
Optimizing Chromatographic Separation with Redosing: Effects on Separation Efficiency of a Model System in Centrifugal Partition Chromatography

Abstract
:1. Introduction
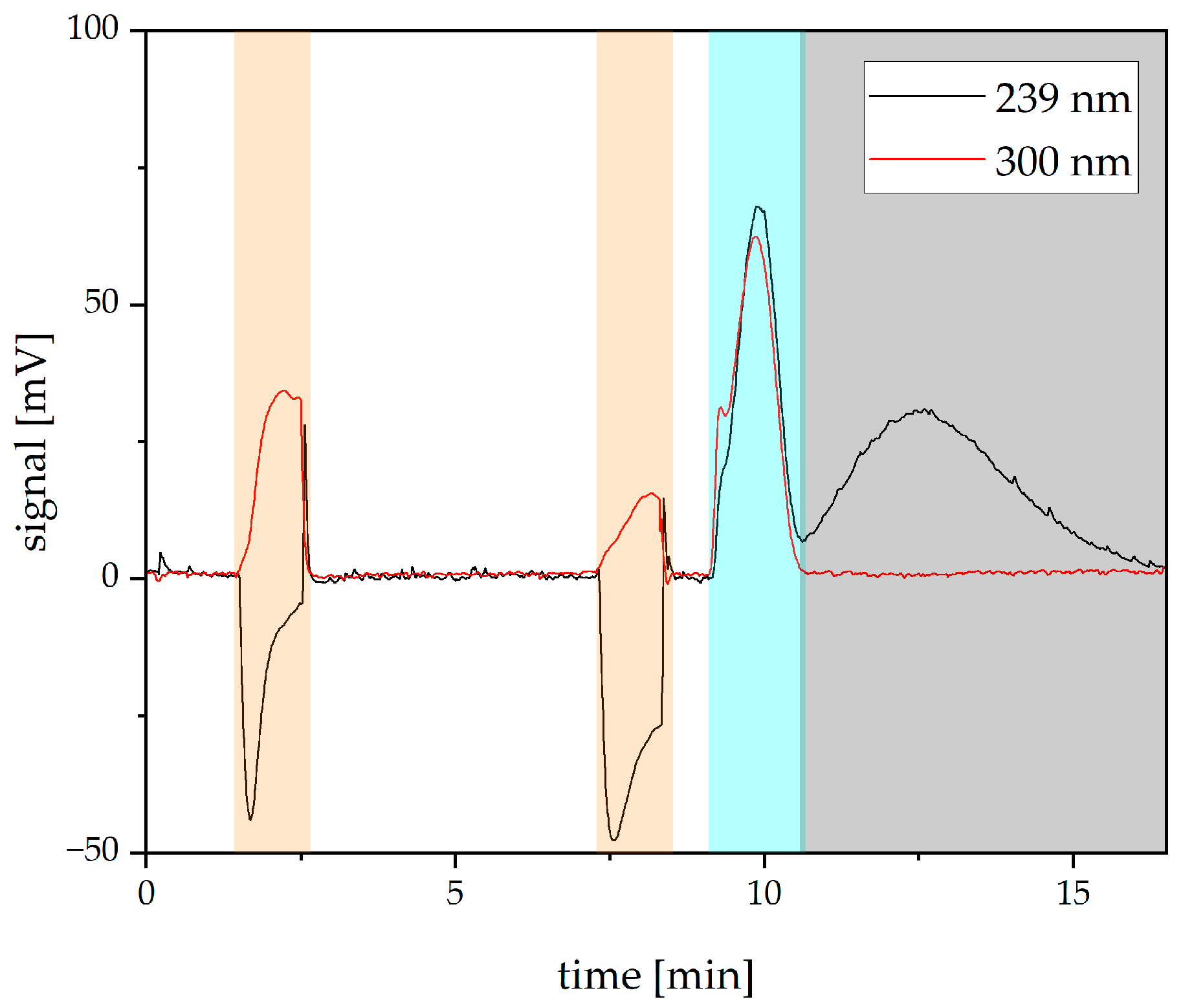
- Separation efficiency with redosing of the stationary phase.The influence of the redosing process on the separation must be addressed. With stabilized retention values over time, the chromatographic resolution should be constant too. Therefore, separation runs without redosing are necessary as a benchmark. Furthermore, potential optimization of the separation should be highlighted.
- Interdependence of redosing and sample injection.It is essential to analyze whether the redosing of the stationary phase and the injection of samples during operation interfere.
2. Materials and Methods
2.1. Phase System and Model System
2.2. Centrifugal Partition Chromatograph
2.3. Influence of Redosing on Resolution
2.4. Optimizing the Resolution
2.5. Separation Experiments with Redosing
2.6. Reference Experiments without Redosing
3. Results and Discussion
3.1. Model System Analysis
3.2. Influence of Redosing on Resolution
3.3. Optimizing the Resolution
3.4. Separation Experiments with Redosing
3.5. Reference Experiments without Redosing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Almeida, M.R.; Ferreira, F.; Domingues, P.; Coutinho, J.A.; Freire, M.G. Towards the purification of IgY from egg yolk by centrifugal partition chromatography. Sep. Purif. Technol. 2022, 299, 121697. [Google Scholar] [CrossRef]
- Kopilovic, B.; Valente, A.I.; Ferreira, A.M.; Almeida, M.R.; Tavares, A.P.M.; Freire, M.G.; Coutinho, J.A.P. Towards the sustainable extraction and purification of non-animal proteins from biomass using alternative solvents. RSC Sustain. 2023, 1, 1314–1331. [Google Scholar] [CrossRef]
- Friesen, J.B.; McAlpine, J.B.; Chen, S.-N.; Pauli, G.F. Countercurrent Separation of Natural Products: An Update. J. Nat. Prod. 2015, 78, 1765–1796. [Google Scholar] [CrossRef]
- Szekely, G.; Zhao, D. (Eds.) Sustainable Separation Engineering: Materials, Techniques and Process Development; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2022; ISBN 9781119740087. [Google Scholar]
- Schmidt-Traub, H.; Schulte, M.; Seidel-Morgenstern, A. (Eds.) Preparative Chromatography, 3rd ed.; Wiley-VCH: Weinheim, Germany, 2020; ISBN 978-3-527-34486-4. [Google Scholar]
- Nioi, C.; Riboul, D.; Destrac, P.; Marty, A.; Marchal, L.; Condoret, J.S. The centrifugal partition reactor, a novel intensified continuous reactor for liquid–liquid enzymatic reactions. Biochem. Eng. J. 2015, 103, 227–233. [Google Scholar] [CrossRef]
- Beschkov, V.N.; Yankov, D. Downstream Processing in Biotechnology; De Gruyter: Berlin, Germany, 2021; ISBN 9783110574111. [Google Scholar]
- Chmiel, H. (Ed.) Bioprozesstechnik, 3rd ed.; Spektrum Akademischer Verlag: Heidelberg, Germany, 2011; ISBN 9783827424778. [Google Scholar]
- Klefentz, H. Industrial Pharmaceutical Biotechnology; WILEY VCH: Weinheim, Germany, 2021; ISBN 9783527318919. [Google Scholar]
- Jerz, G.; Winterhalter, P. The 10th international conference on countercurrent chromatography held at Technische Universität Braunschweig, Braunschweig, Germany, August 1–3, 2018. J. Chromatogr. A 2020, 1617, 460698. [Google Scholar] [CrossRef]
- Berthod, A. Separation and Purification with a Liquid Stationary Phase. Separations 2017, 4, 30. [Google Scholar] [CrossRef]
- Marchal, L.; Legrand, J.; Foucault, A. Centrifugal partition chromatography: A survey of its history, and our recent advances in the field. Chem. Rec. 2003, 3, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Foucault, A.P. (Ed.) Centrifugal Partition Chromatography; Dekker: New York, NY, USA, 1995; ISBN 9780824792572. [Google Scholar]
- Luca, S.V.; Bruesewitz, M.; Minceva, M. Evaluation of advanced liquid–liquid chromatography operating modes for the tetrahydrocannabinol-remediation of hemp extracts. Chem. Eng. Sci. 2023, 282, 119279. [Google Scholar] [CrossRef]
- Eschlwech, F.; Ruedenauer, F.; Leonhardt, S.D.; Minceva, M.; Luca, S.V. Liquid-liquid chromatography separation of hemp terpenes with repellent properties against Hyalomma marginatum: A multi-methodological approach. Ind. Crops Prod. 2023, 197, 116562. [Google Scholar] [CrossRef]
- Berthod, A. (Ed.) Countercurrent Chromatography: The Support-Free Liquid Stationary Phase; Elsevier: Amsterdam, The Netherlands, 2002; ISBN 9780444507372. [Google Scholar]
- Buthmann, F.; Laby, P.; Hamza, D.; Koop, J.; Schembecker, G. Spatially and Temporally Resolved Analysis of Bleeding in a Centrifugal Partition Chromatography Rotor. Separations 2024, 11, 56. [Google Scholar] [CrossRef]
- Meucci, J.; Faure, K.; Mekaoui, N.; Berthod, A. Solvent Selection in Countercurrent Chromatography Using Small-Volume Hydrostatic Columns. LCGC N. Am. 2013, 31, 132–143. [Google Scholar]
- Foucault, A.P.; Frias, E.C.; Bordier, C.G.; Le Goffic, F. Centrifugal Partition Chromatography: Stability of Various Biphasic Systems and Pertinence of the “Stoke’s Model” to Describe the Influence of the Centrifugal Field Upon the Efficiency. J. Liq. Chromatogr. 1994, 17, 1–17. [Google Scholar] [CrossRef]
- Oka, F.; Oka, H.; Ito, Y. Systematic search for suitable two-phase solvent systems for high-speed counter-current chromatography. J. Chromatogr. A 1991, 538, 99–108. [Google Scholar] [CrossRef]
- Chollet, S.; Marchal, L.; Jérémy, M.; Renault, J.-H.; Legrand, J.; Foucault, A. Methodology for optimally sized centrifugal partition chromatography columns. J. Chromatogr. A 2015, 1388, 174–183. [Google Scholar] [CrossRef]
- Adelmann, S. On Hydrodynamics in Centrifugal Partition Chromatography; Dissertation, TU Dortmund University: München, Germany, 2014; ISBN 9783843916110. [Google Scholar]
- Buthmann, F.; Volpert, S.; Koop, J.; Schembecker, G. Prediction of Bleeding via Simulation of Hydrodynamics in Centrifugal Partition Chromatography. Separations 2024, 11, 16. [Google Scholar] [CrossRef]
- Murayama, W.; Kobayashi, T.; Kosuge, Y.; Yano, H.; Nunogaki, Y.; Nunogaki, K. A new centrifugal counter-current chromatograph and its application. J. Chromatogr. A 1982, 239, 643–649. [Google Scholar] [CrossRef]
- van Buel, M.J.; van Halsema, F.E.D.; van der Wielen, L.A.M.; Luyben, K.C.A.M. Flow regimes in centrifugal partition chromatography. AIChE J. 1998, 44, 1356–1362. [Google Scholar] [CrossRef]
- Buthmann, F.; Hohlmann, J.; Volpert, S.; Neuwald, M.; Hamza, D.; Schembecker, G. Counteracting Bleeding in Centrifugal Partition Chromatography: Redosing of the Stationary Phase. Separations 2024, 11, 98. [Google Scholar] [CrossRef]
- Buthmann, F.; Pley, F.; Schembecker, G.; Koop, J. Automated Image Analysis for Retention Determination in Centrifugal Partition Chromatography. Separations 2022, 9, 358. [Google Scholar] [CrossRef]
- Berthod, A.; Hassoun, M.; Ruiz-Angel, M.J. Alkane effect in the Arizona liquid systems used in countercurrent chromatography. Anal. Bioanal. Chem. 2005, 383, 327–340. [Google Scholar] [CrossRef]
- Fromme, A. Systematic Approach towards Solvent System Selection for Ideal Fluid Dynamics in Centrifugal Partition Chromatography; Dissertation, TU Dortmund University; Dr. Hut: München, Germany, 2020; ISBN 9783843948647. [Google Scholar]
- Schwienheer, C. Advances in Centrifugal Purification Techniques for Separating (Bio-)chemical Compounds; Dissertation, TU Dortmund University: München, Germany, 2016; ISBN 9783843929165. [Google Scholar]
- Clarity; Data Apex: Prague, Czech Republic, 2023. Available online: https://www.dataapex.com/downloads/26068/view (accessed on 4 January 2024).
- Moler, C. Matlab; Mathworks: Natick, MA, USA, 2022. Available online: https://de.mathworks.com/help/matlab/ (accessed on 4 January 2024).
- LabVIEW; National Instruments: Austin, TX, USA, 2020. Available online: https://www.ni.com/docs/de-DE/bundle/labview/page/user-manual-welcome.html (accessed on 4 January 2024).
- Abdel-Kader, M.S.; Alam, P.; Alqasoumi, S.I. Densitometric HPTLC method for qualitative, quantitative analysis and stability study of Coenzyme Q10 in pharmaceutical formulations utilizing normal and reversed-phase silica gel plates. Pak. J. Pharm. Sci. 2016, 29, 477–484. [Google Scholar] [PubMed]
- Wang, Y.; Xu, P.-P.; Li, X.-X.; Nie, K.; Tuo, M.-F.; Kong, B.; Chen, J. Monitoring the hydrolyzation of aspirin during the dissolution testing for aspirin delayed-release tablets with a fiber-optic dissolution system. J. Pharm. Anal. 2012, 2, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Bouju, E.; Berthod, A.; Faure, K. Scale-up in centrifugal partition chromatography: The “free-space between peaks” method. J. Chromatogr. A 2015, 1409, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Lorántfy, L.; Rutterschmid, D.; Örkényi, R.; Bakonyi, D.; Faragó, J.; Dargó, G.; Könczöl, Á. Continuous Industrial-Scale Centrifugal Partition Chromatography with Automatic Solvent System Handling: Concept and Instrumentation. Org. Process Res. Dev. 2020, 24, 2676–2688. [Google Scholar] [CrossRef]
- Hopmann, E.; Minceva, M. Separation of a binary mixture by sequential centrifugal partition chromatography. J. Chromatogr. A 2012, 1229, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Hopmann, E.; Goll, J.; Minceva, M. Sequential Centrifugal Partition Chromatography: A New Continuous Chromatographic Technology. Chem. Eng. Technol. 2012, 35, 72–82. [Google Scholar] [CrossRef]
- Goll, J.; Minceva, M. Continuous fractionation of multicomponent mixtures with sequential centrifugal partition chromatography. AIChE J. 2017, 63, 1659–1673. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Device | Manufacturer | Model |
|---|---|---|
| CCD camera | Jai Pulnix, Yokohama, Japan | AccuPIXEL TM 1327 GE |
| chromatograph | Kromaton, Annonay, France | FCPC-A 30506 |
| controller unit | Gardasoft, Cambridge, UK | RTCC 420 |
| detector | Knauer, Berlin, Germany | Smartline DA-UV-detector |
| phase pumps | Knauer, Berlin, Germany | P 4.1S with 50 mL pump head |
| sample pump | Ismatec, Stadtprozelten, Germany | MS-4/12 100 Reglo Digital |
| fractionation valve | Knauer, Berlin, Germany | HighSpeedValve |
| six-port-two-position valves | Knauer, Berlin, Germany | K-6/12/16 |
| Volumetric Flow Rate of Mobile Phase [mL·min−1] | Sf*setpoint |
|---|---|
| 5 | {0.26, 0.35, 0.45, 0.55, 0.65, 0.75, 0.85, 0.9} |
| 10 | {0.26, 0.35, 0.45, 0.55, 0.65, 0.75} |
| 15 | {0.26, 0.35, 0.45, 0.55, 0.65} |
| 20 | {0.26, 0.35, 0.45, 0.55} |
| Redosing → Injection | Injection → Redosing | Normal Separation | |||||
|---|---|---|---|---|---|---|---|
| Vstat [mL] | 0.125 | 0.25 | 0.375 | 0.125 | 0.25 | 0.375 | 0–0.32 |
| R [-] | 0.615 ± 0.032 | 0.638 ± 0.066 | 0.668 ± 0.048 | 0.620 ± 0.041 | 0.624 ± 0.059 | 0.677 ± 0.040 | 0.677 ± 0.037 |
| Volumetric Flow Rate of Mobile Phase [mL·min−1] | Separation Time [min] |
|---|---|
| 5 | |
| 10 | |
| 15 | |
| 20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buthmann, F.; Hohlmann, J.; Neuwald, M.; Schembecker, G. Optimizing Chromatographic Separation with Redosing: Effects on Separation Efficiency of a Model System in Centrifugal Partition Chromatography. Separations 2024, 11, 111. https://doi.org/10.3390/separations11040111
Buthmann F, Hohlmann J, Neuwald M, Schembecker G. Optimizing Chromatographic Separation with Redosing: Effects on Separation Efficiency of a Model System in Centrifugal Partition Chromatography. Separations. 2024; 11(4):111. https://doi.org/10.3390/separations11040111
Chicago/Turabian StyleButhmann, Felix, Jan Hohlmann, Mareen Neuwald, and Gerhard Schembecker. 2024. "Optimizing Chromatographic Separation with Redosing: Effects on Separation Efficiency of a Model System in Centrifugal Partition Chromatography" Separations 11, no. 4: 111. https://doi.org/10.3390/separations11040111