
Gas Chromatography—High-Temperature Proton-Transfer Reaction Mass Spectrometry as a Novel Tool for Forensic Drug Testing


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Samples
2.3. Extraction and Sample Processing
2.4. Instrumental Setup and Settings
2.5. Other Workflows Used
3. Results and Discussion
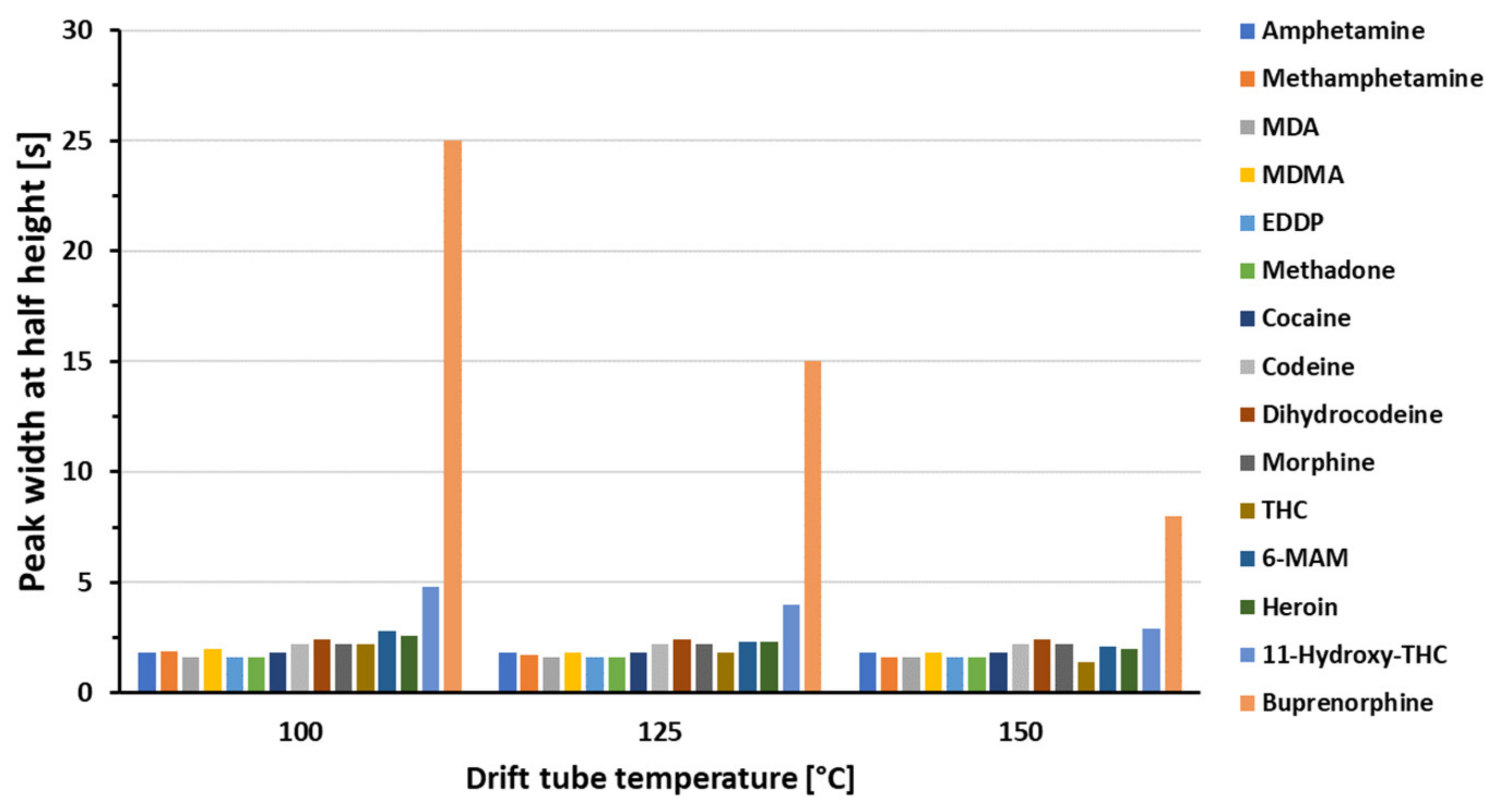
3.1. Optimization of the GC-HT-PTR-MS Setup
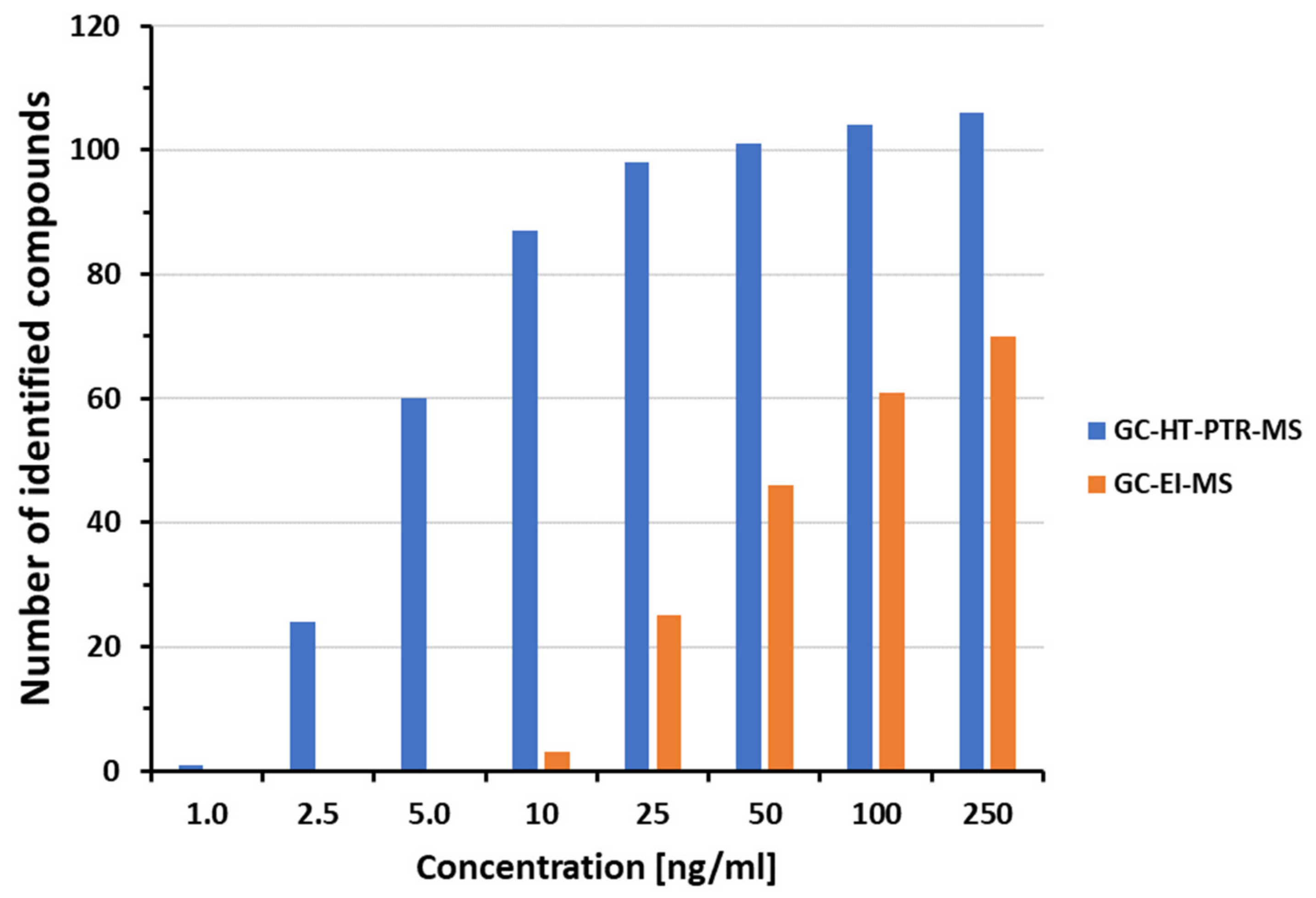
3.2. Forensic Drug Screening
3.3. Quantitative Drug Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blake, R.S.; Monks, P.S. Proton-Transfer Reaction Mass Spectrometry. Chem. Rev. 2009, 109, 861–896. [Google Scholar] [CrossRef] [PubMed]
- Hartungen, E.; Jürschik, S. Proton transfer reaction-mass spectrometry: Fundamentals, recent advances and applications. Eur. Phys. J. Appl. Phys. 2013, 61, 24303. [Google Scholar] [CrossRef]
- Yuan, B.; Koss, A.R. Proton-transfer-reaction mass spectrometry: Applications in atmospheric sciences. Chem. Rev. 2017, 117, 13187–13229. [Google Scholar] [CrossRef] [PubMed]
- Hansel, A.; Jordan, A. Proton transfer reaction mass spectrometry: On-line trace gas analysis at the ppb level. Int. J. Mass Spectrom. Ion Process. 1995, 149, 609–619. [Google Scholar] [CrossRef]
- Pleil, J.D.; Hansel, A. Advances in proton transfer reaction mass spectrometry (PTR-MS): Applications in exhaled breath analysis, food science, and atmospheric chemistry. J. Breath Res. 2019, 13, 039002. [Google Scholar] [CrossRef] [PubMed]
- Romano, A.; Capozzi, V. Proton transfer reaction–mass spectrometry: Online and rapid determination of volatile organic compounds of microbial origin. Appl. Microbiol. Biotechnol. 2015, 99, 3787–3795. [Google Scholar] [CrossRef] [PubMed]
- Winkler, K.; Herbig, J. Real-time metabolic monitoring with proton transfer reaction mass spectrometry. J. Breath Res. 2013, 7, 036006. [Google Scholar] [CrossRef] [PubMed]
- Biasioli, F.; Gasperi, F. PTR-MS monitoring of VOCs and BVOCs in food science and technology. Trends Anal. Chem. 2011, 30, 968–977. [Google Scholar] [CrossRef]
- Piel, F.; Müller, M. Introducing the extended volatility range proton-transfer-reaction mass spectrometer (EVR PTR-MS). Atmos. Meas. Tech. 2021, 14, 1355–1363. [Google Scholar] [CrossRef]
- Mikoviny, T.; Kaser, L. Development and characterization of a high-temperature proton-transfer-reaction mass spectrometer (HT-PTR-MS). Atmos. Meas. Tech. 2010, 3, 537–544. [Google Scholar] [CrossRef]
- Fang, J.; Zhao, H. Atmospheric pressure chemical ionization in gas chromatography-mass spectrometry for the analysis of persistent organic pollutants. Trends Environ. Anal. Chem. 2020, 25, e00076. [Google Scholar] [CrossRef]
- Li, D.-X.; Gan, L. Gas chromatography coupled to atmospheric pressure ionization mass spectrometry (GC-API-MS): Review. Anal. Chim. Acta 2015, 891, 43–61. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, P.; Hartungen, E. A Proton Transfer Reaction-Quadrupole interface Time-Of-Flight Mass Spectrometer (PTR-QiTOF): High speed due to extreme sensitivity. Int. J. Mass Spectrom. 2014, 368, 1–5. [Google Scholar] [CrossRef]
- Stempfer, M.; Reinstadler, V. Analysis of cannabis seizures by non-targeted liquid chromatography-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2021, 205, 114313. [Google Scholar] [CrossRef] [PubMed]
- Reinstadler, V.; Lierheimer, S. A validated workflow for drug detection in oral fluid by non-targeted liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2019, 411, 867–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberacher, H.; Reinstadler, V. Annotating Nontargeted LC-HRMS/MS Data with Two Complementary Tandem Mass Spectral Libraries. Metabolites 2018, 9, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnhard, K.; Gottschall, A. Applying ‘Sequential Windowed Acquisition of All Theoretical Fragment Ion Mass Spectra’ (SWATH) for systematic toxicological analysis with liquid chromatography-high-resolution tandem mass spectrometry. Anal. Bioanal. Chem. 2015, 407, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Oberacher, H.; Schubert, B. Detection and identification of drugs and toxicants in human body fluids by liquid chromatography–tandem mass spectrometry under data-dependent acquisition control and automated database search. Anal. Chim. Acta 2013, 770, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Pitterl, F.; Kob, S. Applying LC with Low-Resolution MS/MS and subsequent library search for reliable compound identification in systematic toxicological analysis. LCGC N. Am. 2017, 35, 23–31. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Calibration Range [ng/mL] | R2 | Maximum Bias [%] | Maximum Relative Standard Deviation [%] |
|---|---|---|---|---|
| Amphetamine | 5–100 | 0.998 | 3.7 | 6.4 |
| Methamphetamine | 5–100 | 0.996 | 4.5 | 8.7 |
| MDMA | 5–100 | 0.997 | 3.9 | 6.0 |
| MDA | 5–100 | 0.999 | −2.8 | 5.6 |
| Cocaine | 5–100 | 0.998 | −3.9 | 8.5 |
| Methadone | 10–250 | 0.996 | −4.5 | 9.8 |
| EDDP | 5–250 | 0.997 | 9.2 | 8.3 |
| THC | 10–250 | 0.998 | −3.9 | 9.2 |
| Morphine | 25–500 | 0.997 | 6.2 | 7.9 |
| 6-AM | 10–250 | 0.993 | 4.3 | 9.5 |
| Dihydrocodeine | 10–250 | 0.997 | −5.1 | 14.1 |
| Sample | Compound | C with GC-HT-PTR-MS [ng/mL] | C with GC-EI-MS [ng/mL] | Difference between Measured Concentrations [%] |
|---|---|---|---|---|
| 1 | Dihydrocodeine | 12.9 | 20 | −35.3 |
| Morphine | 121 | 113 | 7.0 | |
| 2 | THC | 19.3 | 20 | −3.6 |
| 3 | Cocaine | 19.9 | 20 | −0.7 |
| 4 | Cocaine | 121 | 120 | 0.8 |
| THC | 15.5 | 17 | −8.9 | |
| 5 | Cocaine | 30.9 | 31 | −0.3 |
| 6 | EDDP | <LOQ | 4.0 | |
| Methadone | 73.2 | 73 | 0.3 | |
| THC | <LOQ | 4.9 | ||
| 7 | Amphetamine | 58.9 | 57 | 3.4 |
| MDA | 21.1 | 20 | 5.3 | |
| MDMA | 232.3 | 230 | 1.0 | |
| THC | <LOQ | 6.5 | −9.9 | |
| 8 | Cocaine | 15.4 | 17 | −9.1 |
| THC | 18.6 | 20 | −6.9 | |
| 9 | Cocaine | 26.1 | 30 | −13.0 |
| THC | <LOQ | 5.3 | ||
| 10 | Cocaine | 43.9 | 45 | −2.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reinstadler, V.; Gutmann, R.; Pitterl, F.; Winkler, K.; Oberacher, H. Gas Chromatography—High-Temperature Proton-Transfer Reaction Mass Spectrometry as a Novel Tool for Forensic Drug Testing. Separations 2022, 9, 328. https://doi.org/10.3390/separations9110328
Reinstadler V, Gutmann R, Pitterl F, Winkler K, Oberacher H. Gas Chromatography—High-Temperature Proton-Transfer Reaction Mass Spectrometry as a Novel Tool for Forensic Drug Testing. Separations. 2022; 9(11):328. https://doi.org/10.3390/separations9110328
Chicago/Turabian StyleReinstadler, Vera, Rene Gutmann, Florian Pitterl, Klaus Winkler, and Herbert Oberacher. 2022. "Gas Chromatography—High-Temperature Proton-Transfer Reaction Mass Spectrometry as a Novel Tool for Forensic Drug Testing" Separations 9, no. 11: 328. https://doi.org/10.3390/separations9110328