
Nucleophilic Substitution at a Coordinatively Saturated Five-Membered NHC∙Haloborane Centre


1
Inorganic Chemistry and Catalysis Division, CSIR-National Chemical Laboratory, Dr. Homi Bhabha Road, Pashan, Pune 411008, India
2
Academy of Scientific and Innovative Research (AcSIR), Ghaziabad 201002, India
3
Analytical and Environmental Sciences Division and Centralized Instrumentation Facility, CSIR-Central Salt and Marine Chemicals Research Institute, Gijubhai Badheka Marg, Bhavnagar 364002, India
*
Author to whom correspondence should be addressed.
Inorganics 2022, 10(7), 97; https://doi.org/10.3390/inorganics10070097
Submission received: 21 June 2022
/
Revised: 1 July 2022
/
Accepted: 4 July 2022
/
Published: 7 July 2022
(This article belongs to the Special Issue Fifth Element: The Current State of Boron Chemistry)
Abstract
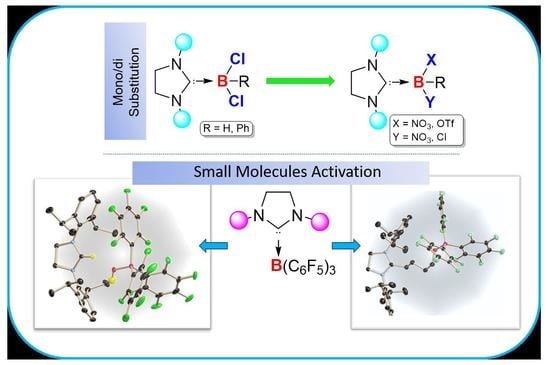
:In this paper, we have used a saturated five-membered N-Heterocyclic carbene (5SIDipp = 1,3-bis-(2,6-diisopropylphenyl)imidazolin-2-ylidine) for the synthesis of SNHC-haloboranes adducts and their further nucleophilic substitutions to put unusual functional groups at the central boron atom. The reaction of 5-SIDipp with RBCl2 yields Lewis-base adducts, 5-SIDipp·RBCl2 [R = H (1), Ph (2)]. The hydrolysis of 1 gives the NHC stabilized boric acid, 5-SIDipp·B(OH)3 (3), selectively. Replacement of chlorine atoms from 1 and 2 with one equivalent of AgOTf led to the formation of 5-SIDipp·HBCl(OTf) (4) and 5-SIDipp·PhBCl(OTf) (5a), where all the substituents on the boron atoms are different. The addition of two equivalents of AgNO3 to 2 leads to the formation of rare di-nitro substituted 5-SIDipp·BPh(NO3)2 (6). Further, the reaction of 5-SIDipp with B(C6F5)3 in tetrahydrofuran and diethyl ether shows a frustrated Lewis pair type small molecule activated products, 7 and 8.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
While less than two decades ago, the N-heterocyclic carbene-borane adducts were conceived rare and exotic, they are now readily accessible owing to rapid synthetic development and constitute an important class of compounds because they display different chemistry from the most existing classes of boron compounds such as boranes or borates [1]. Since the unveiling of the concept that NHC·boranes undergo nucleophilic substitution at boron by the groups of Fensterbank, Lacôte, Malacria, and Curran, the chemistry of these systems has flourished [2,3,4,5,6]. However, these systems largely rely on imidazole-2-ylidene type carbenes. Braunschweig and coworkers recently broadened the range of Lewis base by demonstrating nucleophilic addition and substitution of cyclic alkyl amino carbene (cAAC) BH3 adduct [7]. In our previous works, we have shown the nucleophilic substitution at 5-SIDipp·MeBCl2 (5-SIDipp = 1,3,-bis-(2,6-diisopropylphenyl)imidazolin-2-ylidine) [8]. Further, we have explored the substitution at 6-SIDipp∙BH3 (6-SIDpp = 1,3-di(2,6-diisopropylphenyl) tetrahydropyrimidine-2-ylidene) center and the introduction of rare functional groups such as -OTf, –NO3 [9]. Although the saturated NHCs are more nucleophilic than its unsaturated analogue, the earlier work on the synthesis of more than two dozen of NHC∙borane and haloborane complexes relied on the unsaturated five-membered NHC [1,2,3,4,5,6,10,11,12,13,14,15,16]. Clearly, the extension of unexplored carbenes in NHC·borane chemistry is desirable, as it may lead to the discovery of a range of interesting new applications. Due to our current interest in 5-SIDipp [17,18,19], we have prepared here haloboranes 5SIDipp·BHCl2, 1 and 5SIDipp·BPhCl2, 2 and studied their substitution reactions with AgOTf, AgNO3, and water. Furthermore, we have shown that the combination 5-SIDipp and B(C6F5)3 led to the activation of THF and diethyl ether via frustrated Lewis pair (FLP) way. Our results are reported herein.
2. Results and Discussion
In our previous work, we have reported the first carbene MeBCl2 adduct via salt metathesis procedure [8]. In this work, we have prepared the 5SIDipp·haloborane adducts and shown their reactivities towards nucleophilic substitution. The addition of BHCl2∙dioxane in the solution of 5SIDipp in n-hexane gives the white precipitation of 5-SIDipp·BHCl2, 1 at room temperature (Scheme 1). The precipitate was further dissolved in toluene and dichloromethane to afford colorless crystals of 1 at −36 °C. The 11B NMR spectrum of 1 displays a resonance at 6.9 ppm as a sharp singlet. The backbone four protons appeared at 4.08 ppm in the 1H NMR spectrum. 1 is characterized by single-crystal X-ray diffraction studies (Figure 1). 1 is crystalized in the monoclinic P21/n space group. The B–C bond length in 1 [1.628(7) Å] is in good accordance with the previously reported 5-SIDipp·MeBCl2 [1.6261(19) Å], but considerably longer compared to that in the 5-SIDipp·BH3 [1.593(4) Å] [8]. The increase in the bond length can be ascribed to the enhancement of steric hindrance at the central boron atom. The average B–Cl distance is 1.86 Å, which matches with the previously reported carbene-haloborane adducts (NHC·BCl3, NHC·BRCl2, and NHC·BR2Cl).
The reaction of 1.1 equivalent of PhBCl2 with 5-SIDipp in n-hexane gives an immediate white precipitate formation of 5-SIDipp∙PhBCl2 (2) (Scheme 1). The 11B NMR spectrum of 2 shows one resonance at 1.8 ppm. 2 crystallizes in the monoclinic P21/n space group (Figure 2). The carbene carbon atom C5 is tri-coordinated and features a trigonal-planar geometry, and the boron atom connected with the C5 atom adopts a tetrahedral geometry. The B−CNHC bond distance is 1.6661(10) Å, which is slightly longer in comparison to that in 1 due to the steric congestion of the phenyl group at the boron center. The B–Cl bonds are 1.8992(8) Å and 1.8698(8) Å, which match with the previously reported B–Cl bond distance [8].
Treatment of 1.05 equivalents of water in a dichloromethane solution of 1 hydrolyzes all the B–H and B–Cl bonds and forms NHC-stabilized boric acid, 5-SIDipp∙B(OH)3, 3 (Scheme 2) exclusively. In our earlier work, we have reported the isolation of 6-SIDipp·B(OH)3 as a minor product from the reaction of Br2/H2O with 6-SIDipp·BH3 [9]. Replacement of the chloride and the hydride groups by hydroxide moieties in 3 is accompanied by an upfield shift in the 11B NMR spectrum (−1.6 ppm) from that of 1. 3 crystallizes in the monoclinic P21/c space group (Figure 3). The central boron-carbon distance is 1.650(5) Å, which is marginally shorter compared to that in 6-SIDipp·B(OH)3. The average B– (OH) bond distances are 1.38 Å.
Further, we added silver triflate to a dichloromethane solution of 1 at −78 °C, which replaced one of the labile chlorine atoms by the triflate group (Scheme 2). In the 11B NMR spectrum of 4, the resonance for the central boron atom appears at −3.4 ppm. The resonance at −76.7 ppm in the 19F NMR is characteristic of the triflate group attached to the central boron atom. Colorless crystals of 4 suitable for X-ray diffraction studies were grown from a saturated toluene solution at 4 °C. The constitution of 4 was authenticated by a single-crystal X-ray study (Figure 4). 4 crystallizes in the monoclinic space group P21/n. The relevant bond length and angles are given in the legend of Figure 4. All the four substituents on the central boron atom are different in 4. The central boron atom (B1) lies slightly below the plane of an imidazolinium ring (torsion angles (deg): C(2)−N(1)−C(1)−B(1) = 174.4(2) and C(3)−N(2)−C(1)−B(1) −166.5(2)) and the B−O bond is not orthogonal to the plane of the imidazolinium ring with the torsion angles N1−C1−B1−O1 = −41.6 (2)° and N2−C1−B1−O1 = 148.4 (2)°. The B−OTf bond length is 1.515(2) Å, which is in good agreement with the B−O bond length in our previously reported 5-SIDipp∙BMeOTfCl (B1−O3 1.503(3) Å) [8].
Treatment of one and two equivalents of AgOTf and AgNO3 with 2 in toluene afforded mono-triflate and di-nitro substituted 5-SIDipp·boranes, respectively. Substitution of one and two chlorine atoms from the tetra coordinated boron atom of 2 resulted in the formation of 5-SIDipp·BPhCl(OTf), 5a, and 5-SIDipp-BPh(ONO2)2, 6 (Scheme 3). The functional groups such as nitrate and triflate are rarely found to bind with the boron atom [5,9,20,21,22,23,24,25,26]. 5a crystallizes in the monoclinic P21/n space group (Figure 5). The boron atom lies on a tetrahedral geometry, which can be confirmed from the bond angles around the boron atoms in 5a (C5–B2–Cl1 100.54(11), C5–B2–O1 106.51(13), and C5–B2–C7 118.55(14)). The B–CNHC bond length in 5a (1.648(2) Å) is in well agreement with that in 2. The B–O and B–Cl bonds in 5a is almost orthogonal to the plane (torsion angle: N1–C5–B2–Cl1 76.96(17)°, N2-C5–B2–Cl1 −89.6(2)° and torsion angle: N1–C5–B2–O1 −170.96(14)°, N2–C5–B2–O1 22.5(2)°, respectively). However, the spectroscopic characterization of 5a becomes complicated because of solvent-mediated slow hydrolysis. The only signal in the 11B NMR spectra of the product, 5b appears at 30.9 ppm as a singlet, which is indicative of a three-coordinated boron center, instead of a four-coordinated boron, as expected in 5a. We regrow the crystals from the NMR tube and realized that there is hydrolysis taking place at the B–Cl bond with adventitious water leading to 5-SIDipp stabilized borenium cation, with a triflate as a counter anion (5b). The constitution of 5b rationalizes the resonance at 30.9 ppm in the 11B NMR spectrum. Although the formation of the 5b clearly can be seen from the molecular structure (Figure S2), but due to low-quality data we refrain from discussing its structural parameters. However, even after repeated attempts, we were unable to stop this hydrolysis and hence, could not characterize 5a spectroscopically. The CF3 group of the triflate moiety in 5b resonates at −78.6 ppm in the 19F NMR, which is slightly different from the resonances of triflates bound to the boron atom (−76.7 ppm in 4) and is characteristic of the free triflate anion.
In the 11B|NMR, 6 shows resonance at 4.2 ppm, shifted slight low-field with respect to that in 2 (1.9 ppm), presumably due to electron-withdrawing nature of the ONO2 moieties. The solid-state structure of 6 also was confirmed by X-ray crystal analysis. 6 crystallizes in the monoclinic P21/c space group (Figure 6) and important structural parameters are given in the legend of Figure 6.
The combination of N-heterocyclic carbene and B(C6F5)3 has been exploited in FLP chemistry [27]. In our previous work, we have demonstrated the adduct formation between 5-SIDipp and B(C6F5)3 [8]. We have prepared the adduct in toluene/n-hexane. When we performed the same reaction in THF or diethyl ether, it led to the activation of those etheral solvents. The THF solution of the 5-SIDipp·B(C6F5)3 was kept for 12 h at room temperature, which afforded the zwitterionic species 7 in quantitative yield as a white solid (Scheme 4). The molecular structure of 7 was additionally established by X-ray diffraction analysis (Figure 7). 7 crystallizes in the triclinic P space group. One of the C–O bonds in the THF molecule is cleaved, and as a result, the THF ring becomes acyclic and inserts between the Lewis pairs. Similar to the case for 5-SIDipp·B(C6F5)3, 7 is not stable in solution at room temperature, so we were unable to satisfactorily characterize it by NMR spectroscopy. The 11B NMR resonance at −2.8 ppm is similar to those established for the tetra-coordinated boron compounds. The C5 atom adopts a trigonal planar geometry, which is confirmed by the sum of the bond angles [N1–C5–N2 112.29(15)°, N1–C5–C6 125.21(15)°, N2–C5-C6 122.50(14)°]. The C5–C6 bond distance is marginally shorter compared to the adjacent C–C bond (C5–C6 1.501(2) Å and C6–C7 1.539(2) Å). The boron atom adopts a tetrahedral geometry. The C–O (1.404(2) Å) and the B–O (1.453(2) Å) bond distances are similar to the other previously reported structures [28]. This reactivity was extended to diethyl ether (DEE), which resulted in the isolation of imidazolinium salt with a borate counter-anion (8). This compound presumably results from activation of the C–O bond of the diethyl ether with concomitant elimination of two ethylene molecules (Scheme 4). A signal at 8.9 ppm in the 1H NMR spectrum confirms the presence of an imidazolinium cation. The 11B NMR displays the characteristic resonance at −4.2 ppm, which can be assigned to a tetrahedral [(HO)B(C6F5)3] anion. X-ray crystallographic analysis later confirmed the structure of 8 (Figure 8).
3. Conclusions
In summary, we have prepared 5-SIDipp∙haloboranes adducts, 5-SIDipp∙HBCl2 (1), and 5-SIDipp∙PhBCl2 (2) and shown the selective nucleophilic substitution at the tetra-coordinated boron center to obtain several boranes with rare functional groups such as –ONO2, –OTf, etc. The treatment of one equivalent of AgOTf with 1 and 2 led to the formation of haloboranes, 5-SIDipp∙BHCl(OTf), 4 and 5-SIDipp∙BPhCl(OTf), 5a, respectively, where all three substituents of the boron atom are different. 5a was found to be unstable and undergoes hydrolysis in the presence of adventitious water to give hydroxyborenium cation, 5b. The treatment of two equivalents of AgNO3 forms a rare di-nitrate substituted NHC-coordinated borane (6). The combination of 5-SIDipp and B(C6F5)3 were shown to affect the C–O bond cleavage differently for THF and diethyl ether. As expected, the 5-SIDipp/B(C6F5)3 combination in THF resulted in ring opening of the THF to produce borate 7. In the case of diethyl ether, the rupture of the C–O bond takes place along with the elimination of two molecules of ethylene, leading to the formation of an imidazolinium cation with tris (pentafluorophenyl) hydroxy borate as the counter anion (8).
4. General Procedures and Instrumentation
All manipulations were carried out in an inert atmosphere of argon using standard Schlenk techniques and in argon filled glove box. The solvents, especially toluene, tetrahydrofuran, dichloromethane, and n-hexane were purified by MBRAUN solvent purification system MB SPS-800. Other chemicals were purchased from Sigma Aldrich and TCI Chemicals and were used without further purification. The starting material, 5-SIDipp, was synthesized by using the literature procedure [29]. 1H, 13C, 11B NMR, and 19F spectra were recorded in CDCl3, using Bruker Avance DPX 400, or a Bruker Avance DPX 500 spectrometer. CDCl3 was dried by distillation over CaH2. Chemical shifts (δ) are given in ppm. NMR spectra were referenced to external SiMe4 (1H and 13C), BF3·OEt2 (11B), CFCl3 (19F) respectively.
1: A slightly excess of BHCl2·dioxane (0.20 mL, 0.58 mmol) was added to a 10 mL hexane solution of 5-SIDipp (0.20 g, 0.48 mmol) at room temperature in a Schlenk flask. Stirring the resulted mixture for 2 h at room temperature resulted in the generation of a white precipitate. Colorless crystals of 1 were isolated after keeping the white powder in the mixture of 1 mL dichloromethane and 2 mL toluene solution at −36 °C. Yield = 0.20 g (90%).
1H NMR (400 MHz, 298 K, CDCl3): δ = 1.29 (d, J = 6.88 Hz, 12 H, CH(CH3)2), 1.40 (d, J = 6.63 Hz, 12 H, CH(CH3)2), 3.17 (sept, J = 6.75 Hz, 4 H, CH(CH3)2), 4.08 (s, 4 H, NCH2CH2N), 7.23 (d, J = 7.75 Hz, 4 H, Ar–H), 7.40 (t, J = 7.75 Hz, 2 H, Ar–H) ppm (Figure S1).
13C{1H} NMR (101 MHz, 298 K, CDCl3): δ = 23.4, 26.0, 28.9, 53.1, 124.4, 129.8, 133.2, 146.1 ppm (Figure S2).
2. A slightly excess of BPhCl2·dioxane (0.090 g, 0.58 mmol) was added to a 10 mL hexane solution of 5-SIDipp (0.20 g, 0.48 mmol) at room temperature in a flask. Stirring the resulted mixture for further 2 h at room temperature accessed a white precipitate. Colorless crystals of 2 were isolated after keeping the white powder in the mixture of 1 mL dichloromethane and 2 mL toluene solution. Yield = 0.24 g (90%).
1H NMR (400 MHz, 298 K, CDCl3): δ = 1.42 (d, J = 6.72 Hz, 12 H, CH(CH3)2), 1.51 (d, J = 6.65 Hz, 12 H, CH(CH3)2), 3.39 (sept, J = 6.75 Hz, 4 H, CH(CH3)2), 4.22 (s, 4 H, NCH2CH2N), 6.95 (d, J = 6.88 Hz, 2 H, ortho–H of B-Ph), 7.25 (d, J = 7.75 Hz, 4 H, Ar–H), 7.26 (bs, 1 H, para-H of B–Ph), 7.44 (t, J = 7.73 Hz, 2 H, Ar–H), 7.49 (t, J = 7.88 Hz, meta–H of B–Ph) ppm (Figure S4).
13C{1H} NMR (101 MHz, 298 K, CDCl3): δ = 23.2, 26.2, 28.9, 53.9, 124.2, 129.6, 133.5, 135.2, 145.7 ppm (Figure S5).
3: 1.05 equivalent of water (0.01 g, 0.54 mmol) was added drop by drop to a 15 mL dichloromethane solution of 1 (0.20 g, 0.53 mmol) at room temperature in a flask. Stirring the resulted mixture for further 2 h at room temperature accessed a clear solution. The reaction mixture was concentrated to 5 mL and kept at 4 °C to obtain the colorless crystals of 3. Yield = 0.36 g (80%).
1H NMR (400 MHz, 298 K, CDCl3):δ = 1.31 (d, J = 6.97 Hz, 12 H, CH(CH3)2), 1.35 (d, J = 6.85 Hz, 12 H, CH(CH3)2), 3.07 (sept, J = 6.85 Hz, 4 H, CH(CH3)2), 4.06 (s, 4 H, NCH2CH2N), 7.23 (d, J = 7.70 Hz, 4 H, Ar–H), 7.39 (t, J = 7.75 Hz, 2 H, Ar–H) ppm (Figure S7).
13C{1H} NMR (101 MHz, 298 K, CDCl3): δ = 23.4, 25.4, 28.9, 53.3, 67.9, 124.2, 129.6, 133.1, 146.0 ppm (Figure S8).
4: A DCM solution (20 mL) of 1 (0.47 g, 1 mmol) was added dropwise to a DCM solution (20 mL) of previously weighed AgOTf (0.25 g, 1 mmol) at −78 °C in the absence of light. A white precipitate of AgCl was formed immediately, and it was filtered through frit filtration after the reaction mixture was warmed to room temperature. The colorless toluene solution was concentrated (5 mL) and kept for crystallization at 4 °C, which afforded colorless crystals of 4 after 1−2 day(s). Yield = 0.48 g (82%).
1H NMR (400 MHz, 298 K, CDCl3): δ = 1.31 (d, J = 6.88 Hz, 12 H, CH(CH3)2), 1.40 (d, J = 6.63 Hz, 12 H, CH(CH3)2), 3.13 (sept, J = 7.63 Hz, 4 H, CH(CH3)2), 4.11 (s, 4 H, NCH2CH2N), 7.20 (d, J = 7.38 Hz, 4 H, Ar–H), 7.42 (t, J = 7.63 Hz, 2 H, Ar–H) ppm (Figure S10).
13C{1H} NMR (101 MHz, 298 K, CDCl3): δ = 21.4, 23.2, 25.9, 26.3, 28.9, 53.4, 124.7, 128.2, 130.2, 132.4, 137.8, 145.7, 145.9 ppm (Figure S11).
5b: A toluene solution (20 mL) of 2 (0.55 g, 1 mmol) was added dropwise to a toluene solution (20 mL) of previously weighed AgOTf (0.25 g, 1 mmol) at −30 °C in the absence of light. A white precipitate of AgCl formed immediately, and it was filtered through frit filtration after the reaction mixture was warmed to room temperature. The colorless toluene solution was concentrated (5 mL) and was kept for crystallization at 4 °C, which afforded colorless crystals of 5b after 1 day. Yield = 0.25 g (45%).
1H NMR (400 MHz, 298 K, CDCl3): δ = 1.24 (d, J = 6.88 Hz, 12 H, CH(CH3)2), 1.38 (d, J = 6.63 Hz, 12 H, CH(CH3)2), 2.99 (sept, J = 6.75 Hz, 4 H, CH(CH3)2), 4.59 (s, 4 H, NCH2CH2N), 7.29 (d, J = 7.75 Hz, 4 H, Ar–H), 7.48 (t, J = 7.75 Hz, 2 H, Ar–H), 7.52 (t, J = 7.50 Hz, 2 H, meta-H of B–Ph), 7.61 (t, J = 7.38 Hz, para–H of B–Ph), 8.25 (d, J = 6.75 Hz, 2 H, ortho–H of B–Ph) ppm (Figure S14).
13C{1H} NMR (101 MHz, 298 K, CDCl3): δ = 21.4, 23.8, 25.1, 29.2, 54.7, 125.0, 127.9, 129.0, 135.6, 137.8, 146.2 ppm (Figure S15).
6: A toluene solution (20 mL) of 2 (0.55 g, 1 mmol) was added dropwise to a toluene solution (20 mL) of previously weighed AgNO3 (0.34 g, 2 mmol) at −30 °C in the absence of light. A white precipitate of AgCl was formed slowly, and it was filtered via frit filtration after 6 h. The colorless toluene solution was concentrated (5 mL) and was kept for crystallization at 4 °C, which afforded colorless crystals of 6 after 2 days. Yield = 0.37 g (62%).
1H NMR (400 MHz, 298 K, CDCl3): δ = 1.12 (d, J = 6.75 Hz, 12 H, CH(CH3)2), 1.20 (d, J = 6.75 Hz, 12 H, CH(CH3)2), 3.13 (sept, J = 6.75 Hz, 4 H, CH(CH3)2), 4.15 (s, 4 H, NCH2CH2N), 6.50 (d, J = 6.88 Hz, 2 H, ortho–H of B–Ph), 6.89 (t, J = 7.38 Hz, 2 H, meta–H of B–Ph), 7.01 (t, J = 7.25 Hz, para−H of B–Ph), 7.24 (d, J = 7.88 Hz, 4 H, Ar–H), 7.46 (t, J = 7.75 Hz, 2 H, Ar–H), ppm (Figures S18 and S19).
13C{1H} NMR (101 MHz, 298 K, CDCl3): δ = 22.4, 26.8, 28.8, 53.8, 124.6, 127.1, 131.6, 134.5, 146.2 ppm (Figure S20).
7: A THF solution (5 mL) of 5-SIDipp (0.382 g, 1 mmol) was added dropwise to a THF solution (20 mL) of previously weighed B(C6F5)3 (0.512 g, 1 mmol) at room temperature. The reaction mixture turned to a clear colorless solution immediately and run for 12 h. The solution was dried completely and 3 mL of toluene solution was added to dissolve the white solid product. Colorless crystals of 7 were afforded after keeping the toluene solution for crystallization at 4 °C after 2 days. Yield = 0.45 g (46%). The formation of 7 was accompanied by some other side products, which could not be identified. Hence, we did not have a spectroscopically pure product to record the 1H and 13C NMR.
8: A diethyl ether solution (5 mL) of 5-SIDipp (0.382 g, 1 mmol) was added dropwise to a diethyl ether solution (5 mL) of previously weighed B(C6F5)3 (0.512 g, 1 mmol) at room temperature. The reaction mixture turned to a clear colorless solution immediately and run for 12 h. The solution was dried completely and 3 mL of toluene solution was added to dissolve the white solid product. Colorless crystals of 8 were afforded after keeping the toluene solution for crystallization at 4 °C after 1 day. Yield = 0.78 g (85%).
1H NMR (400 MHz, 298 K, CDCl3): δ = 1.20 (d, J = 6.88 Hz, 12 H, CH(CH3)2), 1.35 (d, J = 6.88 Hz, 12 H, CH(CH3)2), 2.87 (sept, J = 6.75 Hz, 4 H, CH(CH3)2), 4.39 (s, 4 H, NCH2CH2N), 7.29 (d, J = 7.75 Hz, 4 H, Ar–H), 7.54 (t, J = 7.88 Hz, 2 H, Ar–H), 8.90 (s, 1 H, N–CH–N) ppm (Figure S24).
13C{1H} NMR (101 MHz, 298 K, CDCl3): δ = 23.9 24.4, 29.4, 53.6, 60.8 125.2, 128.6, 131.9, 145.6, 160.3 ppm (Figure S25).
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/inorganics10070097/s1. The Supplementary Materials contains structural description of 1–8 and representative NMR spectra. The callouts of each Figure given in the Supporting Information are provided in the main text. (accessed on 19 June). References [30,31,32,33,34] are cited in the Supplementary Materials.
Author Contributions
S.S.S. conceptualized the work, G.K. performed the experiments and characterizations. S.T. performed the single crystal X-ray analysis. S.S.S. and G.K. cowrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
The research was funded by CSIR, Young Scientist Contingency Grant (YSA000726).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All the data reported in the study can be found in the supporting information and from the CCDC repository with accession numbers: 2180189-2180191, 2180197-2180200, 2180457.
Acknowledgments
GK thanks CSIR, India for senior research fellowship (SRF). ST is grateful to AESD&CIF, CSIR-CSMCRI for instrumentation facilities and infrastructure.
Conflicts of Interest
The authors declare no conflict of interests.
References
- Curran, D.P.; Solovyev, A.; MakhloufBrahmi, M.; Fensterbank, L.; Malacria, M.; Lacôte, E. Synthesis and Reactions of N-Heterocyclic Carbene Boranes. Angew. Chem. Int. Ed. 2011, 50, 10294–10317. [Google Scholar] [CrossRef] [PubMed]
- Ueng, S.-H.; MakhloufBrahmi, M.; Derat, É.; Fensterbank, L.; Lacôte, E.; Malacria, M.; Curran, D.P. Complexes of Borane and N-Heterocyclic Carbenes: A New Class of Radical Hydrogen Atom Donor. J. Am. Chem. Soc. 2008, 130, 10082–10083. [Google Scholar] [CrossRef] [PubMed]
- Ueng, S.-H.; Fensterbank, L.; Lacôte, E.; Malacria, M.; Curran, D.P. Radical Deoxygenation of Xanthates and Related Functional Groups with New Minimalist N-Heterocyclic Carbene Boranes. Org. Lett. 2010, 12, 3002–3005. [Google Scholar] [CrossRef] [PubMed]
- MakhloufBrahmi, M.; Monot, J.; Desage-El Murr, M.; Curran, D.P.; Fensterbank, L.; Lacote, E.; Malacria, M. Preparation of NHC Borane Complexes by Lewis Base Exchange with Amine− and Phosphine−Boranes. J. Org. Chem. 2010, 75, 6983–6985. [Google Scholar]
- Solovyev, A.; Chu, Q.; Geib, S.J.; Fensterbank, L.; Malacria, M.; Lacôte, E.; Curran, D.P. Substitution Reactions at Tetracoordinate Boron: Synthesis of N-Heterocyclic Carbene Boranes with Boron-Heteroatom Bonds. J. Am. Chem. Soc. 2010, 132, 15072–15080. [Google Scholar] [CrossRef]
- Dai, W.; Geib, S.J.; Curran, D.P. Reactions of N-Heterocyclic Carbene Boranes with 5-Diazo-2,2-dimethyl-1,3-dioxane-4,6-dione: Synthesis of Mono- and Bis-hydrazonyl NHC-Boranes. J. Org. Chem. 2018, 83, 8775–8779. [Google Scholar] [CrossRef]
- Auerhammer, D.; Arrowsmith, M.; Braunschweig, H.; Dewhurst, R.D.; Jiménez-Halla, J.O.C.; Kupfer, T. Nucleophilic Addition and Substitution at Coordinatively Saturated Boron by Facile 1,2-Hydrogen Shuttling onto a Carbene Donor. Chem. Sci. 2017, 8, 7066–7071. [Google Scholar] [CrossRef] [Green Version]
- Kundu, G.; Pahar, S.; Tothadi, S.; Sen, S.S. Stepwise Nucleophilic Substitution to Access Saturated N-heterocyclic Carbene Halo-boranes with Boron−Methyl Bonds. Organometallics 2020, 39, 4696–4703. [Google Scholar] [CrossRef]
- Kundu, G.; Ajithkumar, V.S.; Raj, K.V.; Vanka, K.; Tothadi, S.; Sen, S.S. Substitution at sp3 Boron of a Six-Membered NHC·BH3: Convenient Access to a Dihydroxyborenium Cation. Chem. Commun. 2022, 58, 3783–3786. [Google Scholar] [CrossRef]
- Bissinger, P.; Braunschweig, H.; Kraft, K.; Kupfer, T. Trapping the Elusive Parent Borylene. Angew. Chem. Int. Ed. 2011, 50, 4704–4707. [Google Scholar] [CrossRef]
- Bissinger, P.; Braunschweig, H.; Damme, A.; Dewhurst, R.D.; Kraft, K.; Kramer, T.; Radacki, K. Base-stabilized boryl and cationic haloborylene complexes of iron. Chem.-Eur. J. 2013, 19, 13402–13407. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Quillian, B.; Wei, P.; Wannere, C.S.; Xie, Y.; King, R.B.; Schaefer, H.F., III; Schleyer, P.V.R.; Robinson, G.H. A Stable Neutral Diborene Containing a B=B Double Bond. J. Am. Chem. Soc. 2007, 129, 12412–12413. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Quillian, B.; Wei, P.; Xie, Y.; Wannere, C.S.; King, R.B.; Schaefer, H.F.; Schleyer, P.v.R.; Robinson, G.H. Planar, Twisted, and Trans-Bent: Conformational Flexibility of Neutral Diborenes. J. Am. Chem. Soc. 2008, 130, 3298–3299. [Google Scholar] [CrossRef] [PubMed]
- Tamm, M.; Lügger, T.; Hahn, E.F. Isocyanide and Ylidene Complexes of Boron: Synthesis and Crystal Structures of (2- (Trimethylsiloxy)phenylisocyanide)−Triphenylborane and (1,2-Dihydrobenzoxazol-2-ylidene)−Triphenylborane. Organometallics 1996, 15, 1251–1256. [Google Scholar] [CrossRef]
- Chase, P.A.; Stephan, D.W. Hydrogen and Amine Activation by a Frustrated Lewis Pair of a Bulky N-Heterocyclic Carbene and B(C6F5)3. Angew. Chem. Int. Ed. 2008, 47, 7433–7437. [Google Scholar] [CrossRef]
- Doddi, A.; Peters, M.; Tamm, M. N-Heterocyclic Carbene Adducts of Main Group Elements and Their Use as Ligands in Transition Metal Chemistry. Chem. Rev. 2019, 119, 6994–7112. [Google Scholar] [CrossRef] [PubMed]
- Pait, M.; Kundu, G.; Tothadi, S.; Karak, S.; Jain, S.; Vanka, K.; Sen, S.S. C–F Bond Activation by a Saturated N-Heterocyclic Carbene: Mesoionic Compound Formation and Adduct Formation with B(C6F5)3. Angew. Chem. Int. Ed. 2019, 58, 2804–2808. [Google Scholar] [CrossRef]
- Kundu, G.; De, S.; Tothadi, S.; Das, A.; Koley, D.; Sen, S.S. Saturated N-Heterocyclic Carbene Based Thiele’s Hydrocarbon with a Tetrafluorophenylene Linker. Chem. Eur. J. 2019, 25, 16533–16537. [Google Scholar] [CrossRef]
- Kundu, G.; Ajithkumar, V.S.; Bisai, M.K.; Tothadi, S.; Das, T.; Vanka, K.; Sen, S.S. Diverse reactivity of carbenes and silylenes towards fluoropyridines. Chem. Commun. 2021, 57, 4428–4431. [Google Scholar] [CrossRef]
- Guibert, C.R.; Marshall, M.D. Synthesis of the Tetranitratoborate Anion. J. Am. Chem. Soc. 1966, 88, 189–190. [Google Scholar] [CrossRef]
- Titova, K.V.; Rosolovskii, V.Y. Tetraalkylammonium Nitratoborates. Bull. Acad. Sci. USSR Div. Chem. Sci. 1970, 19, 2515–2519. [Google Scholar] [CrossRef]
- Titova, K.V.; Rosolovskii, V.Y. Reaction of Nitrates of Monovalent Cations with BCl3. Bull. Acad. Sci. USSR Div. Chem. Sci. 1975, 24, 2246–2248. [Google Scholar] [CrossRef]
- Becker, M.; Schulz, A.; Villinger, A.; Voss, K. Stable Sulfate and Nitrate Borane-Adduct Anions. RSC Adv. 2011, 1, 128–134. [Google Scholar] [CrossRef]
- Zelenov, V.P.; Gorshkov, E.Y.; Zavaruev, M.V.; Dmitrienko, A.O.; Troyan, I.A.; Pivkina, A.N.; Khakimov, D.V.; Pavlikov, A.V. Synthesis and Mutual Transformations of Nitronium tetrakis(nitrooxy)- and tetrakis(2,2,2-trifluoroacetoxy)borates. N. J. Chem. 2020, 44, 13944–13951. [Google Scholar] [CrossRef]
- Hawthorne, M.F.; Mavunkal, I.J.; Knobler, C.B. Electrophilic Reactions of Protonated Closo-B10H102- with Arenes, Alkane Carbon-Hydrogen Bonds, and Triflate Ion Forming Aryl, Alkyl, and Triflate Nido-6-X-B10H13 Derivatives. J. Am. Chem. Soc. 1992, 114, 4427–4429. [Google Scholar] [CrossRef]
- Berkeley, E.R.; Ewing, W.C.; Carroll, P.J.; Sneddon, L.G. Synthesis, Structural Characterization, and Reactivity Studies of 5-CF3SO3-B10H13. Inorg. Chem. 2014, 53, 5348–5358. [Google Scholar] [CrossRef]
- Erker, G. Frustrated Lewis Pairs: Metal-free Hydrogen Activation and More. Angew. Chem. Int. Ed. 2010, 49, 46–76. [Google Scholar]
- Kronig, S.; Theuergarten, E.; Holschumacher, D.; Bannenberg, T.; Daniliuc, C.G.; Jones, P.G.; Tamm, M. Dihydrogen Activation by Frustrated Carbene-Borane Lewis Pairs: An Experimental and Theoretical Study of Carbene Variation. Inorg. Chem. 2011, 50, 7344–7359. [Google Scholar] [CrossRef]
- Filippou, A.C.; Chernov, O.; Schakenburg, G. Chromium Silicon Multiple Bonds: The Chemistry of Terminal N-Heterocyclic Carbene-Stabilized Halosilylidyne Ligands. Chem.-Eur. J. 2011, 17, 13574–13583. [Google Scholar] [CrossRef]
- APEX3, SAINT-Plus and SADABS; Bruker AXS Inc.: Madison, WI, USA, 2006.
- Apex CCD and SAINT v8.30C; Bruker AXS Inc.: Madison, WI, USA, 2013.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krause, L.; Herbst-Irmer, R.; Stalke, D. An empirical correction for the influence of low-energy contamination. J. Appl. Crystallogr. 2015, 48, 1907–1913. [Google Scholar] [CrossRef]
Scheme 1.
Synthesis of 5-SIDipp·RBCl2 adducts 1 and 2.

Figure 1.
The molecular structure of 1 (hydrogen atoms except at the boron atom in 1 are omitted for clarity). Selected bond lengths [Å] or angles [deg]: C5–N1 1.337(5), C5–N2 1.329(6), C25–N1 1.491(6), C26–N2 1.477(6), C5–B1 1.628(7), B1–Cl1 1.865(5), B1–Cl2 1.869(5); N1–C5–N2 109.2(4), N1–C5–B1 128.9(4), N2–C5–B1 121.9(4), Cl1–B1–Cl2 110.1(3).
Figure 1.
The molecular structure of 1 (hydrogen atoms except at the boron atom in 1 are omitted for clarity). Selected bond lengths [Å] or angles [deg]: C5–N1 1.337(5), C5–N2 1.329(6), C25–N1 1.491(6), C26–N2 1.477(6), C5–B1 1.628(7), B1–Cl1 1.865(5), B1–Cl2 1.869(5); N1–C5–N2 109.2(4), N1–C5–B1 128.9(4), N2–C5–B1 121.9(4), Cl1–B1–Cl2 110.1(3).

Figure 2.
The molecular structure of 2. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] or angles [deg]: C5–N1 1.3403(8), C5–N2 1.3443(8), C19–N1 1.4810(9), C18–N2 1.4756(9), C5–B20 1.6661(10), B20–C21 1.6204(10), B20–Cl1 1.8992(8), B20–Cl2 1.8698(8); N1–C5–N2 108.63(6), N1–C5–B20 127.41(6), N2–C5–B20 123.20(6), C5–B20–Cl1 100.88(4), C5–B20–C21 110.99(5), Cl1–B20–Cl2 106.42(4).
Figure 2.
The molecular structure of 2. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] or angles [deg]: C5–N1 1.3403(8), C5–N2 1.3443(8), C19–N1 1.4810(9), C18–N2 1.4756(9), C5–B20 1.6661(10), B20–C21 1.6204(10), B20–Cl1 1.8992(8), B20–Cl2 1.8698(8); N1–C5–N2 108.63(6), N1–C5–B20 127.41(6), N2–C5–B20 123.20(6), C5–B20–Cl1 100.88(4), C5–B20–C21 110.99(5), Cl1–B20–Cl2 106.42(4).

Scheme 2.
Reactivity of 1 with H2O and AgOTf.

Figure 3.
The molecular structure of 3. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] or angles [deg]: N1–C1 1.316(4), N2–C1 1.337(4), C2–N1 1.471(4), C3–N2 1.478(4), C1–B1 1.650(5), B1–O1 1.375(4), B1–O2 1.386(4); N1–C1–N2 113.2(3), N1–C1–B1 125.4(3), N2–C1–B1 125.0(3), C1–B1–O1 110.4(3), C1–B1–O2 108.5(3), O1–B1–O2 111.1(3).
Figure 3.
The molecular structure of 3. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] or angles [deg]: N1–C1 1.316(4), N2–C1 1.337(4), C2–N1 1.471(4), C3–N2 1.478(4), C1–B1 1.650(5), B1–O1 1.375(4), B1–O2 1.386(4); N1–C1–N2 113.2(3), N1–C1–B1 125.4(3), N2–C1–B1 125.0(3), C1–B1–O1 110.4(3), C1–B1–O2 108.5(3), O1–B1–O2 111.1(3).

Figure 4.
The molecular structure of 4. Hydrogen atom expect H1 in 4 are omitted for clarity. Selected bond lengths [Å] or angles [deg]: N1–C1 1.316(4), N2–C1 1.337(4), C2–N1 1.471(4), C3–N2 1.478(4), C1–B1 1.650(5), B1–O1 1.375(4); N1–C1–N2 113.2(3), N1–C1–B1 125.4(3), N2–C1–B1 125.0(3), C1–B1–O1 110.4(3).
Figure 4.
The molecular structure of 4. Hydrogen atom expect H1 in 4 are omitted for clarity. Selected bond lengths [Å] or angles [deg]: N1–C1 1.316(4), N2–C1 1.337(4), C2–N1 1.471(4), C3–N2 1.478(4), C1–B1 1.650(5), B1–O1 1.375(4); N1–C1–N2 113.2(3), N1–C1–B1 125.4(3), N2–C1–B1 125.0(3), C1–B1–O1 110.4(3).

Scheme 3.
Nucleophilic substitution of 2 with AgOTf and AgNO3.

Figure 5.
The molecular structure of 5a. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] or angles [deg]: N1–C5 1.341(2), N2–C5 1.330(2), C23–N1 1.485(2), C24–N2 1.482(2), C5–B2 1.648(2), B2–Cl1 1.8948(19), B2–O1 1.532(2); N1–C5–N2 109.30(14), N1–C5–B2 123.00(14), N2–C5–B2 126.57(14), C5–B2–Cl1 100.54(11), C5–B2–O1 106.51(13), C5–B2–C7 118.55(14).
Figure 5.
The molecular structure of 5a. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] or angles [deg]: N1–C5 1.341(2), N2–C5 1.330(2), C23–N1 1.485(2), C24–N2 1.482(2), C5–B2 1.648(2), B2–Cl1 1.8948(19), B2–O1 1.532(2); N1–C5–N2 109.30(14), N1–C5–B2 123.00(14), N2–C5–B2 126.57(14), C5–B2–Cl1 100.54(11), C5–B2–O1 106.51(13), C5–B2–C7 118.55(14).

Figure 6.
The molecular structure of 6. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] or angles [deg]: N1−C1 1.337(3), C1−N2 1.340(3), N1−C2 1.480(3), N2–C3 1.484(3), C1–B1 1.662(4), B1–O5 1.535(4), B1–O6 1.522(3); N1–C1–N2 112.4(2), C1–N1–C2 112.4(2), C1–N2–C3 111.8(2), C1–B1–O5 101.59(19), C1–B1–O6 109.7(2), O5–B1–O6 111.3(2), C1–B1–C28 118.4(2).
Figure 6.
The molecular structure of 6. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] or angles [deg]: N1−C1 1.337(3), C1−N2 1.340(3), N1−C2 1.480(3), N2–C3 1.484(3), C1–B1 1.662(4), B1–O5 1.535(4), B1–O6 1.522(3); N1–C1–N2 112.4(2), C1–N1–C2 112.4(2), C1–N2–C3 111.8(2), C1–B1–O5 101.59(19), C1–B1–O6 109.7(2), O5–B1–O6 111.3(2), C1–B1–C28 118.4(2).

Scheme 4.
5-SIDipp∙B(C6F5)3 Lewis pair mediated tetrahydrofuran and diethyl ether activation at room temperature.
Scheme 4.
5-SIDipp∙B(C6F5)3 Lewis pair mediated tetrahydrofuran and diethyl ether activation at room temperature.

Figure 7.
The molecular structure of 7. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] or angles [deg]: N2–C5 1.322(2), N1–C5 1.327(2), N1–C22 1.479(2), N2–C21 1.484(2), C5–C6 1.501(2), C6–C7 1.539(2), C9–O1 1.404(2), O1–B10 1.453(2); N1–C5–N2 112.29(15), N1–C5–C6 125.21(15), N2–C5–C6 122.50(14), C9–O1–B10 117.96(13).
Figure 7.
The molecular structure of 7. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] or angles [deg]: N2–C5 1.322(2), N1–C5 1.327(2), N1–C22 1.479(2), N2–C21 1.484(2), C5–C6 1.501(2), C6–C7 1.539(2), C9–O1 1.404(2), O1–B10 1.453(2); N1–C5–N2 112.29(15), N1–C5–C6 125.21(15), N2–C5–C6 122.50(14), C9–O1–B10 117.96(13).

Figure 8.
The molecular structure of 8. Hydrogen atoms except the H5 and H7 are omitted for clarity. Selected bond lengths [Å] or angles [deg]: N1–C5 1.3156(11), N2–C5 1.3174(12), C5–H5 0.950, N1–C27 1.4810(13), N2–C26 1.4817(12), B1–O7 1.4696(12), O7–H1 0.83(2); N1–C5–N2 112.94(8), C27–N1–C5 110.17(8), C26–N2–C5 109.91(8), B1–O7–H7 114.3(15).
Figure 8.
The molecular structure of 8. Hydrogen atoms except the H5 and H7 are omitted for clarity. Selected bond lengths [Å] or angles [deg]: N1–C5 1.3156(11), N2–C5 1.3174(12), C5–H5 0.950, N1–C27 1.4810(13), N2–C26 1.4817(12), B1–O7 1.4696(12), O7–H1 0.83(2); N1–C5–N2 112.94(8), C27–N1–C5 110.17(8), C26–N2–C5 109.91(8), B1–O7–H7 114.3(15).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kundu, G.; Tothadi, S.; Sen, S.S. Nucleophilic Substitution at a Coordinatively Saturated Five-Membered NHC∙Haloborane Centre. Inorganics 2022, 10, 97. https://doi.org/10.3390/inorganics10070097
AMA Style
Kundu G, Tothadi S, Sen SS. Nucleophilic Substitution at a Coordinatively Saturated Five-Membered NHC∙Haloborane Centre. Inorganics. 2022; 10(7):97. https://doi.org/10.3390/inorganics10070097
Chicago/Turabian StyleKundu, Gargi, Srinu Tothadi, and Sakya S. Sen. 2022. "Nucleophilic Substitution at a Coordinatively Saturated Five-Membered NHC∙Haloborane Centre" Inorganics 10, no. 7: 97. https://doi.org/10.3390/inorganics10070097
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.