
New Aspects of Ruthenium-Mediated Polyhedral Contraction of Monocarbollides

1
A.N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, 28, Vavilova St., 119991 Moscow, Russia
2
Higher Engineering School “New Materials and Technologies”, Plekhanov Russian University of Economics, 36, Stremyanny per., 117997 Moscow, Russia
3
Kurnakov Institute of General and Inorganic Chemistry, Russian Academy of Sciences, 31, Leninsky Prosp., 119991 Moscow, Russia
4
Department of Materials Science and Engineering Technology, Institute of Mechanical and Power Engineering Named after V.P. Goryachkin, Russian State Agrarian University-Moscow Timiryazev Agricultural Academy, 49, Timiryazevskaya St., 127550 Moscow, Russia
5
Department of Inorganic Chemistry, Faculty of Science, Peoples Friendship University of Russia (RUDN University), 6, Miklukho-Maklaya St., 117198 Moscow, Russia
*
Author to whom correspondence should be addressed.
Inorganics 2022, 10(10), 158; https://doi.org/10.3390/inorganics10100158
Submission received: 30 August 2022
/
Revised: 21 September 2022
/
Accepted: 23 September 2022
/
Published: 28 September 2022
(This article belongs to the Special Issue Fifth Element: The Current State of Boron Chemistry)
Abstract
:It has been shown that the interaction of tris(triphenylphosphine)ruthenium dichloride RuCl2(PPh3)3 (1) with 10-vertex monocarborane [6-Ph-nido-6-CB9H11]−[Et4N]+ (2) under mild thermolysis conditions is not selective due to the undesired coordination of ruthenium to a phenyl substituent in the carborane and phosphine ligands, giving the series of new classical and non-classical metallacarborane complexes. In contrast, the reaction of 1 and monocarborane [arachno-6-CB9H14]−[Et4N]+ (3) proceeds more selectively with the formation of the only one product, a isocloso-structured metallacarborane. The structures of two ruthenacarboranes were resolved by X-ray diffraction.
1. Introduction
Metallacarboranes are of particular interest due to their unique structures in which the metal atom is covalently bonded to the carborane polyhedron [1,2,3]. In the last decade, they have found a wide range of applications in catalysis [4,5]. In particular, the introduction of substituents into the carborane ligand allows to tune on the electronic, steric, and enantioselective properties of the catalyst. Thus, metallacarboranes showed good catalytic efficiency for amination of carbonyl compounds [6], radical polymerization of methyl methacrylate [7,8,9,10], oxidative coupling of benzoic acids and arenes with internal alkynes [11,12,13,14], carbene transfer reactions [15], as well as hydrogenation, isomerization, and metathesis of olefins [16,17,18]. Very recently, Romero and Teixidor with co-workers demonstrated the use of the cobalt bis(dicarbollide) anion and its derivatives as photocatalysts in various organic transformations [19,20,21].
Another way to modify electronic and steric parameters of metallacarboranes are the polyhedral contraction reactions, which occur with the removal of BH vertices [22,23,24]. However, many such reactions are not clean and selective, which reduces their use in synthesis. In continuation of our studies on the polyhedral contraction [25,26], in this work, we studied reactions of the electron-deficient complex RuCl2(PPh3)3 (1) with 10-vertex monocarboranes [6-Ph-nido-6-CB9H11]−[Et4N]+ (2) and [arachno-6-CB9H14]−[Et4N]+ (3) to compare their outcome and selectivity. It turned out that the presence of phenyl substituent in 2 significantly complicates the polyhedral contraction due to undesirable coordination of this substituent, while carborane 3 reacts smoothly, giving only one ruthenacarborane.
2. Results and Discussion
We found that refluxing of carborane 2 and ruthenium complex 1 in MeOH gives new 10-vertex clusters 4a,b and 5a,b, zwitterionic ruthenium arene complex 6, as well as 8-vertex ruthenacarboranes 7a,b with a total yield of ~25% (Scheme 1). We have previously shown that a similar reaction of 2 with the osmium complex OsCl2(PPh3)3 is more selective and gives the isocloso-{OsCB8} cluster as the only stable product [25].
According to the X-ray diffraction data (Figure 1), diruthenacarborane 1-Ph-2-[5,9-exo-RuClPPh3(μ,η6-C6H5PPh2)]-5,9-(μ-H)2-closo-2,1-RuCB8H5(OMe) (4a) has the geometry of a bicapped tetragonal antiprism and belongs to the classical 10-vertex clusters (22 skeletal electrons). The Ru(1) atom coordinates one chloride and two triphenylphosphine ligands, forming an exo-RuClPPh3(PPh2-μ,η6-C6H5)]+ fragment, which is bonded to the carborane frame via two B-H···Ru bonds involving boron atoms located in the α- and β-positions relative to the carbon atom. Notably, the formation of 4a is accompanied by the incorporation of methoxy to the 6-positioned boron atom and the loss of one boron vertex. Probably, the methoxylation of carborane cage is the first step of the polyhedral contraction, and methanol plays a key role in this fascinating process. For example, Stone with co-workers showed that the reaction of 2 with [ReBr(thf)2(CO)3] in non-alcoholic solvent (THF) leads to 11-vertex {ReCB9} species without loss of BH vertexes [27]. Moreover, we recently found that polyhedral contraction can occur not only simultaneously upon coordination with a metal atom, but also upon simple boiling of a preliminarily prepared metallacarborane in methanol [26].
A similar structure was proposed for 1-Ph-2-[5,9-exo-RuClPPh3(μ,η6-C6H5PPh2)]-5,9-(μ-H)2-closo-2,1-RuCB8H4(OMe)2 (4b), which has two methoxy-substituents in the carborane ligand in contrast to 4a. Thus, the 1H NMR spectra of 4a,b contain signals of MeO groups with δ 3.88 ppm (4a) and 3.46, 3.86 ppm (4b) with a relative intensity of 3H. Unfortunately, the exact position of the substituents in 4b has not been determined, since we were not able to obtain single crystals suitable for X-ray crystallography.
The 1H NMR spectra of 4a,b also contain a set of broad high-field signals corresponding to the bridging protons of B-H···Ru groups (δ −3.34, −16.34 and −3.30, −15.83 ppm for 4a and 4b, respectively). It can be seen that the chemical shifts in the bridging H atoms differ greatly. A similar phenomenon was previously observed in “three-bridged” exonido-osmacarboranes exonido-5,6,10-[Cl(Ph3P)2Os]-5,6,10-(μ-H)3-10-H-7-R-8-R1-7,8-C2B9H6 (R, R1 = H, Alk) [28], where it was rigorously proven that the downfield B-H···Os bond signal corresponds to the bridging hydrogen atom that occupies the trans-position relative to the Os-Cl bond in the octahedral environment of the metal atom. By analogy, the signals B-H···Ru(1) with δ −16.34 and −15.83 ppm in 1H NMR spectra of 4a,b were attributed to H(9) atoms, and the signals with δ −3.34 and −3.30 ppm were attributed to H(5).
In addition, the 1H NMR spectra of both complexes contain signals characteristic of a coordinated arene ligand: a set of four well-resolved multiplets in the range δ 6.0−4.0 ppm. Each of these signals corresponds to one proton; the fifth signal overlaps with proton signals of uncoordinated phenyl rings. The downfield shift in the signal of one of the hydrogen atoms of the bridging μ,η6-phenyl ligand can be explained by its participation in the intramolecular Ph-o-H···Cl hydrogen bond. According to X-ray diffraction data for complex 4a, the distance between H(23A) and the Cl atom (2.48 Å) is shorter than the sum of the corresponding Van der Waal radii (2.86 Å [29]) and the C-H···Cl angle is 141.4° (average values are given for two independent molecules). Such μ,η6-coordination of triphenylphosphine ligands is known for a number of complexes [30]. Among them, several crystallographically studied clusters belonging to polyhedral boron-containing complexes are described: PMe2Ph-μ-η6(Ru)-η1(Pt)-(C6H5PPh2)-closo-PtRuB9H9 [31] and μ,η6-C6H5PPh2)]-7,11-(μ-H)2-closo-2,1-RuCB10H8R (R = H, 6-OMe, 3-OMe) [32], in which the phosphine ligand on the one metal acts as an η6-coordinated ligand with respect to another metal atom.
Other products of the reaction discussed are mononuclear 10-vertex isocloso complexes: 1,3-(PPh3)2-1-H-1-Cl-2-Ph-4-OMe-isocloso-1,2-RuCB8H6 (5a) and 1-PPh3-1-H-1-Cl-2-Ph-4-OMe-isocloso-1,2-RuCB8H5(OMe)(PPh3) (5b), which belong to electron-deficient 2n clusters (20 skeletal electrons). We used label “isocloso” for these compounds to emphasize their non-conventional structure, which is quite distinct from the cluster geometry of classically structured closo 10-vertex metallacarbaboranes [33]. Similar to 4a,b, the formation of 5a,b is also accompanied by the deboronation of the carborane framework and the methoxylation process, which leads to a mixture of mono- and dimethoxy-substituted derivatives. It should be noted that monomethoxy substituted compound 5a is indefinitly stable and does not transform into 5b upon boiling in methanol.
According to X-ray diffraction analysis (Figure 2), complex 5a has the geometry of a 10-vertex polyhedron with the {CB8} carborane ligand substituted with the MeO group and triphenylphosphine at the boron atoms. The ruthenium atom coordinates the six-membered {CB5} open face of carborane in the hexahapta chair conformation. The metallacarborane cage has idealized C3v symmetry with the ruthenium atom in the axial position. In the molecule, there are three short distances from the Ru(1) atom to the C(2), B(3), and B(4) atoms with a cluster coordination number of 4 [2.188(14), 2.063(13), 2.090(14) Å) and three longer distances to B(5), B(6), and B(7) atoms with a coordination number of 5 [2.448(14), 2.364(11), 2.514(14) Å].
The 11B NMR spectra of complexes 5a and 5b contain three groups of signals in the range δ −30–+83 ppm. The signals of the first group are in a low field (δ from +64 to +83 ppm) and, in accordance with [34], are assigned to B(3) and B(4) atoms, which have a low coordination number equal to 4. Since one of the signals is present as a singlet, it can be concluded that the boron atom [B(3) or B(4)] corresponding to this signal has a MeO substituent. The evidence that in complexes 5a and 5b one of the PPh3 groups is also bound to the boron atom is the presence of doublet splitting with 1J(P,B) = 160 and 138 Hz, respectively, in one of the signals in the 11B/11B{1H} NMR spectra of these compounds. Despite the presence of two different PPh3 groups in complexes 5a and 5b (at the metal atom and in the carborane ligand), in the 1H NMR spectra, the signal of the terminal hydride appears as a broadened doublet with a spin–spin coupling constant 2J(P,H) = 63 and 43 Hz, respectively. However, in the 1H{11B} NMR spectrum of complex 5a (with complete decoupling from 11B), the hydride signal already has the form of a doublet of doublets, and the long-range spin–spin coupling constant 3J(P,H) = 16 Hz can be observed experimentally. The position of the triphenylphosphine substituent as well as the second methoxy group in 5b is not defined.
The zwitterionic mononuclear complex 6-[η6-C6H5RuH(PPh3)2-nido-6-CB9H11 (6) was also isolated in the reaction in low yield. The structure of complex 6 was determined from the 1H and 31P{1H} NMR spectra. According to the Cs symmetry, the 11B NMR spectrum of complex 6 contains a set of six signals with a relative intensity of 2:1:2:2:1:1. In the 31P{1H} NMR spectrum, there is one singlet signal from two equivalent PPh3 groups with δ 53.8 ppm. In the 1H NMR spectrum, a high-field broad signal was found corresponding to two bridging (B-H-B) hydrogen atoms of carborane (δ −3.17 ppm) and a terminal hydride triplet signal [δ −9.95 ppm, J(P,H) = 38 Hz].
The ruthenium atom η6-coordinates the phenyl group of the carborane, as well as one hydride and two triphenylphosphine ligands. At the same time, the carborane ligand in 6 retains two B–H–B bridging hydrogen atoms in the same position as in the starting carborane 2, i.e., it formally has a negative charge. Taking into account that the ruthenium atom coordinates only one hydride ligand, it should be concluded that complex 6 is zwitterionic, and the metal atom has a formal oxidation state of +2. A similar example of zwitterionic metallacarboranes, 6-[η6-C6H5RuH(PPh3)2-nido-7,9-C2B9H11, is described in the literature [35].
Among the reaction products, another group of metallacarboranes with an extraordinary structure was found, which belongs to rare 8-vertex capped closo-clusters with a boron vertex built over a triangular face. Two compounds of this type were isolated by column chromatography: capped closo-l-Ph-2,2-(PPh3)2-2-H-3,8-(OMe)2-6-R-2,1-RuCB6H3 [R = H (7a), R = OMe (7b)], which are products of a deeper degradation (the loss of two boron vertices) of the initial carborane 2.
Structure of compounds 7a,b was confirmed using the 1H, 31P{1H}, 11B/11B{1H} NMR and mass spectroscopic data by analogy with the carbon-unsubstituted analogue capped closo-2,2-(PPh3)2-2-H-3,6,8-(OMe)3-2,1-RuCB6H4 (Table 1), whose structure was unambiguously confirmed previously by X-ray diffraction [36].
In the 1H NMR spectrum of complex 7b, two of the three MeO groups appear as a single signal (δ 2.8 ppm) with an intensity of 6H, which indicates their equivalence and, therefore, the location at the B(3) and B(6) (both are in the α-position relative to the carbon atom). Taking into account the preservation of symmetry in this complex, the third MeO-group should be bound to the B(8) atom, which is located symmetrically over the Ru(2)-B(4)-B(5) face. In the 1H NMR spectrum, the signal of the terminal hydride has the form of a broadened triplet, while in the 31P{1H} NMR spectrum, only one singlet signal (δ 48.3 ppm) from two equivalent PPh3 ligands bound to the metal atom is observed. In the 11B{1H} NMR spectrum of 7b, in accordance with the Cs symmetry, a set of four signals with a relative intensity of 1:2:1:2 is observed. Extremely weak field signal with δ 70.1 ppm confirms the presence of a coordinatively unsaturated B(8) atom in the complex.
Complex 7a with two MeO groups has C1 symmetry. Therefore, in the 11B NMR spectrum, six signals appear, each with an intensity of 1B. Two of these signals are singlets, which indicates two MeO substituents associated with the carborane ligand. Here, as in spectrum of 7b, one signal from the B(8) atom appears in a much weaker field than the others (the assignment of some of them was performed by analogy with complex 7b). The 31P{1H} NMR spectrum of 7a contains two doublet signals with δ 46.2 and 46.0 ppm with 2J(P,P) = 10 Hz. Accordingly, in the 1H NMR spectrum, the hydride signal appears as a broadened triplet with 2J(P,H) = 24 Hz.
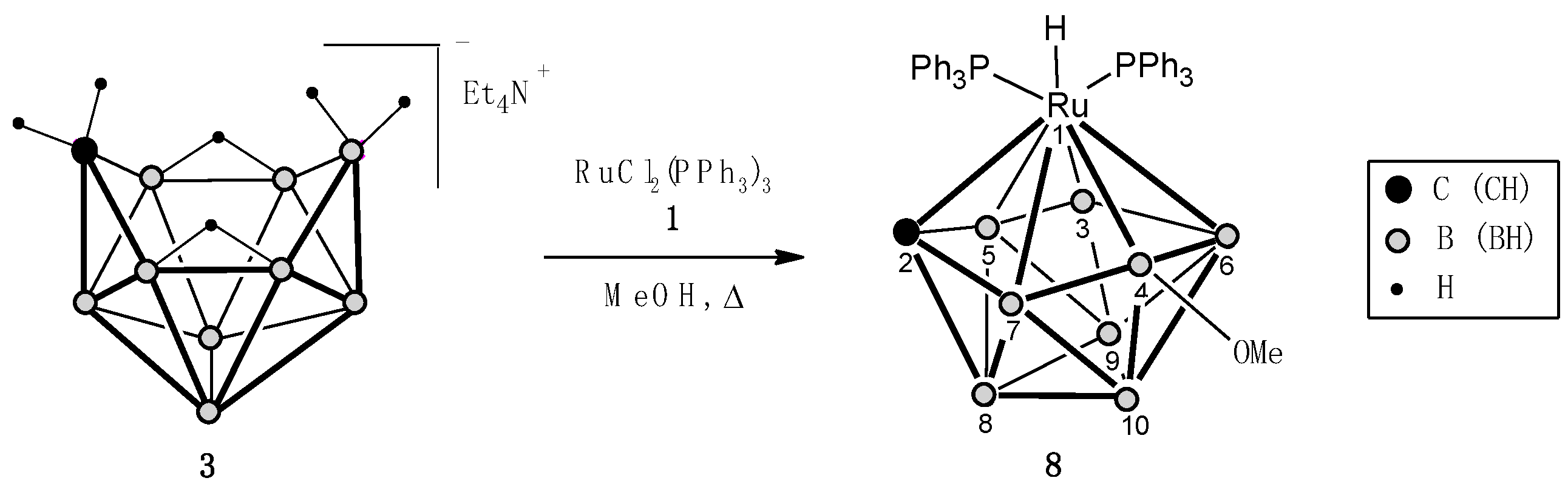
In contrast to the previous reaction, RuCl2(PPh3)3 under similar conditions selectively reacts with monocarbon carborane [arachno-6-CB9H14]−[Et4N]+ (3), forming 10-vertex ruthenacarborane 1,1-(PPh3)2-1-H-3-OMe-isocloso-1,2-RuCB8H7 (8) as the only stable product in 26% yield (Scheme 2). The higher selectivity in this case can be explained by the absence of phenyl substituent in 3, which significantly complicates the reactions due to the strong coordination affinity of ruthenium to arene ligands [37].
The structure of 8 was established on the basis of the 1H, 31P{1H}, and 11B/11B{1H} NMR spectra. The 1H NMR spectrum contains a signal of the hydrogen atom of the CH group of the carborane ligand (δ 7.83 ppm), which is a doublet with 2J(P,H) = 10 Hz, and one singlet signal of the MeO group (δ 4.03 ppm). The hydride signal with δ −5.30 ppm appears as a broadened doublet of doublets with 2J(Pa,H) = 22 Hz and 2J(Pb,H) = 46 Hz. In the 31P{1H} NMR spectrum, one of the two signals of the phosphorus atoms of triphenylphosphine ligands is a broadened singlet (δ 49.2 ppm), and the second is a doublet (δ 35.9 ppm) with 2JAB = 15 Hz, probably due to the trans-location of the first ligand relative to one of the boron atoms of the carborane fragment. In the 11B NMR spectra of complex 8, as well as in the spectra of ruthenacarboranes 5a,b, there is a broad singlet signal (δ 83.1 ppm) with doubled intensity, which we attributed to two coordinatively unsaturated atoms B(3) and B(4). Since the remaining signals in the spectrum have the form of doublets with their usual spin–spin coupling constant 1J(B,H) in the range of 130–145 Hz, it can be concluded that the MeO group is located precisely at one of these boron atoms, B(3) or B(4) (the positions are equivalent).
3. Materials and Methods
3.1. General Procedures
The reaction described above was carried out using standard Schlenk equipment under an atmosphere of dry argon. All solvents, including those used for column chromatography as eluents, were dried under appropriate drying agents and distilled under argon prior to use. The reactions products were isolated by column chromatography and purified by crystallization in air. Chromatography was performed on silica gel (Merck, 70–230 mesh). The NMR spectra were obtained with a Bruker AMX-400 spectrometer (1H, 400.13 MHz; 31P, 161.98 MHz; 11B, 128.33 MHz) using TMS as an internal reference and 85% H3PO4 and BF3·Et2O as the external references, respectively. Microanalyses were performed at the Analytical Laboratory of the Institute of Organoelement Compounds of the RAS. The starting material, tris(triphenylphosphine)ruthenium dichloride (1) was prepared by methods published elsewhere [38]. Tetraethylammonium 6-phenyl-nido-6-carbadecaborate (2) and tetraethylammonium arachno-6-carbadecaborate (3) were obtained from the laboratory of Prof. Kennedy J.D. (University of Leeds, Leeds, UK).
3.2. Reaction of RuCl2(PPh3)3 (1) with Tetraethylammonium 6-Phenyl-Nido-6-Carbadecaborate (2)
A solution of complex 1 (1 g, 1.04 mmol) in 50 mL of benzene was added dropwise over 2 h to a stirred solution of nido-carborane 2 (0.38 g, 1.16 mmol) in 50 mL of anhydrous methanol under argon at low boiling. The use of toluene as a co-solvent instead of benzene leads to a significant decrease in the yield of products. The solution was refluxed with stirring for another 3 h, while the color of the reaction mixture changed from brown to dark red. After cooling the reaction mixture and removing the solvent, the solid residue was placed on a silica gel column 63–210 μm, eluting with CH2Cl2. The first light yellow fraction containing PPh3, 6, and 7a,b, and the next two red fractions, 4a, 5a and 4b, 5b. The mixture of products from the light yellow fraction was separated again on a column of silica gel in benzene, washing off, successively, 7a, 7b, and 6. The products from the two red fractions were also chromatographed on a column of silica gel using CH2C12/n-hexane (2:1) as the eluent; from the first red fraction, an orange substance 5a and a red substance 4a were obtained; from the second red fraction, an orange product 5b and a red product 4b. The complexes were finally purified by recrystallization from a CH2C12/n-hexane solvent mixture.
1-Ph-2-[5,9-exo-RuClPPh3(μ,η6-C6H5PPh2)]-5,9-(μ-H)2-closo-2,1-RuCB8H5(OMe)(4a). Yield 64 mg, 6%. IR, ν/cm−1: 2546 (B-H). 1H NMR (400.13 MHz, CD2Cl2): δ 7.83, 7.60–7.18, 6.84 m, 30H + 1H (HPh); 5.91 t, 1H, J = 6 Hz (η-Ph); 5.53 t, 1H, J = 6 Hz (η-Ph); 5.05 t, 1H, J = 6 Hz (η-Ph); 4.16 t, 1H, J = 6 Hz (η-Ph); 3.88 s, 3H (OMe); −3.34 br m, 1H (H-5); −16.34 br m, 1H (H-9). 31P{1H} NMR (161.98 MHz, CD2Cl2): δ 64.1 d, 1P, 2JAB = 32 Hz; 59.2 d, 1P, 2JAB = 32 Hz. 11B NMR (128.33 MHz, CD2Cl2): δ 42.5 s, 1B; 16.6 d, 1B, 1J(B,H) = 137 Hz; 10.9 d, 2B, 1J(B,H) = 145 Hz; 8.6 d, 1B, 1J(B,H) = 137 Hz; −6.5 d, 1B, 1J(B,H) = 143 Hz; −20.2 d, 2B, 1J(B,H) = 131 Hz. For C44H45B8ClOP2Ru2 (975.86) calculated (%): C, 54.15; H, 4.65; B, 8.86; P, 6.35. Found (%): C, 54.05; H, 4.52; B, 8.97; P, 6.28.
1-Ph-2-[5,9-exo-RuClPPh3(μ,η6-C6H5PPh2)]-5,9-(μ-H)2-closo-2,1-RuCB8H4(OMe)2(4b). Yield 54 mg, 5%. IR, ν/cm−1: 2541 (B-H). 1H NMR (400.13 MHz, CD2Cl2): δ 7.83–7.15, 6.94 m, 30H + 1H (HPh); 5.76 t, 1H, J = 6 Hz (η-Ph); 5.44 t, 1H, J = 6 Hz (η-Ph); 4.88 t, 1H, J = 6 Hz (η-Ph); 3.98 t, 1H, J = 6 Hz (η-Ph); 3.86 s, 3H (OMe); 3.46 s, 3H (OMe); −3.30 br m, 1H (H-5); −15.83 br m, 1H (H-9). 31P{1H} NMR (161.98 MHz, CD2Cl2): δ 63.1 d, 1P, 2JAB = 32 Hz; 58.3 d, 1P, 2JAB = 32 Hz. 11B NMR (128.33 MHz, CD2Cl2): δ 38.0 br s, 1B; 27.5 br s, 1B; 4.8 br s, 2B; −12.4 br s, 1B; −22.6 br s, 2B; −31.4 br s, 1B. For C45H47B8ClO2P2Ru2 (1005.88) calculated (%): C, 53.73; H, 4.71; B, 8.60; P, 6.16. Found (%): C, 53.54; H, 4.69; B, 8.71; P, 6.21.
1,3-(PPh3)2-1-H-1-Cl-2-Ph-4-OMe-isocloso-1,2-RuCB8H6 (5a). Yield 35 mg, 4%. IR, ν/cm−1: 2531 (B-H); 1989 (Ru-H). 1H{11B} NMR (400.13 MHz, CD2Cl2): δ 7.92–7.06 m, 6.03 t, 28H + 2H (HPh); 3.64 br s, 1H (BH); 3.44 br s, 1H (BH); 3.16 br s, 1H (BH); 2.99 s, 3H (OMe); 2.85 br s, 1H (BH); 2.69 br s, 1H (BH); 0.00 br s, 1H (BH); −5.44 dd, 1H, 2J(P,H) = 16 Hz, 2J(P,H) = 63 Hz (HRu). 31P{1H} NMR (161.98 MHz, CD2Cl2): δ 50.1 s, 1P (Ru-PPh3); 8.4 q, 1P, 1J(P,B) = 160 Hz (B-PPh3). 11B NMR (128.33 MHz, CD2Cl2): δ 82.9 s, 1B (B-4); 64.5 d, 1B, 1J(B,P) = 160 Hz (B-3); 14.1 d, 1B, 1J(B,H) = 141 Hz (B-9); 12.5 d, 1B, 1J(B,H) = 137 Hz (B-10); 1.7 d, 1B, 1J(B,H) = 146 Hz (B-8); −6.9 d, 1B, 1J(B,H) = 118 Hz (B-7); −9.8 d, 1B, 1J(B,H) = 128 Hz (B-5); −21.8 d, 1B, 1J(B,H) = 131 Hz (B-6). 13C{1H} NMR (100.51 MHz, CD2Cl2): δ 169.8 br s (Ccarb); 147.5, 136.0−126.6, 123.5, 122.9 (CPh); 57.8 s (OMe). For C44H45B8ClOP2Ru (874.79) calculated (%): C, 60.41; H, 5.18; B, 9.89; P, 7.08. Found (%): C, 60.36; H, 5.22; B, 10.02; P, 6.98.
1-PPh3-1-H-1-Cl-2-Ph-4-OMe-isocloso-1,2-RuCB8H5(OMe)(PPh3)(5b). Yield 24 mg, 3%. IR, ν/cm−1: 2536 (B-H); 1999 (Ru-H). 1H NMR (400.13 MHz, CD2Cl2): δ 7.53-7.23 m, 35H (HPh); 4.15 s, 3H (OMe); 3.71 s, 3H (OMe); −3.11 br d, 1H, 2J(H,P) = 43 Hz (HRu). 31P{1H} NMR (161.98 MHz, CD2Cl2): δ 45.9 s, 1P (Ru-P); 5.3 br q, 1P (B-P). 11B NMR (128.33 MHz, CD2Cl2): δ 79.4 s, 1B (B-4); 73.5 d, 1B, 1J(B,H) = 96 Hz (B-3); 16.6 d, 1B, 1J(B,H) = 122 Hz [B-8 (9 or 10)]; 13.4 s, 1B [B-5 (6 or 7)]; 8.41 d, 2B, 1J(B,H) = 112 Hz [B-9,10 (8,10 or 8,9)]; −26.7 d, 1B, 1J(B,H) = 151 Hz [B-6 (7 or 5)]; −29.8 d, 1B, 1J(B,P) = 138 Hz [B-7 (5 or 6)]. For C45H47B8ClO2P2Ru (904.81) calculated (%): C, 59.73; H, 5.24; B, 9.56; P, 6.85. Found (%): C, 60.01; H, 5.21; B, 9.58; P, 6.76. m/z 904.2 [M]+.
6-[(η6-C6H5)RuH(PPh3)2]-nido-CB9H11(6). Yield 14 mg, 2%. IR, ν/cm−1: 2534 (B-H); 1985 (Ru-H). 1H NMR (400.13 MHz, CD2Cl2): δ 7.34−7.16 m, 30H (PPh3); 6.13 t, 1H, 3J(H,H) = 5.5 Hz (Hpara, η-Ph); 5.05 d, 2H, 3J(H,H) = 6.5 Hz (Hortho, η-Ph); 4.85 t, 2H, 3J(H,H) = 6 Hz (Hmeta, η-Ph); −3.17 br s, 2H (μ-H); −8.95 t, 1H, 2J(H,P) = 38 Hz (HRu). 31P{1H} NMR (161.98 MHz, CD2Cl2): δ 53.8 s, 2P (PPh3). 11B NMR (128.33 MHz, CD2Cl2): δ 4.0 d, 2B, 1J(B,H) = 142 Hz; 2.1 d, 1B, 1J(B,H) = 80 Hz; −3.2 d, 2B, 1J(B,H) = 141 Hz; −11.5 d, 2B, 1J(B,H) = 115 Hz; −25.7 d, 1B, 1J(B,H) = 158 Hz; −35.6 d, 1B, 1J(B,H) = 143 Hz. For C43H37B9P2Ru (824.15) calculated (%): C, 62.67; H, 5.75; B, 11.81; P, 7.52. Found (%): C, 62.61; H, 5.71; B, 11.92; P, 7.48. m/z 824.3 [M]+.
Capped closo-1-Ph-2,2-(PPh3)2-2-H-3,8-(OMe)2-2,1-RuCB6H4(7a). Yield 24 mg, 3%. IR, ν/cm−1: 2522 (B-H); 2050 (Ru-H). 1H NMR (400.13 MHz, CD2Cl2): δ7.30−6.97 m, 35H (HPh); 4.24 s, 3H (OMe); 2.96 s, 3H (OMe); −9.84 br t, 1H, 2J(H,P) = 24 Hz (HRu). 31P{1H} NMR (161.98 MHz, CD2Cl2): δ 46.2 d, 1P, 2JAB = 10 Hz; 46.0 d, 1P, 2JAB = 10 Hz. 11B NMR (160.46 MHz, CD2Cl2): δ 67.5 s, 1B (B-8); 33.7 s, 1B (B-3); 24.1 br s, 1B [B-6 (7)]; 14.0 d, 1B, 1J(B,H) = 147 Hz [B-7 (6)]; -26.3 d, 1B, 1J(B,H) = 149 Hz [B-4 (5)]; −30.1 d, 1B, 1J(B,H) = 147 Hz [B-5 (4)]. For C45H46B6O2P2Ru (846.73) calculated (%): C, 63.83; H, 5.48; B, 7.66; P, 7.32. Found (%): C, 63.75; H, 5.52; B, 7.51; P, 7.16. m/z 847.3 [M]+.
Capped closo-1-Ph-2,2-(PPh3)2-2-H-3,6,8-(OMe)3-2,1-RuCB6H3(7b). Yield 10 mg, 1%. IR, ν/cm−1: 2517 (B-H); 2043 (Ru-H). 1H NMR (400.13 MHz, CD2Cl2): δ 7.24−6.96 m, 6.48 d, 35H (HPh); 4.27 s, 3H (OMe); 2.80 s, 6H (OMe); −10.43 br t, 1H, 2J(H,P) = 24 Hz (HRu). 31P{1H} NMR (161.98 MHz, CD2Cl2): δ 48.3 s, 2P. 11B{1H} NMR (128.33 MHz, CD2Cl2): δ 70.1 s, 1B (B-8); 32.8 s, 2B (B-3,6); 14.1 s, 1B (B-7); −32.4 s, 2B (B-4,5). For C46H48B6O3P2Ru (876.76) calculated (%): C, 63.02; H, 5.52; B, 7.40; P, 7.07. Found (%): C, 63.38; H, 5.49; B, 7.48; P, 7.34. Found: m/z 876.4 [M]+.
3.3. Reaction of RuCl2(PPh3)3 (1) with Tetraethylammonium Arachno-6-Carbadecaborate (3)
Complex 1 (0.5 g, 0.52 mmol) was added to a stirred solution of arachno-carborane 3 (0.16 g, 0.63 mmol) in 15 mL of absolute methanol under argon. The mixture was refluxed with stirring for 14 h. Gradually, the precipitate turned from brown to yellow. The reaction mixture was cooled, and after removing the solvent, the resulting oily residue was chromatographed through a silica-gel column (63–210 μm) in a mixture of solvents CH2Cl2/n-hexane (2:1). After crystallization from a mixture of solvents CH2Cl2/n-hexane, yellow crystals of complex 8 were obtained.
1,1-(PPh3)2-1-H-3-OMe-isocloso-1,2-RuCB8H7(8). Yield 0.11 g, 26%. IR, ν/cm−1: 2538 (B-H); 1990 (Ru-H). 1H NMR (400.13 MHz, CD2Cl2): δ 7.83 br d, 1H, 3J(H,P) = 10 Hz (CHcarb); 7.40−7.16 m, 30H (Ph); 4.03 s, 3H (OMe); −5.30 br dd, 1H, 2J(Pa,H) = 22 Hz, 2J(Pb,H) = 46 Hz (HRu). 31P{1H} NMR (161.98 MHz, CD2Cl2): δ 49.2 br s, 1P; 35.9 d, 1P, 2JAB = 15 Hz. 11B NMR (128.33 MHz, CD2Cl2): δ 83.1 br s, 2B (B-3,4); 16.6 d, 1B, 1J(B,H) = 137 Hz [B-8 (9 or 10)]; 10.9 d, 1B, 1J(B,H) = 145 Hz [B-9 (10 or 8)]; 8.6 d, 1B, 1J(B,H) = 137 Hz [B-10 (8 or 9)]; −6.5 d, 1B, 1J(B,H) = 143 Hz [B-5 (6 or 7)]; −20.2 d, 2B, 1J(B,H) = 131 Hz [B-6,7 (5,7 or 5,6)]. For C38H42B8OP2Ru (764.25) calculated (%): C, 59.72; H, 5.54; B, 11.32; P, 8.11. Found (%): C, 59.48; H, 5.58; B, 11.50; P, 8.32.
3.4. X-ray Diffraction Study of 4a and 5a
The X-ray single crystal data were collected using Mo Kα radiation (λ = 0.71073 Å) on Bruker SMART 1000 diffractometer equipped with the Cryostream (Oxford Cryosystems) open-flow nitrogen cryostat. The structures were solved by direct methods and refined by the full-matrix least squares technique against F2 with anisotropic thermal parameters for all non-hydrogen atoms using the SHELXL software [39]. Hydrogen atoms of the carborane ligands, as well hydride ligand in 5a, were located from the Fourier syntheses. The remaining hydrogen atoms were placed geometrically and included in the structure factors calculation in the riding motion approximation. Crystal data and parameters of the refinements are listed in Table 2. Crystallographic data for the structures have been deposited with the Cambridge Crystallographic Data Centre as supplementary publications CCDC 2201207–2201208 and can be found in the Supplementary Materials.
4. Conclusions
In summary, we showed that the interaction of arachno-carborane 3 with ruthenium complex 1 leads to the selective formation of a 10-vertex 20-electron isocloso-cluster as the only stable product, whereas an analogous reaction of the phenyl substituted nido-carborane 2 produces a complex mixture of cluster compounds. For example, a number of products with the (η6-Ph)Ru motif were isolated along with other 8- and 10-vertex clusters, and this can be explained by the strong coordination affinity of ruthenium to arene ligands. By contrast, we have shown previously that the related osmium complex OsCl2(PPh3)3 provides a more selective polyhedral contraction of monocarbollides [25,26].
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/inorganics10100158/s1, crystallographic data for 4a and 5a.
Author Contributions
Conceptualization, V.E.K. and D.A.L.; methodology, D.A.L.; investigation, V.E.K. and M.V.T.; data curation, V.E.K.; X-ray crystallography, F.M.D.; writing—original draft preparation, V.E.K.; writing—review and editing, F.M.D. and D.A.L. All authors have read and agreed to the published version of the manuscript.
Funding
Generous support by the Ministry of Science and Higher Education of the Russian Federation (Contract/agreement No. 075-00697-22-00) is gratefully acknowledged.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Accession codes 2201207 (4a) and 2201208 (5a) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing [email protected], or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44-1223-336033.
Acknowledgments
We would like to dedicate this work to the memory of Igor T Chizhevsky, who has been the main protagonist of the metallacarborane chemistry in Russia. The single-crystal X-ray diffraction analysis was performed using the equipment of the JRC PMR IGIC RAS.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hosmane, N.S. (Ed.) Boron Science: New Technologies and Applications; CRC Press: Boca Raton, FL, USA, 2011. [Google Scholar]
- Grimes, R.N. Carboranes, 3rd ed.; Academic Press: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Kar, S.; Pradhan, A.N.; Ghosh, S. Comprehensive Organometallic Chemistry IV; Parkin, G., O’Hare, D., Meyer, K., Eds.; Elsevier Science: Amsterdam, The Netherlands, 2022; Volume 9, pp. 263–369. [Google Scholar]
- Loginov, D.A.; Konoplev, V.E. Oxidative coupling of benzoic acids with alkynes: Catalyst design and selectivity. J. Organomet. Chem. 2018, 867, 14–24. [Google Scholar] [CrossRef]
- Vinogradov, M.M.; Loginov, D.A. Rhoda- and iridacarborane halide complexes: Synthesis, structure and application in homogeneous catalysis. J. Organomet. Chem. 2020, 910, 121135. [Google Scholar] [CrossRef]
- Molotkov, A.P.; Vinogradov, M.M.; Moskovets, A.P.; Chusova, O.; Timofeev, S.V.; Fastovskiy, V.A.; Nelyubina, Y.V.; Pavlov, A.A.; Chusov, D.A.; Loginov, D.A. Iridium halide complexes [1,1-X2-8-SMe2-1,2,8-IrC2B9H10]2 (X = Cl, Br, I): Synthesis, reactivity and catalytic activity. Eur. J. Inorg. Chem. 2017, 2017, 4635–4644. [Google Scholar] [CrossRef]
- Grishin, I.D.; Knyazeva, N.A.; Penkal, A.M. Novel ruthenium(II) and (III) carborane complexes with diphosphine ligands and their application in radical polymerization. Russ. Chem. Bull. 2020, 69, 1520–1529. [Google Scholar] [CrossRef]
- Kaltenberg, A.A.; Somov, N.V.; Malysheva, Y.B.; Knyazeva, N.A.; Piskunov, A.V.; Grishin, I.D. Novel carborane complexes of ruthenium with tridentate phosphine ligands: Synthesis and application in atom transfer radical polymerization. J. Organomet. Chem. 2020, 917, 121291. [Google Scholar] [CrossRef]
- Grishin, D.F.; Grishin, I.D. Modern trends in controlled synthesis of functional polymers: Fundamental aspects and practical applications. Russ. Chem. Rev. 2021, 90, 231–264. [Google Scholar] [CrossRef]
- Grishin, I.D.; Zimina, A.M.; Anufriev, S.A.; Knyazeva, N.A.; Piskunov, A.V.; Dolgushin, F.M.; Sivaev, I.B. Synthesis and Catalytic Properties of Novel Ruthenacarboranes Based on nido-[5-Me-7,8-C2B9H10]2− and nido-[5,6-Me2-7,8-C2B9H9]2− Dicarbollide Ligands. Catalysts 2021, 11, 1409. [Google Scholar] [CrossRef]
- Loginov, D.A.; Belova, A.O.; Vologzhanina, A.V.; Kudinov, A.R. Cationic iridacarboranes [3-(arene)-3,1,2-IrC2B9H11]+ and [3-(MeCN)3-3,1,2-IrC2B9H11]+: Synthesis, reactivity, and bonding. Catalysis of oxidative coupling of benzoic acid with alkynes. J. Organomet. Chem. 2015, 793, 232–240. [Google Scholar] [CrossRef]
- Loginov, D.A.; Muratov, D.V.; Nelyubina, Y.V.; Laskova, J.; Kudinov, A.R. µ-Borole triple-decker complexes as catalysts for oxidative couplingof benzoic acid with alkynes. Structure of a hybridrhodacyclopentadienyl/borole triple-decker complex. J. Mol. Catal. A Chem. 2017, 426, 393–397. [Google Scholar] [CrossRef]
- Molotkov, A.P.; Timofeev, S.V.; Loginov, D.A. Trimethylammonium-containing rhodacarborane [(9-NMe3-7,8-C2B9H10)RhCl2]2 as a catalyst for the annulation of arylcarboxylic acids with alkynes. Russ. Chem. Bull. 2021, 70, 1922–1926. [Google Scholar] [CrossRef]
- Kharitonov, V.B.; Muratov, D.V.; Nelyubina, Y.V.; Loginov, D.A. Formation of a Naphthalene Framework by Rhodium(III)-Catalyzed Double C–H Functionalization of Arenes with Alkynes: Impact of a Supporting Ligand and an Acid Additive. Synthesis 2022. [Google Scholar] [CrossRef]
- Wang, L.; Perveen, S.; Ouyang, Y.; Zhang, S.; Jiao, J.; He, G.; Nie, Y.; Li, P. Well-Defined, Versatile and Recyclable Half-Sandwich Nickelacarborane Catalyst for Selective Carbene-Transfer Reactions. Chem. Eur. J. 2021, 27, 5754–5760. [Google Scholar] [CrossRef] [PubMed]
- Alekseev, L.S.; Lyubimov, S.E.; Dolgushin, F.M.; Novikov, V.V.; Davankov, V.A.; Chizhevsky, I.T. New Rhodacarborane−Phosphoramidite Catalyst System for Enantioselective Hydrogenation of Functionalized Olefins and Molecular Structure of the Chiral Catalyst Precursor [3,3-{(S)-PipPhos}2-3-H-1,2-(o-xylylene)-closo-3,1,2-RhC2B9H9]. Organometallics 2011, 30, 1942–1950. [Google Scholar] [CrossRef]
- Xie, Q.; Wang, T.; Wu, S.; Guo, W.; Zhang, H. A stable ruthenium complex bearing a 1,2-dicarbadodecaborane(12)-1,2-dithiolate ligand and its activation for olefin metathesis. J. Organomet. Chem. 2018, 865, 95–99. [Google Scholar] [CrossRef]
- Chan, A.P.Y.; Parkinson, J.A.; Rosair, G.M.; Welch, A.J. Bis(phosphine)hydridorhodacarborane derivatives of 1,1′-bis(ortho-carborane) and their catalysis of alkene isomerization and the hydrosilylation of acetophenone. Inorg. Chem. 2020, 59, 2011–2023. [Google Scholar] [CrossRef]
- Guerrero, I.; Kelemen, Z.; Viñas, C.; Romero, I.; Teixidor, F. Metallacarboranes as Photoredox Catalysts in Water. Chem. Eur. J. 2020, 26, 5027–5036. [Google Scholar] [CrossRef]
- Guerrero, I.; Viñas, C.; Romero, I.; Teixidor, F. A stand-alone cobalt bis(dicarbollide) photoredox catalyst epoxidates alkenes in water at extremely low catalyst load. Green Chem. 2021, 23, 10123–10131. [Google Scholar] [CrossRef]
- Guerrero, I.; Viñas, C.; Fontrodona, X.; Romero, I.; Teixidor, F. Aqueous Persistent Noncovalent Ion-Pair Cooperative Coupling in a Ruthenium Cobaltabis(dicarbollide) System as a Highly Efficient Photoredox Oxidation Catalyst. Inorg. Chem. 2021, 60, 8898–8907. [Google Scholar] [CrossRef]
- Jones, C.J.; Francis, J.N.; Hawthorne, M.F. New 10- and 11-atom polyhedral metallocarboranes prepared by polyhedral contraction. J. Am. Chem. Soc. 1972, 94, 8391–8399. [Google Scholar] [CrossRef]
- Hanusa, T.P.; Huffman, J.C.; Curtis, T.L.; Todd, L.J. Synthesis of (π-arene)metallacarbaboranes containing ruthenium or osmium and a (π-cyclohexadienyl)cobaltacarbaborane. Crystal structure of 2,5,6-(η-C6H6)RuC2B7H11. Inorg. Chem. 1985, 24, 787–792. [Google Scholar] [CrossRef]
- Buckner, S.W.; Fischer, M.J.; Jelliss, P.A.; Luo, R.; Minteer, S.D.; Rath, N.P.; Siemarczuk, A. Dual Fluorescence from an Isonido ReIII Rhenacarborane Phosphine Complex, [7,10-μ-H-7-CO-7,7-(PPh3)2-isonido-7,8,9-ReC2B7H9]. Inorg. Chem. 2006, 45, 7339–7347. [Google Scholar] [CrossRef] [PubMed]
- Konoplev, V.E.; Pisareva, I.V.; Vorontsov, E.V.; Dolgushin, F.M.; Franken, A.; Kennedy, J.D.; Chizhevsky, I.T. Ten-vertex osmamonocarbaboranes via arachno and nido {CB9} monocarbaboranes. Polyhedral contraction promoted by [OsCl2(PPh3)3] in MeOH and the crystal and molecular structure of [1-H-1,1-(PPh3)2-2-Ph-3-(OMe)-isocloso-1,2-OsCB8H7]. Inorg. Chem. Commun. 2003, 6, 1454–1458. [Google Scholar] [CrossRef]
- Konoplev, V.E.; Dolgushin, F.M.; Loginov, D.A.; Tachaev, M.V. Step-by-step polyhedral contraction of the osmacarborane framework {OsCB10} → {OsCB9} → {OsCB8}. J. Organomet. Chem. 2021, 949, 121948. [Google Scholar] [CrossRef]
- Du, S.; Kautz, J.A.; McGrath, T.D.; Stone, F.G.A. Synthesis and Structure of the Novel 11-Vertex Rhenacarborane Dianion [1,1,1-(CO)3-2-Ph-closo-1,2-ReCB9H9]2− and Its Reactivity toward Cationic Transition Metal Fragments. Organometallics 2003, 22, 2842–2850. [Google Scholar] [CrossRef]
- Kolomnikova, G.D.; Petrovskii, P.V.; Sorokin, P.V.; Dolgushin, F.M.; Yanovsky, A.I.; Chizhevsky, I.T. Synthesis, structure and isomerism of “three-bridge” exo-nido-osmacarborane clusters. Rus. Chem. Bull. 2001, 50, 706–715. [Google Scholar] [CrossRef]
- Rowland, R.S.; Taylor, R. Intermolecular Nonbonded Contact Distances in Organic Crystal Structures: Comparison with Distances Expected from van der Waals Radii. J. Phys. Chem. 1996, 100, 7384–7391. [Google Scholar] [CrossRef]
- Grimes, R.N.; Sinn, E.; Pipal, J.R. Tetracarbon metallacarboranes. 9. New types of nido cage geometry. Crystal and molecular structures of [(C6H5)2PCH2]2Ni(CH3)4C4B8H8 (isomer 1) and (η5-C5H5)Co(CH3)4C4B7H7 (isomer 2). Inorg. Chem. 1980, 19, 2087–2095. [Google Scholar] [CrossRef]
- Ditzel, E.J.; Fontaine, X.L.R.; Greenwood, N.N.; Kennedy, J.D.; Sisan, Z.; Štíbr, B.; Thornton-Pett, M. An unusual direct conversion of metallaboranes to metallacarbaboranes and the isolation of a novel isoarachno twelve-vertex cluster compound. J. Chem. Soc. Chem. Commun. 1990, 1741–1743. [Google Scholar] [CrossRef]
- Pisareva, I.V.; Konoplev, V.E.; Petrovskii, P.V.; Vorontsov, E.V.; Dolgushin, F.M.; Yanovsky, A.I.; Chizhevsky, I.T. Synthesis and characterization of novel monocarbollide exo-closo-(π-arene)biruthenacarboranes [(PPh3)mClRu(η6-C6H5R)Ru′CB10H11-n(OMe)n] (where R = H, m = 2, n = 1; R = μ-PPh2, m = 1, n = 0, 1). Inorg. Chem. 2004, 43, 6228–6237. [Google Scholar] [CrossRef]
- Beckett, M.A.; Brellochs, B.; Chizhevsky, I.T.; Damhus, T.; Hellwich, K.-H.; Kennedy, J.D.; Laitinen, R.; Powella, W.H.; Rabinovich, D.; Viñas, C.; et al. Nomenclature for boranes and related species (IUPAC Recommendations 2019). Pure Appl. Chem. 2020, 92, 355–381. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, J.D. NMR in Inorganic and Organometallic Chemistry. In Multinuclear NMR; Mason, J., Ed.; Plenum: London, UK; New York, NY, USA, 1987; p. 221. [Google Scholar]
- Chizhevsky, I.T.; Yanovsky, A.I.; Struchkov, Y.T. Semi-sandwich platinum metals metallacarboranes derived from nido-C2B9H12−: Chemistry and structural studies. J. Organomet. Chem. 1997, 51, 51–63. [Google Scholar] [CrossRef]
- Pisareva, I.V.; Dolgushin, F.M.; Yanovsky, A.I.; Balagurova, E.V.; Petrovskii, P.V.; Chizhevsky, I.T. The first 8-vertex monocarbon metallacarborane with an “isolated” boron-capped pentagonal bipyramidal cage. Synthesis and molecular structure of capped closo-2,2-(Ph3P)2-2-H-3,6,8-(MeO)3-RuCB6H4. Inorg.Chem. 2001, 40, 5318–5319. [Google Scholar] [CrossRef] [PubMed]
- Perekalin, D.S.; Kudinov, A.R. Cyclopentadienyl ruthenium complexes with naphthalene and other polycyclic aromatic ligands. Coord. Chem. Rev. 2014, 276, 153–173. [Google Scholar] [CrossRef]
- Stephenson, T.A.; Wilkinson, G.J. New complexes of ruthenium (II) and (III) with triphenylphosphine, triphenylarsine, trichlorostannate, pyridine and other ligands. J. Inorg. Nucl. Chem. 1966, 28, 945–956. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
Scheme 1.
Reaction of RuCl2(PPh3)3 (1) with tetraethylammonium 6-phenyl-nido-6-carbadecaborate (2).

Figure 1.
Molecular structure of the complex 4a (only the hydrogen atoms of the carborane and η6-phenyl moieties are shown; thermal ellipsoids are drawn at the 30% probability level). Selected interatomic distances (Å) for two independent molecules A/B are: Ru(1)···Ru(2) 2.9261(6)/2.9101(6), Ru(1)-Cl(1) 2.418(1)/2.430(1), Ru(1)-P(1) 2.224(1)/2.241(1), Ru(1)-P(2) 2.273(1)/2.279(1), Ru(1)…B(9) 2.234(4)/2.242(4), Ru(1)…B(5) 2.372(4)/2.356(4), Ru(1)-H(5) 2.00(3)/2.04(3), Ru(1)-H(9) 1.67(4)/1.56(4), Ru(2)-C(1) 2.083(3)/2.101(3), Ru(2)-B(3,5,6,9) 2.231(4)-2.306(4), Ru(2)-C(18-23) 2.221(3)-2.303(4), P(1)-C(18) 1.844(4)/1.837(4), C(1)-C(12) 1.484(5)/1.498(5).
Figure 1.
Molecular structure of the complex 4a (only the hydrogen atoms of the carborane and η6-phenyl moieties are shown; thermal ellipsoids are drawn at the 30% probability level). Selected interatomic distances (Å) for two independent molecules A/B are: Ru(1)···Ru(2) 2.9261(6)/2.9101(6), Ru(1)-Cl(1) 2.418(1)/2.430(1), Ru(1)-P(1) 2.224(1)/2.241(1), Ru(1)-P(2) 2.273(1)/2.279(1), Ru(1)…B(9) 2.234(4)/2.242(4), Ru(1)…B(5) 2.372(4)/2.356(4), Ru(1)-H(5) 2.00(3)/2.04(3), Ru(1)-H(9) 1.67(4)/1.56(4), Ru(2)-C(1) 2.083(3)/2.101(3), Ru(2)-B(3,5,6,9) 2.231(4)-2.306(4), Ru(2)-C(18-23) 2.221(3)-2.303(4), P(1)-C(18) 1.844(4)/1.837(4), C(1)-C(12) 1.484(5)/1.498(5).
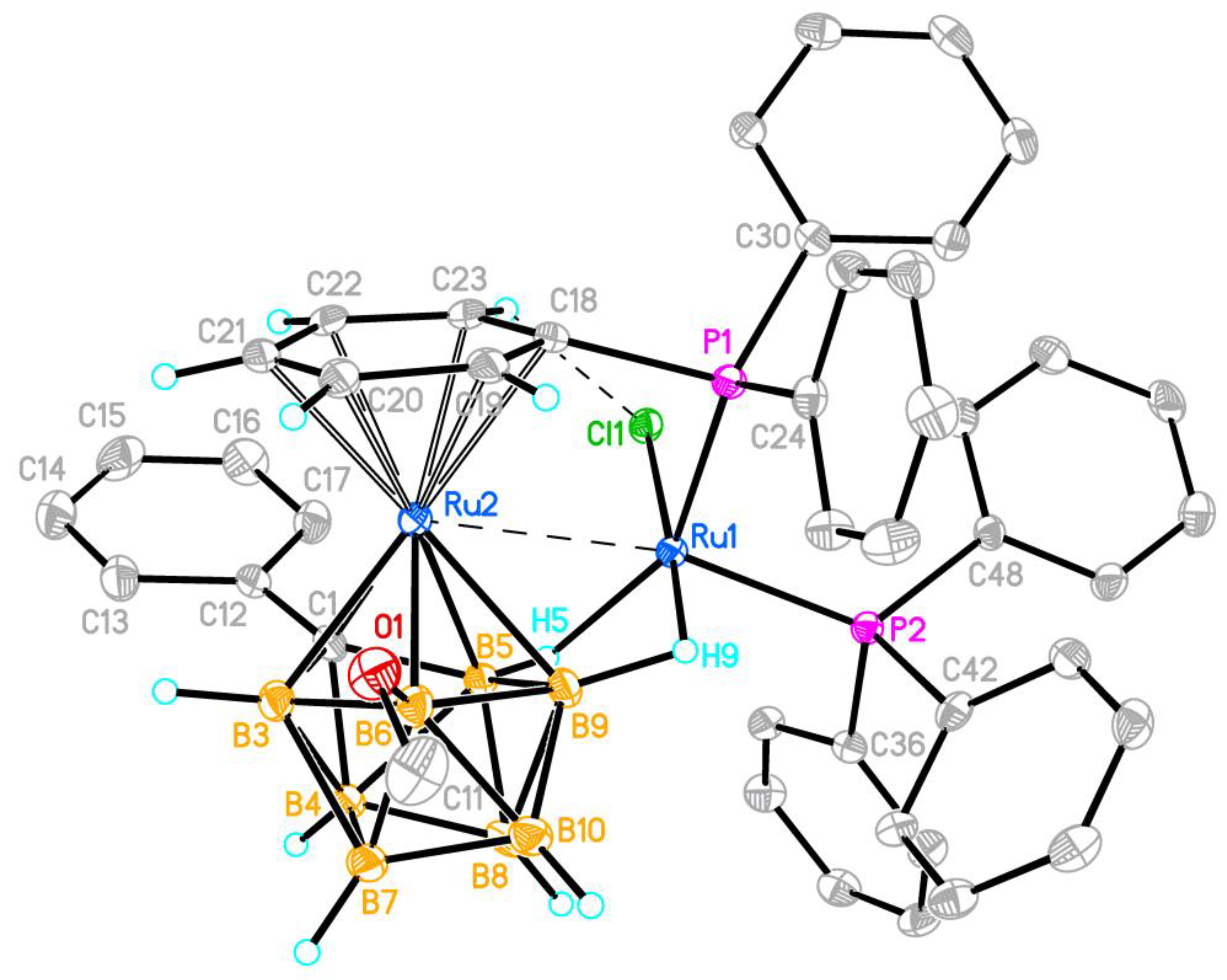
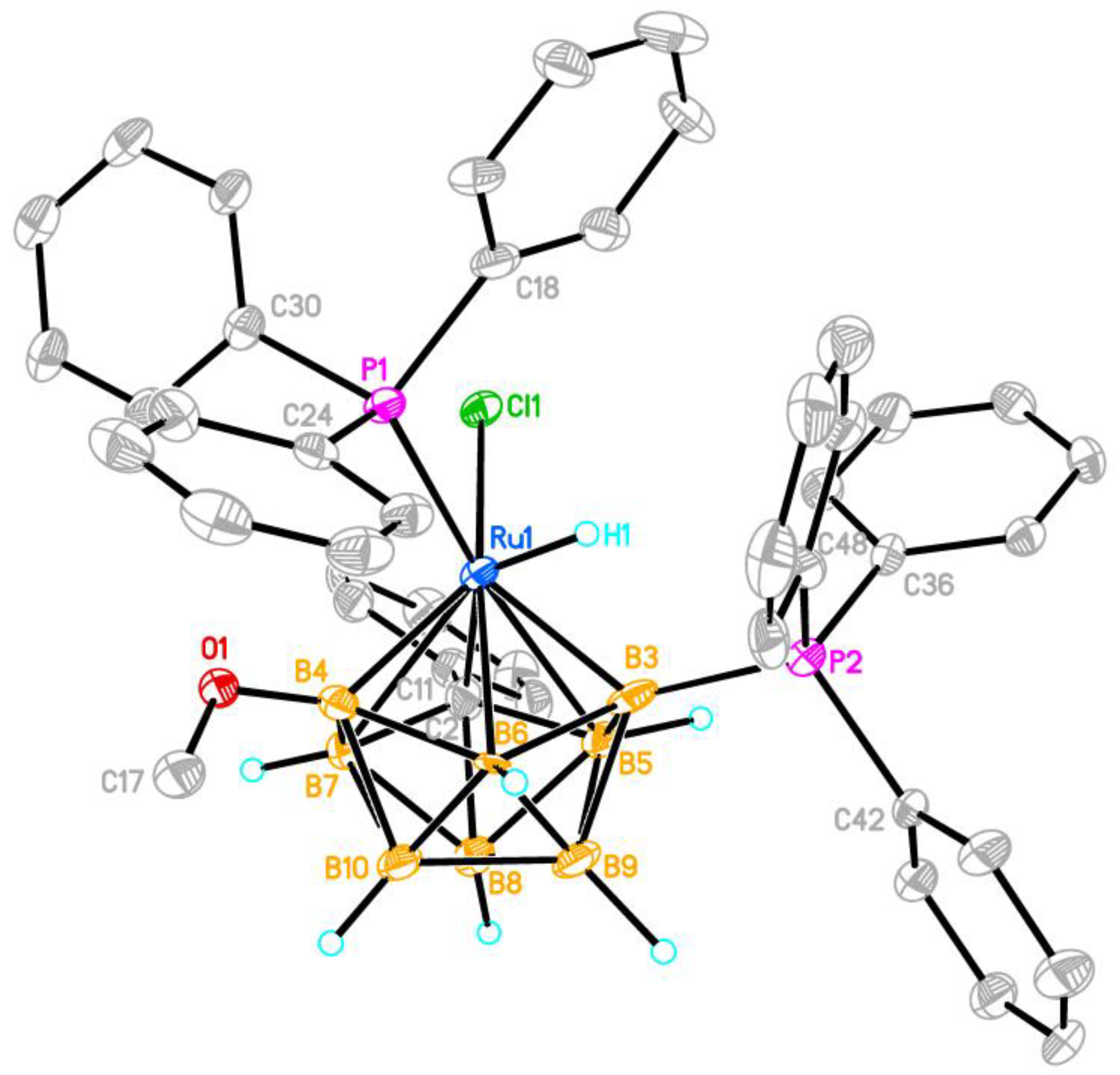
Figure 2.
Molecular structure of the complex 5a (only the hydrogen atoms of the hydride ligand and carborane moiety are shown; thermal ellipsoids are drawn at the 30% probability level). Selected interatomic distances (Å) are: Ru(1)-Cl(1) 2.411(3), Ru(1)-P(1) 2.351(3), Ru(1)-H(1) 1.34, Ru(1)-C(2) 2.181(11), Ru(1)-B(7) 2.516(12), Ru(1)-B(5) 2.449(11), Ru(1)-B(4) 2.095(11), Ru(1)-B(3) 2.072(11), Ru(1)-B(6) 2.364(9), P(2)-B(3) 1.889(13), C(2)-C(11) 1.478(15).
Figure 2.
Molecular structure of the complex 5a (only the hydrogen atoms of the hydride ligand and carborane moiety are shown; thermal ellipsoids are drawn at the 30% probability level). Selected interatomic distances (Å) are: Ru(1)-Cl(1) 2.411(3), Ru(1)-P(1) 2.351(3), Ru(1)-H(1) 1.34, Ru(1)-C(2) 2.181(11), Ru(1)-B(7) 2.516(12), Ru(1)-B(5) 2.449(11), Ru(1)-B(4) 2.095(11), Ru(1)-B(3) 2.072(11), Ru(1)-B(6) 2.364(9), P(2)-B(3) 1.889(13), C(2)-C(11) 1.478(15).
Scheme 2.
Reaction of RuCl2(PPh3)3 (1) with tetraethylammonium arachno-6-carbadecaborate (3).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Selected NMR Data for 7a,b and capped closo-2,2-(PPh3)2-2-H-3,6,8-(OMe)3-2,1-RuCB6H4.
| NMR | 7a | 7b | (PPh3)2HRuCB6H4(OMe)3 [36] | |
|---|---|---|---|---|
| 1H | OMe | 4.24 s (3H); 2.96 s (3H) | 4.27 s (3H); 2.80 s (6H) | 4.17 s (3H); 3.05 s (6H) |
| HRu | −9.84 br t (1H), 2J(H,P) = 24 Hz | −10.43 br t (1H), 2J (H,P) = 24 Hz | −9.02 br t (1H), 2J(H,P) = 20.7 Hz | |
| 31P{1H} | PPh3 | 46.2 d (1P), 2J = 10 Hz; 46.0 d (1P), 2J = 10 Hz | 48.3 s (2P) | 49.4 s (2P) |
| 11B{1H} | B-8 | 67.5 s (1B) | 70.1 s (1B) | 68.9 s (1B) |
| B-3,6 | 33.7 s (1B); 24.1 s (1B) | 32.8 s (2B) | 33.7 s (2B) | |
| B-7 | 14.0 s (1B) | 14.1 s (1B) | 12.8 s (1B) | |
| B-4,5 | −26.3 (1B); −30.1 (1B) | −32.4 s (2B) | −31.8 s (2B) |
Table 2.
Crystal data, data collection and structure refinement parameters for 4a and 5a.
| Compound | 4a | 5a |
|---|---|---|
| CCDC No. | 2201207 | 2201208 |
| Empirical formula | C44H45B8ClOP2Ru2 | C44H45B8ClOP2Ru · 2(CH2Cl2) |
| Molecular weight | 975.81 | 1044.59 |
| Temperature (K) | 120(2) | 110(2) |
| Crystal system | triclinic | monoclinic |
| Space group | P21/c | |
| a (Å) | 10.560(2) | 18.467(11) |
| b (Å) | 20.205(4) | 15.515(9) |
| c (Å) | 22.352(4) | 18.611(11) |
| α (deg) | 114.814(4) | 90 |
| β (deg) | 101.570(4) | 100.170(11) |
| γ (deg) | 90.753(4) | 90 |
| V (Å3) | 4215.4(14) | 5249(5) |
| Z | 4 | 4 |
| Dcalc (g cm−3) | 1.538 | 1.322 |
| linear absorption μ (cm−1) | 8.92 | 6.47 |
| Tmin/Tmax | 0.659/0.862 | 0.560/0.928 |
| 2θmax (deg) | 58 | 52 |
| Reflections collected | 54553 | 45233 |
| Independent reflections (Rint) | 22374 (0.0329) | 10275 (0.1263) |
| Observed reflections (I > 2σ(I)) | 15232 | 5005 |
| Number of parameters | 1102 | 602 |
| R1 (on F for I > 2σ(I)) a | 0.0462 | 0.0987 |
| wR2 (on F2 for all data) b | 0.1000 | 0.2767 |
| GOOF | 1.075 | 1.009 |
| Largest diff. peak/hole (e Å−3) | 1.792/−0.643 | 2.931/−1.039 |
a R1 = Σ ||Fo| − |Fc||/Σ|Fo|, b wR2 = {Σ[w(Fo2 – Fc2)2]/Σw(Fo2)2}1/2.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Loginov, D.A.; Dolgushin, F.M.; Konoplev, V.E.; Tachaev, M.V. New Aspects of Ruthenium-Mediated Polyhedral Contraction of Monocarbollides. Inorganics 2022, 10, 158. https://doi.org/10.3390/inorganics10100158
AMA Style
Loginov DA, Dolgushin FM, Konoplev VE, Tachaev MV. New Aspects of Ruthenium-Mediated Polyhedral Contraction of Monocarbollides. Inorganics. 2022; 10(10):158. https://doi.org/10.3390/inorganics10100158
Chicago/Turabian StyleLoginov, Dmitry A., Fedor M. Dolgushin, Vitalii E. Konoplev, and Maxim V. Tachaev. 2022. "New Aspects of Ruthenium-Mediated Polyhedral Contraction of Monocarbollides" Inorganics 10, no. 10: 158. https://doi.org/10.3390/inorganics10100158
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.