
Utility of Atmospheric-Pressure Chemical Ionization and Photoionization Mass Spectrometry in Bottom-Up Proteomics

 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagent
2.2. BSA Digest
2.3. Peptide Standards
2.4. Mass Spectrometry
2.5. Infusion Experiments and Flow Injection Analysis
2.6. Liquid Chromatography-Tandem Mass Spectrometry
3. Results and Discussion
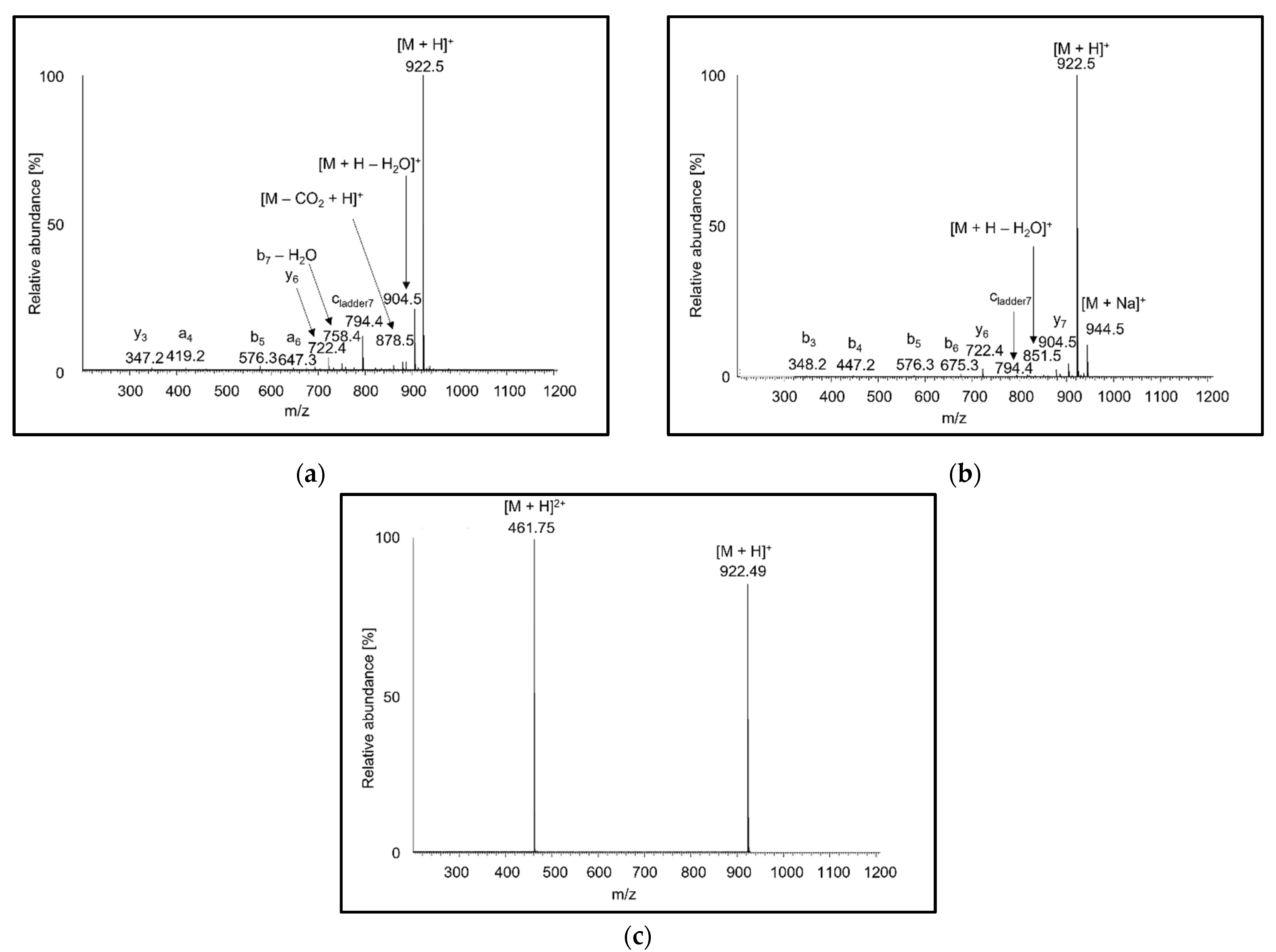
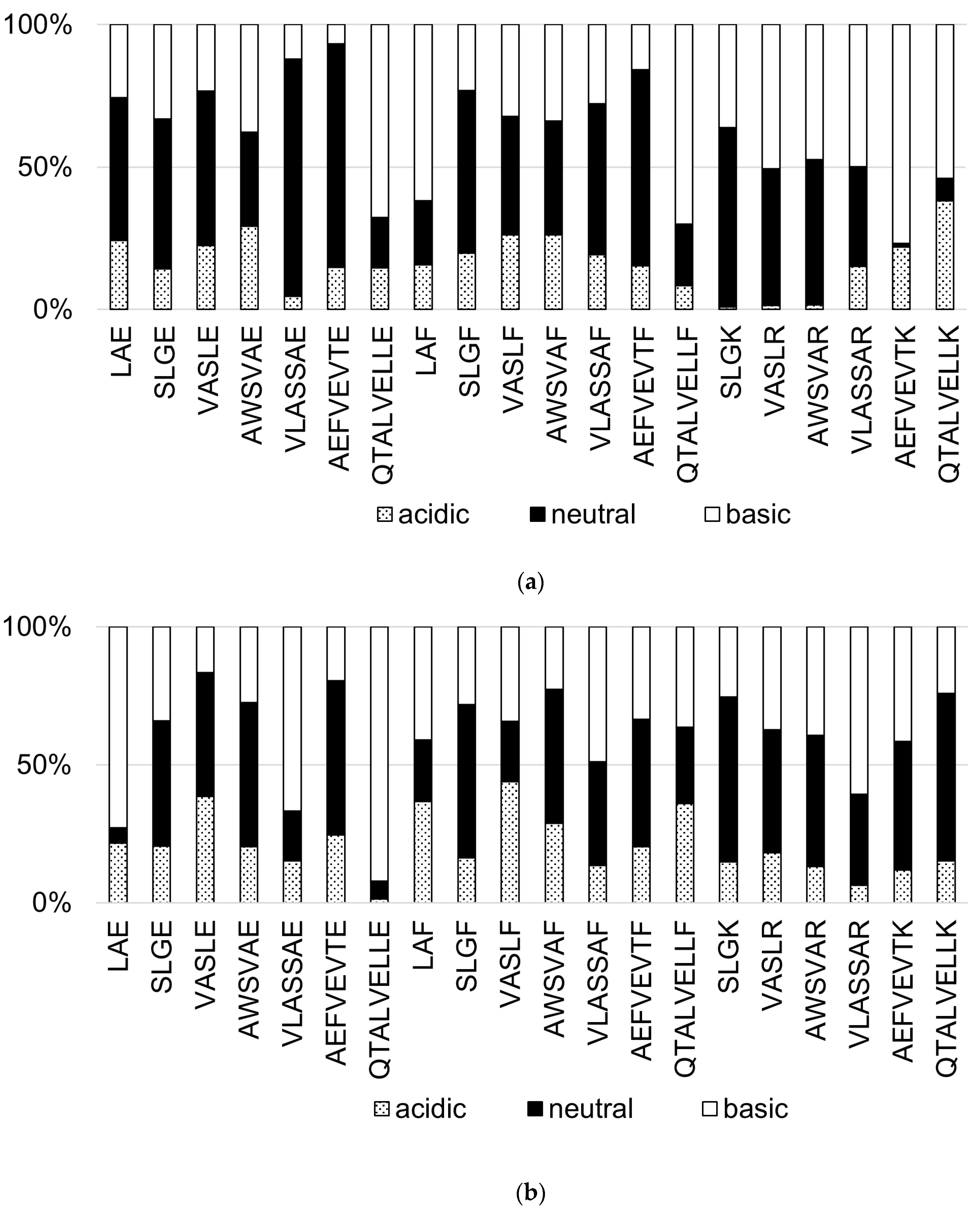
3.1. Mass Spectra of Peptide Standards
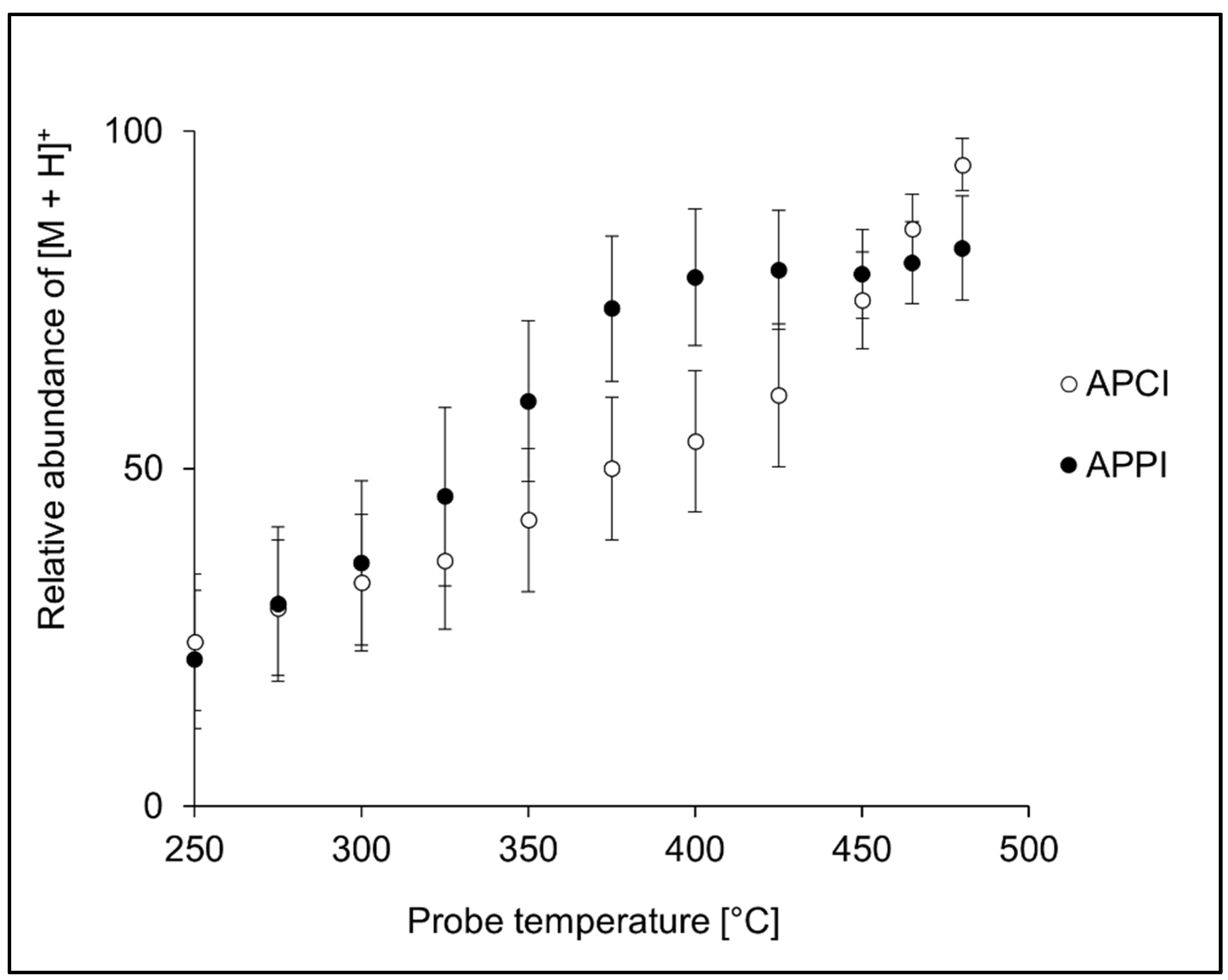
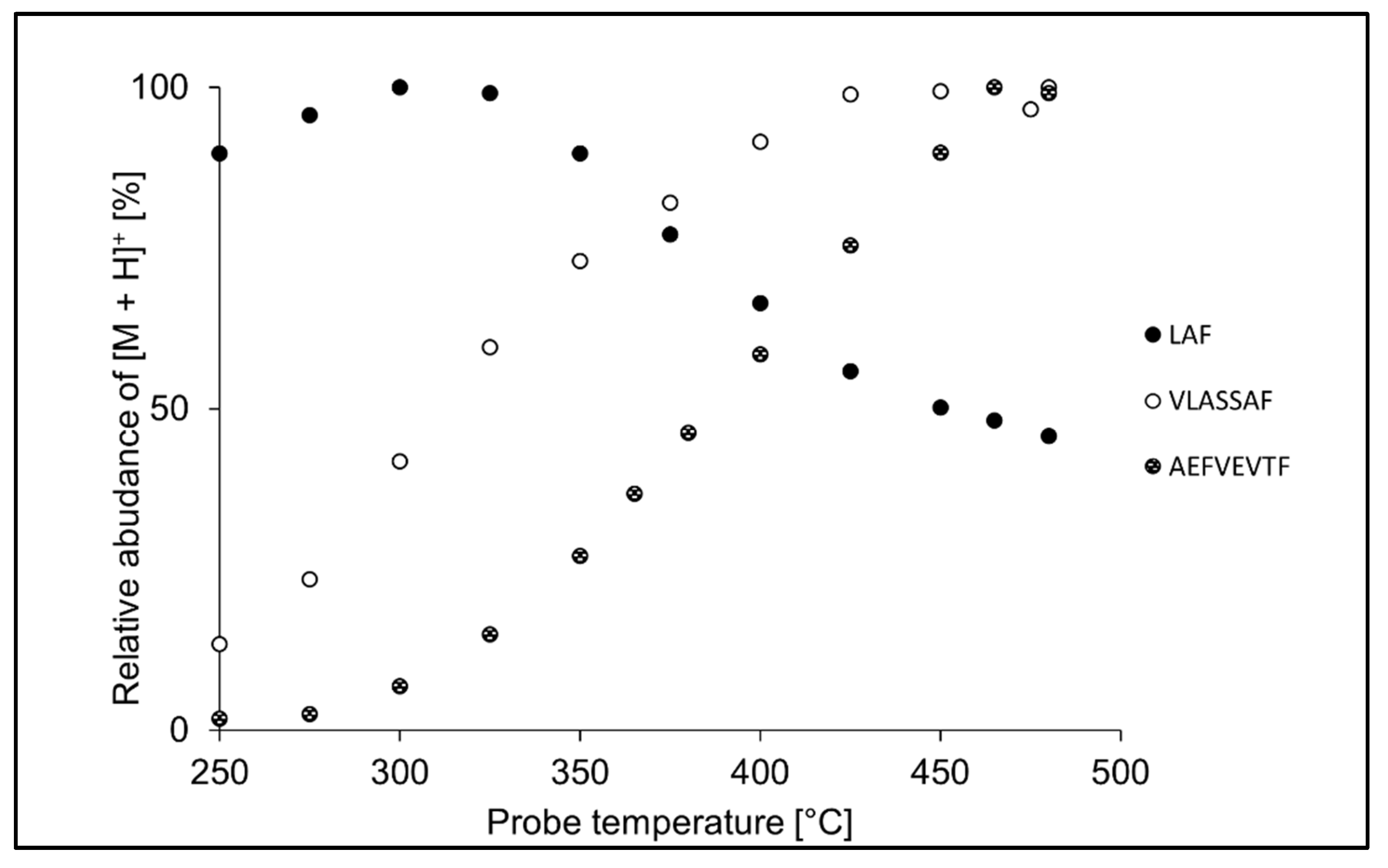
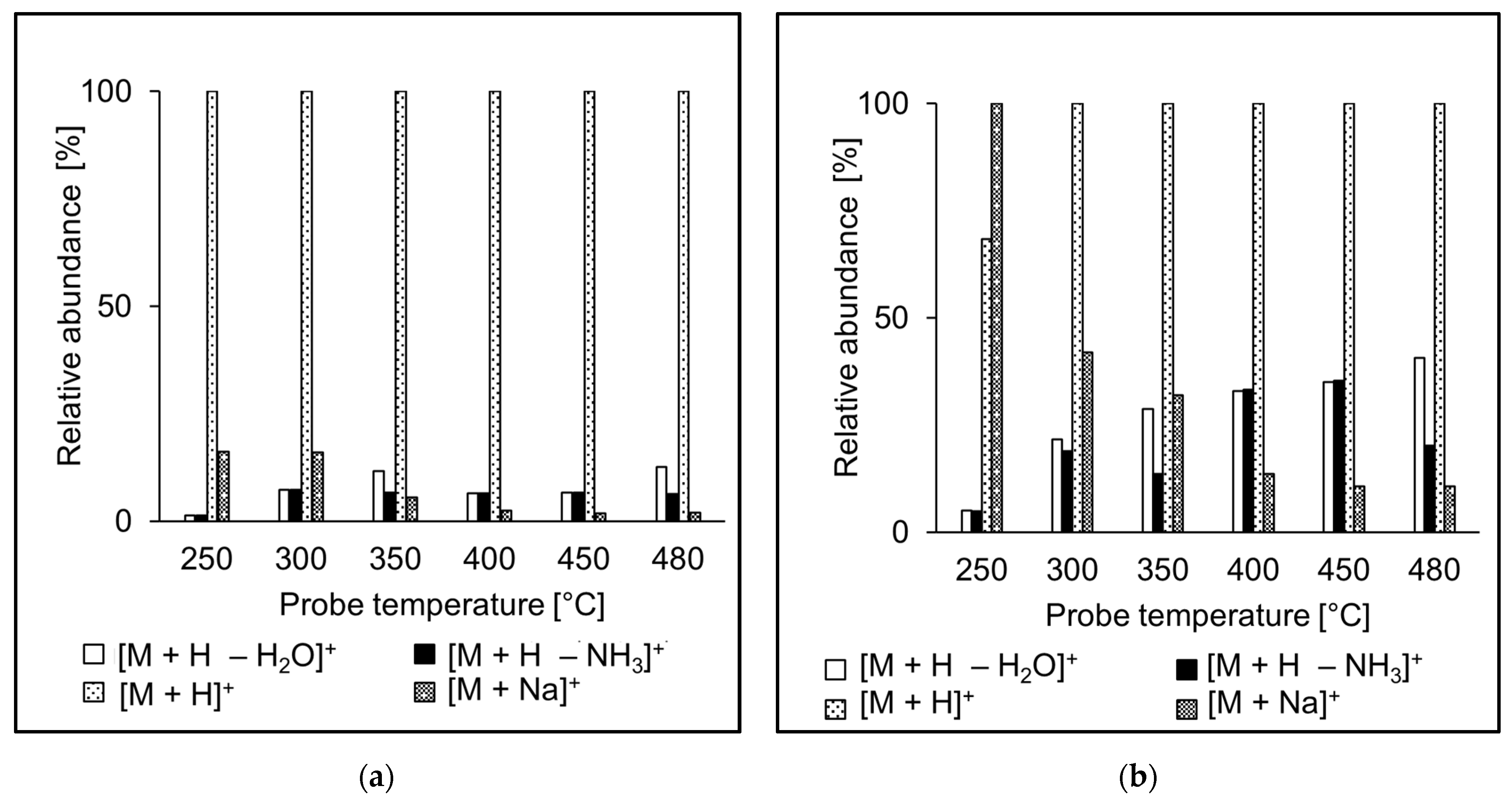
3.2. Ion Source Probe Temperature
3.3. Effect of the Mobile Phase pH
3.4. Effect of Dopant in APPI
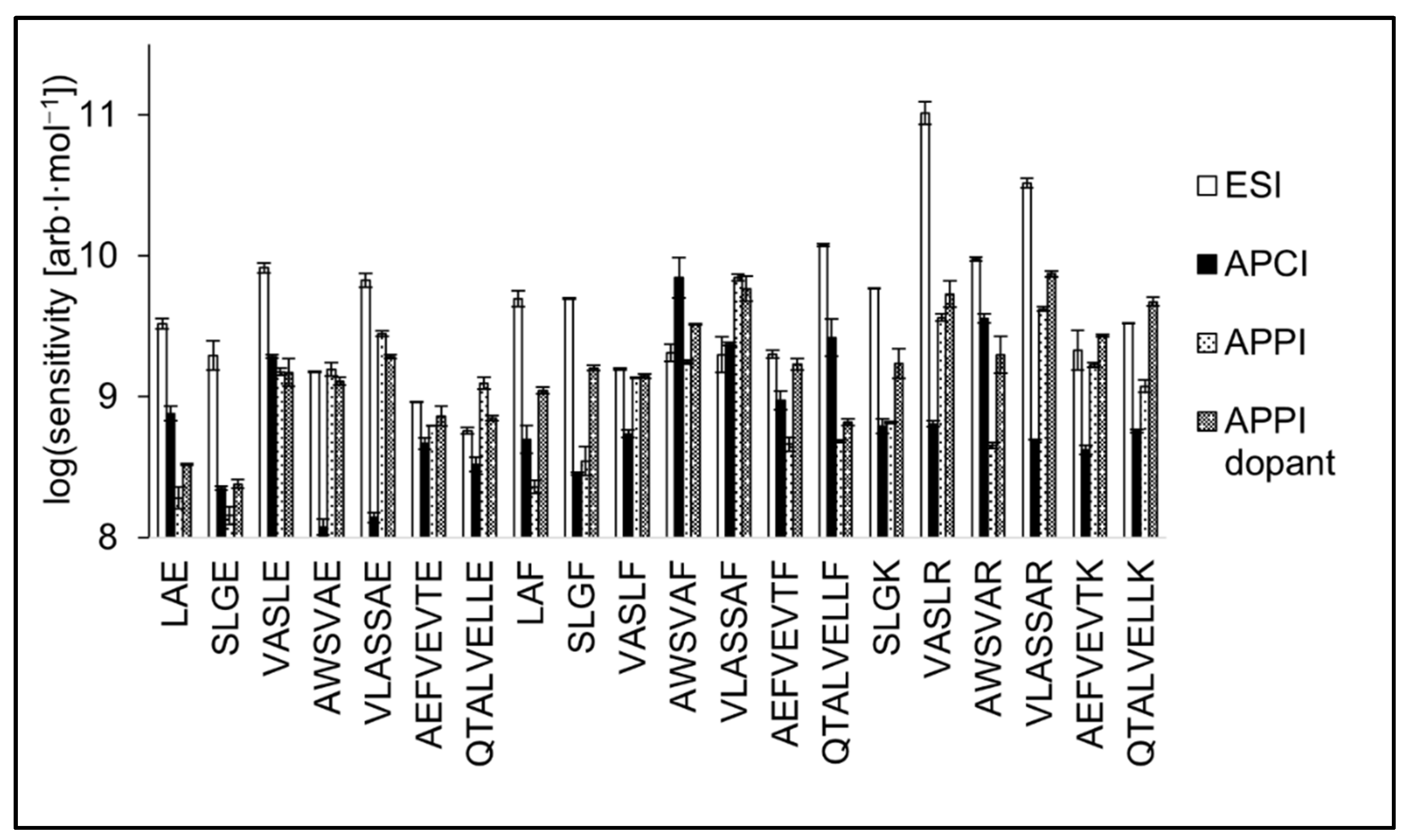
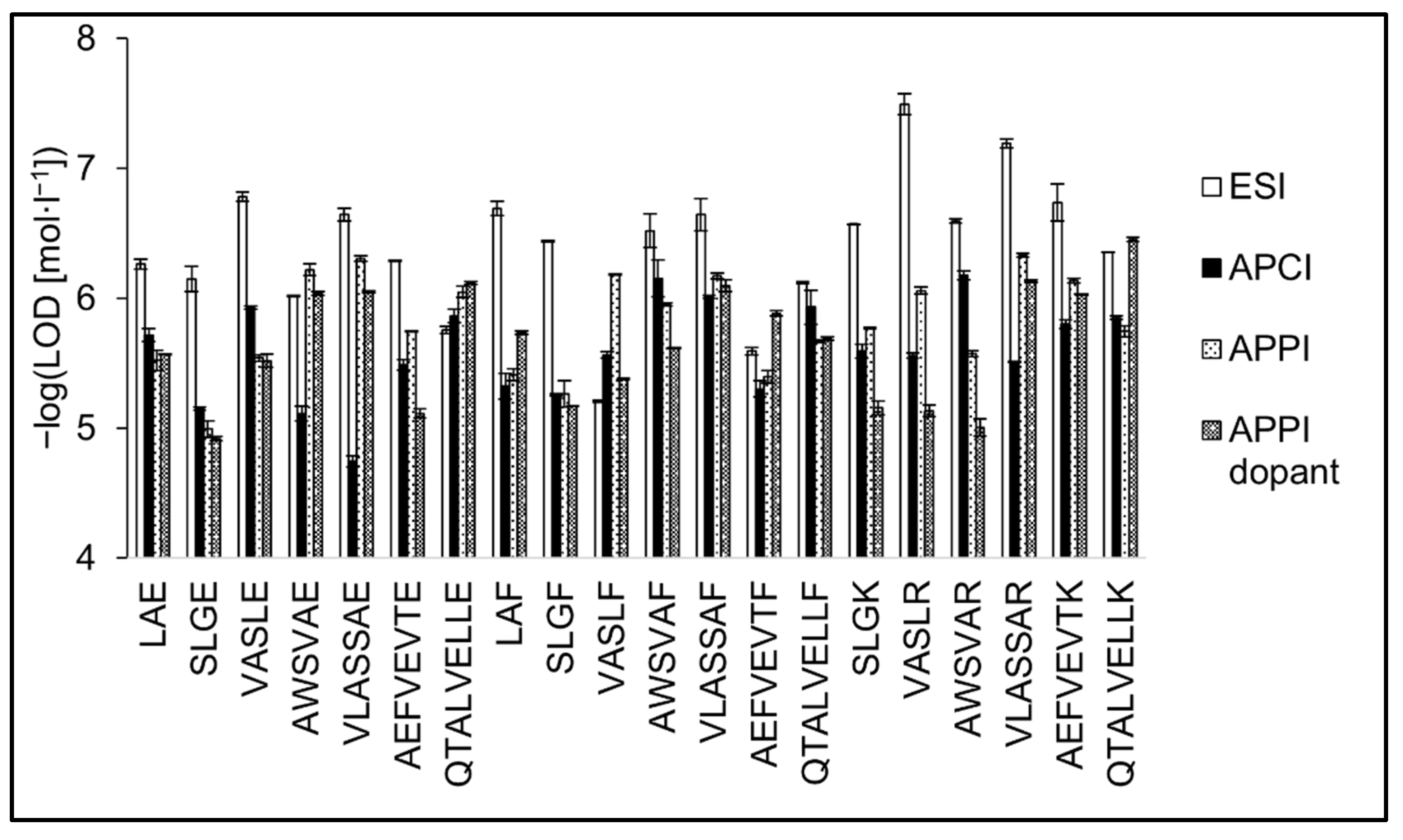
3.5. Analytical Figures of Merit
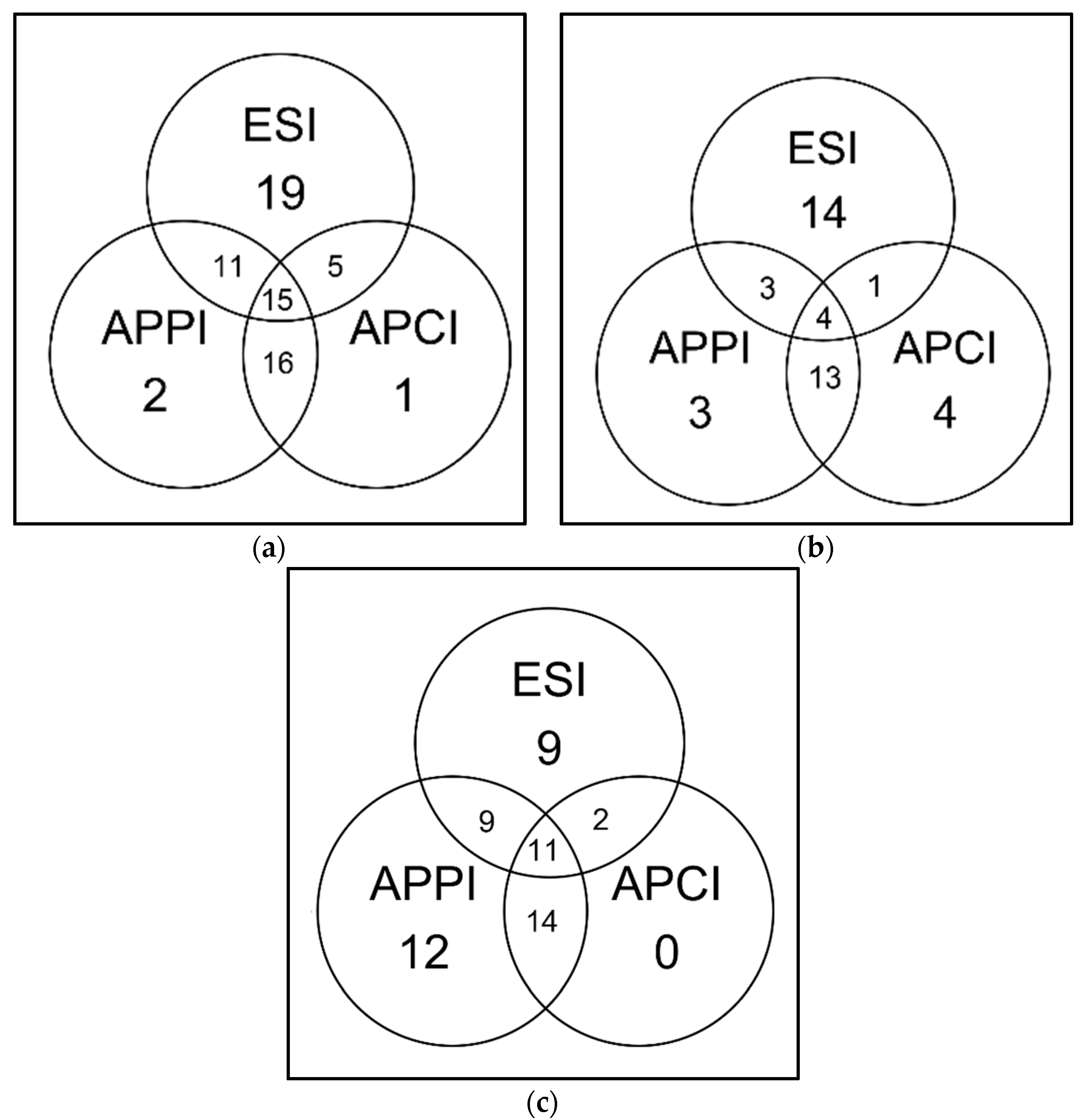
3.6. Chromatographic Analysis of BSA Digest
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Biemann, K.; Gapp, F.; Seibl, J. Application of Mass Spectrometry to Structure Problems. I. Amino Acid Sequence in Peptides. J. Am. Chem. Soc. 1959, 81, 2274–2275. [Google Scholar] [CrossRef]
- Morris, H.R.; Panico, M.; Barber, M.; Bordoli, R.S.; Sedgwick, R.D.; Tyler, A. Fast Atom Bombardment: A New Mass Spectrometric Method for Peptide Sequence Analysis. Biochem. Biophys. Res. Commun. 1981, 101, 623–631. [Google Scholar] [CrossRef]
- Przybylski, M.; Lüderwald, I.; Kraas, E.; Voelter, W.; Nelson, S.D. Field Desorption Mass Spectra of Gastrine Peptides and Glutathione Derivatives. Z. Naturforsch.-Sect. B J. Chem. Sci. 1979, 34, 736–743. [Google Scholar] [CrossRef] [Green Version]
- Mudgett, M.; Bowen, D.V.; Field, F.H.; Kindt, T.J. Peptide Sequencing: The Utility of Chemical Ionization Mass Spectrometry. Biol. Mass Spectrom. 1977, 4, 159–171. [Google Scholar] [CrossRef]
- Hunt, D.F.; Yates, J.R.; Shabanowitz, J.; Winston, S.; Hauer, C.R. Protein Sequencing by Tandem Mass Spectrometry. Proc. Natl. Acad. Sci. USA 1986, 83, 6233–6237. [Google Scholar] [CrossRef] [Green Version]
- Carr, S.A.; Biemann, K. Identification of Posttranslationally Modified Amino Acids in Proteins by Mass Spectrometry. Methods Enzymol. 1984, 106, 29–58. [Google Scholar] [CrossRef]
- Naylor, S.; Findeis, A.F.; Gibson, B.W.; Williams, D.H. An Approach toward the Complete FAB Analysis of Enzymic Digests of Peptides and Proteins. J. Am. Chem. Soc. 1986, 108, 6359–6363. [Google Scholar] [CrossRef]
- Yergey, A.L.; Edmonds, C.G.; Lewis, I.A.S.; Vestal, M.L. Applications of LC/MS to Amino Acids, Peptides, and Proteins. In Liquid Chromatography/Mass Spectrometry; Hercules, D., Ed.; Springer Science + Business Media: New York, NY, USA, 1990; pp. 137–145. [Google Scholar]
- Jones, D.S.; Krolik, S.T.; Aplin, R. Analysis of Nonderivatised Peptides by Thermospray Using a Magnetic Sector Mass Spectrometer D. Rapid Commun. Mass Spectrom. 1987, 1, 67–68. [Google Scholar] [CrossRef]
- Kilpatrick, G.; Lewis, I.A.S.; Smith, J.F. The Reduction of Collision Induced Dissociation Effects in Thermospray Sources on Sector Instruments. Biomed. Environ. Mass Spectrom. 1987, 14, 155–159. [Google Scholar] [CrossRef]
- Yamashita, M.; Fenn, J.B. Electrospray Ion Source. Another Variation on the Free-Jet Theme. J. Phys. Chem. 1984, 88, 4451–4459. [Google Scholar] [CrossRef]
- Karas, M.; Hillenkamp, F. Laser Desorption Ionization of Proteins with Molecular Masses Exceeding 10 000 Daltons. Anal. Chem. 1988, 60, 2299–2301. [Google Scholar] [CrossRef] [PubMed]
- Nadler, W.M.; Waidelich, D.; Kerner, A.; Hanke, S.; Berg, R.; Trumpp, A.; Rösli, C. MALDI versus ESI: The Impact of the Ion Source on Peptide Identification. J. Proteome Res. 2017, 16, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Waki, H.; Ido, Y.; Akita, S.; Yoshida, Y.; Yoshida, T.; Matsuo, T. Protein and Polymer Analyses up to m/z 100 000 by Laser Ionization Time-of-Flight Mass Spectrometry. Rapid Commun. Mass Spectrom. 1988, 2, 151–153. [Google Scholar] [CrossRef]
- Shen, Z.; Thomas, J.J.; Averbuj, C.; Broo, K.M.; Engelhard, M.; Crowell, J.E.; Finn, M.G.; Siuzdak, G. Porous Silicon as a Versatile Platform for Laser Desorption/Ionization Mass Spectrometry. Anal. Chem. 2001, 73, 612–619. [Google Scholar] [CrossRef]
- Abdelhamid, H.N.; Wu, H.F. Facile Synthesis of Nano Silver Ferrite (AgFeO2) Modified with Chitosan Applied for Biothiol Separation. Mater. Sci. Eng. C 2014, 45, 438–445. [Google Scholar] [CrossRef]
- Woods, A.G.; Darie, C.C. Advancements of Mass Spectrometry in Biomedical Research, 2nd ed; Springer: Cham, Switzerland, 2019; ISBN 10:3030159507. [Google Scholar]
- Bagag, A.; Jault, J.M.; Sidahmed-Adrar, N.; Réfrégiers, M.; Giuliani, A.; Le Naour, F. Characterization of Hydrophobic Peptides in the Presence of Detergent by Photoionization Mass Spectrometry. PLoS ONE 2013, 8, e79033. [Google Scholar] [CrossRef]
- Xu, N.; Lin, Y.; Hofstadler, S.A.; Matson, D.; Call, C.J.; Smith, R.D. A Microfabricated Dialysis Device for Sample Cleanup in Electrospray Ionization Mass Spectrometry. Anal. Chem. 1998, 70, 3553–3556. [Google Scholar] [CrossRef]
- Lei, J.; Chen, D.A.; Regnier, F.E. Rapid Verification of Disulfide Linkages in Recombinant Human Growth Hormone by Tandem Column Tryptic Mapping. J. Chromatogr. A 1998, 808, 121–131. [Google Scholar] [CrossRef]
- Waliczek, M.; Kijewska, M.; Rudowska, M.; Setner, B.; Stefanowicz, P.; Szewczuk, Z. Peptides Labeled with Pyridinium Salts for Sensitive Detection and Sequencing by Electrospray Tandem Mass Spectrometry. Sci. Rep. 2016, 6, 37720. [Google Scholar] [CrossRef]
- Rebane, R.; Rodima, T.; Kütt, A.; Herodes, K. Development of Amino Acid Derivatization Reagents for Liquid Chromatography Electrospray Ionization Mass Spectrometric Analysis and Ionization Efficiency Measurements. J. Chromatogr. A 2015, 1390, 62–70. [Google Scholar] [CrossRef]
- DeGraan-Weber, N.; Ward, S.A.; Reilly, J.P. A Novel Triethylphosphonium Charge Tag on Peptides: Synthesis, Derivatization, and Fragmentation. J. Am. Soc. Mass Spectrom. 2017, 28, 1889–1900. [Google Scholar] [CrossRef] [PubMed]
- Bagag, A.; Giuliani, A.; Réfrégiers, M.; Le Naour, F. Atmospheric Pressure Photoionization Study of Post-Translational Modifications: The Case of Palmitoylation. Int. J. Mass Spectrom. 2012, 328–329, 23–27. [Google Scholar] [CrossRef]
- Ruiz, J.; García, C.; Del Carmen Díaz, M.; Cava, R.; Florencio Tejeda, J.; Ventanas, J. Dry Cured Iberian Ham Non-Volatile Components as Affected by the Length of the Curing Process. Food Res. Int. 1999, 32, 643–651. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Yasuda, K.; Kodama, H. Measurement of Iminodipeptides in the Serum of Patients with Prolidase Deficiency Using Liquid Chromatography-Mass Spectrometry. Clin. Chem. Lab. Med. 1994, 32, 113–118. [Google Scholar] [CrossRef] [Green Version]
- Cristoni, S.; Bernardi, L.R.; Biunno, I.; Guidugli, F. Analysis of Peptides Using Partial (No Discharge) Atmospheric Pressure Chemical Ionization Conditions with Ion Trap Mass Spectrometry. Rapid Commun. Mass Spectrom. 2002, 16, 1686–1691. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.J.; Armstrong, D.W. Analysis of Native Amino Acid and Peptide Enantiomers by High-Performance Liquid Chromatography/Atmospheric Pressure Chemical Ionization Mass Spectrometry. J. Mass Spectrom. 2004, 39, 177–187. [Google Scholar] [CrossRef]
- Shen, Y.; Han, C.; Chen, J.; Wang, X. Analysis of Cyclic Peptides in Pseudostellaria Heterophylla (Miq.) Pax by HPLC-APCI-MS. Chromatographia 2007, 66, 319–323. [Google Scholar] [CrossRef]
- Han, H.; Xia, Y.; Yang, M.; McLuckey, S.A. Rapidly Alternating Transmission Mode Electron-Transfer Dissociation and Collisional Activation for the Characterization of Polypeptide Ions. Anal. Chem. 2008, 80, 3492–3497. [Google Scholar] [CrossRef] [Green Version]
- McLuckey, S.A.; Han, H.; Xia, Y. Method and Apparatus for Collisional Activation of Polypeptide Ions. U.S. Patent Application No. 7,829,851, 9 November 2008. [Google Scholar]
- Wei, Y.; Bian, C.; Ouyang, Z.; Xu, W. A Pulsed Pinhole Atmospheric Pressure Interface for Simplified Mass Spectrometry Instrumentation with Enhanced Sensitivity. Rapid Commun. Mass Spectrom. 2015, 29, 701–706. [Google Scholar] [CrossRef]
- Bose, U.; Hodson, M.; Shaw, P.; Fuerst, J.; Hewavitharana, A. Two Peptides, Cycloaspeptide A and Nazumamide A from a Sponge Associated Marine Actinobacterium salinispora Sp. Nat. Prod. Commun. 2014, 9, 545–546. [Google Scholar] [CrossRef] [Green Version]
- Bagag, A.; Giuliani, A.; Laprévote, O. Atmospheric Pressure Photoionization of Peptides. Int. J. Mass Spectrom. 2011, 299, 1–4. [Google Scholar] [CrossRef]
- Delobel, A.; Halgand, F.; Laffranchise-Gosse, B.; Snijders, H.; Laprévote, O. Characterization of Hydrophobic Peptides by Atmospheric Pressure Photoionization-Mass Spectrometry and Tandem Mass Spectrometry. Anal. Chem. 2003, 75, 5961–5968. [Google Scholar] [CrossRef]
- Debois, A.D.; Giuliani, A.; Laprévote, O. Fragmentation Induced in Atmospheric Pressure Photoionization of Peptides. J. Mass Spectrom. 2006, 41, 1554–1560. [Google Scholar] [CrossRef] [PubMed]
- Ross, M.S.; Wong, C.S. Comparison of Electrospray Ionization, Atmospheric Pressure Photoionization, and Anion Attachment Atmospheric Pressure Photoionization for the Analysis of Hexabromocyclododecane Enantiomers in Environmental Samples. J. Chromatogr. A 2010, 1217, 7855–7863. [Google Scholar] [CrossRef]
- Robb, D.B.; Rogalski, J.C.; Kast, J.; Blades, M.W. A New Ion Source and Procedures for Atmospheric Pressure-Electron Capture Dissociation of Peptides. J. Am. Soc. Mass Spectrom. 2011, 22, 1699–1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLuckey, S.A.; Liang, X.; Xiu, Y. Method and Apparatus for Transmission Mode Ion/Ion Dissociation. U.S. Patent Application No. 11/998,306, 25 April 2008. [Google Scholar]
- Méjean, M.; Giuliani, A.; Brunelle, A.; Touboul, D. Exploring the Peptide Fragmentation Mechanisms under Atmospheric Pressure Photoionization Using Tunable VUV Synchrotron Radiation. Int. J. Mass Spectrom. 2015, 379, 80–86. [Google Scholar] [CrossRef]
- Hockaday, W.C.; Purcell, J.M.; Marshall, A.G.; Baldock, J.A.; Hatcher, P.G. Electrospray and Photoionization Mass Spectrometry for the Characterization of Organic Matter in Natural Waters: A Qualitative Assessment. Limnol. Oceanogr. Methods 2009, 7, 81–95. [Google Scholar] [CrossRef] [Green Version]
- Robb, D.B.; Blades, M.W. An Electropneumatic-Heated Nebulizer for Enhancing Spray Ionization in PhotoSpray Atmospheric Pressure Photoionization Sources for Liquid Chromatography/Mass Spectrometry. Rapid Commun. Mass Spectrom. 2009, 23, 3394–3400. [Google Scholar] [CrossRef]
- Taylor, L.C.E.; Johnson, R.L.; Raso, R. Open Access Atmospheric Pressure Chemical Ionization Mass Spectrometry for Routine Sample Analyses. J. Am. Soc. Mass Spectrom. 1995, 6, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Horning, E.C.; Horning, M.G.; Carroll, D.I.; Dzidic, I.; Stillwell, R.N. New Picogram Detection System Based on a Mass Spectrometer with an External Ionization Source at Atmospheric Pressure. Anal. Chem. 1973, 45, 936–943. [Google Scholar] [CrossRef]
- Robb, D.B.; Covey, T.R.; Bruins, A.P. Atmospheric Pressure Photoionization: An Ionization Method for Liquid Chromatography-Mass Spectrometry. Anal. Chem. 2000, 72, 3653–3659. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Yang, S.; Wang, P.G. Matrix Effects and Application of Matrix Effect Factor. Bioanalysis 2017, 9, 1839–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.; Xia, Y.; McLuckey, S.A. Alternately Pulsed Nanoelectrospray Ionization/Atmospheric Pressure Chemical Ionization for Ion/Ion Reactions in an Electrodynamic Ion Trap. Anal. Chem. 2006, 78, 3208–3212. [Google Scholar] [CrossRef] [Green Version]
- Syrstad, E.A.; Tureček, F. Toward a General Mechanism of Electron Capture Dissociation. J. Am. Soc. Mass Spectrom. 2005, 16, 208–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawicka, A.; Skurski, P.; Hudgins, R.R.; Simons, J. Model Calculations Relevant to Disulfide Bond Cleavage via Electron Capture Influenced by Positively Charged Groups. J. Phys. Chem. B 2003, 107, 13505–13511. [Google Scholar] [CrossRef] [Green Version]
- Zubarev, R.A.; Kruger, N.A.; Fridriksson, E.K.; Lewis, M.A.; Horn, D.M.; Carpenter, B.K.; McLafferty, F.W. Electron Capture Dissociation of Gaseous Multiply-Charged Proteins Is Favored at Disulfide Bonds and Other Sites of High Hydrogen Atom Affinity. J. Am. Chem. Soc. 1999, 121, 2857–2862. [Google Scholar] [CrossRef]
- Giuliani, A.; Giorgetta, J.L.; Ricaud, J.P.; Jamme, F.; Rouam, V.; Wien, F.; Laprévote, O.; Réfrégiers, M. Atmospheric Pressure Photoionization Using Tunable VUV Synchrotron Radiation. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2012, 279, 114–117. [Google Scholar] [CrossRef]
- Tsiatsiani, L.; Heck, A.J.R. Proteomics beyond Trypsin. FEBS J. 2015, 282, 2612–2626. [Google Scholar] [CrossRef]
- Giansanti, P.; Tsiatsiani, L.; Low, T.Y.; Heck, A.J.R. Six Alternative Proteases for Mass Spectrometry-Based Proteomics beyond Trypsin. Nat. Protoc. 2016, 11, 993–1006. [Google Scholar] [CrossRef]
- Erde, J.; Loo, R.R.O.; Loo, J.A. Improving Proteome Coverage and Sample Recovery with Enhanced FASP (EFASP) for Quantitative Proteomic Experiments. Methods Mol. Biol. 2017, 1550, 11–18. [Google Scholar] [CrossRef]
- Vrkoslav, V.; Muck, A.; Brown, J.M.; Hubálek, M.; Cvačka, J. The Matrix-Assisted Laser Desorption/Ionisation in-Source Decay of Peptides Using Ion Mobility Enabled Quadrupole-Time-of-Flight Mass Spectrometry. Rapid Commun. Mass Spectrom. 2018, 32, 2099–2105. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.S.; Cui, W.; Reilly, J.P. Fragmentation of Singly Charged Peptide Ions by Photodissociation at λ = 157 Nm. Angew. Chem.-Int. Ed. 2004, 43, 4791–4794. [Google Scholar] [CrossRef]
- Yuan, M.; Namikoshi, M.; Otsuki, A.; Watanabe, M.F.; Rinehart, K.L. Electrospray Ionization Mass Spectrometric Analysis of Microcystins, Cyclic Heptapeptide Hepatotoxins: Modulation of Charge States and [M + H]+ to [M + Na]+ Ratio. J. Am. Soc. Mass Spectrom. 1999, 10, 1138–1151. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Cole, R.B. Mechanistic Interpretation of the Dependence of Charge State Distributions on Analyte Concentrations in Electrospray Ionization Mass Spectrometry. Anal. Chem. 1995, 67, 2892–2900. [Google Scholar] [CrossRef]
- Schaberg, A.; Wroblowski, R.; Goertz, R. Comparative Study of the Thermal Decomposition Behaviour of Different Amino Acids and Peptides. J. Phys. Conf. Ser. 2018, 1107, 032013. [Google Scholar] [CrossRef]
- Basile, F.; Zhang, S.; Kandar, S.K.; Lu, L. Mass Spectrometry Characterization of the Thermal Decomposition/Digestion (TDD) at Cysteine in Peptides and Proteins in the Condensed Phase. J. Am. Soc. Mass Spectrom. 2011, 22, 1926–1940. [Google Scholar] [CrossRef] [Green Version]
- Raffaelli, A.; Saba, A. Atmospheric Pressure Photoionization Mass Spectrometry. Mass Spectrom. Rev. 2003, 22, 318–331. [Google Scholar] [CrossRef]
- Gronert, S.; Simpson, D.C.; Conner, K.M. A Reevaluation of Computed Proton Affinities for the Common α-Amino Acids. J. Am. Soc. Mass Spectrom. 2009, 20, 2116–2123. [Google Scholar] [CrossRef] [Green Version]
- Robb, D.B.; Blades, M.W. State-of-the-Art in Atmospheric Pressure Photoionization for LC/MS. Anal. Chim. Acta 2008, 627, 34–49. [Google Scholar] [CrossRef]
- Wilm, M.; Shevchenko, A.; Houthaeve, T.; Breit, S.; Schweigerer, L.; Fotsis, T.; Mann, M. Femtomole Sequencing of Proteins from Polyacrylamide Gels by Nano-Electrospray Mass Spectrometry. Nature 1996, 379, 466–469. [Google Scholar] [CrossRef]
- Oosterkamp, A.J.; Gelpí, E.; Abian, J. Quantitative Peptide Bioanalysis Using Column-Switching Nano Liquid Chromatography/Mass Spectrometry. J. Mass Spectrom. 1998, 33, 976–983. [Google Scholar] [CrossRef]
- Emmett, M.R.; Caprioli, R.M. Micro-Electrospray Mass Spectrometry: Ultra-High-Sensitivity Analysis of Peptides and Proteins. J. Am. Soc. Mass Spectrom. 1994, 5, 605–613. [Google Scholar] [CrossRef] [Green Version]
- Kruve, A.; Haapala, M.; Saarela, V.; Franssila, S.; Kostiainen, R.; Kotiaho, T.; Ketola, R.A. Feasibility of Capillary Liquid Chromatography-Microchip-Atmospheric Pressure Photoionization-Mass Spectrometry for Pesticide Analysis in Tomato. Anal. Chim. Acta 2011, 696, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Östman, P.; Marttila, S.J.; Kotiaho, T.; Franssila, S.; Kostiainen, R. Microchip Atmospheric Pressure Chemical Ionization Source for Mass Spectrometry. Anal. Chem. 2004, 76, 6659–6664. [Google Scholar] [CrossRef]
- Saarela, V.; Haapala, M.; Kostiainen, R.; Kotiaho, T.; Franssila, S. Glass Microfabricated Nebulizer Chip for Mass Spectrometry. Lab Chip 2007, 7, 644–646. [Google Scholar] [CrossRef]
- Kauppila, T.J.; Östman, P.; Marttila, S.; Ketola, R.A.; Kotiaho, T.; Franssila, S.; Kostiainen, R. Atmospheric Pressure Photoionization-Mass Spectrometry with a Microchip Heated Nebulizer. Anal. Chem. 2004, 76, 6797–6801. [Google Scholar] [CrossRef]
- Strmeň, T.; Vrkoslav, V.; Pačes, O.; Cvačka, J. Evaluation of an Ion Source with a Tubular Nebulizer for Microflow Atmospheric Pressure Chemical Ionization. Mon. Chem. 2018, 149, 987–994. [Google Scholar] [CrossRef]
- Vrkoslav, V.; Rumlová, B.; Strmeň, T.; Nekvasilová, P.; Šulc, M.; Cvačka, J. Applicability of Low-Flow Atmospheric Pressure Chemical Ionization and Photoionization Mass Spectrometry with a Microfabricated Nebulizer for Neutral Lipids. Rapid Commun. Mass Spectrom. 2018, 32, 639–648. [Google Scholar] [CrossRef]
- Tan, A.; Fanaras, J.C. Use of High-PH (Basic/Alkaline) Mobile Phases for LC–MS or LC–MS/MS Bioanalysis. Biomed. Chromatogr. 2019, 33, e4409. [Google Scholar] [CrossRef] [Green Version]
- Kostiainen, R.; Kauppila, T.J. Effect of Eluent on the Ionization Process in Liquid Chromatography-Mass Spectrometry. J. Chromatogr. A 2009, 1216, 685–699. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | APCI | APPI | APPI Dopant 1 | ESI |
|---|---|---|---|---|
| LOD [pmol] | LOD [pmol] | LOD [pmol] | LOD [pmol] | |
| LAE | 4.8 ± 0.6 | 7.5 ± 1.4 | 6.8 ± 0.1 | 1.4 ± 0.1 |
| SLGE | 17.6 ± 0.9 | 25.4 ± 3.7 | 30.0 ± 2.1 | 1.8 ± 0.4 |
| VASLE | 3.0 ± 0.1 | 7.2 ± 0.4 | 7.6 ± 1.7 | 0.4 ± 0.0 |
| AWSVAE | 19.3 ± 3.4 | 1.5 ± 0.2 | 2.3 ± 0.2 | 2.4 ± 0.2 |
| VLASSAE | 45.0 ± 4.4 | 1.2 ± 0.1 | 2.2 ± 0.1 | 0.6 ± 0.1 |
| AEFVEVTE | 8.1 ± 0.7 | 4.5 ± 0.0 | 19.2 ± 3.1 | 1.3 ± 0.0 |
| QTALVELLE | 3.4 ± 2.8 | 2.2 ± 0.2 | 1.9 ± 0.1 | 4.4 ± 0.3 |
| LAF | 11.9 ± 2.7 | 9.7 ± 1.0 | 4.6 ± 0.3 | 0.5 ± 0.1 |
| SLGF | 13.8 ± 0.3 | 13.6 ± 3.2 | 16.8 ± 0.3 | 0.9 ± 0.0 |
| VASLF | 6.8 ± 0.3 | 1.6 ± 0.0 | 10.4 ± 0.4 | 15.6 ± 0.0 |
| AWSVAF | 1.8 ± 0.6 | 2.8 ± 0.0 | 6.0 ± 0.1 | 0.8 ± 0.2 |
| VLASSAF | 2.4 ± 0.1 | 1.7 ± 0.0 | 2.0 ± 0.4 | 0.6 ± 0.3 |
| AEFVEVTF | 12.5 ± 1.9 | 10.1 ± 1.1 | 3.3 ± 0.4 | 6.4 ± 0.4 |
| QTALVELLF | 2.9 ± 0.9 | 5.3 ± 0.1 | 5.1 ± 0.3 | 1.9 ± 0.0 |
| SLGK | 6.4 ± 0.8 | 4.3 ± 0.0 | 17.4 ± 4.3 | 0.7 ± 0.0 |
| VASLR | 6.9 ± 0.3 | 2.2 ± 0.1 | 18.4 ± 3.9 | 0.1 ± 0.0 |
| AWSVAR | 1.7 ± 0.1 | 6.7 ± 0.3 | 24.7 ± 8.0 | 0.6 ± 0.0 |
| VLASSAR | 7.7 ± 0.1 | 1.2 ± 0.0 | 1.8 ± 0.1 | 0.2 ± 0.0 |
| AEFVEVTK | 4.0 ± 0.3 | 1.8 ± 0.1 | 2.4 ± 0.0 | 0.5 ± 0.2 |
| QTALVELLK | 3.5 ± 0.1 | 4.5 ± 0.5 | 0.9 ± 0.1 | 1.1 ± 0.0 |
| Mobile Phase | Sequence Coverage [%] | ||
|---|---|---|---|
| APCI | APPI//Dopant APPI 1 | ESI | |
| ACN (0.1% FA)/H2O (0.1% FA)//+acetone | 7 | 38//10 | 65 |
| MeOH (0.1% FA)/H2O (0.1% FA)//+acetone | 15 | 37//10 | 66 |
| ACN/H2O//+acetone | 17 | 43//14 | 62 |
| MeOH/H2O//+acetone | 25 | 41//18 | 63 |
| ACN/10 mmol·L−1 CH3COONH4 in H2O, adjusted pH = 9 (NH4OH)//+acetone | - 2 | 52//39 | 64 |
| MeOH/10 mmol·L−1 CH3COONH4 in H2O, adjusted pH = 9 (NH4OH) | 13 | - 3 | 66 |
| ACN/H2O adjusted pH = 9 (NH4OH)//+acetone | 14 | 57//36 | 68 |
| MeOH/H2O adjusted pH = 9 (NH4OH) | 21 | - 3 | 68 |
| ACN/10 mmol·L−1 CH3COONH4 in H2O//+acetone | - 2 | 44//21 | 65 |
| MeOH/10 mmol·L−1 CH3COONH4 in H2O | 13 | - 3 | 68 |
| Mobile Phase | Ion Source | Protease | Sequence Coverage [%] |
|---|---|---|---|
| MeOH/H2O | APCI | trypsin | 27 |
| chymotrypsin | 18 | ||
| Glu-C | 22 | ||
| ACN/H2O adjusted pH = 9 (NH4OH) | APPI | trypsin | 46 |
| chymotrypsin Glu-C | 23 48 | ||
| ACN (0.1% FA)/H2O (0.1% FA) | ESI | trypsin | 70 |
| chymotrypsin Glu-C | 36 35 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sedláčková, S.; Hubálek, M.; Vrkoslav, V.; Blechová, M.; Cvačka, J. Utility of Atmospheric-Pressure Chemical Ionization and Photoionization Mass Spectrometry in Bottom-Up Proteomics. Separations 2022, 9, 42. https://doi.org/10.3390/separations9020042
Sedláčková S, Hubálek M, Vrkoslav V, Blechová M, Cvačka J. Utility of Atmospheric-Pressure Chemical Ionization and Photoionization Mass Spectrometry in Bottom-Up Proteomics. Separations. 2022; 9(2):42. https://doi.org/10.3390/separations9020042
Chicago/Turabian StyleSedláčková, Simona, Martin Hubálek, Vladimír Vrkoslav, Miroslava Blechová, and Josef Cvačka. 2022. "Utility of Atmospheric-Pressure Chemical Ionization and Photoionization Mass Spectrometry in Bottom-Up Proteomics" Separations 9, no. 2: 42. https://doi.org/10.3390/separations9020042