
Copper Complexes of Silicon Pyridine-2-olates RSi(pyO)3 (R = Me, Ph, Bn, Allyl) and Ph2Si(pyO)2

1
Institute of Inorganic Chemistry, TU Bergakademie Freiberg, D-09596 Freiberg, Germany
2
Institute of Resource Ecology, Helmholtz-Zentrum Dresden-Rossendorf eV, D-01328 Dresden, Germany
3
Institute of Analytical Chemistry, TU Bergakademie Freiberg, D-09596 Freiberg, Germany
*
Author to whom correspondence should be addressed.
Inorganics 2023, 11(1), 2; https://doi.org/10.3390/inorganics11010002
Submission received: 6 December 2022
/
Revised: 15 December 2022
/
Accepted: 16 December 2022
/
Published: 21 December 2022
(This article belongs to the Special Issue Metal Complexes with N-donor Ligands)
Abstract
:The organosilicon pyridine-2-olates 1a–1d (RSi(pyO)3, R = Me (a), Ph (b), Bn (c), Allyl (d); pyO = pyridine-2-olate) may serve as tripodal ligands toward CuCl with formation of complexes of the type RSi(μ2-pyO)3CuCl (2a–2d). In addition, for R = Allyl, formation of the more stable isomer 2d′ (κO-pyO)Si(μ2-pyO)2(μ2-Allyl)CuCl was observed. In the presence of dry air (as a source of oxygen), reactions of 1a–1d and CuCl afforded Cu(II) complexes RSi(μ2-pyO)4CuCl (3a–3d); 3a–3c in good yield, and 3d only as a side product. Reaction of Ph2Si(pyO)2 (4) and CuCl in equimolar ratio afforded, depending on reaction conditions, a series of (CuCl)n-ladder-type oligonuclear Cu(I) complexes Ph2Si(μ2-pyO)2(CuCl)n(μ2-pyO)2SiPh2 (n = 2 (52), 3 (53), 4 (54)). In all of the above compounds, the pyO group is Si–O bound and, in the case of μ2 coordination, Cu–N bound. All new compounds (1c, 1d, 2b, 2c, 2d, 2d′, 3b, 3c, 3d, 52, 53, 54) were characterized by single-crystal X-ray diffraction, and further characterization includes solution 1H, 13C, 29Si NMR spectroscopy (1c, 1d, 2b, 2c, 2d’, 53, 54), solid-state 29Si (2b, 2c, 2d′, 53, 54) and 63Cu NMR spectroscopy (2c, 2d′) as well as computational analyses of the isomerization of the couple 2d, 2d′.
1. Introduction
The facets of silicon transition metal (TM) coordination chemistry span a wide range of research fields because of the variety of binding modes encountered. Figure 1 gives some illustrative examples. Low-valent silicon species such as silylenes (in I [1]), intramolecularly donor-stabilized silylenes (in II [2]) and others (e.g., in III [3]) with their Si-located lone pair may act as lone pair donor ligands in the TM coordination sphere. In terms of agostic interaction, Si–Si bonds of tetravalent silicon compounds (such as disilanes, compound IV) may also serve as electron pair donors toward d-block elements [4]. Tetravalent silicon species may give rise to a wide range of silyl TM complexes, in which the Si atom has coordination number 4 (e.g., compounds V [5] and VI [6]). Bridging coordination of silyl anions with two or more TM atoms (e.g., in VII [5], VIII [7], IX [8], X [9]) is one way of achieving higher Si coordination numbers. Furthermore, the Lewis acidic Si site in some tetravalent silicon compounds may serve as a lone pair acceptor toward electron rich TM sites, thus furnishing higher-coordinate tetravalent Si compounds (e.g., Si coordination numbers 5 and 6) with formally dative TM→Si bond in the Si coordination sphere. Hence, silicon in Si–TM-compounds covers the full range of L-, X-, Z-type ligand characteristics [10].
Whereas higher-coordinate silicon compounds have fascinated chemists for many decades [11,12,13], TM→Si coordination chemistry represents a special and rather young topic of the coordination chemistry of higher-coordinate silicon compounds. Figure 2 illustrates examples of this class of compounds. The utilization of buttressing ligands was key to support weak attractive d10-TM→Si interactions and allowed Grobe et al. to observe 3J(31P-19F) coupling in NMR spectra of compound XI [14]. Some further examples of related complexes have been reported by the same group [15,16]. In these compounds, crystallographic analyses revealed TM–Si-distances slightly below the sum of the van der Waals radii. Over the course of the past two decades, further buttressing ligand systems have given rise to the successful preparation of a greater variety of higher-coordinate Si complexes with a TM→Si motif (e.g., compounds XII [17], XIII [18], XIV [19] and XV [20]). In some cases, TM–Si bond lengths approach the sum of covalent radii. A common feature of the buttresses used is their 1,3-ambidentate donor nature. One donor site is designed to bind to Si (as a kinetically inert Si–C bond or as a rather hard donor anion), whereas the second site represents a rather soft donor, thus being more attractive toward late transition metals. Last but not least, the chelate effect of the ambidentate group (formation of a four-membered chelate ring) is likely to be over-compensated by the advantage of formation of a five-membered ring system when including an additional atom (i.e., a TM atom). Our recent studies, which utilized pyridine-2-olate as a buttressing motif in silane MeSi(pyO)3 (1a), gave access to a Cu(I) complex MeSi(μ2-pyO)3CuCl (2a) in a deliberate manner. Furthermore, oxidative decomposition (upon access of traces of air) gave rise to the formation of small amounts of Cu(II) compound MeSi(μ2-pyO)4CuCl (3a), X-ray crystallographic analysis of which delivered the first proof of Cu(II)→Si coordination [20]. In the current study, we address tasks which arose from findings in this pioneering work: (1) variation of the hydrocarbyl substituent R in Cu(I) compounds of the type RSi(μ2-pyO)3CuCl and exploration of a route toward deliberate synthesis of Cu(II) compounds of type RSi(μ2-pyO)4CuCl to elucidate potential influence of the R–Si bond on the trans-disposed Cu→Si coordination, and (2) replacing one pyO buttress by another hydrocarbyl group to elucidate the influence of the more open system on the Cu···Si atom distance.
2. Results and Discussion
2.1. Syntheses and Characterization of Cu(I) Compounds RSi(μ2-pyO)3CuCl
2.1.1. Syntheses of RSi(μ2-pyO)3CuCl
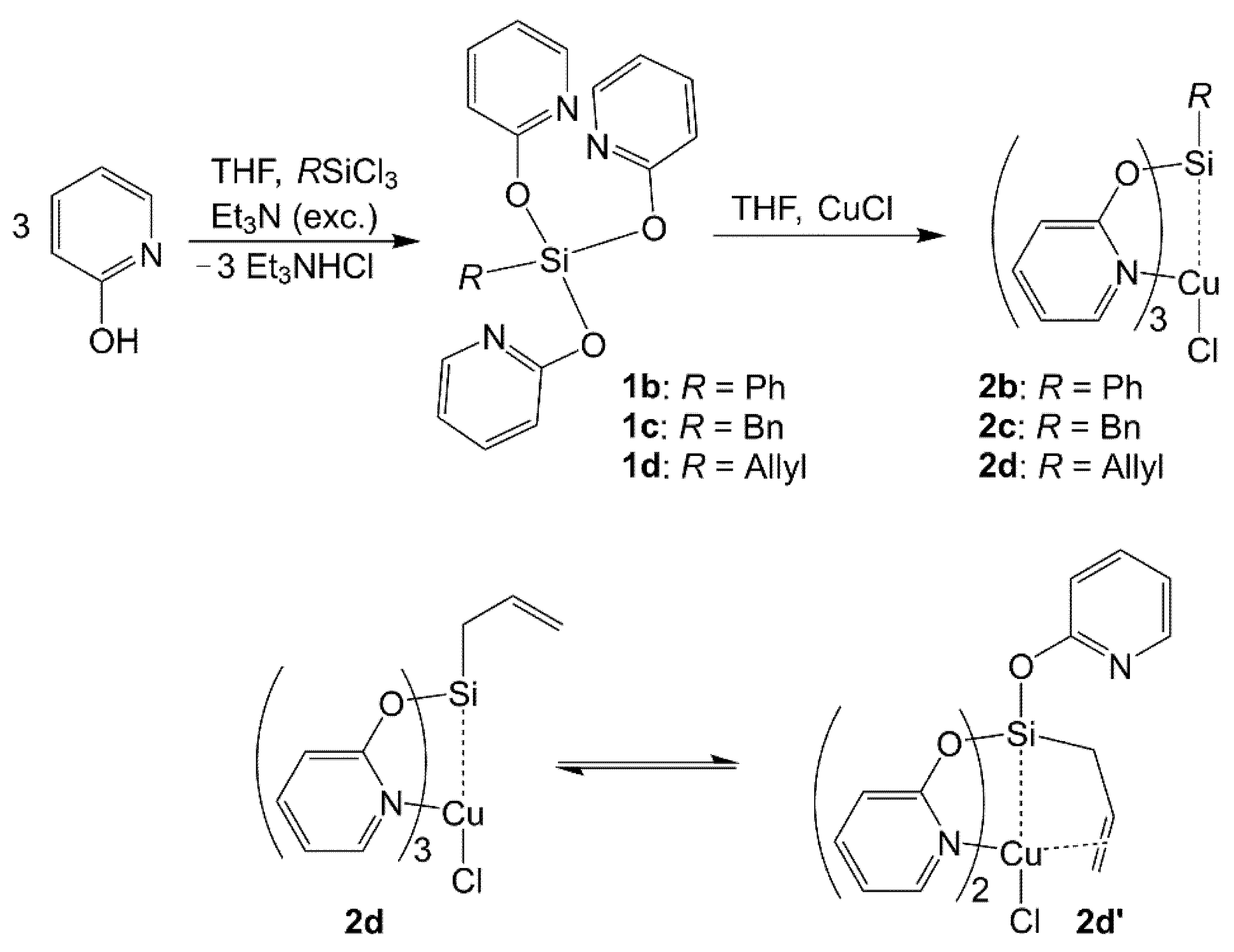
In studies of silatranes of the type RSi(OCH2CH2)3N, the Si-bound hydrocarbyl substituent exerts influence on the through-cage N→Si dative bond. For silatranes with R = Me, Ph (two modifications), Allyl, Si-N-distances of 2.16 [21], 2.16, 2.13 [22,23] and 2.14 Å [24], respectively, were found. With higher electronegativity of the hydrocarbyl group, a shorter trans-disposed N–Si bond is observed. Thus, in order to probe the response of the trans-disposed potential donor atom (Cu(I) in our study), the literature example silane 1a (R = Me) was supplemented by silanes RSi(pyO)3 with R = Ph (1b) [25], Bn (1c) and Allyl (1d). (Note: For this study we decided to vary the Si-bound substituent within hydrocarbyl groups only as other substituents, which are less kinetically inert at Si, may undergo substituent scrambling with pyO groups as shown for the pyridine-2-olate/-2-thiolate system [26].) Syntheses of 1a [20] and 1b [25] are reported in the literature, and silanes 1c and 1d were prepared in an analogous manner (Scheme 1). Their synthesis protocols, 1H, 13C, 29Si NMR data as well as molecular structures (determined by single-crystal X-ray diffraction, see Table A1) are included in the Supplementary Materials.
As for the conversion of 1a and CuCl in THF with formation of 2a [20], the analogous reactions of silanes 1b, 1c and 1d afforded the desired CuCl-complexes 2b, 2c and 2d, respectively (Scheme 1). Compounds 2b (upon diffusion of n-pentane into the THF solution) and 2c (from the reaction mixture in THF) crystallized within a few days and were isolated in good yields. Compound 2d, however, initially separated as an oil (upon diffusion of n-pentane into the THF solution), which turned into a yellow crystalline solid of 2d (a crystal of which was manually extracted for XRD analysis, see Section 2.1.2). Upon further storage in the presence of the supernatant, the yellow solid recrystallized spontaneously to give colorless crystals of isomer 2d′ (see Section 2.1.3 and Section 2.1.4). Further attempts at repeated syntheses of 2d failed to give solid 2d but afforded either an oily product or crystals of isomer 2d′. Thus, solid-state characterization of 2d is limited to single-crystal XRD. For further characterization in solution (see Section 2.1.5), 2d′ was used.
2.1.2. Molecular Structures of RSi(μ2-pyO)3CuCl (R = Me (2a), Ph (2b), Bn (2c), Allyl (2d))
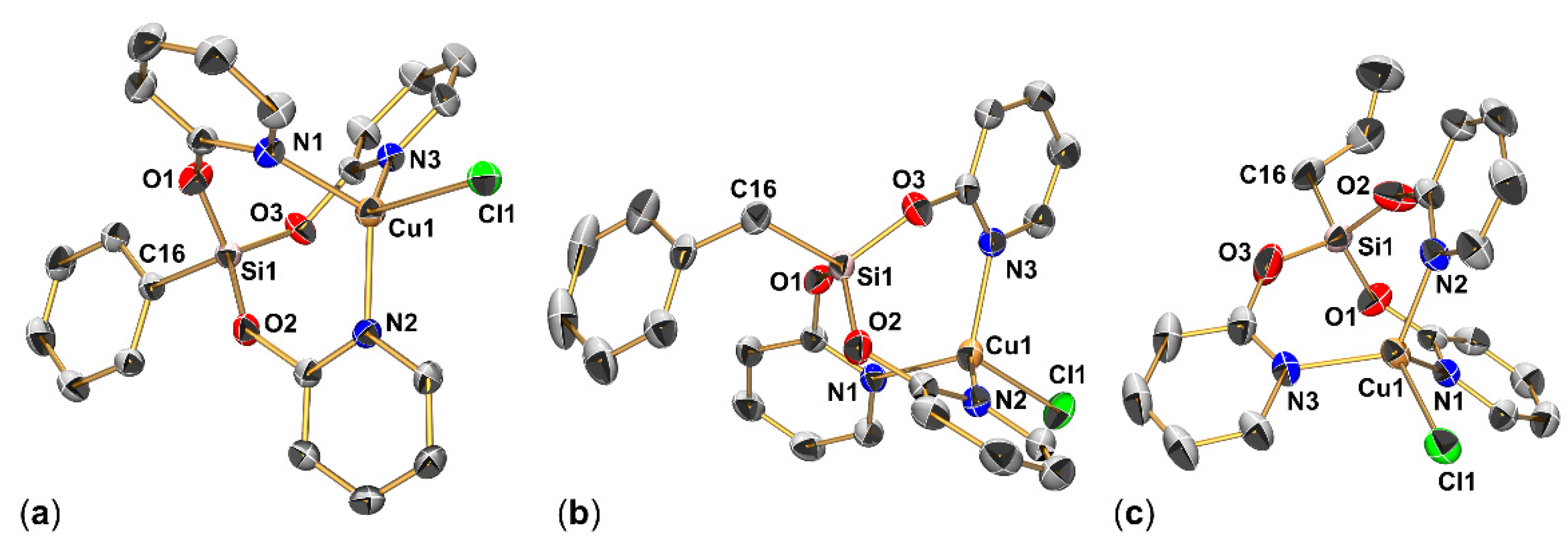
The syntheses of compounds 2b, 2c and 2d (the latter by chance) afforded crystals of the respective compound which were suitable for single-crystal X-ray diffraction analysis (Table A1 and Table A2, Figure 3). Compound 2b crystallized as a THF solvate (2b)2·THF, the other two compounds without a solvent of crystallization. In principle, their molecular structures resemble that of pioneer 2a [20], i.e., a cage formed by three pyO buttresses (Si–O- and Cu–N-bound) with the bridgehead atoms and their next substituent (C–Si···Cu–Cl) in a rather linear arrangement. For comparison, selected sets of corresponding interatomic distances and angles of 2b, 2c and 2d are listed in Table 1.
In principle, bond lengths of corresponding bonds are very similar within this set of four related compounds. The slightly shorter Si–O bonds in 2d are simulated by the disorder of the AllylSiO3 moiety in the crystal structure (unresolved disorder effects reflected in the rather large thermal displacement ellipsoids of the O atoms). Interestingly, the Cu···Si distances are also very similar. They vary within a very narrow range (0.03 Å) and do not show any systematic trend with respect to the different Si-bound hydrocarbyl substituents. Within the groups of O–Si–O and N–Cu–N angles, however, noticeable variability is observed. Whereas in 2c both sets span a rather narrow range (O–Si–O 109.9(1)–112.2(1)°, N–Cu–N 111.4(1)–113.1(1)°), the different N–Cu–N angles in 2b (103.6(1)–119.5(1)°) indicate flexibility of these cage structures, especially in the Cu coordination sphere. With respect to the distortion of the tetrahedral Cu coordination sphere, the geometry index τ4′ [27] (2a: 0.93; 2b: 0.87, 2c: 0.95; 2d: 0.89) also reflects noticeable differences between these related molecules in their solid-state structures. (τ4′ = [(β − α)/(360° − θ)] + [(180° − β)/(180° − θ)] with α, β being the two greatest valence angles, β > α, θ = ideal tetrahedral angle).
2.1.3. Molecular Structure of (κO-pyO)Si(μ2-pyO)2(Allyl)CuCl (2d′)
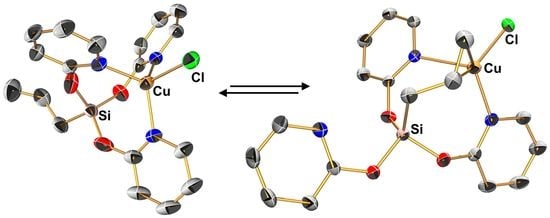
Compound 2d’ crystallizes in the triclinic space group P with one molecule in the asymmetric unit (Figure 4, Table A2). The Cu···Si distance (ca. 3.3 Å) is similar to that in compounds 2a, 2b, 2c and 2d. The sum of angles around the Cu-capped tetrahedral face at Si1 (spanned by O1, O2, C16) amounts to 335.3°, thus exhibits some widening with respect to an ideal tetrahedral face. Slightly more widening, however, is observed for the face spanned by O2, O3, C16 (sum of angles 337.1°), caused by N3, which caps this face. The Cu coordination sphere can be interpreted as highly distorted pseudo-tetrahedral (with respect to the entity C17C18 occupying one ligand site), which is close to trigonal-pyramidal. With respect to the latter geometry, the ligand sites Cl1, N1 and (C17=C18) represent the base (the cis-angles about Cu1 spanned by those atoms amount to 352.6°), N2 the apex with N2-Cu1-X angles (X = N1, Cl1, C17, C18) ranging from 95.3 to 104.0°. The different coordination sites occupied by N1 and N2 are reflected by the different Cu–N bond lengths, with Cu1–N2 being 0.3 Å longer. Both the release of donor atom N3 in favor of Cu-allyl-coordination and the positioning of the olefinic donor entity (C17=C18) in the basal trigonal coordination plane while donor site N2 occupies a more distant axial coordination site speak for allyl groups as competitive donor arms with respect to pyridine-2-olate groups.
In principle, η2-coordination of an allylic C=C bond to Cu(I) is a known motif. Crystallographic characterization of Cu(I) halide complexes of allyl silanes, however, is limited to few examples, a CuCl complex of Si(Allyl)4 [28] and a CuI complex of Me2Si(Allyl)(2-py), which forms dimer [Me2Si(μ2-2-py)(μ2-Allyl)Cu(μ2-I)2Cu(μ2-2-py)(μ2-Allyl)SiMe2] [29]. In the latter case, a 2-pyridyl group serves as an additional bridging ligand between Si and Cu, and the Cu atom is situated in a pseudo-tetrahedral (N,C=C,I,I)-coordination sphere. This pyridyl-supported CuI-complex of an allyl silane was found to be an allyl transfer reagent when reacted with benzaldehyde in the presence of CsF, whereas a mixture of Me3Si(Allyl), CuI, Benzaldehyde and CsF did not result in allyl transfer. Thus, complexes such as 2d or 2d′ bear potential for further exploration as allyl transfer reagents.
2.1.4. Computational Analysis of the Isomerization of 2d and 2d’
The finding of the crystallization of 2d by chance and the preferred crystallization of its isomer 2d′ gave rise to the question as to the relative stability of this pair of isomers and to their potentially facile interconversion in solution. With respect to THF as the solvent used for synthesis, the molecular structures of 2d and 2d′ were optimized at the RKS PBE0 B3BJ ZORA-def2-TZVPP level of theory with a solvent model (COSMO) of THF environment applied. A Gibbs free energy difference of 2.6 kcal·mol−1 in favor of the formation of allyl complex 2d′ was found (at 293 K).
In order to find a potential transition state for the interconversion of 2d and 2d′, two scenarios of ligand dissociation-association were considered: (a) Indicated by the molecular structure of 2d′ in the solid state (Figure 4), the two buttressing pyO ligands create an Si(μ2-pyO)2Cu eight-membered ring system, which is bridged by the allyl group. The aryl ring of the dangling pyO moiety is positioned on the same side of this ring as the allyl bridge, and thus the N atom may approach the Cu atom on the same side of the Si(μ2-pyO)2Cu eight-membered ring where the allyl group leaves. (b) Alternatively, reorientation of the dangling pyO N atom and backside attack at Cu were taken into consideration. A nudged elastic band (NEB) analysis failed to identify a transition state for scenario (a) but predicted a possible interconversion in accordance with scenario (b). This transition proceeds with initial dissociation of one ligand arm (Allyl C=C moiety or pyO N atom, depending on the direction of the conversion), followed by planarization and inversion of the Cu(N,N,Cl) coordination sphere with simultaneous bending of the Si(pyO)(Allyl) moiety to the opposite side of the eight-membered Si(μ2-pyO)2Cu ring, finalized by Cu-coordination of the respective other bridging ligand (cf. Figure S30 in the Supplementary Materials). For this scenario, a transition state with an energetic barrier of 10.6 kcal·mol−1 (from 2d′) or 8.0 kcal·mol−1 (from 2d) (values correspond to Gibbs free energy at 293 K) was identified (Figure 5). The energy found for this transition state already proves that the interconversion of 2d and 2d′ should be facile at room temperature. Nonetheless, we cannot claim that this simple monomolecular scenario would be the predominant mechanism. Alternative, more complex scenarios (e.g., with Cu-ligand dissociation supported by Cu(μ2-Cl)2Cu dimerization or by coordination-dissociation effects of discrete solvent molecules) might also play reasonable roles and could potentially further lower the activation barrier.
2.1.5. NMR Spectroscopic Analyses of 2b, 2c and 2d′
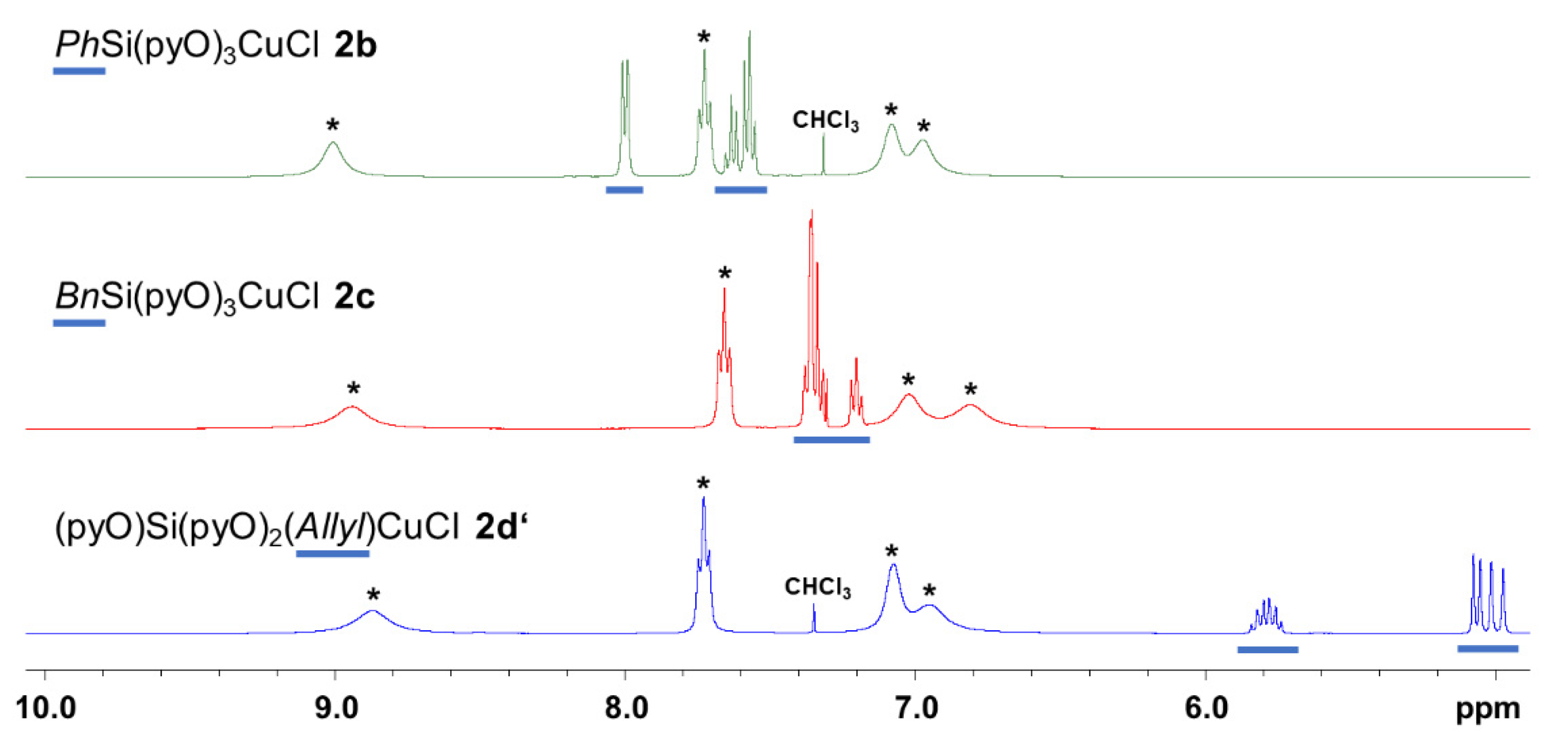
In CDCl3 solution at room temperature, compounds 2b, 2c and 2d′ give rise to 1H and 13C NMR spectra, which exhibit rather sharp signals of the nuclei of the respective different Si-bound hydrocarbyl substituents and very broad signals of the nuclei of the pyO groups (Figure 6 and Figure 7). With respect to the latter, both the similar broadening and the similar chemical shifts of corresponding signals of the three different compounds indicate their similar conformational situation in solution. Thus, we probed whether the pyO signal broadening arises from rapid exchange (close to coalescence) of chemically different pyO ligands within one molecule or from ligand–metal dissociation/association processes (in this case, Cu–N bond dissociation and formation) within a symmetrical molecule with (on the NMR time scale) equivalent pyO ligands. For this purpose, we recorded 1H (Figure 8) and 13C NMR spectra (cf. Figure S16 in the the Supplementary Materials) of compound 2c as representative examples at lower temperatures. At lower temperatures, systematic sharpening of signals with a resultant single set of signals for the pyO moieties (four 1H signals, five 13C signals) was observed. We attribute the signal broadening at room temperature to predominant ligand–metal dissociation/association processes within a symmetrical molecule. This is in accordance with the molecular structures encountered within compounds 2a, 2b, 2c and 2d in the crystal structures. The relevance of allyl coordination at Cu (in case of 2d′ in solution) is particularly indicated by the noticeable upfield shift of the allyl group’s olefinic 13C NMR signals upon Cu complexation (1d: δ = 131.1 and 115.6 ppm, 2d′: δ = 117.4 and 107.1 ppm), whereas 1H NMR shifts of corresponding signals of these two compounds are similar, and even the 3JHH couplings of the -CH=CH2 moiety show only little effect (1d: 17.1 and 10.1 Hz, 2d’: 16.3 and 9.7 Hz for trans- and cis-3JHH coupling, respectively).
A common feature of CDCl3 solutions of the herein discussed group of silanes RSi(pyO)3 (1a, 1b, 1c, 1d) and their corresponding Cu(I) complexes RSi(μ2-pyO)3CuCl is a systematic upfield shift of the 29Si NMR signal upon CuCl complexation (Table 2). In cases where R = Me, Ph, Bn, this shift is ca. −17 ppm. For the couple with R = Allyl, this upfield shift is less pronounced (−12 ppm). This was unexpected, and it hints at different structural features in solutions of 2d. In the solid state, 29Si NMR spectroscopy of 2a, 2b and 2c essentially confirmed the chemical shift encountered with CDCl3 solutions (slightly more or less shielded Si nuclei). In case of 2d′, however, the signal emerged close to the shift of silane 1d. Hence, we interpret the 29Si NMR shift observed with CDCl3 solutions of 2d/2d’ as an average chemical shift which, in a dynamic equilibrium, has contributions of a propeller-type molecule 2d (AllylSi(μ2-pyO)3CuCl coordination), causing a notable upfield shift of the signal, and contributions of its isomer 2d’ ((κO-pyO)Si(μ2-pyO)2(μ2-Allyl)CuCl coordination), which do not cause notable upfield shift of the 29Si signal with respect to silane 1d. Thus, even though the 1H and 13C NMR signal broadening and shift ranges observed for pyO groups of 2d′ in CDCl3 solution indicate predominance of an essentially symmetrical molecule (2d), the 29Si shift characteristics hint at contributions of a pyO vs. allyl coordination exchange (i.e., the relevance of contributions of isomer 2d’ also in CDCl3 solution).
As the Cu···Si distance in compound 2d′ (3.30 Å) is similar to that in 2c (3.23 Å), and the effect of Si hypercoordination by Cu(I) should be similar, the noticeable 29Si NMR shift differences between 2c and 2d′ give rise to the assumption that the upfield shift encountered with CuCl complexation is only in part associated with Si hypercoordination by Cu···Si coordination. Polarization of the pyO ligands upon N···Cu coordination (and resultant changes in Si–O bonding) may play a major role. In order to probe this hypothesis, we calculated the 29Si NMR shifts of compound 2b (which was chosen because of the rather rigid hydrocarbyl substituent at Si) and a hypothetical tripodal LiCl complex of silane 1b (PhSi(μ2-pyO)3LiCl, 2bLi), which lacks a potential fifth lone pair donor site but still features a mono-cation to polarize the three pyO moieties (via N···Li coordination). For compound 2b, the computational approach (COSMO model with chloroform solvent environment) predicted a 29Si NMR shift of −80.8 ppm, which essentially resembles the experimental value. For the LiCl substitute 2bLi, the same theoretical model predicted a 29Si NMR shift of −75.7 ppm (which is also noticeably upfield shifted vs. −57.7 ppm for silane 1b at the same level of theory), thus pointing at polarization via N···cation coordination as an important influence on 29Si NMR shifts in complexes of silicon pyridine-2-olates.
For further solid-state characterization of these compounds, 63Cu MAS NMR spectra were recorded for 2c and 2d′ (Figure 9). From the three complexes with Cu(N,N,N,Cl) coordination sphere, compound 2c was selected because of the only weak distortion of the Cu coordination sphere (in contrast to 2b) and the presence of only one molecule in the crystallographic asymmetric unit (in contrast to 2a).
The spectra were externally referenced to CuCl, which was set at δiso(CuCl) = −332 ppm [30]. This shift is also in accordance with that of solution state experiments, in which a suspension of CuCl had been used (and set at −331 ppm) [31]. This way, the CuCl chemical shift is in agreement with the IUPAC suggested zero-point reference for 63Cu solution NMR spectra (where a saturated solution of [(MeCN)4Cu][ClO4] in MeCN with ≤10% C6D6 is used as reference) [32]. Note: In other reports, CuCl was used as zero-point reference [33,34]. In general, compounds 2c and 2d′ produce 63Cu MAS NMR spectra in which the isotropic chemical shift falls into a range of +100–+200 ppm (with CuCl set at −332 ppm). This is in accordance with Cu chemical shifts reported for other compounds close to the tetrahedral Cu coordination sphere such as [(PhCN)4Cu][BF4] (510 ppm relative to CuCl at 0 ppm) and tetracoordinate Cu in BrCu(PPh3) (210 ppm relative to CuCl at 0 ppm) [34]. The most striking difference between the 63Cu spectra of 2c and 2d′ is the signal pattern, which is strongly influenced by the tensor of chemical shift anisotropy (CSA), quadrupolar coupling and residual dipolar coupling (the latter is caused by Cl in particular and by the N atoms to a minor extent). As expected, the compound with the more symmetric Cu coordination sphere (close to tetrahedral, 2c) produces a rather narrow spinning side band spectrum, and the shape of the individual bands reflects the effects of quadrupolar coupling. In this case, a simulation [35] allowed for the extraction of the parameters of the chemical shift (δiso = 128 ppm) and of quadrupolar coupling (CQ = 4.85 MHz with an asymmetry parameter η = 0.38). In spite of the mixed donor atom situation in 2c, the quadrupolar coupling is still similar to that in the more symmetrical Cu coordination sphere of [(PhCN)4Cu][BF4] (CQ = 4.1 MHz [34]). In compound 2d′, the effects of the electric field gradient (EFG) remain moderate and the signal pattern is dominated by the CSA tensor. The spinning side band spectrum resulting therefrom indicates a noticeably wider span of the CSA for compound 2d′ than for 2c, which is in accordance with the different donor moieties in and the lower symmetry of the Cu coordination sphere of 2d′.
2.2. Syntheses and Crystallographic Characterization of Cu(II) Compounds RSi(μ2-pyO)4CuCl (R = Me (3a), Ph (3b), Bn (3c), Allyl (3d))
In our previous report [20], we presented the first example of a paddlewheel complex in which Cu(II) is a potential donor atom in the coordination sphere of a hexacoordinate Si atom, compound MeSi(μ2-pyO)4CuCl (3a). The Cu···Si distance (2.919(1) Å) indicated attraction of the two atoms, and a natural localized molecular orbital (NLMO) analysis indicated some polarization of one of the Cu atom’s d-orbitals toward Si. As in this initial study compound 3a was obtained only in small amounts as a decomposition product of Cu(I) complex 2a, we now aimed to establish a deliberate synthesis route of this class of compounds and to prepare some analogs of this series for comparison of their molecular structures with regard to the response of the Cu(II)–Si-interaction to the different trans-disposed hydrocarbyl substituents. Starting from silanes RSi(pyO)3 (1a–d) and CuCl, the oxygen content of dry air (in a close to stoichiometric manner) was used as an oxidant, and excess silane needed to be employed as a source of additional pyO ligands and a scavenger of oxide formed in the redox reaction. Scheme 2 gives an impression of the targeted reaction. (Note: For the by-products of oxide capture, siloxanes, only a symbolic example is drawn. The portfolio of siloxanes formed has not been characterized.) For that purpose, the respective amounts of silane 1 and CuCl (in approximate stoichiometric ratio 3:2) were placed in a Schlenk flask under a dry argon atmosphere and dissolved into a small amount of THF, whereafter the calculated amount of air (which had been stored in a flask over anhydrous CaCl2 for at least one week) was added to the gas phase in the Schlenk flask via syringe. Syntheses of 3a, 3b and 3c were successful. Within a few days, dark blue crystals of the desired product formed in good yield. In case of conversion of 1d and CuCl, however, a brown precipitate formed in the blue solution. The crystals of 3b and 3c were of good quality for X-ray diffraction analysis (Figure 10, Table A3). Some crystals of 3d (not in a deliberate manner to determine yield and purity, but of sufficient quality for single-crystal X-ray diffraction analysis) were obtained along the initial “by chance” method, i.e., allowing access of traces of air to one of the synthesis batches of 2d, where silane 1d and CuCl were used in an approximately 1:1 stoichiometric ratio. For comparison of the molecular structures, selected atom distances and angles are listed in Table 3.
The four paddlewheel shaped compounds of type 3 exhibit essentially the same cage structure, in which Si and Cu atom are situated in square-based pyramidal coordination spheres with basal (O)4 and (N)4 coordination, respectively, and axial monodentate substituents (hydrocarbyl and Cl, respectively). The sets of Si–O and Cu–N bond lengths do not exhibit any systematic trends of deviations, and Cu···Si distances only vary in a narrow range (2.89–2.92 Å). In contrast to complexes of type 2, the variation of Cu···Si distances in compounds 3 hints at systematic response of this interatomic interaction to the different Si-bound hydrocarbyl substituents, as the Si-Ph derivative 3b features the shortest Cu···Si contact, the Si-Me compound 3a the longest.
The crystal structure of compound 3b is reminiscent of the structures of the paddlewheel complexes PhSb(μ2-pyO)4Ru(CO) and PhSb(μ2-pyO)4RuCl [36], which crystallize in the same orthorhombic space group Pbca with isomorphous unit cell parameters. In the former case, some essential molecular metrics adhere to the same proportions (distance between opposite pyO-C4 atoms in the paddlewheel 9.42 and 9.46 Å, distance between Cl atom and Ph-p-C atom 9.94 Å for 3b; distance between opposite pyO-C4 atoms in the paddlewheel 9.86 and 9.87 Å, distance between carbonyl O atom and Ph-p-C atom 10.41 Å for PhSb(μ2-pyO)4Ru(CO)). In the latter case, the distance between Cl atom and Ph-p-C atom (9.86 Å) is similar to that in 3b, whereas the distances between opposite pyO-C4 atoms in the paddlewheel (9.81 and 9.82 Å) are similar to those in PhSb(μ2-pyO)4Ru(CO).
2.3. Reaction of Ph2Si(pyO)2 and CuCl
For a comparison of CuCl complexation with a pyO functionalized silane of the type R2Si(pyO)2, the diphenyl derivative Ph2Si(pyO)2 (4) [26] was chosen as its aryl groups may support crystallization (and thus support solid-state characterization of the products). Furthermore, the phenyl ring still offers an alternative ligand motif through coordination of its π-electron system to Cu(I) (if required). Benzene rings have already proven capable of coordinating Cu(I). Even in rather simple compounds obtained from CuCl and ZrCl4, benzene itself may serve as a ligand in the Cu(I) coordination sphere [37,38].
Reaction of 4 and CuCl in THF proceeded instantly with dissolution of CuCl. Within a few hours, crystallization commenced and produced a mixture of yellow and colorless crystals of sufficient quality for single-crystal X-ray diffraction analysis. The former were identified as compound [Ph2Si(μ2-pyO)2(CuCl)2(μ2-pyO)2SiPh2] (52), which adheres to the stoichiometry of 4 and CuCl used (Scheme 3, Table A4, Figure 11). The latter were identified as THF solvate of compound [Ph2Si(μ2-pyO)2(CuCl)4(μ2-pyO)2SiPh2] (54), which features excessive CuCl in its molecular structure in a distorted ladder-type motif (Table A4, Figure 12). Repeated reaction of 4 and CuCl (even with use of excess of silane 4) failed to produce 52 but delivered the THF solvate of 54 as the exclusive solid product in good yield. This observation hints at the co-existence of various species (such as 4, 52 and 54) in solution in equilibrium and solvent-driven crystallization of 54. Therefore, the synthesis was repeated in chloroform as an alternative solvent. In spite of the slight excess of 4 used, another CuCl-excess complex crystallized. In this case, a CHCl3 solvate of compound [Ph2Si(μ2-pyO)2(CuCl)3(μ2-pyO)2SiPh2] (53) crystallized, which also features the excessive CuCl in its molecular structure in a ladder-type arrangement (Table A4, Figure 11).
In the molecules of 52, 53 and 54 the Cu···Si atom distances range between 3.52 and 3.64 Å. These distances are noticeably longer than those in Cu(I) compounds 2a–2d (ca. 3.2 Å) or compound 2d′ (3.3 Å). The Si atom merely plays a role as a backbone building block, which connects the two Cu-coordinating pyO moieties. The Si-bound phenyl groups remain at the periphery of the molecules and do not establish coordination with the Cu atoms. A common feature of the Si coordination spheres of 52, 53 and 54 is the widening of the O–Si–O angles (both the C–Si–C and O–Si–O angles are wider than the tetrahedral angle). With respect to the central (CuCl)n parts, only the well-ordered structures of 52 and 53 will be taken into consideration for discussion. The pyO-chelated Cu atoms are located in highly distorted tetrahedral coordination spheres, which feature wide N–Cu–N angles (ranging from 123° to 136°) and Cl–Cu–Cl angles in the range of 95–100°. The former can be interpreted as a result of the small bond angles within the four-membered Cu2Cl2-rings. In the three compounds case, bridging Cu–Cl–Cu-coordination is the favored motif to accomplish tetracoordination of the (pyO)2-chelated Cu atoms, and in 53 and 54, the central Cu sites in the (CuCl)n ladder motifs remain tricoordinate. The resultant Cu2Cl2-rings can be planar (as in the case of 52, resulting from a crystallographically imposed center of inversion) and may also deviate noticeably from planarity as one of the two Cu2Cl2-rings in 53. In the latter compound, Cu1, Cl1, Cu2 and Cl2 are still close to planarity (the angle between the planes Cu1Cl1Cu2 and Cu1Cl2Cu2 is 8.02(7)°), whereas the angle between the planes Cu2Cl2Cu3 and Cu2Cl3Cu3 is 40.27(5)°. Even in the planar Cu2Cl2 ring in 52, this ring is of rather low symmetry as it features asymmetric Cu–Cl–Cu bridges (CuCl bond lengths of 2.301(1) and 2.624(1) Å). In contrast, in the less symmetric molecule 53, the Cu2Cl2 rings may exhibit symmetric (Cu1–Cl2 2.533(2), Cu2–Cl2 2.527(2) Å) and less symmetric Cu–Cl–Cu bridges (Cu1–Cl1 2.450(2), Cu2–Cl1 2.158(2) Å). These selected examples already indicate very high flexibility of (CuCl)n cores in their multinuclear complexes, and ladder-type complexes thereof with terminal Cu-complexation by chelating ligands are just one among other motifs: The portfolio of literature-known complexes with (CuCl)n cores offers examples for further motifs (Figure 13), e.g., with n = 1 and 2 within one molecule (XVI [39]), n = 3 ladder-type with ligands bridging Cu-positions 1 and 3 (XVII [40]) or with monodentate ligands at Cu in positions 1 and 3 (XVIII [41]), n = 3 cyclic motif with two Cu atoms chelated (XIX [42]) or three Cu atoms chelated (XX [43]) and (CuCl)4-cubes with additional ligands at Cu (e.g., XXI and XXII, which feature N-donor ligands [44]). Ligands capable of monodentate bridging (such as thiourea derivatives), which adopt the bridging role of the Cl atom, enhance the portfolio of complexes with (CuCl)n cores even further, e.g., in compound XXIII [45].
The variety of coordination motifs accessible for CuCl-oligomers is manifold, and lowering the number of donor arms in a ligating system (e.g., going from silane 2b to 4 by replacing one pyO unit by Ph) opens coordination sites at Cu for CuCl-coordination. The selection of compounds 5n (n = 2, 3, 4), which were obtained from 1:1 stoichiometric reactions of 4 and CuCl, hints at a greater variety of species in the reaction solutions and solubility driven crystallization of the one or the other species. This hampered characterization in solution. The two compounds (53 and 54), which were isolated as pure products, decomposed upon dissolution in CDCl3. In both cases, a yellow solution and a beige precipitate formed, and both solutions produced essentially identical 1H, 13C and 29Si NMR spectra. In the 29Si spectrum, a single signal at −43.9 ppm indicates the formation of a new tetracoordinate Si-compound rather than the release of ligand 4. Silane 4 in CDCl3 produces a signal at −33.1 ppm [26]. In addition, the solids 53·(CHCl3) (δiso = −32.9, −33.5 ppm) and 54·(THF)2 (δiso = −32.4 ppm) produce 29Si NMR signals close to the 29Si chemical shift of compound 4. The broad signals of the pyO moieties in the 1H and 13C spectra hint at rapid exchange processes. The flexibility of these ligand systems (of compound 4, and most likely the same is true for related silanes) poses challenges for the deliberate synthesis of simple complexes of a desired stoichiometry and for the characterization of their CuCl complexes in solution.
3. Materials and Methods
3.1. General Considerations
Starting materials 2-hydroxypyridine (ABCR, Karlsruhe, Germany, 98%), benzyltrichlorosilane (ABCR, Karlsruhe, Germany, 98%) and allyltrichlorosilane were used as received without further purification. Silanes MeSi(pyO)3 (1a) [20], PhSi(pyO)3 (1b) [25] and Ph2Si(pyO)2 (4) [26] as well as CuCl [46] have been available from our previous studies. THF, diethyl ether and triethylamine were distilled from sodium/benzophenone and kept under argon atmosphere. n-Pentane (Th.Geyer, Renningen, Germany, >99%), chloroform, stabilized with amylenes (Honeywell, Seelze, Germany, ≥99.5%) and CDCl3 (Deutero, Kastellaun, Germany, 99.8%) were stored over activated molecular sieves (3 Å) for at least 7 days and used without further purification. All reactions were carried out under an atmosphere of dry argon utilizing standard Schlenk techniques. (The silanes and Cu complexes reported in this paper are sensitive toward hydrolysis, and the Cu(I) complexes are sensitive toward oxidation.) Solution NMR spectra (1H, 13C, 29Si) (cf. Figures S3–S23 in the Supplementary Materials) were recorded on Bruker Avance III 500 MHz and Bruker Nanobay 400 MHz spectrometers. 1H, 13C and 29Si chemical shifts are reported relative to Me4Si (0 ppm) as the internal reference. 1H and 13C NMR signals were assigned in accordance with mutual coupling patterns (in case of 1H) and according to the shifts of corresponding 1H or 13C NMR signals in related compounds MeSi(pyO)3 [20], Ph2Si(pyO)2 [26] and PhP(pyO)2 [47]. Furthermore, 1H-13C-HSQC NMR spectra of compounds 1c and 2c were recorded to confirm signal assignments of corresponding pyO 13C signals within groups of compounds RSi(pyO)3 and RSi(μ2-pyO)3CuCl, respectively. 29Si CP/MAS NMR spectra (cf. Figures S24–S29 in the Supplementary Materials) were recorded on a Bruker Avance 400 WB spectrometer using 4 mm zirconia (ZrO2) rotors and an MAS frequency of υrot = 5 kHz. The chemical shift is reported relative to Me4Si (0 ppm) and was referenced externally to octakistrimethylsiloxyoctasilsesquioxane Q8M8 (the most upfield signal of its Q4 groups at δiso = −109 ppm). 63Cu MAS NMR spectra were recorded on a Bruker Avance Neo 700 SB spectrometer using 3.2 mm zirconia (ZrO2) rotors and MAS frequencies of up to υrot = 23 kHz. The chemical shift is reported relative to and referenced with a sample of CuCl (at δiso = −332 ppm). Elemental analyses were performed on an Elementar Vario MICRO cube. For single-crystal X-ray diffraction analyses, crystals were selected under an inert oil and mounted on a glass capillary (which was coated with silicone grease). Diffraction data were collected on a Stoe IPDS-2/2T diffractometer (STOE, Darmstadt, Germany) using Mo Kα-radiation. Data integration and absorption correction were performed with the STOE softwares XArea and XShape, respectively. The structures were solved by direct methods using SHELXS-97 or SHELXT and refined with the full-matrix least-squares methods of F2 against all reflections with SHELXL-2014/7 or SHELXL-2018/3 [48,49,50,51,52]. All non-hydrogen atoms were anisotropically refined, and hydrogen atoms were isotropically refined in idealized position (riding model). For details of data collection and refinement (incl. the use of SQUEEZE in the refinement of the structure of 54), see Appendix A, Table A1, Table A2, Table A3 and Table A4. Graphics of molecular structures were generated with ORTEP-3 [53,54] and POV-Ray 3.7 [55]. CCDC 2,222,519 (52), 2,222,520 (1d), 2,222,521 (2c), 2,222,522 (3b), 2,222,523 ((54)·(THF)2), 2,222,524 (3c), 2,222,525 (1c), 2,222,526 (3d), 2,222,527 (2d’), 2,222,528(2d), 2,222,529 ((2b)2·(THF)), and 2,222,530 ((53)·(CHCl3)), contain the supplementary crystal data for this article. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via https://www.ccdc.cam.ac.uk/structures/ (accessed on 27 November 2022).
All geometry optimizations and NEB (Nudged Elastic Band Method) calculations were carried out with ORCA 5.0.3 [56] using the restricted PBE0 functional with relativistically recontracted Karlsruhe basis sets ZORA-def2-TZVPP [57,58] (for all atoms), the scalar relativistic ZORA Hamiltonian [59,60], atom-pairwise dispersion correction with the Becke–Johnson damping scheme (D3BJ) [61,62] and COSMO solvation (THF: ε = 7.58, rsolv = 3.18; CHCl3: ε = 4.8, rsolv = 3.17). VeryTightSCF and slowconv options were applied and the DEFGRID3 was used with a radial integration accuracy of 10 for copper and silicon for all calculations. Calculations were started from the molecular structures obtained by single-crystal X-ray diffraction analysis and isomers were created by modifying these structures. Numerical frequency calculations were performed to prove convergence at the local minimum after geometry optimization and to obtain the Gibbs free energy (293.15 K). On the final structures, single-point calculations were performed with a restricted B2T-PLYP functional with relativistically recontracted Karlsruhe basis sets ZORA-def2-TZVPP [57,58] (for all atoms) and by utilizing the AutoAux generation procedure [63], relaxed MP2 densities, the scalar relativistic ZORA Hamiltonian [59,60], atom-pairwise dispersion correction with the Becke–Johnson damping scheme (D3BJ) [61,62] and COSMO solvation (THF). The transition state search was started from the geometry of step five of the NEB calculation and confirmed with numerical frequency calculations. NMR calculations were performed using ORCA 5.0.3 [56] with GIAO formalism using the restricted PBE0 functional with relativistically recontracted Karlsruhe basis sets ZORA-def2-TZVPP [57,58] (for all atoms), the scalar relativistic ZORA Hamiltonian [59,60], atom-pairwise dispersion correction with the Becke–Johnson damping scheme (D3BJ) [61,62] and COSMO solvation (CHCl3: ε = 4.8, rsolv = 3.17). NMR chemical shifts were referenced against tetramethylsilane. Graphics of the optimized molecular structures were generated using Chemcraft [64].
3.2. Syntheses and Characterization
Compound 2b (PhSi(μ2-pyO)3CuCl)2·(THF) (C46H42Cl2Cu2N6O7Si2). Under dry argon atmosphere, a Schlenk flask was charged with magnetic stirring bar, silane 1b (PhSi(pyO)3·0.5(THF), 1.025 g, 2.23 mmol) and CuCl (0.195 g, 1.97 mmol), whereafter THF (3 mL) was added. The resultant mixture was stirred in a water bath (40 °C) for few minutes to afford a clear light yellow (slightly greenish-yellow) solution. Upon cooling to room temperature, this Schlenk flask was connected to another flask with n-pentane (5 mL) for gas phase diffusion of pentane into the solution. The product crystallized within 1 d, whereupon the supernatant was decanted, and the solid product was washed with a mixture of THF (2 mL) and n-pentane (1 mL) and briefly dried in vacuum. Yield: 0.97 g (0.928 mmol, 94%) of (2b)2·(THF). Elemental analysis indicated loss of some solvent upon sample preparation. Calculated for C21H17ClCuN3O3Si·0.3(THF) (508.09 g·mol−1): C, 52.48%; H, 3.85%; N, 8.27%; found C, 52.5%; H, 3.9%; N, 8.3%. 1H NMR (CDCl3): δ (ppm) 9.01 (br, 3H, H6), 8.00 (m, 2H, Ph-o), 7.72 (m, br, 3H, H4), 7.66–7.60 (m, 1H, Ph-p), 7.60–7.54 (m, 2H, Ph-m), 7.08 (br, 3H, H5), 6.97 (br, 3H, H3); 13C{1H} NMR (CDCl3): δ (ppm) 157.6 (C2), 149.2 (C6), 140.3 (C4), 134.3 (Ph-o), 131.8 (Ph-p), 128.4 (Ph-m), 128.4 (Ph-i), 119.8 (C5), 114.2 (C3); 29Si{1H} NMR (CDCl3): δ (ppm) −80.9; (CP/MAS): δiso (ppm) −82.3.
Compound 2c BnSi(μ2-pyO)3CuCl (C22H19ClCuN3O3Si). Under dry argon atmosphere, a Schlenk flask was charged with magnetic stirring bar, silane 1c (0.49 g, 1.22 mmol) and CuCl (0.115 g, 1.16 mmol), whereafter THF (1.5 mL) was added. The resultant mixture was briefly stirred at room temperature until complete dissolution of CuCl was achieved (some crystals of 1c still remained undissolved) to afford a light yellow (slightly greenish-yellow) solution, which was then stored undisturbed at room temperature. Crystallization of 2c commenced within some minutes, and the initially undissolved silane 1c dissolved during crystallization of 2c. After 4 d, the supernatant was decanted, and the coarse crystalline yellow product was dried in vacuum. Yield: 0.50 g (1.00 mmol, 86%) of 2c. Elemental analysis calculated for C22H19ClCuN3O3Si (500.49 g·mol−1): C, 52.80%; H, 3.83%; N, 8.40%; found C, 52.6%; H, 4.0%; N, 8.4%. 1H NMR (CDCl3): δ (ppm) 8.94 (br, 3H, H6), 7.66 (m, br, 3H, H4), 7.40–7.29 (mm, 4H, Ph-o/m), 7.24–7.17 (m, 1H, Ph-p), 7.02 (br, 3H, H5), 6.81 (br, 3H, H3), 2.76 (s, 2H, CH2); 13C{1H} NMR (CDCl3): δ (ppm) 157.5 (C2), 149.1 (C6), 140.1 (C4), 135.4 (Ph-i), 128.9, 128.6 (Ph-o/m), 125.6 (Ph-p), 119.6 (C5), 114.0 (C3), 22.2 (CH2); 29Si{1H} NMR (CDCl3): δ (ppm) −72.1; (CP/MAS): δiso (ppm) −67.6.
Compounds 2d/2d′ AllylSi(μ2-pyO)3CuCl/(κO-pyO)Si(μ2-pyO)2(μ2-Allyl)CuCl (C18H17ClCuN3O3Si). Under a dry argon atmosphere, a Schlenk flask was charged with a magnetic stirring bar, silane 1d (0.45 g, 1.28 mmol) and CuCl (0.114 g, 1.15 mmol), whereafter THF (3 mL) was added. The resultant mixture was briefly stirred at room temperature until complete dissolution of CuCl was achieved to afford a light yellow (slightly greenish-yellow) solution, which was passed through a syringe filter to remove some turbidity before n-pentane (6 mL) was added. From this mixture, the product separated as a yellow oil. Storage at 5 °C and then at room temperature afforded crystalline 2d′ as a colorless solid, which was isolated from the supernatant by decantation, washed with a mixture of THF (1 mL) and n-pentane (2 mL) and dried in vacuum. Yield: 0.43 g (0.95 mmol, 83%) of 2d′. Elemental analysis calculated for C18H17ClCuN3O3Si (450.42 g·mol−1): C, 48.00%; H, 3.80%; N, 9.33%; found C, 47.89%; H, 4.32%; N, 9.33%. (In a similar synthesis, the yellow oil solidified with formation of crystals of 2d, which were suitable for single-crystal X-ray diffraction analysis, but then re-crystallized in the mother liquor with formation of 2d’ before 2d had been isolated as a pure solid.) 1H NMR (CDCl3): δ (ppm) 8.87 (br, 3H, H6), 7.73 (m, br, 3H, H4), 7.08 (br, 3H, H5), 6.95 (br, 3H, H3), 5,79 (m, 1H, =CH), 5.06 (m, 1H, incl. 3JHH(cis) 9.7 Hz, =CH2), 4.99 (m, 1H, incl. 3JHH(trans) 16.3 Hz, =CH2), 2.21 (d, 2H, 7.6 Hz, CH2); 13C{1H} NMR (CDCl3): δ (ppm) 158.4 (C2), 148.7 (C6), 140.4 (C4), 119.6 (C5), 117.4 (Allyl =CH), 114.2 (C3), 107.1 (Allyl =CH2), 20.5 (CH2); 29Si{1H} NMR (CDCl3): δ (ppm) −65.4; (CP/MAS): δiso 2d′ (ppm) −52.6.
Compound 3a MeSi(μ2-pyO)4CuCl (C21H19ClCuN4O4Si). Under a dry argon atmosphere, a Schlenk flask (volume ca. 50 mL) was charged with magnetic stirring bar, silane 1a (MeSi(pyO)3, 229 mg, 0.70 mmol) and CuCl (46 mg, 0.46 mmol), whereafter THF (2 mL) was added. The resultant mixture was stirred in a water bath (45 °C) for few minutes to afford a clear light yellow (slightly greenish-yellow) solution. Upon cooling to room temperature, dry air (12 mL, corresponding to ca. 0.105 mmol of O2) was added into the lower half of gas phase of this Schlenk flask via syringe through a septum, and excess argon was allowed to leave through an oil trap before the flask was closed. Within minutes, the color of the solution turned deep blue, and formation of dark blue crystals commenced within a few hours. The contents were stored undisturbed at room temperature overnight, whereupon the supernatant was decanted, and the solid product was washed with THF (0.5 mL) and dried in vacuum. Yield: 156 mg (0.30 mmol, 73%) of 3a. Elemental analysis calculated for C21H19ClCuN4O4Si (518.48 g·mol−1): C, 48.65%; H, 3.69%; N, 10.81%; found C, 48.80%; H, 4.02%; N, 10.55%.
Compound 3b PhSi(μ2-pyO)4CuCl (C26H21ClCuN4O4Si). Under a dry argon atmosphere, a Schlenk flask (volume ca. 50 mL) was charged with magnetic stirring bar, silane 1b (PhSi(pyO)3·0.5(THF), 288 mg, 0.63 mmol) and CuCl (42 mg, 0.42 mmol), whereafter THF (2 mL) was added. The resultant mixture was stirred in a water bath (35 °C) for few minutes to afford a clear light yellow (slightly greenish-yellow) solution. Upon cooling to room temperature, dry air (12 mL, corresponding to ca. 0.105 mmol of O2) was added into the lower half of gas phase of this Schlenk flask via syringe through a septum, and excess argon was allowed to leave through an oil trap before the flask was closed. Within minutes, the color of the solution turned deep blue, and formation of dark blue crystals commenced within few hours. The contents were stored undisturbed at room temperature overnight, whereupon the supernatant was decanted, and the solid product was dried in vacuum. Yield: 171 mg (0.29 mmol, 71%) of 3b. Elemental analysis calculated for C26H21ClCuN4O4Si (580.55 g·mol−1): C, 53.79%; H, 3.65%; N, 9.65%; found C, 53.95%; H, 3.96%; N, 9.33%.
Compound 3c BnSi(μ2-pyO)4CuCl (C27H23ClCuN4O4Si). Under a dry argon atmosphere, a Schlenk flask (volume ca. 80 mL) was charged with magnetic stirring bar, silane 1c (BnSi(pyO)3, 390 mg, 0.97 mmol) and CuCl (65 mg, 0.66 mmol), whereafter THF (3 mL) was added. The resultant mixture was briefly stirred at room temperature to afford a clear light yellow (slightly greenish-yellow) solution. Thereafter, dry air (18.5 mL, corresponding to ca. 0.162 mmol of O2) was added into the lower half of gas phase of this Schlenk flask via syringe through a septum, and excess argon was allowed to leave through an oil trap before the flask was closed. Within minutes, the color of the solution turned deep blue, and formation of yellow crystals of 2c and of dark blue crystals of 3c commenced within few hours. The contents were stored undisturbed at room temperature for 4 d to allow for dissolution and reaction of 2c and final crystallization of 3c. Thereafter, the supernatant was decanted, and the solid product was washed with THF (1 mL) and dried in vacuum. Yield: 290 mg (0.49 mmol, 74%) of 3c. Elemental analysis calculated for C27H23ClCuN4O4Si (594.58 g·mol−1): C, 54.54%; H, 3.90%; N, 9.42%; found C, 54.44%; H, 3.80%; N, 9.34%.
Compounds 52 Ph2Si(μ2-pyO)2(CuCl)2(μ2-pyO)2SiPh2 (C44H36Cl2Cu2N4O4Si2) and (54)·(THF)2 Ph2Si(μ2-pyO)2(CuCl)4(μ2-pyO)2SiPh2·(THF)2 (C52H52Cl4Cu4N4O6Si2). Under a dry argon atmosphere, a Schlenk flask was charged with magnetic stirring bar, silane 4 (0.308 g, 0.83 mmol) and CuCl (82 mg, 0.83 mmol), whereafter THF (1 mL) was added. The resultant mixture was briefly stirred at room temperature until complete dissolution of CuCl was achieved to afford a light yellow (slightly greenish-yellow) solution, which was connected to another flask with 3 mL of diethyl ether for gas phase diffusion. In the course of ether diffusion, yellow crystals and colorless crystals (of 52 and (54)·(THF)2, respectively) formed simultaneously. Judged by observation, the product mixture contained near equal amounts of the two products. They were suitable for single-crystal X-ray diffraction analysis. In a repeated attempt, using a slight excess of silane 4 to suppress formation of the CuCl-rich by-product (54)·(THF)2 (batch size: 1.20 g, 3.24 mmol of 4; 280 mg, 2.83 mmol of CuCl, 3 mL of THF, 5 mL of diethyl ether), colorless crystals of (54)·(THF)2 were obtained exclusively. After 2 d, the supernatant was removed by decantation, the crystals were washed with 2 × 2 mL of a mixture (1:1) of THF and diethyl ether and briefly dried in vacuum. Yield: 0.88 g (0.687 mmol, 97%) of (54)·(THF)2. Elemental analysis indicated loss of solvent during sample preparation, the composition is close to solvent free: Calculated for C44H36Cl4Cu4N4O4Si2 (1145.01 g·mol−1): C, 46.48%; H, 3.19%; N, 4.93%; found C, 46.07%; H, 3.40%; N, 4.81%. 29Si{1H} NMR (CP/MAS): δiso (ppm) −32.4.
Compound (53)·(CHCl3) Ph2Si(μ2-pyO)2(CuCl)3(μ2-pyO)2SiPh2·(CHCl3) (C45H37Cl6Cu3N4O4Si2). Under dry argon atmosphere, a Schlenk flask was charged with magnetic stirring bar, silane 4 (0.735 g, 1.98 mmol) and CuCl (180 mg, 1.82 mmol), whereafter chloroform (2 mL) was added. The resultant mixture was briefly stirred at room temperature until complete dissolution of CuCl was achieved to afford a light yellow (slightly greenish-yellow) solution, which was connected to another flask with 5 mL of diethyl ether for gas phase diffusion. During ether diffusion, colorless crystals of (53)·(CHCl3) formed. After 1 week, the supernatant was removed by decantation, the crystals were washed with 2 mL of a mixture (1:1) of chloroform and diethyl ether and briefly dried in vacuum. Yield: 0.45 g (0.389 mmol, 64%) of (53)·(CHCl3). Elemental analysis indicated loss of solvent during sample preparation, the composition corresponds to chloroform content of 0.4 CHCl3: Calculated for C44H36Cl3Cu3N4O4Si2 0.4(CHCl3) (1085.70 g·mol−1): C, 49.12%; H, 3.38%; N, 5.16%; found C, 49.05%; H, 3.57%; N, 5.15%. 29Si{1H} NMR (CP/MAS): δiso (ppm) −32.9, −33.5.
4. Conclusions
With the successful syntheses of further Cu(I) complexes of the type RSi(μ2-pyO)3CuCl (2b: R = Ph, 2c: R = Bn) we have shown that silanes of the type RSi(pyO)3 are suitable tridentate N-donor ligands for complex formation with Cu(I). If R = Allyl, this hydrocarbyl group serves as an alternative ligand site, and complex AllylSi(μ2-pyO)3CuCl (2d) isomerizes to the more stable isomer (κO-pyO)Si(μ2-pyO)2(μ2-Allyl)CuCl (2d’). The inherent allyl group activation by Cu(I) coordination of the latter asks for further exploration of 2d’ as an allyl transfer reagent. As shown by 29Si and 63Cu solid-state NMR spectroscopy for the couple of 2c and 2d’, the switch from (pyO)3- to (pyO)2(Allyl)-Cu-coordination causes noticeable differences in the 29Si isotropic chemical shift and in the 63Cu chemical shift anisotropy.
A deliberate synthesis of paddlewheel compounds of the type RSi(μ2-pyO)4CuCl (3a: R = Me, 3b: R = Ph, 3c: R = Bn) has been established. The use of dry air (in stoichiometric ratio with respect to oxygen contained therein) and of excess of the respective silane RSi(pyO)3 to account for pyO-group delivery and oxide trapping proved successful. The corresponding allyl compound 3d (AllylSi(μ2-pyO)4CuCl), however, was obtained in small amounts only.
Crystal structure analyses of the series 2a–2d and 3a–3d indicated that the Cu···Si distance is not significantly responsive to the Si-bound hydrocarbyl group in series 2, whereas a trend of slight Si···Cu approach with increasing electronegativity of R is indicated in series 3. Especially in the series 2, the Cu···Si atom distances of ca. 3.2 Å should be interpreted as a close spatial proximity without noticeable effects of Si hypercoordination. Computational comparison of the 29Si NMR shifts of PhSi(pyO)3 (1b), PhSi(μ2-pyO)3CuCl (2b) and hypothetical compound PhSi(μ2-pyO)3LiCl (2bLi) indicated that the upfield shift of the Si NMR signal in compounds 2 relative to those of the corresponding silane 1 mainly arises from polarization through coordination of a cation in the (pyO)3 clamp, not necessarily from lone pair donation of this cation toward Si.
In solution, the ligands in compounds of the type 2 are very mobile, even in the absence of competing donor arms (such as Allyl) their 1H and 13C NMR spectra exhibit signs of exchange processes in CDCl3 solution (broad signals of pyO-based H and C atoms). Upon replacing one of the three pyO buttresses by a non-coordinating group, i.e., going to silane Ph2Si(pyO)2 (4), CuCl-coordination of such a silane involves Cu(μ2-Cl)2Cu bridging. The variety of ladder-type (CuCl)n-complexes obtained (with n = 2, 3, 4 for compounds 52, 53 and 54, respectively) in spite of using compound 4 in excess demonstrates the preference given to this Cu(μ2-Cl)2Cu coordination mode and points at the challenges (in deliberate syntheses and isolation and characterization of pure products) arising therefrom for further studies.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/inorganics11010002/s1, Crystallographic data for the compounds reported in this paper (in CIF format) and a document containing the following: Details of syntheses and NMR spectroscopic data of BnSi(pyO)3 (1c) and AllylSi(pyO)3 (1d); graphics of molecular structures of BnSi(pyO)3 (1c) and AllylSi(pyO)3 (1d) in their crystal structures (Figures S1 and S2) and selected bond lengths and angles (Tables S1–S4); graphics of solution state NMR spectra (1H, 13C{1H} and 29Si{1H}) of CDCl3 solutions of compounds 1c, 1d, (2b)2·(THF), 2c, 2d′, (53)·(CHCl3) and (54)·(THF)2 (Figures S3–S23); graphics of 29Si CP/MAS NMR spectra of (2b)2·(THF), 2c, 2d′, (53)·(CHCl3) and (54)·(THF)2 (Figures S24–S29); graphical representation of the NEB scan of the interconversion of isomers 2d, 2d′ (Figure S30); graphics of optimized molecular structures and total energies (Figures S31–S33) as well as atomic coordinates (Tables S5–S7) of compounds 2d, 2d′ and the transition state (TS) of their interconversion; graphics of optimized molecular structures and total energies (Figures S34–S37) as well as atomic coordinates (Tables S8–S11) of tetramethylsilane and of compounds 1b, 2b, 2bLi which were used for calculation of their 29Si NMR shifts.
Author Contributions
Conceptualization, J.W.; investigation, A.S., R.G., E.B. and J.W.; writing—original draft preparation, J.W.; writing—review and editing, R.G., E.B. and J.W.; visualization, R.G. and J.W.; supervision, J.W. All authors have read and agreed to the published version of the manuscript.
Funding
The authors are grateful for computing time at the High-Performance Computing Cluster at TU Bergakademie Freiberg, which was funded by Deutsche Forschungsgemeinschaft (DFG)—397252409.
Data Availability Statement
Not applicable.
Acknowledgments
The authors are grateful to the students Sara Braun and Paul Daßler for the syntheses of some samples of 1c, 1d and 2c, to Beate Kutzner (TU Bergakademie Freiberg, Institut für Anorganische Chemie) for solution NMR service and to Franziska Gründler and Mareike Weigel (TU Bergakademie Freiberg, Institut für Anorganische Chemie) for elemental microanalysis service.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table A1.
Crystallographic data from data collection and refinement for 1c, 1d and (2b)2 (THF).
| Parameter | 1c | 1d 1 | (2b)2 (THF) |
|---|---|---|---|
| Formula | C22H19N3O3Si | C18H17N3S3Si | C46H42Cl2Cu2N6O7Si2 |
| Mr | 401.49 | 351.43 | 1045.04 |
| T(K) | 180(2) | 180(2) | 200(2) |
| λ(Å) | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | monoclinic | triclinic | monoclinic |
| Space group | P21/c | P | P21/n |
| a(Å) | 10.6121(6) | 9.7675(8) | 9.1764(4) |
| b(Å) | 17.1874(9) | 10.6315(8) | 18.4453(10) |
| c(Å) | 11.2970(6) | 17.7868(16) | 13.3241(5) |
| α(°) | 90 | 78.951(6) | 90 |
| β(°) | 92.385(4) | 88.972(7) | 91.991(3) |
| γ(°) | 90 | 77.779(6) | 90 |
| V(Å3) | 2058.72(19) | 1771.2(3) | 2253.89(18) |
| Z | 4 | 4 | 2 |
| ρcalc(g·cm−1) | 1.30 | 1.32 | 1.54 |
| μMoKα (mm−1) | 0.1 | 0.2 | 1.2 |
| F(000) | 840 | 736 | 1072 |
| θmax(°), Rint | 28.0, 0.0309 | 25.0, 0.0560 | 28.0, 0.0395 |
| Completeness | 99.9% | 99.8% | 99.9% |
| Reflns collected | 24,896 | 16,879 | 23,599 |
| Reflns unique | 4961 | 4242 | 5437 |
| Restraints | 0 | 20 | 4 |
| Parameters | 263 | 477 | 316 |
| GoF | 1.118 | 1.025 | 1.068 |
| R1, wR2 [I > 2σ(I)] | 0.0423, 0.1032 | 0.0435, 0.0905 | 0.0341, 0.0770 |
| R1, wR2 (all data) | 0.0613, 0.1176 | 0.0794, 0.1011 | 0.0507, 0.0848 |
| Largest peak/hole (e·Å−3) | 0.28, −0.36 | 0.19, −0.26 | 0.39, −0.37 |
1 The asymmetric unit consists of two molecules of this compound. Both molecules feature disordered allyl groups, which were refined with site occupancies of 0.917(5), 0.083(5) and 0.750(8), 0.260(8) for molecule 1 and 2, respectively.
Table A2.
Crystallographic data from data collection and refinement for 2c, 2d and 2d′.
| Parameter | 2c 1 | 2d 2 | 2d′ |
|---|---|---|---|
| Formula | C22H19ClCuN3O3Si | C18H17ClCuN3O3Si | C18H17ClCuN3O3Si |
| Mr | 500.48 | 450.42 | 450.42 |
| T(K) | 200(2) | 200(2) | 180(2) |
| λ(Å) | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | orthorhombic | monoclinic | triclinic |
| Space group | Pca21 | P21/c | P |
| a(Å) | 15.3617(7) | 9.0369(2) | 8.7413(3) |
| b(Å) | 8.6072(5) | 23.5711(6) | 10.4028(3) |
| c(Å) | 16.3936(7) | 9.6583(2) | 10.7168(3) |
| α(°) | 90 | 90 | 80.052(3) |
| β(°) | 90 | 108.839(2) | 80.903(2) |
| γ(°) | 90 | 90 | 89.273(3) |
| V(Å3) | 2167.58(18) | 1947.10(8) | 947.69(5) |
| Z | 4 | 4 | 2 |
| ρcalc(g·cm−1) | 1.53 | 1.54 | 1.58 |
| μMoKα (mm−1) | 1.2 | 1.3 | 1.4 |
| F(000) | 1024 | 920 | 460 |
| θmax(°), Rint | 28.0, 0.0273 | 28.0, 0.0375 | 28.0, 0.0299 |
| Completeness | 99.9% | 100% | 100% |
| Reflns collected | 20,601 | 32,981 | 19,166 |
| Reflns unique | 5100 | 4701 | 4570 |
| Restraints | 1 | 27 | 0 |
| Parameters | 280 | 285 | 244 |
| GoF | 1.087 | 1.081 | 1.101 |
| R1, wR2 [I > 2σ(I)] | 0.0235, 0.0557 | 0.0336, 0.0790 | 0.0303, 0.0732 |
| R1, wR2 (all data) | 0.0276, 0.0583 | 0.0414, 0.0826 | 0.0353, 0.0753 |
| Largest peak/hole (e·Å−3) | 0.22, −0.24 | 0.51, −0.32 | 0.31, −0.43 |
1 The absolute structure parameter χFlack of this non-centrosymmetric structure refined to −0.010(5). 2 The allyl group of this compound is disordered over two positions, and this disorder causes a disorder of the Si(pyO)3 moiety. Thus, except for the CuCl moiety, the molecule has been refined disordered over two sites with site occupancies 0.901(4) and 0.099(4). For the sake of a stable refinement, the pyridine rings of the part of low occupancy were included in the refinement as ideal hexagons (using the AFIX66 instruction).
Table A3.
Crystallographic data from data collection and refinement for 3b, 3c and 3d.
| Parameter | 3b | 3c | 3d 1 |
|---|---|---|---|
| Formula | C26H21ClCuN4O4Si | C27H23ClCuN4O4Si | C23H21ClCuN4O4Si |
| Mr | 580.55 | 594.57 | 544.52 |
| T(K) | 200(2) | 200(2) | 200(2) |
| λ(Å) | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | orthorhombic | monoclinic | monoclinic |
| Space group | Pbca | P21/c | P21/c |
| a(Å) | 15.7556(8) | 15.8215(9) | 15.2140(3) |
| b(Å) | 18.2565(7) | 9.8221(4) | 9.6459(2) |
| c(Å) | 17.2013(6) | 17.4885(10) | 17.0865(3) |
| α(°) | 90 | 90 | 90 |
| β(°) | 90 | 109.961(4) | 108.677(1) |
| γ(°) | 90 | 90 | 90 |
| V(Å3) | 4947.8(4) | 2554.5(2) | 2375.44(8) |
| Z | 8 | 4 | 4 |
| ρcalc(g·cm−1) | 1.56 | 1.55 | 1.52 |
| μMoKα (mm−1) | 1.1 | 1.0 | 1.1 |
| F(000) | 2376 | 1220 | 1116 |
| θmax(°), Rint | 28.0, 0.0550 | 26.0, 0.0312 | 26.0, 0.0677 |
| Completeness | 99.6% | 99.8% | 100% |
| Reflns collected | 26,190 | 26,505 | 33,151 |
| Reflns unique | 5951 | 5012 | 4670 |
| Restraints | 0 | 0 | 2 |
| Parameters | 334 | 343 | 326 |
| GoF | 1.011 | 1.081 | 1.075 |
| R1, wR2 [I > 2σ(I)] | 0.0356, 0.0787 | 0.0291, 0.0704 | 0.0367, 0.0769 |
| R1, wR2 (all data) | 0.0656, 0.0877 | 0.0412, 0.0747 | 0.0517, 0.0817 |
| Largest peak/hole (e·Å−3) | 0.48, −0.61 | 0.38, −0.41 | 0.31, −0.44 |
1 The allyl group of this compound is disordered over two positions and has been refined with site occupancies 0.685(12) and 0.315(12).
Table A4.
Crystallographic data from data collection and refinement for 52, (54)·(THF)2 and (53)·(CHCl3).
Table A4.
Crystallographic data from data collection and refinement for 52, (54)·(THF)2 and (53)·(CHCl3).
| Parameter | 52 | (54) (THF)2 1 | (53) (CHCl3) 2 |
|---|---|---|---|
| Formula | C44H36Cl2Cu2N4O4Si2 | C52H52Cl4Cu4N4O6Si2 | C45H37Cl6Cu3N4O4Si2 |
| Mr | 938.93 | 1281.11 | 1157.28 |
| T(K) | 180(2) | 180(2) | 180(2) |
| λ(Å) | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | triclinic | triclinic | monoclinic |
| Space group | P | P | Pn |
| a(Å) | 9.6434(6) | 9.6236(3) | 8.9766(2) |
| b(Å) | 10.4351(6) | 12.6110(4) | 19.8415(3) |
| c(Å) | 12.0659(7) | 12.8592(4) | 13.5256(3) |
| α(°) | 100.595(5) | 110.726(2) | 90 |
| β(°) | 110.075(5) | 107.692(2) | 92.846(2) |
| γ(°) | 106.546(5) | 98.836(2) | 90 |
| V(Å3) | 1038.34(12) | 1329.53(8) | 2406.06(8) |
| Z | 1 | 1 | 2 |
| ρcalc(g·cm−1) | 1.50 | 1.60 | 1.60 |
| μMoKα (mm−1) | 1.3 | 1.9 | 1.7 |
| F(000) | 480 | 652 | 1168 |
| θmax(°), Rint | 28.0, 0.0293 | 28.0, 0.0387 | 28.0, 0.0547 |
| Completeness | 100% | 100% | 99.9% |
| Reflns collected | 16,743 | 25,647 | 52,253 |
| Reflns unique | 5010 | 6424 | 11,597 |
| Restraints | 0 | 0 | 32 |
| Parameters | 262 | 316 | 603 |
| GoF | 1.072 | 1.038 | 1.086 |
| R1, wR2 [I > 2σ(I)] | 0.0334, 0.0838 | 0.0346, 0.0833 | 0.0330, 0.0697 |
| R1, wR2 (all data) | 0.0391, 0.0865 | 0.0469, 0.0876 | 0.0418, 0.0737 |
| Largest peak/hole (e·Å−3) | 0.90, −0.45 | 0.40, −0.48 | 0.25, −0.46 |
1 This structure features two heavy disorders: (1) The [Ph2Si(pyO)2(CuCl)2]2 molecule is located on a crystallographically imposed center of inversion, but the central (CuCl)4 moiety does not adhere to inversion symmetry. Thus, the entire (CuCl)4 moiety was modeled with site occupancy 0.5, and the center of inversion generates its alternative orientation within the peripheral Ph2Si(pyO)2 clamps. (2) The structure features solvent of crystallization (THF), which was found severely disordered and could not be refined to satisfactory extent. Thus, the data set was treated with SQUEEZE as implemented in PLATON [65,66,67]. This procedure detected, per unit cell, solvent accessible volume of 271 Å3 and contributions of 78 electrons therein (in accordance with 80 electrons for the two THF molecules per unit cell, which have been omitted from refinement). 2 The absolute structure parameter χFlack of this non-centrosymmetric structure refined to −0.004(5). The molecule of solvent of crystallization (chloroform) is disordered and was refined in two positions with site occupancies 0.35(3) and 0.65(3).
References
- Avent, A.G.; Gehrhus, B.; Hitchcock, P.B.; Lappert, M.F.; Maciejewski, H. Synthesis and characterisation of bis(amino)silylene–nickel(0), –palladium(II), –platinum(0), –platinum(II) and copper(I) complexes. J. Organomet. Chem. 2003, 686, 321–331. [Google Scholar] [CrossRef]
- Paesch, A.N.; Kreyenschmidt, A.-K.; Herbst-Irmer, R.; Stalke, D. Side-Arm Functionalized Silylene Copper(I) Complexes in Catalysis. Inorg. Chem. 2019, 58, 7000–7009. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wang, Y.; Xie, Y.; Wei, P.; Gilliard, R.J., Jr.; Schwartz, N.A.; Schaefer, H.F., III; von Schleyer, R.P.; Robinson, G.H. Dynamic Complexation of Copper(I) Chloride by Carbene-Stabilized Disilicon. Chem. Eur. J. 2014, 20, 9208–9211. [Google Scholar] [CrossRef]
- Gualco, P.; Amgoune, A.; Miqueu, K.; Ladeira, S.; Bourissou, D. A Crystalline σ Complex of Copper. J. Am. Chem. Soc. 2011, 133, 4257–4259. [Google Scholar] [CrossRef] [PubMed]
- Plotzitzka, J.; Kleeberg, C. [(NHC)CuI−ER3 ] Complexes (ER3 = SiMe2Ph, SiPh3, SnMe3): From Linear, Mononuclear Complexes to Polynuclear Complexes with Ultrashort CuI···CuI Distances. Inorg. Chem. 2016, 55, 4813–4823. [Google Scholar] [CrossRef] [PubMed]
- Plotzitzka, J.; Kleeberg, C. [(18-C-6)K][(N≡C)CuI−SiMe2Ph], a Potassium Silylcyanocuprate as a Catalyst Model for Silylation Reactions with Silylboranes: Syntheses, Structures, and Catalytic Properties. Inorg. Chem. 2017, 56, 6671–6680. [Google Scholar] [CrossRef]
- Klett, J.; Klinkhammer, K.W.; Niemeyer, M. Ligand Exchange between Arylcopper Compounds and Bis(hypersilyl)tin or Bis(hypersilyl)lead: Synthesis and Characterization of Hypersilylcopper and a Stannanediyl Complex with a Cu−Sn Bond. Chem. Eur. J. 1999, 5, 2531–2536. [Google Scholar] [CrossRef]
- Frogley, B.J.; Hill, A.F.; Sharma, M.; Sinha, A.; Ward, J.S. Semi-bridging σ-silyls as Z-type ligands. Chem. Commun. 2020, 56, 3532–3535. [Google Scholar] [CrossRef]
- Nova, A.; Suh, H.-W.; Schmeier, T.J.; Guard, L.M.; Eisenstein, O.; Hazari, N.; Maseras, F. An Unusual Example of Hypervalent Silicon: A Five-Coordinate Silyl Group Bridging Two Palladium or Nickel Centers through a Nonsymmetrical Four-Center Two-Electron Bond. Angew. Chem. Int. Ed. 2014, 53, 1103–1108. [Google Scholar] [CrossRef]
- Green, M.L.H. A new approach to the formal classification of covalent compounds of the elements. J. Organomet. Chem. 1995, 500, 127–148. [Google Scholar] [CrossRef]
- Chuit, C.; Corriu, R.J.P.; Reye, C.; Young, J.C. Reactivity of Penta- and Hexacoordinate Silicon Compounds and Their Role as Reaction Intermediates. Chem. Rev. 1993, 93, 1371–1448. [Google Scholar] [CrossRef]
- Wagler, J.; Böhme, U.; Kroke, E. Higher Coordinated Molecular Silicon Compounds. Struct. Bond. 2014, 155, 29–105. [Google Scholar] [CrossRef]
- Lemière, G.; Millanvois, A.; Ollivier, C.; Fensterbank, L. A Parisian Vision of the Chemistry of Hypercoordinated Silicon Derivatives. Chem. Rec. 2021, 21, 1119–1129. [Google Scholar] [CrossRef] [PubMed]
- Grobe, J.; Wehmschulte, R.; Krebs, B.; Läge, M. Alternativ-Liganden. XXXII Neue Tetraphosphan-Nickelkomplexe mit Tripod-Liganden des Typs XM’(OCH2PMe2)n(CH2CH2PR2)3-n (M‘ = Si, Ge; n = 0–3). Z. Anorg. Allg. Chem. 1995, 621, 583–596. [Google Scholar] [CrossRef]
- Grobe, J.; Krummen, N.; Wehmschulte, R.; Krebs, B.; Läge, M. Alternativ-Liganden. XXXI Nickelcarbonylkomplexe mit Tripod-Liganden des Typs XM’(OCH2PMe2)n(CH2CH2PR2)3-n (M‘ = Si, Ge; n = 0–3). Z. Anorg. Allg. Chem. 1994, 620, 1645–1658. [Google Scholar] [CrossRef]
- Grobe, J.; Lütke-Brochtrup, K.; Krebs, B.; Läge, M.; Niemeyer, H.-H.; Würthwein, E.-U. Alternativ-Liganden XXXVIII. Neue Versuche zur Synthese von Pd(0)- und Pt(0)-Komplexen des Tripod-Phosphanliganden FSi(CH2CH2PMe2)3. Z. Naturforsch. 2007, 62, 55–65. [Google Scholar] [CrossRef]
- Gualco, P.; Lin, T.-P.; Sircoglou, M.; Mercy, M.; Ladeira, S.; Bouhadir, G.; Pérez, L.M.; Amgoune, A.; Maron, L.; Gabbaï, F.P.; et al. Gold–Silane and Gold–Stannane Complexes: Saturated Molecules as σ-Acceptor Ligands. Angew. Chem. Int. Ed. 2009, 48, 9892–9895. [Google Scholar] [CrossRef]
- Wagler, J.; Brendler, E. Metallasilatranes: Palladium(II) and Platinum(II) as Lone-Pair Donors to Silicon(IV). Angew. Chem. Int. Ed. 2010, 49, 624–627. [Google Scholar] [CrossRef]
- Wahlicht, S.; Brendler, E.; Heine, T.; Zhechkov, L.; Wagler, J. 7-Azaindol-1-yl(organo)silanes and Their PdCl2 Complexes: Pd-Capped Tetrahedral Silicon Coordination Spheres and Paddlewheels with a Pd-Si Axis. Organometallics 2014, 33, 2479–2488. [Google Scholar] [CrossRef]
- Ehrlich, L.; Gericke, R.; Brendler, E.; Wagler, J. (2-Pyridyloxy)silanes as Ligands in Transition Metal Coordination Chemistry. Inorganics 2018, 6, 119. [Google Scholar] [CrossRef]
- Lyssenko, K.A.; Korlyukov, A.A.; Antipin, M.Y.; Knyazev, S.P.; Kirin, V.N.; Alexeev, N.V.; Chernyshev, E.A. The nature of the intramolecular transannular Si···N interaction in crystalline 1-methylsilatrane, as found from X-ray diffraction data. Mendeleev Commun. 2000, 10, 88–90. [Google Scholar] [CrossRef]
- Párkányi, L.; Simon, K.; Nagy, J. Crystal and molecular structure of [beta]-1-phenylsilatrane, C12H17NSi. Acta Crystallogr. B 1974, 30, 2328–2332. [Google Scholar] [CrossRef]
- Párkányi, L.; Nagy, J.; Simon, K. Crystal and molecular structures of γ-1-phenylsilatrane: Some structural features of silatranes. J. Organomet. Chem. 1975, 101, 11–18. [Google Scholar] [CrossRef]
- White, J.M.; Jones, S. Low-temperature structure of allyl silatrane. Acta Crystallogr. C 1999, 55, 962–963. [Google Scholar] [CrossRef]
- Kuß, S.; Brendler, E.; Wagler, J. Molecular Structures of the Pyridine-2-olates PhE(pyO)3 (E = Si, Ge, Sn) − [4+3]-Coordination at Si, Ge vs. Heptacoordination at Sn. Crystals 2022, 12, 1802. [Google Scholar] [CrossRef]
- Seidel, A.; Weigel, M.; Ehrlich, L.; Gericke, R.; Brendler, E.; Wagler, J. Molecular Structures of the Silicon Pyridine-2-(thi)olates Me3Si(pyX), Me2Si(pyX)2 and Ph2Si(pyX)2 (py = 2-Pyridyl, X = O, S), and Their Intra- and Intermolecular Ligand Exchange in Solution. Crystals 2022, 12, 1054. [Google Scholar] [CrossRef]
- Okuniewski, A.; Rosiak, D.; Chojnaki, J.; Becker, B. Coordination polymers and molecular structures among complexes of mercury(II) halides with selected 1-benzoylthioureas. Polyhedron 2015, 90, 47–57. [Google Scholar] [CrossRef]
- Dużak, T.; Kinzhybalo, V.; Ślepokura, K.; Olijnyk, V. Tetraallylsilane π−Complexation: Synthesis and Structure of [Cu5Cl5(CH2−CH=CH2)4Si]. Z. Anorg. Allg. Chem. 2009, 635, 2324–2327. [Google Scholar] [CrossRef]
- Kamei, T.; Fujita, K.; Itami, K.; Yoshida, J. Copper-Catalyzed Allylation of Carbonyl Derivatives Using Allyl(2-pyridyl)silanes. Org. Lett. 2005, 7, 4725–4728. [Google Scholar] [CrossRef]
- Shi, C.; Xi, X.; Hou, Z.; Liu, E.; Wang, W.; Jin, S.; Wu, Y.; Wu, G. Atomic-Level Characterization of Dynamics of Copper Ions in CuAgSe. J. Phys. Chem. C 2016, 120, 3229–3234. [Google Scholar] [CrossRef]
- Endo, K.; Yamamoto, K.; Deguchi, K.; Matsushita, K. 63Cu High Resolution NMR of Cu(I) Halides in Aqueous Solution and Suspension. Bull. Chem. Soc. Jpn. 1987, 60, 2803–2807. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.K.; Becker, E.D.; Cabral de Menzenes, S.M.; Goodfellow, R.; Granger, P. Nuclear spin properties and conventions for the chemical shift. Pure Appl. Chem. 2001, 73, 1795–1818. [Google Scholar] [CrossRef]
- Sakida, S.; Kato, N.; Kawamoto, Y. 63Cu magic angle spinning (MAS) nuclear magnetic resonance (NMR) study of CuX crystals (X = Cl, Br, and I) and CuX-based glasses (X = Cl, Br, and I). Mat. Res. Bull. 2002, 37, 2263–2274. [Google Scholar] [CrossRef]
- Tang, J.A.; Ellis, B.D.; Warren, T.H.; Hanna, J.V.; Macdonald, C.L.B.; Schurko, R.W. Solid-State 63Cu and 65Cu NMR Spectroscopy of Inorganic and Organometallic Copper(I) Complexes. J. Am. Chem. Soc. 2007, 129, 13049–13065. [Google Scholar] [CrossRef]
- Massiot, D.; Fayon, F.; Capron, M.; King, I.; Le Calvlé, S.; Alonso, B.; Durand, J.-O.; Bujoli, B.; Gan, Z.; Hoatson, G. Modeling one- and two-dimensional Solid State NMR spectra. Magn. Reson. Chem. 2002, 40, 70–76. [Google Scholar] [CrossRef]
- Gericke, R.; Wagler, J. Coordination and Electrochemical Switching on Paddle-Wheel Complexes Containing an As−Ru or a Sb−Ru Axis. Inorg. Chem. 2021, 60, 18122–18132. [Google Scholar] [CrossRef]
- Dattelbaum, A.M.; Martin, J.D. Benzene−Copper(I) Coordination in a Bimetallic Chain Complex. Inorg. Chem. 1999, 38, 6200–6205. [Google Scholar] [CrossRef] [PubMed]
- Dattelbaum, A.M.; Martin, J.D. Benzene and ethylene binding to copper(I)–ziconium(IV) chloride materials: The crystal structure and solid-state reactivity of ((bz)2Cu)2Zr2Cl10 · bz. Polyhedron 2006, 25, 349–359. [Google Scholar] [CrossRef]
- Daly, S.; Haddow, M.F.; Orpen, A.G.; Rolls, G.T.A.; Wass, D.F.; Wingad, R.L. Copper(I) Diphosphine Catalysts for C-N Bond Formation: Synthesis, Structure, and Ligand Effects. Organometallics 2008, 27, 3196–3202. [Google Scholar] [CrossRef]
- Chen, B.-L.; Mok, K.-F.; Ng, S.-C. Synthesis, crystal structures and dynamic NMR studies of novel trinuclear copper(I) halide complexes with 2,5-bis[(diphenylphosphino)-methyl]thiophene. J. Chem. Soc., Dalton Trans. 1998, 2861–2866. [Google Scholar] [CrossRef]
- Collins, L.R.; Lowe, J.P.; Mahon, M.F.; Poulten, R.C.; Whittlesey, M.K. Copper Diamidocarbene Complexes: Characterization of Monomeric to Tetrameric Species. Inorg. Chem. 2014, 53, 2699–2707. [Google Scholar] [CrossRef] [PubMed]
- Crestani, M.G.; Manbeck, G.F.; Brennessel, W.W.; McCormick, T.M.; Eisenberg, R. Synthesis and Characterization of Neutral Luminescent Diphosphine Pyrrole- and Indole-Aldimine Copper(I) Complexes. Inorg. Chem. 2011, 50, 7172–7188. [Google Scholar] [CrossRef] [PubMed]
- Köhn, R.D.; Laudo, L.D.; Pan, Z.; Speiser, F.; Kociok-Köhn, G. Triangular tricopper(I) clusters supported by donor-substituted triazacyclohexanes. Dalton Trans. 2009, 4556–4568. [Google Scholar] [CrossRef] [PubMed]
- Dyason, J.C.; Healy, P.C.; Engelhardt, L.M.; Pakawatchai, C.; Patrick, V.A.; Raston, C.L.; White, A.H. Lewis-base adducts of Group 1B metal(I) compounds. Part 16. Synthesis, structure, and solid-state phosphorus-31 nuclear magnetic resonance spectra of some novel [Cu4X4L4](X = halogen, L = N, P base) ‘cubane’ clusters. J. Chem. Soc. Dalton Trans. 1985, 14, 831–838. [Google Scholar] [CrossRef] [Green Version]
- Noor, A.; Shahzad, A.; Khan, E.; Tahir, M.N.; Khan, G.S.; ur Rashid, A.; Said, M. Polynuclear Cu(I) and Ag(I) Complexes of 1,3-Diisobutyl Thiourea, Synthesis, Crystal Structure and Antioxidant Potentials. Inorganics 2022, 10, 185. [Google Scholar] [CrossRef]
- Kämpfe, A.; Brendler, E.; Kroke, E.; Wagler, J. Tp*Cu(I)–CN–SiL2–NC–Cu(I)Tp*—A hexacoordinate Si-complex as connector for redox active metals via π-conjugated ligands. Dalton Trans. 2015, 44, 4744–4750. [Google Scholar] [CrossRef] [Green Version]
- Gericke, R.; Wagler, J. Ruthenium complexes of phosphino derivatives of carboxylic amides: Synthesis and characterization of tridentate P,E2 and tetradentate P,E3 (E = N,O) ligands and their reactivity towards [RuCl2(PPh3)3]. Polyhedron 2017, 125, 57–67. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Program for the Solution of Crystal Structures; SHELXS-97; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Program for the Refinement of Crystal Structures; SHELXL-2014/7; University of Göttingen: Göttingen, Germany, 2014. [Google Scholar]
- Sheldrick, G.M. Program for the Refinement of Crystal Structures; SHELXL-2018/3; University of Göttingen: Göttingen, Germany, 2018. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Farrugia, L.J. ORTEP-3 for windows—A version of ORTEP-III with a graphical user interface (GUI). J. Appl. Crystallogr. 1997, 30, 565. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- POV-RAY, (Version 3.7). Trademark of Persistence of Vision Raytracer Pty. Hallam Oaks Pty. Ltd.: Williamstown, Australia, 1991–2013. Available online: http://www.povray.org/download/(accessed on 28 June 2021).
- Neese, F. Software update: The ORCA program system—Version 5.0. WIREs Comput. Mol. Sci. 2022, 8, e1606. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Pantazis, D.A.; Neese, F. All-electron basis sets for heavy elements. WIREs Comput. Mol. Sci. 2014, 4, 363–374. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597. [Google Scholar] [CrossRef]
- Van Wüllen, C. Molecular density functional calculations in the regular relativistic approximation: Method, application to coinage metal diatomics, hydrides, fluorides and chlorides, and comparison with first-order relativistic calculations. J. Chem. Phys. 1998, 109, 392. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Stoychev, G.L.; Auer, A.A.; Neese, F. Automatic Generation of Auxiliary Basis Sets. J. Chem. Theory Comput. 2017, 13, 554–562. [Google Scholar] [CrossRef]
- Chemcraft, Version 1.8 (Build 164); 2016. Available online: http://www.chemcraftprog.com/ (accessed on 19 September 2015).
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Cryst. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. D 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. C 2015, 71, 9–18. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Selected examples of Si-Cu (I–VIII) and other Si-TM compounds (IX,X). For reasons of clarity of presentation, formal lone pair donation by L-type ligands (such as phosphanes, carbenes and silylenes) is indicated by a dative bond arrow, whereas X-type bonds (for example of silyl groups, regardless of two- or multi-center bonding thereof) are indicated by a regular bond dash.
Figure 1.
Selected examples of Si-Cu (I–VIII) and other Si-TM compounds (IX,X). For reasons of clarity of presentation, formal lone pair donation by L-type ligands (such as phosphanes, carbenes and silylenes) is indicated by a dative bond arrow, whereas X-type bonds (for example of silyl groups, regardless of two- or multi-center bonding thereof) are indicated by a regular bond dash.

Figure 2.
Selected examples of penta- and hexacoordinate Si compounds, which feature a TM atom as a formal lone pair donor in the Si coordination sphere (XI–XV) and compounds which serve as the starting point for the current study (compounds 1a–3a). For reasons of clarity of presentation, formal lone pair donation by L-type ligands (such as phosphanes) is indicated by a dative bond arrow, whereas X-type bonds (for example to alkyl groups) are indicated by a regular bond dash. Formal charges are included where necessary. Note: For the molecules which feature ambidentate buttressing ligands with both donor atoms involved in π-delocalized systems, further resonance forms may be drawn.
Figure 2.
Selected examples of penta- and hexacoordinate Si compounds, which feature a TM atom as a formal lone pair donor in the Si coordination sphere (XI–XV) and compounds which serve as the starting point for the current study (compounds 1a–3a). For reasons of clarity of presentation, formal lone pair donation by L-type ligands (such as phosphanes) is indicated by a dative bond arrow, whereas X-type bonds (for example to alkyl groups) are indicated by a regular bond dash. Formal charges are included where necessary. Note: For the molecules which feature ambidentate buttressing ligands with both donor atoms involved in π-delocalized systems, further resonance forms may be drawn.

Scheme 1.
Generic scheme of the syntheses of silanes RSi(pyO)3, (1b [25], 1c, 1d) and of Cu(I) complexes thereof (2b–2d), as well as isomerization of 2d into 2d′.
Scheme 1.
Generic scheme of the syntheses of silanes RSi(pyO)3, (1b [25], 1c, 1d) and of Cu(I) complexes thereof (2b–2d), as well as isomerization of 2d into 2d′.

Figure 3.
Molecular structures of (a) 2b (in the crystal structure of the solvate (2b)2·THF), (b) 2c and (c) 2d with thermal displacement ellipsoids at the 50% probability level and labels of selected non-hydrogen atoms. Selected interatomic distances and angles are listed in Table 1. For compound 2d, only the predominant part (site occupancy 0.9) of the disordered molecule is depicted. Hydrogen atoms are omitted for clarity.
Figure 3.
Molecular structures of (a) 2b (in the crystal structure of the solvate (2b)2·THF), (b) 2c and (c) 2d with thermal displacement ellipsoids at the 50% probability level and labels of selected non-hydrogen atoms. Selected interatomic distances and angles are listed in Table 1. For compound 2d, only the predominant part (site occupancy 0.9) of the disordered molecule is depicted. Hydrogen atoms are omitted for clarity.

Figure 4.
Molecular structure of 2d′ with thermal displacement ellipsoids at the 50% probability level and labels of selected non-hydrogen atoms. Hydrogen atoms are omitted for clarity. Selected interatomic distances (Å) and angles (deg): Cu1–Cl1 2.270(1), Cu1···Si1 3.302(1), Cu1–N1 2.047(2), Cu1–N2 2.347(2), Cu1–C17 2.095(2), Cu1–C18 2.077(2), Si1–O1 1.638(2), Si1–O2 1.637(2), Si1–O3 1.642(2), Si1···N3 2.852(2), Si1–C16 1.847(2), C16–C17 1.504(3), C17–C18 1.365(3), O1–Si1–O2 106.83(7), O1–Si1–O3 97.87(7), O2–Si1–O3 107.06(7), O1–Si1–C16 113.37(9), O2–Si1–C16 115.10(8), Cl1–Cu1–N1 102.05(5), Cl1–Cu1–N2 95.29(4), N1–Cu1–N2 104.01(6).
Figure 4.
Molecular structure of 2d′ with thermal displacement ellipsoids at the 50% probability level and labels of selected non-hydrogen atoms. Hydrogen atoms are omitted for clarity. Selected interatomic distances (Å) and angles (deg): Cu1–Cl1 2.270(1), Cu1···Si1 3.302(1), Cu1–N1 2.047(2), Cu1–N2 2.347(2), Cu1–C17 2.095(2), Cu1–C18 2.077(2), Si1–O1 1.638(2), Si1–O2 1.637(2), Si1–O3 1.642(2), Si1···N3 2.852(2), Si1–C16 1.847(2), C16–C17 1.504(3), C17–C18 1.365(3), O1–Si1–O2 106.83(7), O1–Si1–O3 97.87(7), O2–Si1–O3 107.06(7), O1–Si1–C16 113.37(9), O2–Si1–C16 115.10(8), Cl1–Cu1–N1 102.05(5), Cl1–Cu1–N2 95.29(4), N1–Cu1–N2 104.01(6).

Figure 5.
Optimized molecular structures of 2d′, 2d and the transition state (TS) for their interconversion with their relative Gibbs free energy (in kcal⋅mol−1) in parentheses.
Figure 5.
Optimized molecular structures of 2d′, 2d and the transition state (TS) for their interconversion with their relative Gibbs free energy (in kcal⋅mol−1) in parentheses.

Figure 6.
Aryl and olefin section of the 1H NMR spectra of (from top to bottom) 2b, 2c and 2d′ (as solutions in CDCl3, all referenced to internal SiMe4 at 0 ppm, cf. Figures S9, S12 and S17 in the Supplementary Materials) recorded at 400 MHz. The asterisks (*) indicate corresponding signals of their pyO moieties, the underbars indicate the relevant 1H signals of the different hydrocarbyl substituents Ph, Bn and Allyl, respectively.
Figure 6.
Aryl and olefin section of the 1H NMR spectra of (from top to bottom) 2b, 2c and 2d′ (as solutions in CDCl3, all referenced to internal SiMe4 at 0 ppm, cf. Figures S9, S12 and S17 in the Supplementary Materials) recorded at 400 MHz. The asterisks (*) indicate corresponding signals of their pyO moieties, the underbars indicate the relevant 1H signals of the different hydrocarbyl substituents Ph, Bn and Allyl, respectively.

Figure 7.
Aryl and olefin section of the 13C NMR spectra of (from top to bottom) 2b, 2c and 2d′ (as solutions in CDCl3, all referenced to internal SiMe4 at 0 ppm, cf. Figures S10, S13 and S18 in the Supplementary Materials). The asterisks (*) indicate corresponding signals of their pyO moieties, the underbars indicate the relevant 13C signals of the different hydrocarbyl substituents Ph, Bn and Allyl, respectively.
Figure 7.
Aryl and olefin section of the 13C NMR spectra of (from top to bottom) 2b, 2c and 2d′ (as solutions in CDCl3, all referenced to internal SiMe4 at 0 ppm, cf. Figures S10, S13 and S18 in the Supplementary Materials). The asterisks (*) indicate corresponding signals of their pyO moieties, the underbars indicate the relevant 13C signals of the different hydrocarbyl substituents Ph, Bn and Allyl, respectively.

Figure 8.
Aryl section of the 1H NMR spectra of 2c (as solutions in CDCl3, all referenced to internal SiMe4 at 0 ppm) at different temperatures, recorded at 500 MHz.
Figure 8.
Aryl section of the 1H NMR spectra of 2c (as solutions in CDCl3, all referenced to internal SiMe4 at 0 ppm) at different temperatures, recorded at 500 MHz.

Figure 9.
63Cu MAS NMR spectra of 2c and 2d′ recorded at υrot = 20 kHz and 23 kHz, respectively. The central band in the spectrum of 2c indicates the range of the location of δiso. In case of 2d′, an additional spectrum was recorded at a different spinning frequency to identify the band at 148 ppm as the region in which δiso is located.
Figure 9.
63Cu MAS NMR spectra of 2c and 2d′ recorded at υrot = 20 kHz and 23 kHz, respectively. The central band in the spectrum of 2c indicates the range of the location of δiso. In case of 2d′, an additional spectrum was recorded at a different spinning frequency to identify the band at 148 ppm as the region in which δiso is located.

Scheme 2.
Generic scheme of the syntheses of Cu(II) complexes (3a, 3b, 3c, 3d) from silanes RSi(pyO)3, (1a, 1b, 1c, 1d, respectively) and dry air.
Scheme 2.
Generic scheme of the syntheses of Cu(II) complexes (3a, 3b, 3c, 3d) from silanes RSi(pyO)3, (1a, 1b, 1c, 1d, respectively) and dry air.

Figure 10.
Molecular structures of (a) 3b, (b) 3c and (c) 3d with thermal displacement ellipsoids at the 50% probability level and labels of selected non-hydrogen atoms. Hydrogen atoms are omitted for clarity. Selected interatomic distances and angles are listed in Table 3.
Figure 10.
Molecular structures of (a) 3b, (b) 3c and (c) 3d with thermal displacement ellipsoids at the 50% probability level and labels of selected non-hydrogen atoms. Hydrogen atoms are omitted for clarity. Selected interatomic distances and angles are listed in Table 3.

Scheme 3.
Reaction of silane Ph2Si(pyO)2 (4) and CuCl with formation of compound 52 and sketches of products 53 and 54 obtained from this reaction.
Scheme 3.
Reaction of silane Ph2Si(pyO)2 (4) and CuCl with formation of compound 52 and sketches of products 53 and 54 obtained from this reaction.

Figure 11.
Molecular structures of (a) 52 and (b) 53 in its chloroform solvate 53·(CHCl3) with thermal displacement ellipsoids at the 50% probability level and labels of selected non-hydrogen atoms. Hydrogen atoms are omitted for clarity. Selected interatomic distances (Å) and angles (deg) for 52: Cu1–Cl1 2.301(1), Cu1–Cl1* 2.624(1), Cu1–N1 2.059(2), Cu1–N2 1.996(2), Si1–O1 1.644(2), Si1–O2 1.648(2), Cl1–Cu1–Cl1* 100.33(2), Cu1–Cl1–Cu1* 79.67(2), N1–Cu1–N2 122.71(6), O1–Si1–O2 111.90(7), C11–Si1–C17 114.45(8); 53: Cu1–Cl1 2.4499(13), Cu1–Cl2 2.5331(11), Cu2–Cl1 2.1578(13), Cu2–Cl2 2.5274(13), Cu2–Cl3 2.1685(12), Cu3–Cl2 2.5043(11), Cu3–Cl3 2.5512(12), Cu1–N1 1.990(4), Cu1–N2 2.000(4), Cu3–N3 1.986(3), Cu3–N4 1.994(3), Si1–O1 1.646(3), Si1–O2 1.650(3), Si2–O3 1.649(3), Si2–O4 1.648(3), Cl1–Cu1–Cl2 99.50(4), Cl2–Cu3–Cl3 94.96(4), N1–Cu1–N2 131.81(14), N3–Cu3–N4 136.28(14), O1–Si1–O2 109.99(15), C11–Si1–C17 116.62(19), O3–Si2–O4 109.94(15), C33–Si2–C39 118.73(19).
Figure 11.
Molecular structures of (a) 52 and (b) 53 in its chloroform solvate 53·(CHCl3) with thermal displacement ellipsoids at the 50% probability level and labels of selected non-hydrogen atoms. Hydrogen atoms are omitted for clarity. Selected interatomic distances (Å) and angles (deg) for 52: Cu1–Cl1 2.301(1), Cu1–Cl1* 2.624(1), Cu1–N1 2.059(2), Cu1–N2 1.996(2), Si1–O1 1.644(2), Si1–O2 1.648(2), Cl1–Cu1–Cl1* 100.33(2), Cu1–Cl1–Cu1* 79.67(2), N1–Cu1–N2 122.71(6), O1–Si1–O2 111.90(7), C11–Si1–C17 114.45(8); 53: Cu1–Cl1 2.4499(13), Cu1–Cl2 2.5331(11), Cu2–Cl1 2.1578(13), Cu2–Cl2 2.5274(13), Cu2–Cl3 2.1685(12), Cu3–Cl2 2.5043(11), Cu3–Cl3 2.5512(12), Cu1–N1 1.990(4), Cu1–N2 2.000(4), Cu3–N3 1.986(3), Cu3–N4 1.994(3), Si1–O1 1.646(3), Si1–O2 1.650(3), Si2–O3 1.649(3), Si2–O4 1.648(3), Cl1–Cu1–Cl2 99.50(4), Cl2–Cu3–Cl3 94.96(4), N1–Cu1–N2 131.81(14), N3–Cu3–N4 136.28(14), O1–Si1–O2 109.99(15), C11–Si1–C17 116.62(19), O3–Si2–O4 109.94(15), C33–Si2–C39 118.73(19).

Figure 12.
Molecular structure of 54 in its THF solvate 54·(THF)2 with thermal displacement ellipsoids at the 50% probability level and labels of selected non-hydrogen atoms. Section (a) shows the asymmetric unit, section (b) shows the central part of the molecule with positions of the Cu and Cl atoms, which result from disorder by symmetry. For each group of two Cu or Cl atoms, the atom labels are located next to the atom which is part of the asymmetric unit. (The molecule is located on a center of inversion. Only the periphery, the two Ph2Si(pyO)2 motifs, adhere to this symmetry, whereas the central Cu4Cl4 part appears disordered and its Cu and Cl atoms can be refined in two positions. As there had been no superstructure reflections in the data set, which would hint at a larger unit cell, and refinement in space group P1 did not eliminate this disorder, a selected set of the Cu and Cl atoms was chosen as a possible actual asymmetric unit.) Selected interatomic distances (Å) and angles (deg) for 54: Si1–O1 1.649(2), Si1–O2 1.650(2), O1–Si1–O2 111.92(7), C11–Si1–C17 113.58(8).
Figure 12.
Molecular structure of 54 in its THF solvate 54·(THF)2 with thermal displacement ellipsoids at the 50% probability level and labels of selected non-hydrogen atoms. Section (a) shows the asymmetric unit, section (b) shows the central part of the molecule with positions of the Cu and Cl atoms, which result from disorder by symmetry. For each group of two Cu or Cl atoms, the atom labels are located next to the atom which is part of the asymmetric unit. (The molecule is located on a center of inversion. Only the periphery, the two Ph2Si(pyO)2 motifs, adhere to this symmetry, whereas the central Cu4Cl4 part appears disordered and its Cu and Cl atoms can be refined in two positions. As there had been no superstructure reflections in the data set, which would hint at a larger unit cell, and refinement in space group P1 did not eliminate this disorder, a selected set of the Cu and Cl atoms was chosen as a possible actual asymmetric unit.) Selected interatomic distances (Å) and angles (deg) for 54: Si1–O1 1.649(2), Si1–O2 1.650(2), O1–Si1–O2 111.92(7), C11–Si1–C17 113.58(8).

Figure 13.
Selected examples of oligonuclear complexes with (CuCl)n cores (n = 1, 2, 3, 4; XVI–XXII) as well as an example of a complex with bridged (CuCl)-motifs (XXIII). For reasons of clarity of presentation, formal lone pair donation by L-type ligands (such as phosphanes) is indicated by a dative bond arrow, whereas X-type bonds (for example CuCl, even in case of bridging bonds of mixed L,X-type) are indicated by a regular bond dash.
Figure 13.
Selected examples of oligonuclear complexes with (CuCl)n cores (n = 1, 2, 3, 4; XVI–XXII) as well as an example of a complex with bridged (CuCl)-motifs (XXIII). For reasons of clarity of presentation, formal lone pair donation by L-type ligands (such as phosphanes) is indicated by a dative bond arrow, whereas X-type bonds (for example CuCl, even in case of bridging bonds of mixed L,X-type) are indicated by a regular bond dash.

Table 1.
Selected atom distances (Å) and bond angles (deg) in compounds 2b, 2c and 2d in their crystal structures (as well as some corresponding parameters of one molecule of 2a [20], the crystal structure of which features two independent molecules).
Table 1.
Selected atom distances (Å) and bond angles (deg) in compounds 2b, 2c and 2d in their crystal structures (as well as some corresponding parameters of one molecule of 2a [20], the crystal structure of which features two independent molecules).
| 2a | 2b | 2c | 2d | |
|---|---|---|---|---|
| Si1–O1 | 1.626(2) | 1.633(2) | 1.628(2) | 1.621(2) |
| Si1–O2 | 1.629(2) | 1.625(2) | 1.626(2) | 1.613(2) |
| Si1–O3 | 1.618(2) | 1.638(2) | 1.635(2) | 1.612(2) |
| Si1–C16 | 1.834(3) | 1.841(2) | 1.855(3) | 1.845(2) |
| Cu1–N1 | 2.038(2) | 2.060(2) | 2.037(2) | 2.061(2) |
| Cu1–N2 | 2.039(2) | 2.016(2) | 2.049(2) | 2.033(2) |
| Cu1–N3 | 2.023(2) | 2.055(2) | 2.073(2) | 2.021(2) |
| Cu1–Cl1 | 2.361(1) | 2.359(1) | 2.334(1) | 2.349(1) |
| Cu1···Si1 | 3.204(1) | 3.211(1) | 3.227(1) | 3.196(1) |
| O1–Si1–O2 | 111.41(13) | 113.18(9) | 112.21(10) | 110.99(12) |
| O1–Si1–O3 | 113.91(12) | 108.96(9) | 109.94(10) | 111.99(13) |
| O2–Si1–O3 | 111.89(12) | 113.92(9) | 110.54(11) | 114.26(14) |
| N1–Cu1–N2 | 112.39(8) | 115.97(7) | 111.40(9) | 108.02(11) |
| N1–Cu1–N3 | 113.67(8) | 103.62(7) | 111.64(8) | 113.16(11) |
| N2–Cu1–N3 | 114.46(8) | 119.47(7) | 113.13(9) | 118.75(12) |
| Cl1–Cu1···Si1 | 177.35(2) | 179.13(2) | 177.70(3) | 179.60(2) |
| Cu1···Si1–C16 | 177.55(13) | 174.58(7) | 174.01(9) | 178.09(10) |
Table 2.
29Si NMR shifts (in ppm relative to SiMe4) of silanes 1a [20], 1b [25], 1c, 1d and of the Cu(I) complexes thereof (2a [20], 2b, 2c, 2d′). The shift difference between silane (1) and Cu(I) complex (2) (both in CDCl3) is Δδ 29Si (1 vs. 2) = δ 29Si(2)–δ 29Si(1). The shift difference between Cu(I) complexes (2) in CDCl3 and in solid state is Δδ 29Si (2) = δiso 29Si(2)[CP/MAS]–δ 29Si(2) [CDCl3].
Table 2.
29Si NMR shifts (in ppm relative to SiMe4) of silanes 1a [20], 1b [25], 1c, 1d and of the Cu(I) complexes thereof (2a [20], 2b, 2c, 2d′). The shift difference between silane (1) and Cu(I) complex (2) (both in CDCl3) is Δδ 29Si (1 vs. 2) = δ 29Si(2)–δ 29Si(1). The shift difference between Cu(I) complexes (2) in CDCl3 and in solid state is Δδ 29Si (2) = δiso 29Si(2)[CP/MAS]–δ 29Si(2) [CDCl3].
| R–Si = | δ 29Si (RSi(pyO)3) [CDCl3] 1 | δ 29Si (RSi(pyO)3CuCl) [CDCl3] 2 | Δδ 29Si (1vs2) | δiso 29Si (RSi(pyO)3CuCl) [CP/MAS] 2 | Δδ 29Si (2) |
|---|---|---|---|---|---|
| Me–Si a | −46.5 [20] | −64.1 [20] | −17.6 | −70.0 [20] −71.4 [20] | −5.9 −7.3 |
| Ph–Si b | −64.7 [25] | −80.9 | −16.2 | −82.3 | −1.4 |
| Bn–Si c | −54.7 | −72.1 | −17.4 | −67.6 | +4.5 |
| Allyl–Si d/d′ | −53.4 | −65.4 | −12.0 | −52.6 | +12.8 |
Table 3.
Selected atom distances (Å) and bond angles (deg) in compounds 3b, 3c and 3d in their crystal structures (as well as some corresponding parameters of 3a [20]).
Table 3.
Selected atom distances (Å) and bond angles (deg) in compounds 3b, 3c and 3d in their crystal structures (as well as some corresponding parameters of 3a [20]).
| 3a | 3b | 3c | 3d | |
|---|---|---|---|---|
| Si1–O1 | 1.753(2) | 1.765(2) | 1.744(2) | 1.742(2) |
| Si1–O2 | 1.757(2) | 1.733(2) | 1.750(2) | 1.757(2) |
| Si1–O3 | 1.753(2) | 1.764(2) | 1.756(2) | 1.752(2) |
| Si1–O4 | 1.755(2) | 1.746(2) | 1.746(2) | 1.746(2) |
| Si1–C21 | 1.847(2) | 1.861(2) | 1.870(2) | 1.857(3) |
| Cu1–N1 | 2.023(2) | 2.047(2) | 2.042(2) | 2.028(2) |
| Cu1–N2 | 2.055(2) | 2.029(2) | 2.057(2) | 2.035(2) |
| Cu1–N3 | 2.025(2) | 2.038(2) | 2.055(2) | 2.052(2) |
| Cu1–N4 | 2.049(2) | 2.034(2) | 2.044(2) | 2.019(2) |
| Cu1–Cl1 | 2.403(1) | 2.389(1) | 2.403(1) | 2.395(1) |
| Cu1···Si1 | 2.919(1) | 2.888(1) | 2.915(1) | 2.897(1) |
| O1–Si1–O3 | 154.22(6) | 157.60(8) | 154.98(8) | 155.82(9) |
| O2–Si1–O4 | 155.83(6) | 154.21(8) | 154.14(8) | 153.49(9) |
| N1–Cu1–N3 | 159.32(5) | 159.23(8) | 158.15(7) | 157.76(9) |
| N2–Cu1–N4 | 158.66(5) | 160.00(8) | 159.05(7) | 159.26(9) |
| Cl1–Cu1···Si1 | 178.24(2) | 177.27(2) | 179.43(2) | 178.15(3) |
| Cu1···Si1–C21 | 177.51(6) | 177.99(8) | 176.47(8) | 177.16(11) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Seidel, A.; Gericke, R.; Brendler, E.; Wagler, J. Copper Complexes of Silicon Pyridine-2-olates RSi(pyO)3 (R = Me, Ph, Bn, Allyl) and Ph2Si(pyO)2. Inorganics 2023, 11, 2. https://doi.org/10.3390/inorganics11010002
AMA Style
Seidel A, Gericke R, Brendler E, Wagler J. Copper Complexes of Silicon Pyridine-2-olates RSi(pyO)3 (R = Me, Ph, Bn, Allyl) and Ph2Si(pyO)2. Inorganics. 2023; 11(1):2. https://doi.org/10.3390/inorganics11010002
Chicago/Turabian StyleSeidel, Anne, Robert Gericke, Erica Brendler, and Jörg Wagler. 2023. "Copper Complexes of Silicon Pyridine-2-olates RSi(pyO)3 (R = Me, Ph, Bn, Allyl) and Ph2Si(pyO)2" Inorganics 11, no. 1: 2. https://doi.org/10.3390/inorganics11010002
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.