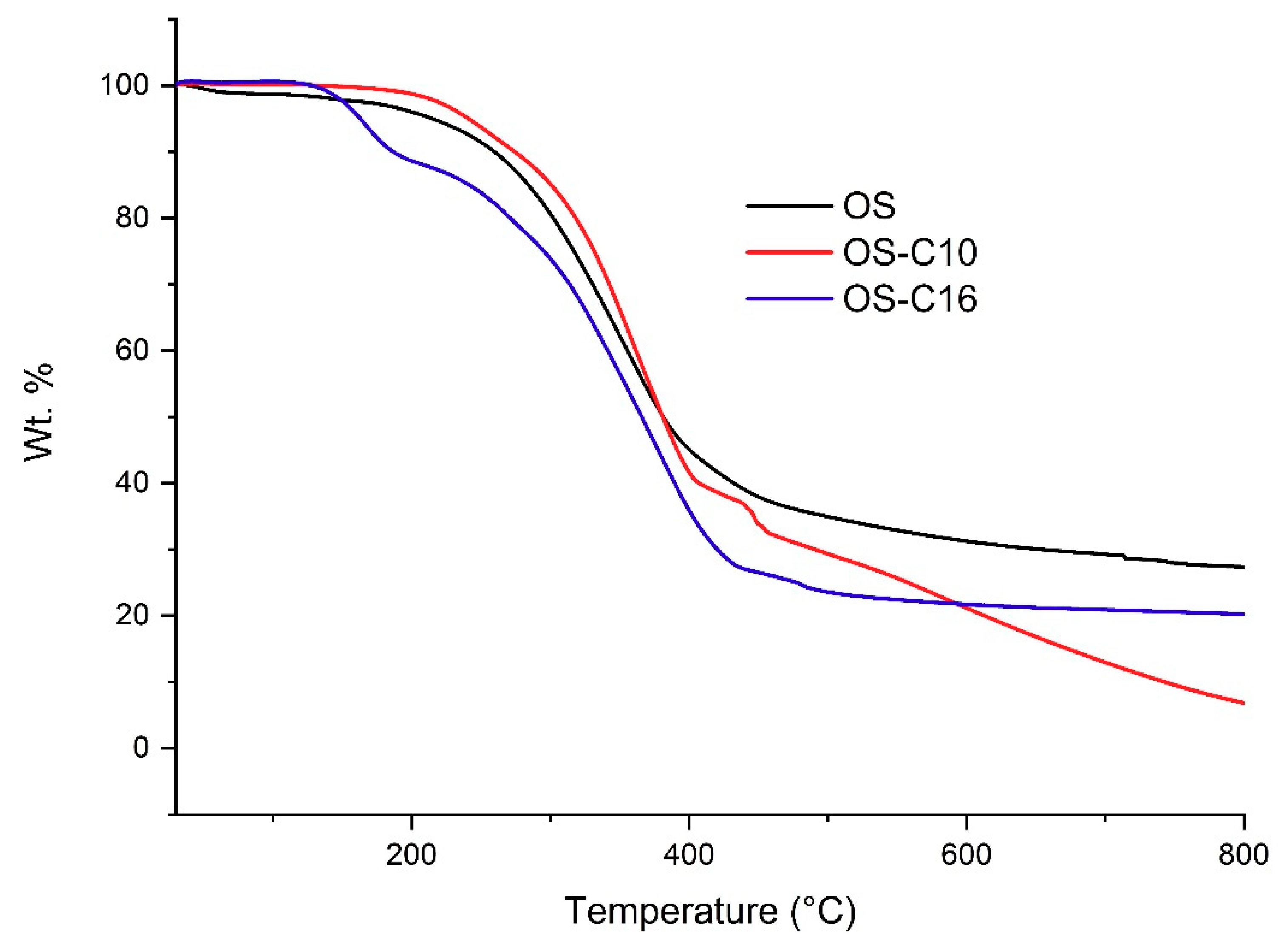
Figure 6.
Thermal decomposition graphs obtained under nitrogen atmosphere of nonfunctionalized and etherified Kraft lignin.
Figure 6.
Thermal decomposition graphs obtained under nitrogen atmosphere of nonfunctionalized and etherified Kraft lignin.
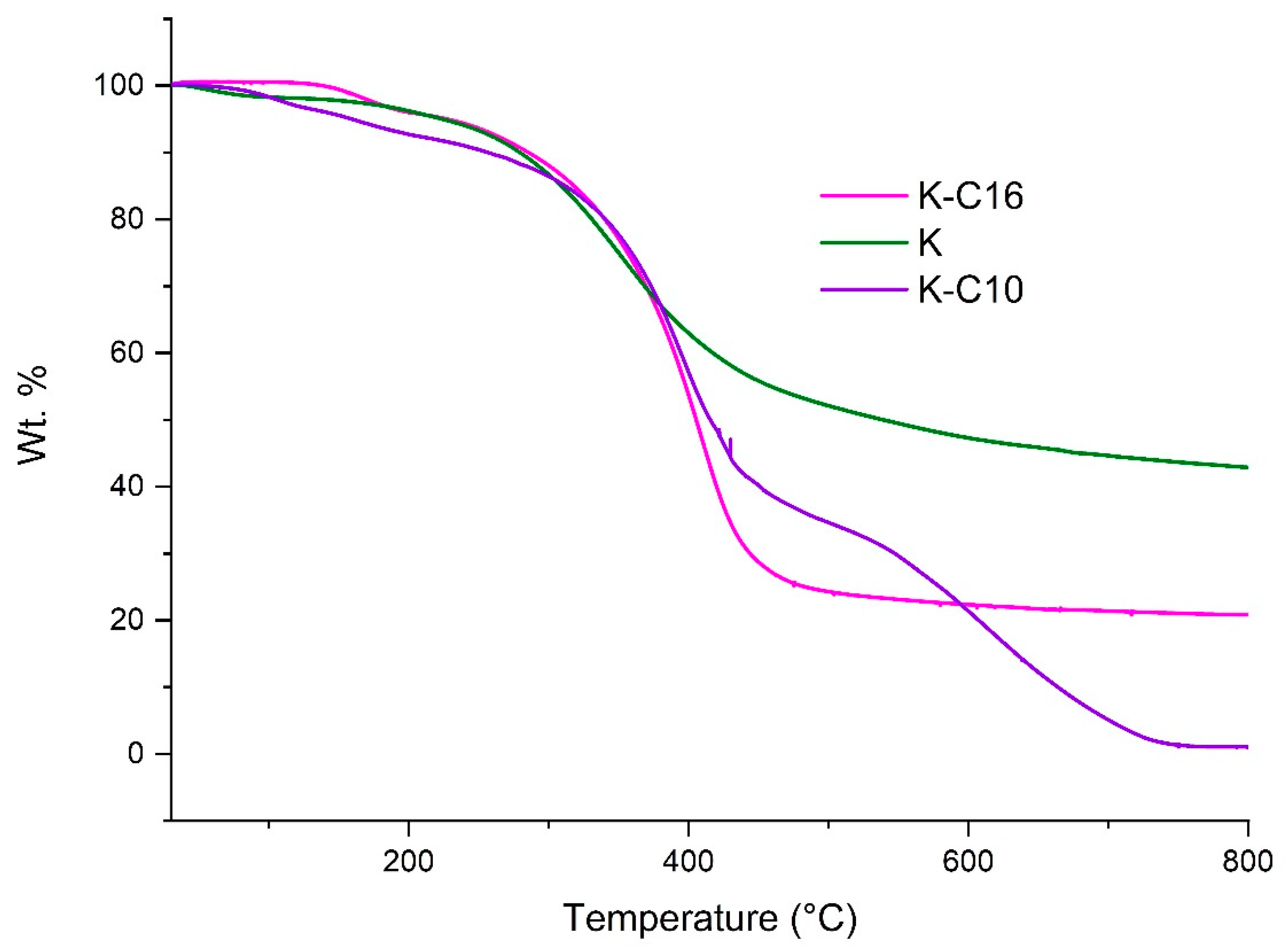
Figure 7.
DSC analysis of functionalized lignins indicating glass transition (Tg) and crystallization (Tc) temperatures.
Figure 7.
DSC analysis of functionalized lignins indicating glass transition (Tg) and crystallization (Tc) temperatures.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
31P NMR (Nuclear Magnetic Resonance) data.
| Lignin | Total Phenol (mmol g−1) | Carboxyl (mmol g−1) | Aliphatic (mmol g−1) |
|---|---|---|---|
| Organosolv | 2.50 | 0.19 | 3.00 |
| Kraft | 3.92 | 0.35 | 2.18 |
| Wave Number (cm−1) | IR Band Assignments |
|---|---|
| 3427–3442 | O-H stretching in aliphatic and phenolic -OH |
| 2924–2938 | C-H stretching in methyl groups |
| 2842–2854 | C-H stretch in methylene groups |
| 1711–1730 | C=O stretching in unconjugated ketones and carboxyl groups; saturated esters |
| 1661 | Stretching of C=O conjugated to aromatic rings (conjugated carbonyl) |
| 1590–1598 | Aromatic skeletal ring vibration (S > G) + C=O stretch |
| 1506–1514 | Aromatic skeletal ring vibrations (G > S) |
| 1452–1466 | C-H asymmetric deformations in methyl and methylene groups |
| 1419–1427 | Aromatic skeletal ring vibrations |
| 1367–1378 | Aliphatic C-H symmetric deformation in methyl (not methoxyl) + O-H deformation in phenols |
| 1328–1330 | S ring breathing vibration + G ring substituted in position 5 |
| 1264–1269 | G ring breathing vibration and C-O stretching |
| 1217–1235 | C-O stretching in phenols and ethers |
| 1122–1149 | C-H stretching in G ring and Aromatic C–H in plane deformation (S) |
| 1082 | C-O stretch of secondary alcohols and aliphatic ethers |
| 1034 | Aromatic C-H in-plane deformations in G units + C-O deformations in primary alcohols |
| 835/855 | Aromatic C-H out-of-plane deformation (only in GS and H lignin types) |
Table 3.
Molecular weight values for non-functionalized organosolv (OS) and Kraft (K) lignin, as well as for their derivatives after etherification.
Table 3.
Molecular weight values for non-functionalized organosolv (OS) and Kraft (K) lignin, as well as for their derivatives after etherification.
| Lignin | Number Average Molecular Weight, Mn (Da) | Weight Average Molecular Weight, Mw (Da) | Polydispersity Index |
|---|---|---|---|
| OS | 1757 | 4603 | 2.62 |
| OS-C10 | 1859 | 3719 | 2.00 |
| OS-C16 | 2661 | 6219 | 2.34 |
| K | 1924 | 7759 | 4.03 |
| K-C10 | 3452 | 11,664 | 3.38 |
| K-C16 | 3493 | 10,925 | 3.13 |
Table 4.
Twt% values of ether-functionalized organosolv (OS) and Kraft (K) lignin samples from the TGA plot.
Table 4.
Twt% values of ether-functionalized organosolv (OS) and Kraft (K) lignin samples from the TGA plot.
| Sample | T0.5% (°C) | T1% (°C) | T2% (°C) |
|---|---|---|---|
| OS | 50 | 65 | 143 |
| OS-C10 | 174 | 193 | 213 |
| OS-C16 | 135 | 140 | 148 |
| K | 54 | 66 | 133 |
| K-C16 | 148 | 156 | 168 |
| K-C10 | 75 | 88 | 104 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bhattacharyya, S.; Matsakas, L.; Rova, U.; Christakopoulos, P. Melt Stable Functionalized Organosolv and Kraft Lignin Thermoplastic. Processes 2020, 8, 1108. https://doi.org/10.3390/pr8091108
AMA Style
Bhattacharyya S, Matsakas L, Rova U, Christakopoulos P. Melt Stable Functionalized Organosolv and Kraft Lignin Thermoplastic. Processes. 2020; 8(9):1108. https://doi.org/10.3390/pr8091108
Chicago/Turabian StyleBhattacharyya, Shubhankar, Leonidas Matsakas, Ulrika Rova, and Paul Christakopoulos. 2020. "Melt Stable Functionalized Organosolv and Kraft Lignin Thermoplastic" Processes 8, no. 9: 1108. https://doi.org/10.3390/pr8091108
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.