
Oxo-Stabilised Phosphonium Ylides as Hydrogen Bond Acceptors
by
 , ,
, ,
R. Alan Aitken
*,
Lee P. Cleghorn
,
Graham Dawson
,
Ian P. Gray
,
Anna Lashtabeg
and
Alexandra M. Z. Slawin
EaStCHEM School of Chemistry, University of St. Andrews, North Haugh, St. Andrews, Fife KY16 9ST, UK
*
Author to whom correspondence should be addressed.
Inorganics 2023, 11(2), 50; https://doi.org/10.3390/inorganics11020050
Submission received: 15 December 2022
/
Revised: 28 December 2022
/
Accepted: 14 January 2023
/
Published: 18 January 2023
(This article belongs to the Special Issue Bond Activation and Catalysis Using Main-Group Elements)
Abstract
:Oxo-stabilised phosphonium ylides are found to form crystalline hydrogen-bonded adducts with aromatic carboxylic acids, as confirmed by X-ray diffraction. There is also strong hydrogen bonding in solution as indicated by 13C NMR spectroscopy and this confirmed adduct formation with acetic acid, benzamide, thiobenzamide, benzyl alcohol, benzenesulfinic acid and diphenylphosphinic acid. The X-ray structure of the benzamide adduct was also determined, showing a hydrogen-bonded dimeric structure. A bis(stabilised ylide) was also prepared and is found to form a complex hydrogen-bonded adduct with benzoic acid, ethanol and water.
1. Introduction
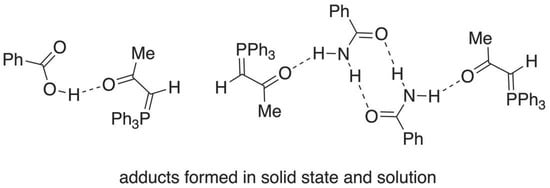
The ability of triphenylphosphine oxide 1 (Figure 1) to act as a hydrogen bonding acceptor is well known, with crystalline complexes being reported with hydroquinone and chlorinated analogues as early as 1959 [1]. More recently, Ph3PO has been used as a “crystallisation aid” for compounds containing a range of hydrogen bond donor functions, including N-acylsulfonamides, phenols, alcohols, amides [2] and acyclic imides [3]. Crystalline hydrogen-bonded adducts have also been characterised between 1 and a wide range of carboxylic acids including oxalic [4], adamantanecarboxylic, terephthalic and phthalic [5], dimethylmalonic [6], trichloroacetic [7], 3-chlorobenzoic acids [8] and agrochemically important phenoxyacetic acid derivatives such as 4-chloro-2-methylphenoxyacetic acid (MCPA) [9]. Adducts with diphenylmethanol [10] and triphenylmethanol [11] have also been reported. In contrast, the hydrogen bonding acceptor ability of the formally vinylogous oxo-stabilised phosphonium ylides 2 has hardly been examined.
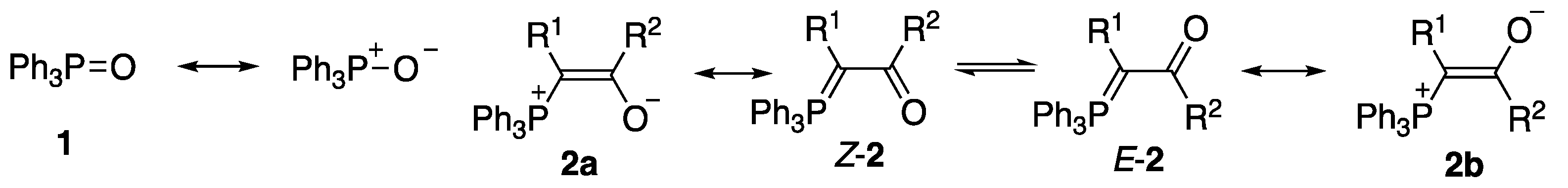
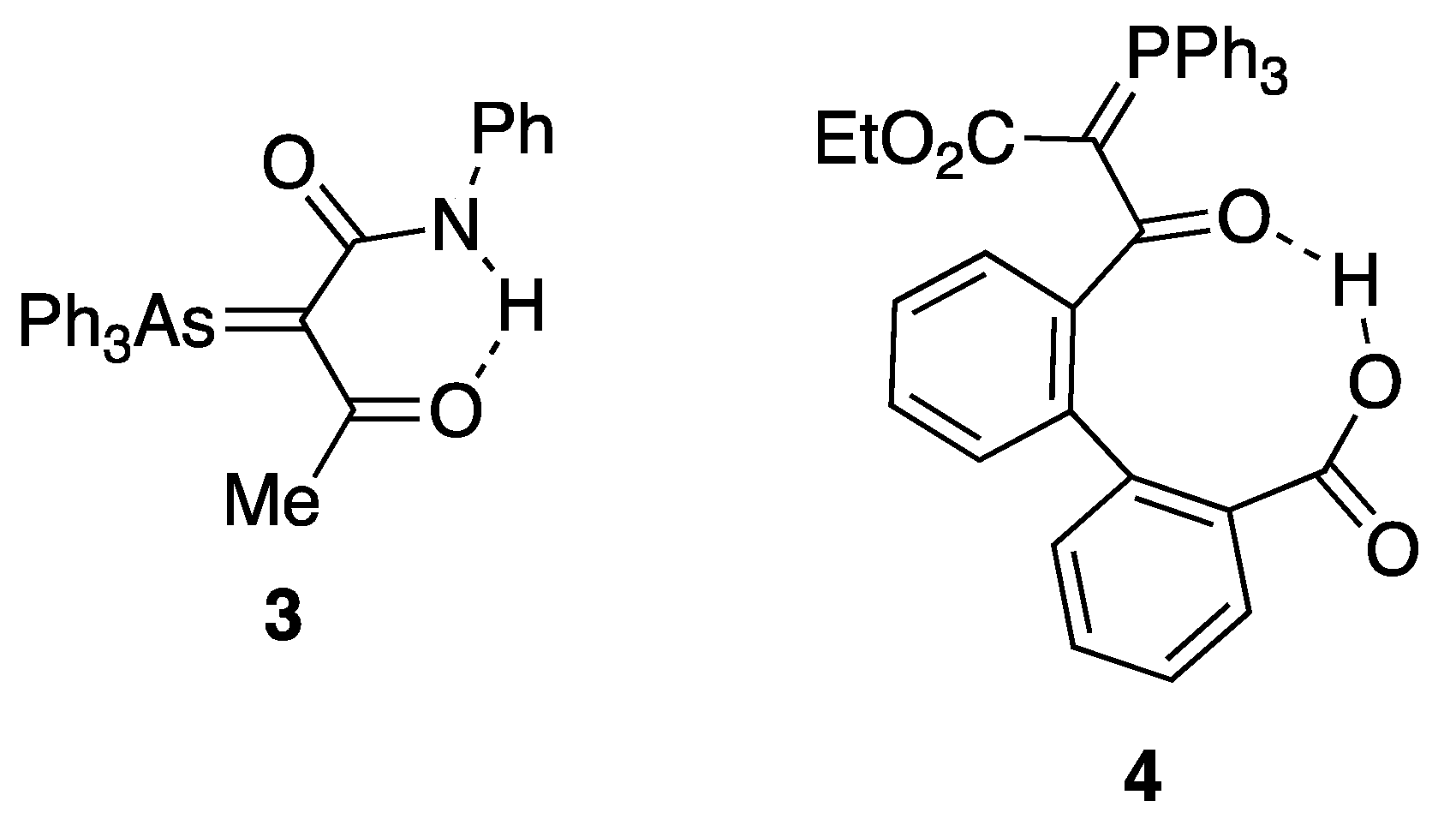
We have previously surveyed the known X-ray structures of such ylides [12,13], and while this revealed a well-defined pattern of E vs. Z relative arrangement of the phosphorus and oxygen atoms depending upon the nature of R1 and R2 (i.e., forms 2a and 2b, Figure 1), we are only aware of two previous cases of hydrogen bonding in such ylides, both intramolecular. The first involves the acetyl(phenylamido)arsonium ylide 3 (Figure 2) [14,15], where there is an S(6) [16] interaction between the NH and acetyl C=O, and the second is the biphenylcarboxylic acid containing phosphonium ylide 4 where there is an S(9) interaction between the carboxylic acid hydrogen and the ylide-conjugated carbonyl [17]. There seems to be no good reason why a similar interaction should not be possible in an intermolecular mode.
Although the behaviour of oxo-stabilised ylides 2 as intermolecular hydrogen-bond acceptors is essentially unexplored until now, such compounds do exhibit a rich coordination chemistry, acting as ligands binding through C or O or both atoms with a range of transition metals [18,19]. Crystalline adducts have also been formed with main group Lewis acidic halides such as HgCl2, HgBr2 and HgI2 [20], Me3SnCl [21] and TiCl4, ZrCl4 and HfCl4 [22]. The formation of complexes between bis(oxo-stabilised ylides) and organotin chlorides Me3SnCl, Me2SnCl2, Ph3SnCl and PhSnCl3 [23] as well as HgCl2 and PdCl2 [24] has also been reported.
In this paper, we describe for the first time the formation and characterisation, both in solution and in the solid state, of hydrogen-bonded adducts between oxo-stabilised phosphonium ylides of type 2 and carboxylic acids, benzamide, thiobenzamide, benzyl alcohol, a sulfinic acid and a phosphinic acid.
2. Results and Discussion
2.1. Initial Discovery
Some time ago, we described the formation and structure of the highly delocalised 3-triphenylphosphoranylidenecyclopentene-1,2-dione 7 in a condensation reaction between methyl triphenylphosphoranylidenepyruvate 5 and the furan-2,3-dione 6 (Scheme 1) [25]. The latter was prepared by dehydration of benzoylpyruvic acid 8, but on one occasion, this dehydration failed, and we unwittingly heated a mixture of 5 and 8 in toluene to give, upon evaporation, a new crystalline material. This was initially suspected to be the ionic salt formed by protonation of the ylide function by the carboxylic acid. However, the structure, as determined by X-ray diffraction, showed it to be a hydrogen-bonded 1:1 adduct with both intra- and intermolecular O–H…O=C hydrogen bonds (Figure 3).
2.2. Scope of the Stabilised Ylide—Carboxylic Acid Interaction
Although discovered fortuitously in the case of 9, it seemed likely that the formation of hydrogen-bonded adducts could be more general, and indeed, this proved to be the case with ylide 5 forming a 1:1 adduct 10 with benzoic acid, and the much simpler ylide triphenylphosphoranylideneacetone also forming a benzoic acid adduct 11 (Figure 5) whose structure could again be confirmed by X-ray diffraction (Figure 6 and Figure 7, Table 1). Again, in this case, we have two independent but closely similar adducts in the crystal.
Comparison of the molecular dimensions of the ylide in adduct 11 with those of the ylide on its own [13,27], as well as its adducts with Me3SnCl [21] and TiCl4 [22] (Figure 8), shows that complexation results in lengthening of P=C and C=O with concomitant shortening of the intervening C–C bond and the effect increases along the series with hydrogen bonding to benzoic acid having less of an effect than bonding to Me3SnCl which in turn is less than to TiCl4.
Although it was initially expected that such adducts would exist only in the solid state and that they would dissociate upon dissolving, careful analysis of the NMR spectra showed that this was not the case. When the 13C NMR data obtained by dissolving an adduct such as 10 or 11 in CDCl3 were compared with those of the individual components, significant differences were noted in the signals due to nuclei around the site of hydrogen bonding (see Supplementary Materials). Specifically, the signal for C=O of the carboxylic acid in the adducts came at 2–4 ppm lower chemical shift than for the pure acid, while the P=C ylide signal came at 2–5 ppm higher chemical shift in the adducts as compared to the pure ylides. This then allowed rapid screening of a range of simple ylides and aromatic carboxylic acids with adducts 12–18 (Figure 5) being obtained. The data are summarised in Table 2.
In most of these cases, well-defined crystalline adducts were formed by heating equimolar amounts of the components in toluene under reflux for 5 min, evaporation and recrystallisation of the residue from ethanol, and the NMR data were obtained by dissolving these in CDCl3. For adducts 12, 16, and 18 only, we were unable to isolate the pure adduct in crystalline form, but the solution interaction could still be observed by directly preparing a 0.1 M solution of the two components in CDCl3.
It was of interest to examine the solvent dependence of the solution interaction, and this was done in the case of 11 by running 13C NMR spectra of the two components together and individually in a range of solvents. The results (Table 3) show that the shift in the ylide carbon drops off steadily with increasing solvent polarity indicating a progressive weakening of the interaction, while the carboxylic acid shifts do not show any meaningful trend.
2.3. Expanding the Range of Hydrogen Bond Donors
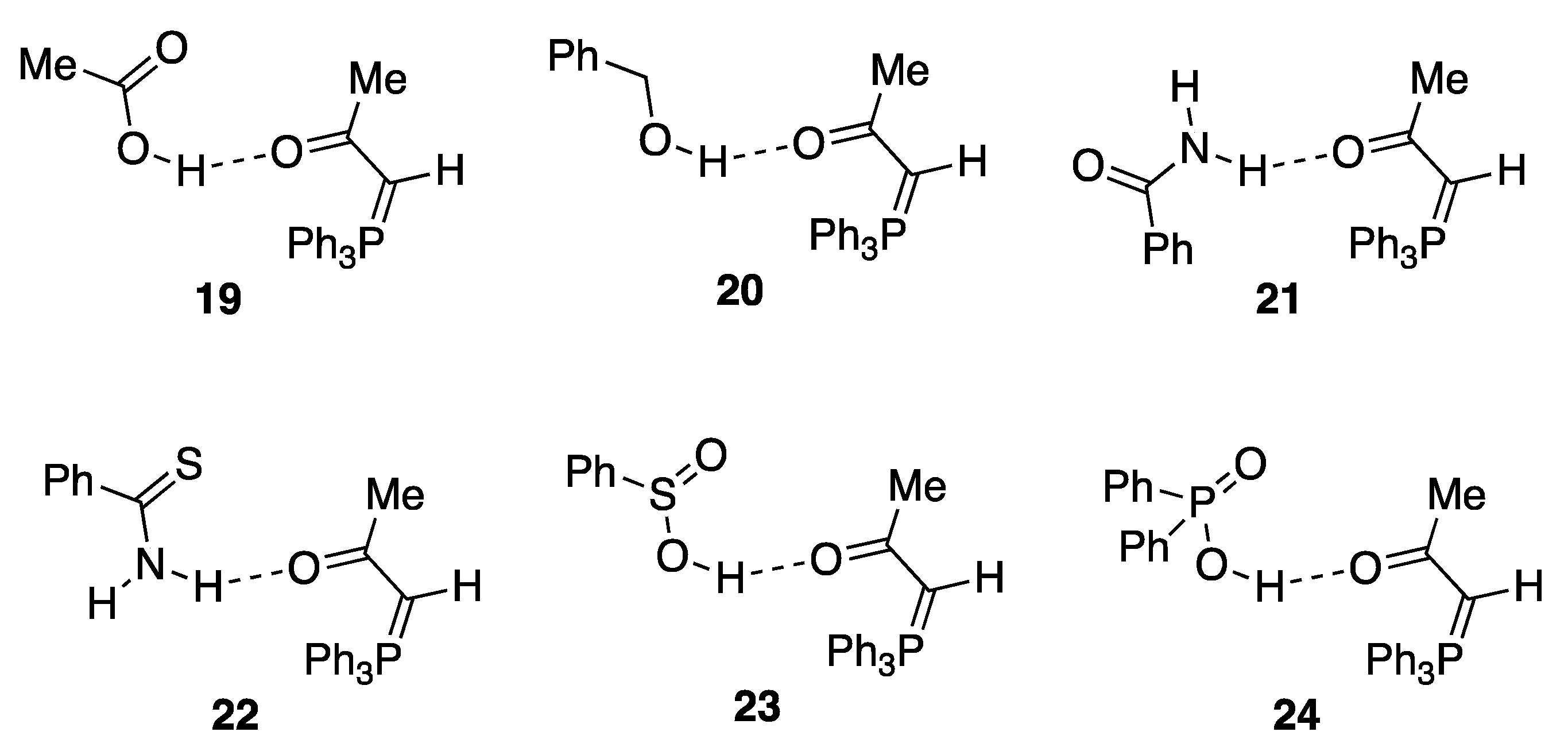
Using the 13C chemical shift of the ylide carbon as a diagnostic parameter, the solution interaction of triphenylphosphoranylideneacetone with a range of potential hydrogen bond donors was investigated by preparing 0.1 M solutions of the two components in CDCl3. The results (Table 4) show clear evidence of adduct formation for acetic acid, benzyl alcohol, benzamide, thiobenzamide, benzenesulfinic acid, and diphenylphosphinic acid with the suggested structures 19–24 shown in Figure 9. For benzylamine the minimal change in δC indicated no adduct formation while for benzenesulfonic acid proton transfer occurred to give the ionic salt: a phosphonium sulfonate.
In the case of benzamide and thiobenzamide, the adducts 21 and 22 could be isolated in crystalline form by following the same procedure as used for aromatic carboxylic acid adducts. These gave the expected analytical and spectroscopic data and for 21 the structure was confirmed by X-ray diffraction (Figure 10 and Figure 11, Table 1). As shown, the crystal structure involves C=O…NH hydrogen bonded pairs of benzamide molecules which are then each bonded by the remaining NH to C=O of the ylide (Figure 11). The hydrogen bonding parameters given in Table 1 are within the expected range.
2.4. Synthesis and Properties of a Bis(Stabilised Ylide)
It occurred to us that the hydrogen bonding interactions revealed by these studies could form the basis of molecular recognition and for this a bis(ylide) with carbonyl acceptor groups at a fixed distance apart and orientation would be ideal. We therefore prepared the meta-isomer 26 by reaction of benzene-1,3-dicarbonyl chloride 25 with 4 equiv. of Ph3P=CH2 (Scheme 2). This was obtained as a high melting point solid from which it was difficult to remove the last traces of solvent, but the NMR data were in full agreement with the expected values. It might be noted that we prepared the para isomer of 26 some years ago and confirmed its structure by X-ray diffraction, while attempts to form the corresponding ortho isomer resulted in an unexpected rearrangement [28].
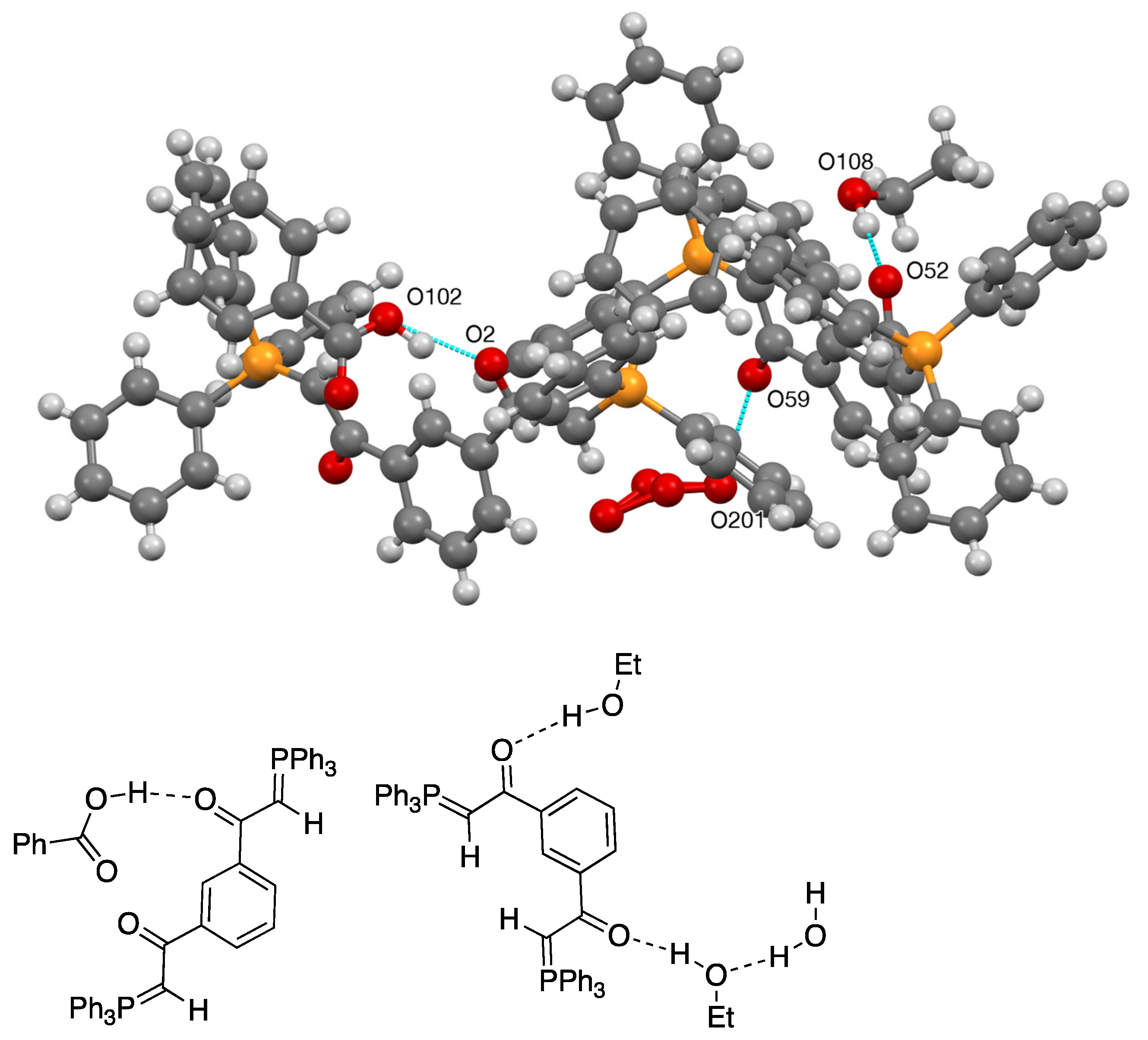
Treating a solution of 26 with benzoic acid in ethanol led to the deposition of crystals 27, whose composition could only be determined by X-ray diffraction as containing two molecules of 26, one of them bonded to a single benzoic acid and the other to two molecules of ethanol and one of water with disorder in the region of one EtOH/H2O (Figure 12 and Figure 13, Table 1).
The ready formation of this unexpectedly complex adduct shows that while bis(ylides) such as 26 have significant hydrogen bond acceptor properties, these can be difficult to control, and the practical use of such compounds for molecular recognition will require further detailed studies.
3. Experimental Section
3.1. General Experimental Details
NMR spectra were recorded on solutions in CDCl3 unless otherwise stated using a Varian Gemini 2000 instrument and chemical shifts are given in ppm to high frequency from Me4Si (H, C) or H3PO4 (P) with coupling constants J in Hz. IR spectra were recorded using the ATR technique on a Shimadzu IRAffinity 1S instrument. Melting points were recorded on a Gallenkamp 50W melting point apparatus or a Reichert hot-stage microscope. Elemental analysis was performed using a Carlo-Erba 1106 elemental analyser.
3.2. Preparation of Adducts with Methyl Triphenylphosphoranylidenepyruvate
3.2.1. Methyl Triphenylphosphoranylidenepyruvate—Benzoylpyruvic Acid Adduct 9
A solution of methyl triphenylphosphoranylidenepyruvate 5 (0.72 g, 2 mmol) and benzoylpyruvic acid 8 (0.42 g, 2 mmol) in toluene (40 mL) was heated under reflux for 5 min and then evaporated. Recrystallisation of the residue from ethanol gave colourless crystals (0.74 g, 65%), mp 140–141 °C.
3.2.2. Methyl Triphenylphosphoranylidenepyruvate—Benzoic Acid Adduct 10
A solution of methyl triphenylphosphoranylidenepyruvate 5 (0.72 g, 2 mmol) and benzoic acid (0.24 g, 2 mmol) in toluene (40 mL) was heated under reflux for 5 min and then evaporated. Recrystallisation of the residue from ethanol gave yellow crystals (0.21 g, 21%), mp 139–140 °C. Found: C, 72.6; H, 4.7. C29H25O5P requires C, 71.9; H, 5.2%. IR (cm−1): 1721, 1681, 1582, 1534, 1438, 1313, 1237, 1107, 851, 753, 722, 648. 1H NMR (300 MHz, CDCl3): δ 8.12 (2H, d, J 9), 7.83–7.25 (18H, m), 4.93 (1H, d, JH–P 23), 3.81 (3H, s). 13C NMR (75 MHz, CDCl3) δ 173.4 (d, J 5, P=CHCO), 170.2 (CO2H), 165.9 (d, J 22, CO2Me), 133.1 (d, J 11, PPh C-2), 132.9 (PhCO2H C-4), 132.6 (d, J 2, PPh C-4), 130.3 (PhCO2H C-1), 129.9 (2CH, PhCO2H), 129.0 (d, J 13, PPh C-3), 128.1 (2CH, PhCO2H), 124.7 (d, J 92, PPh C-1), 58.8 (d, J 107, P=CH), 52.2 (OMe). 31P NMR (121 MHz, CDCl3) δ + 17.3.
3.3. Preparation of Adducts with Triphenylphosphoranylideneacetone
3.3.1. Triphenylphosphoranylideneacetone—Benzoic Acid Adduct 11
A solution of triphenylphosphoranylideneacetone (0.63 g, 2 mmol) and benzoic acid (0.24 g, 2 mmol) in toluene (40 mL) was heated under reflux for 5 min and then evaporated. Recrystallisation of the residue from ethanol gave colourless crystals (0.38 g, 44%), mp 132–133 °C. Found: C, 76.4; H, 5.7. C28H25O3P requires C, 76.4; H, 5.7%. IR (cm−1): 1701, 1437, 1107, 856, 798, 746, 715, 692. 1H NMR (300 MHz, CDCl3): δ 8.06 (2H, m), 7.75–7.16 (18H, m), 6.00 (1H, br s), 2.19 (3H, d, J 2, COMe). 13C NMR (75 MHz, CDCl3) δ 190.4 (COMe), 169.6 (CO2H), 133.8 (PhCO2H C-4), 133.2 (d, J 10, PPh C-2), 132.4 (d, J 2, PPh C-4), 129.7 (2CH, PhCO2H), 129.2 (PhCO2H C-1), 129.0 (d, J 12, PPh C-3), 127.9 (2CH, PhCO2H), 125.8 (d, J 91, PPh C-1), 56.3 (d, J 105, P=CH), 28.0 (d, J 14, Me). 31P NMR (121 MHz, CDCl3) δ + 14.8.
3.3.2. Triphenylphosphoranylideneacetone—Phenylacetic Acid Adduct 13
A solution of triphenylphosphoranylideneacetone (0.63 g, 2 mmol) and phenylacetic acid (0.27 g, 2 mmol) in toluene (40 mL) was heated under reflux for 5 min and then evaporated. Recrystallisation of the residue from ethanol gave yellow crystals (0.69 g, 77%), mp 98–100 °C. Found: C, 76.4; H, 6.3. C29H27O3P requires C, 76.6; H, 6.0%. IR (cm−1): 1700, 1315, 1231, 1105, 996, 857, 749, 692. 1H NMR (300 MHz, CDCl3): δ 8.65 (1H, br s), 7.73–7.15 (20H, m), 3.48 (2H, s), 2.15 (3H, d, J 2, COMe). 13C NMR (75 MHz, CDCl3) δ 190.2 (COMe), 174.6 (CO2H), 135.7 (PhCH2CO2H C-1), 133.0 (d, J 10, PPh C-2), 132.5 (d, J 2, PPh C-4), 129.3 (2CH, PhCH2CO2H), 129.0 (d, J 12, PPh C-3), 128.1 (2CH, PhCH2CO2H), 126.2 (PhCH2CO2H C-4), 125.3 (d, J 91, PPh C-1), 56.1 (d, J 103, P=CH), 42.3 (CH2), 26.8 (d, J 14, Me). 31P NMR (121 MHz, CDCl3) δ + 15.0.
3.3.3. Triphenylphosphoranylideneacetone—Diphenylacetic Acid Adduct 14
A solution of triphenylphosphoranylideneacetone (0.63 g, 2 mmol) and diphenylacetic acid (0.42 g, 2 mmol) in toluene (40 mL) was heated under reflux for 5 min and then evaporated. Recrystallisation of the residue from ethanol gave colourless crystals (0.88 g, 84%), mp 99–100 °C. Found: C, 79.2; H, 5.8. C35H31O3P requires C, 79.2; H, 5.9%. IR (cm−1): 1698, 1112, 996, 746, 716. 1H NMR (300 MHz, CDCl3): δ 8.10 (1H, br s), 7.75–7.11 (25H, m), 4.93 (1H, s), 2.14 (3H, s, COMe). 13C NMR (75 MHz, CDCl3) δ 190.5 (COMe), 175.2 (CO2H), 140.2 (Ph2CHCO2H C-1), 133.2 (d, J 10, PPh C-2), 132.5 (d, J 2, PPh C-4), 129.0 (d, J 12, PPh C-3), 128.9 (4CH, Ph2CHCO2H), 128.2 (4CH, Ph2CHCO2H), 126.5 (Ph2CHCO2H C-4), 125.3 (d, J 91, PPh C-1), 58.2 (Ph2CH), 56.0 (d, J 100, P=CH), 26.8 (d, J 15, Me). 31P NMR (121 MHz, CDCl3) δ + 15.0.
3.3.4. Triphenylphosphoranylideneacetone—2-Phenylbutyric Acid Adduct 15
A solution of triphenylphosphoranylideneacetone (0.63 g, 2 mmol) and 2-phenylbutyric acid (0.33 g, 2 mmol) in toluene (40 mL) was heated under reflux for 5 min and then evaporated. Recrystallisation of the residue from ethanol gave colourless crystals (0.47 g, 49%), mp 113–114 °C. Found: C, 77.1; H, 6.2. C31H31O3P requires C, 77.2; H, 6.5%. IR (cm−1): 1708, 1575, 1106, 860, 719, 699. 1H NMR (300 MHz, CDCl3): δ 7.78–7.20 (21H, m), 3.36 (1H, t, J 7), 2.10 (3H, s), 2.08 (1H, m), 1.72 (1H, m), 0.85 (3H, t, J 7). 13C NMR (75 MHz, CDCl3) δ 190.6 (COMe), 177.0 (CO2H), 140.2 (PhCH(Et)CO2H C-1), 133.1 (d, J 10, PPh C-2), 132.4 (d, J 2, PPh C-4), 129.0 (d, J 12, PPh C-3), 128.20 (2CH, PhCH(Et)CO2H), 128.16 (2CH, PhCH(Et)CO2H), 126.6 (PhCH(Et)CO2H C-4), 125.9 (d, J 91, PPh C-1), 55.4 (d, J 105, P=CH), 54.0 (CH-CO2H), 27.1 (d, J 15, Me), 26.8 (CH2), 12.3 (CH3). 31P NMR (121 MHz, CDCl3) δ + 14.9.
3.4. Preparation of Adducts with Triphenylphosphoranylideneacetophenone
Triphenylphosphoranylideneacetophenone—Diphenylacetic Acid Adduct 17
A solution of triphenylphosphoranylideneacetophenone (0.70 g, 2 mmol) and diphenylacetic acid (0.42 g, 2 mmol) in toluene (40 mL) was heated under reflux for 5 min and then evaporated. Recrystallisation of the residue from ethanol gave orange crystals (0.87 g, 77%), mp 123–124 °C. Found: C, 80.8; H, 5.5. C40H33O3P requires C, 81.1; H, 5.6%. IR (cm−1): 1746, 1587, 1437, 1205, 1200, 881, 745, 697, 634. 1H NMR (300 MHz, CDCl3): δ 8.02–7.18 (31H, m), 4.95 (1H, s). 13C NMR (75 MHz, CDCl3) δ 185.8 (COPh), 174.9 (CO2H), 140.1 (d, J 10, COPh C-1), 139.6 (Ph2CHCO2H C-1), 133.2 (d, J 10, PPh C-2), 132.4 (PPh C-4), 129.9 (COPh C-4), 128.9 (d, J 12, PPh C-3), 128.8 (4CH, Ph2CHCO2H), 128.1 (4CH, Ph2CHCO2H), 127.8 (COPh C-2), 127.3 (COPh C-3), 126.5 (Ph2CHCO2H C-4), 125.5 (d, J 91, PPh C-1), 57.6 (Ph2CH), 52.5 (d, J 106, P=CH). 31P NMR (121 MHz, CDCl3) δ + 18.0.
3.5. Preparation of Adducts with 1-Phenyl-1-triphenylphosphoranylidenepropan-2-One
1-Phenyl-1-triphenylphosphoranylidenepropan-2-One—Benzoic Acid Adduct 12
A solution of 1-phenyl-1-triphenylphosphoranylidenepropan-2-one (39 mg, 0.1 mmol) and benzoic acid (12.2 mg, 0.1 mmol) in CDCl3 was analysed by 13C NMR (75 MHz, CDCl3): δ 187.0 (COPh), 169.7 (CO2H), 137.8 (d, J 13, P=C-Ph C-1), 134.4 (d, J 5, P=C-Ph C-2,6), 133.7 (d, J 9, PPh C-2), 132.2 (PhCO2H C-4), 132.0 (PPh C-4), 131.8 (2C, PhCO2H),129.8 (PhCO2H C-1), 128.6 (d, J 13, PPh C-3), 127.9 (2CH, PhCO2H), 127.7 (2C, P=C-Ph), 125.7 (d, J 91, PPh C-1), 125.7 (P=C-Ph C-4), 75.1 (d, J 118, P=C), 24.3 (Me). All signals are within ±2 ppm of those for the separate components except 169.7 (free acid 172.6) and 75.1 (free ylide 71.0).
3.6. Formation of Solution Adducts with Triphenylphosphoranylideneacetone
For this, 0.1 M solutions of triphenylphosphoranylideneacetone and the potential donor in CDCl3 were prepared, and 13C NMR spectra were obtained. All signals for the ylide carbons in the mixture were within ±2 ppm of those for the pure ylide except those for P=C, which were shifted as shown in Table 4. This provided evidence for the formation of adducts between the ylide and acetic acid 19, benzyl alcohol 20, benzamide 21, thiobenzamide 22, benzenesulfinic acid 23, and diphenylphosphinic acid 24
3.7. Preparation of Adducts with Triphenylphosphoranylideneacetone
3.7.1. Triphenylphosphoranylideneacetone—Benzamide Adduct 21
A solution of triphenylphosphoranylideneacetone (0.79 g, 2.5 mmol) and benzamide (0.30 g, 2.5 mmol) in ethanol (5 mL) was heated to the boil for 5 min and allowed to cool. After 18 h, the solid that had formed was filtered off to give 21 (0.82 g, 75%) as colourless crystals, mp 151–153 °C. Found: C, 76.5; H, 6.0; N, 3.2. C28H26NO2P requires C, 76.5; H, 6.0; N, 3.2%. IR (cm−1) 1686, 1629, 1576, 1508, 1153, 1110, 986, 869, 752, 722, 694, 637. 1H NMR (300 MHz, CDCl3) δ 7.90–7.80 (2H, m), 7.70–7.35 (18H, m), 6.65 (1H, br s), 5.72 (1H, br s), 3.73 (1H, br d, J 23), 2.09 (3H, d, J 2). 1H NMR (300 MHz, CDCl3) δ 190.7 (CO), 169.4 (CONH2), 133.4 (C), 132.9 (d, J 10, PPh C-2), 131.9 (d, J 3, PPh C-4), 131.5 (CH), 128.7 (d, J 12, PPh C-3), 128.2 (2CH), 127.5 (2CH), 126.9 (d, J 90, PPh C-1), 52.2 (d, J 107, P=CH), 28.3 (d, J 16, CH3). 31P NMR (121 MHz, CDCl3) δ + 15.5.
3.7.2. Triphenylphosphoranylideneacetone—Thiobenzamide Adduct 22
A solution of triphenylphosphoranylideneacetone (0.79 g, 2.5 mmol) and thiobenzamide (0.34 g, 2.5 mmol) in ethanol (2 mL) was heated to the boil for 5 min and allowed to cool. After 10 days, the solid that had formed was filtered off to give 22 (0.73 g, 65%) as colourless crystals, mp 140–141 °C. Found: C, 73.6; H, 5.8; N, 3.0. C28H26NOPS requires C, 73.8; H, 5.8; N, 3.1%. IR (cm−1) 1508, 1437, 1396, 1110, 866, 750, 717, 692. 1H NMR (300 MHz, CDCl3) δ 8.40 (1H, br s), 7.90–7.80 (2H, m), 7.72 (1H, br s), 7.70–7.35 (18H, m), 3.80 (1H, br d, J 23), 2.09 (3H, d, J 2). 1H NMR (300 MHz, CDCl3) δ 201.7 (CS), 190.3 (CO), 138.7 (C), 132.8 (d, J 10, PPh C-2), 132.1 (d, J 3, PPh C-4), 131.3 (CH), 128.8 (d, J 12, PPh C-3), 127.8 (2CH), 127.5 (2CH), 126.4 (d, J 90, PPh C-1), 53.6 (d, J 108, P=CH), 28.0 (d, J 16, CH3). 31P NMR (121 MHz, CDCl3) δ + 15.5.
3.8. Preparation of Adducts with 1,3-Bis(triphenylphosphoranylideneacetyl)benzene 26
3.8.1. 1,3-Bis(triphenylphosphoranylideneacetyl)benzene 26
A suspension of methyltriphenylphosphonium bromide (17.6 g, 50 mmol) in dry THF (150 mL) was stirred under nitrogen while n-butyllithium (2.5M in hexane, 20 mL, 50 mmol) was added slowly. The mixture was stirred for 20 min, and then a solution of isophthaloyl dichloride 25 (2.5 g, 12.5 mmol) in THF (20 mL) was added. After stirring for 18 h, the mixture was poured into water and extracted with ethyl acetate (3 × 50 mL). Drying and evaporation followed by recrystallisation from EtOAc/CH2Cl2 gave the product 26 (6.2 g, 73%) as pale-yellow crystals, mp > 300 °C. Found: C, 79.8; H, 4.8. C46H36O2P2 requires C, 80.9; H, 5.3%. IR (cm−1) 1585, 1508, 1466, 1436, 1107, 870, 750, 732, 693. 1H NMR (300 MHz, CDCl3): δ 8.60 (1H, t, J 1.6), 8.01 (2H, dt, J 7.5, 1.6), 7.75–7.68 (12H, m), 7.60–7.40 (18H, m), 7.35 (1H, t, J 7.5), 4.56 (2H, br s). 13C NMR (75 MHz, CDCl3) δ 184.8 (CO), 140.5 (d, J 15, 2 C–CO), 133.2 (d, J 10, PPh C-2), 131.9 (PPh C-4), 128.8 (d, J 12, PPh C-3), 128.1 (2CH), 127.2 (CH), 127.1 (d, J 91, PPh C-1), 125.4 (C), 50.8 (d, J 110, P=C). 31P NMR (121 MHz, CDCl3) δ + 17.8.
3.8.2. 1,3-Bis(triphenylphosphoranylideneacetyl)benzene—Benzoic Acid Adduct 27
The bis(ylide) 26 (0.34 g, 0.5 mmol) and benzoic acid (0.12 g, 1.0 mmol) were dissolved with warming in ethanol (2 mL), and the mixture was allowed to cool and stored at RT until an adduct (0.29 g, 72%) was obtained as colourless crystals, mp 154–156 °C. These were found by X-ray diffraction to be an adduct of formula (26)2•PhCO2H•(EtOH)2•H2O. Found: C, 77.4; H, 5.9. C103H92O9P4 requires C, 77.4; H, 5.8%.
3.9. X-ray Structure Determination of Adducts
Data were collected using graphite monochromated Mo Kα radiation λ = 0.71073 Å. The data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/getstructures. The structure was solved by direct methods and refined by full-matrix least-squares against F2 (SHELXL, Version 2018/3 [33]).
3.9.1. Methyl Triphenylphosphoranylidenepyruvate—Benzoylpyruvic Acid Adduct 9
Crystal data for C32H27O7P, M = 554.51 g mol−1, colourless prism, crystal dimensions 0.19 × 0.10 × 0.10 mm, triclinic, space group P-1 (No. 2), a = 10.4270(10), b = 10.4963(10), c = 29.347(3) Å, α = 80.680(2), β = 88.251(2), γ = 63.914(2)°, V = 2843.6(5) Å3, Z = 4, Dcalc = 1.295 g cm−3, T = 293 K, R1 = 0.0645, Rw2 = 0.1166 for 2698 reflections with I > 2σ(I), and 738 variables. Data were deposited at the Cambridge Crystallographic Data Centre as CCDC 2169369.
3.9.2. Triphenylphosphoranylideneacetone—Benzoic Acid Adduct 11
Crystal data for C28H25O3P, M = 440.45 g mol−1, colourless prism, crystal dimensions 0.10 × 0.10 × 0.10 mm, orthorhombic, space group Pna21 (No. 33), a = 17.924(4), b = 9.364(2), c = 28.710(6) Å, V = 4819.1(17) Å3, Z = 8, Dcalc = 1.214 g cm−3, T = 293 K, R1 = 0.0794, Rw2 = 0.1556 for 8656 reflections with I > 2σ(I), and 586 variables. Data were deposited at the Cambridge Crystallographic Data Centre as CCDC 2169371.
3.9.3. Triphenylphosphoranylideneacetone—Benzamide Adduct 21
Crystal data for C28H26NO2P, M = 439.47 g mol−1, colourless prism, crystal dimensions 0.15 × 0.10 × 0.10 mm, monoclinic, space group P21/n (No. 14), a = 10.9414(3), b = 15.8906(6), c = 14.1127(3) Å, β = 100.9000(10)°, V = 2409.44(12) Å3, Z = 4, Dcalc = 1.211 g cm−3, T = 293 K, R1 = 0.0384, Rw2 = 0.0937 for 2370 reflections with I > 2σ(I), and 298 variables. Data were deposited at the Cambridge Crystallographic Data Centre as CCDC 2169368.
3.9.4. 1,3-Bis(triphenylphosphoranylideneacetyl)benzene—Benzoic Acid Adduct 27
Crystal data for C101H88O9P4, M = 1569.70 g mol−1, colourless prism, crystal dimensions 0.14 × 0.10 × 0.03 mm, triclinic, space group P-1 (No. 2), a = 15.212(2), b = 17.837(2), c = 18.156(2) Å, α = 100.322(2), β = 107.860(2), γ = 110.549(2)°, V = 4157.0(9) Å3, Z = 2, Dcalc = 1.254 g cm−3, T = 125 K, R1 = 0.060, Rw2 = 0.1578 for 9039 reflections with I > 2σ(I), and 1056 variables. Data were deposited at the Cambridge Crystallographic Data Centre as CCDC 2169370.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/inorganics11020050/s1, Figures S1–S16: selected NMR spectra; cif and check-cif files for 9, 11, 21 and 27.
Author Contributions
L.P.C., G.D., I.P.G. and A.L. carried out the experimental work; A.M.Z.S. collected the X-ray data and solved the structures; R.A.A. designed the study, analysed the data, and wrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The X-ray data has been deposited at CCDC as noted above.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ramirez, F.; Dershowitz, S. Crystalline complexes of the phosphoryl group with polyphenols. J. Org. Chem. 1959, 24, 704–705. [Google Scholar] [CrossRef]
- Etter, M.C.; Baures, P.W. Triphenylphosphine oxide as a crystallization aid. J. Am. Chem. Soc. 1988, 110, 639–640. [Google Scholar] [CrossRef]
- Etter, M.C.; Reutzel, S.M. Hydrogen bond directed cocrystallization and molecular recognition properties of acyclic imides. J. Am. Chem. Soc. 1991, 113, 2586–2598. [Google Scholar] [CrossRef]
- Thierbach, D.; Huber, F. Darstellung sowie Krystall- und Molekülstruktur von 2 Ph3PO.(COOH)2. Z. Anorg. Allg. Chem. 1981, 477, 101–107. [Google Scholar] [CrossRef]
- Smith, G.; Lynch, D.E.; Byriel, K.A.; Kennard, C.H.L. Molecular cocrystals of carboxylic acids Part 24: Cocrystals involving triphenylphosphine oxide: Structures of unique adduct hydrates of triphenylphosphine oxide with adamantane carboxylic acid and terephthalic acid and the anhydrous adduct with o-phthalic acid. Z. Krystallogr. 1997, 212, 130–134. [Google Scholar] [CrossRef]
- Declerq, J.P.; Germain, G.; Putzeys, J.P.; Rona, S.; van Meerssche, M. Dimethylmalonic acid-triphenylphosphine oxide, C41H38O6P2. Cryst. Struct. Commun. 1974, 3, 579–582. [Google Scholar]
- Golic, L.; Kaucic, V. Triphenylphosphine oxide-trichloroacetic acid, (C6H5)3PO.HOOC-CCl3. Cryst. Struct. Commun. 1976, 5, 319–324. [Google Scholar]
- Al-Farhan, K.A. Triphenylphosphine oxide—3-chlorobenzoic acid (1/1). Acta Crystallogr. Sect. C 2003, 59, 179–180. [Google Scholar] [CrossRef]
- Lynch, D.E.; Smith, G.; Byriel, K.A.; Kennard, C.H.L.; Whittaker, A.K.; Hanna, J.V. Molecular cocrystals of carboxylic acids XVII. Spectral characterization of the adducts of triphenylphosphine oxide with substituted phenoxyacetic acids and the crystal structure of the 1:1 adduct with (4-chloro-2-methylphenoxy)acetic acid. Aust. J. Chem. 1994, 47, 1401–1411. [Google Scholar] [CrossRef]
- Lariucci, C.; de Almeida Santos, R.H.; Lechat, J.R. Structure of the 1:1 adduct formed by diphenylmethanol with triphenylphosphine oxide. Acta Crystallogr. Sect. C 1986, 42, 1825–1828. [Google Scholar] [CrossRef]
- Steiner, T. Triphenylmethanol-triphenylphosphine oxide (1/1). Acta Crystallogr. Sect. C 2000, 56, 1033–1034. [Google Scholar] [CrossRef] [Green Version]
- Aitken, R.A.; Karodia, N.; Lightfoot, P. The solid state conformation of oxo stabilised ylides: X-ray structures of four new polyoxo phosphorus ylides. J. Chem. Soc. Perkin Trans. 2 2000, 333–340. [Google Scholar] [CrossRef]
- Aitken, R.A.; Boubalouta, Y.; Chang, D.; Cleghorn, L.P.; Gray, I.P.; Karodia, N.; Reid, E.J.; Slawin, A.M.Z. The value of 2Jp–CO as a diagnostic parameter for the structure and reactivity of carbonyl-stabilised phosphonium ylides. Tetrahedron 2017, 73, 6275–6285. [Google Scholar] [CrossRef]
- Gosney, I.; Lloyd, D. Preparation and properties of some stable arsonium ylides. Tetrahedron 1973, 29, 1697–1710. [Google Scholar] [CrossRef]
- Ferguson, G.; Gosney, I.; Lloyd, D.; Ruhl, B.L. Crystal and molecular structures of some triphenylarsonium acetylylides. J. Chem. Res. (S) 1987, 8, 260–261. [Google Scholar]
- Etter, M.C.; MacDonald, J.C.; Bernstein, J. Graph-set analysis of hydrogen-bond patterns in organic crystals. Acta Crystallogr. Sect. B. 1990, 46, 256–262. [Google Scholar] [CrossRef]
- Abell, A.D.; Morris, K.B.; McKee, V. A single-crystal X-ray analysis of a novel intramolecular hydrogen-bonded biphenyl phosphorane. A3ust. J. Chem. 1990, 43, 765–771. [Google Scholar] [CrossRef]
- Falvello, L.R.; Fernández, S.; Navarro, R.; Pascual, I.; Urriolabeitia, E.P. Oxygen vs. carbon coordination of α-keto-stabilized phosphorus ylides Ph3P=C(H)COR (R = Me, Ph or OMe) to palldium(II) cationic complexes. J. Chem. Soc. Dalton Trans. 1997, 763–771. [Google Scholar] [CrossRef]
- Navarro, R.; Urriolabeitia, E.P. α-Stabilized phosphoylides as versatile multifunctional ligands. J. Chem. Soc. Dalton Trans. 1999, 4111–4122. [Google Scholar] [CrossRef]
- Laavanya, P.; Venkatasubramanian, U.; Panchanatheswaran, K.; Krause Bauer, J.A. Subtlety in the reactivity of a diketo ylide towards mercuric halides: The unprecedented O-coordination of α-acetyl-α-benzoylmethylenetriphenylphosphorane to Hg(II). Chem. Commun. 2001, 1660–1661. [Google Scholar] [CrossRef]
- Buckle, J.; Harrison, P.G.; King, T.J.; Richards, J.A. Structural studies in main-group chemistry. Part IX. Crystal structure of chlorotrimethyl(triphenylphosphoranylideneacetone)tin(IV). J. Chem. Soc. Dalton Trans. 1975, 1552–1556. [Google Scholar] [CrossRef]
- Albanese, J.A.; Staley, D.L.; Rheingold, A.L.; Burmeister, J.L. Phosphorus ylides as hard donor ligands: Synthesis and characterization of MCl4(ylide-O)(THF) (M = Ti, Zr, Hf; ylide = (acetylmethylene)triphenylphosphorane, (benzoylmethylene)triphenylphosphorane). Molecular structure of trans-((Acetylmethylene)triphenylphosphorane-O)(tetrahydrofuran)tetrachlorotitanium(IV)-tetrahydrofuran. Inorg. Chem. 1990, 29, 2209–2213. [Google Scholar] [CrossRef]
- Sanehi, R.; Bansal, R.K.; Mehrotra, R.C. Organotin(IV) chloride complexes of bis-β-ketophosphonium ylides. J. Organomet. Chem. 1986, 303, 351–360. [Google Scholar] [CrossRef]
- Sanehi, R.; Bansal, R.K.; Mehrotra, R.C. Preparation & characterization of some new bisphosphonium ylides & their palladium(II) & mercury(II) complexes. Indian J. Chem A 1985, 24, 398–402. [Google Scholar]
- Aitken, R.A.; Kozminykh, E.N.; Kozminykh, V.O.; Lightfoot, P. Solution and solid state structure of highly delocalised 5-triphenylphosphoranylidenecyclopentene-3,4-diones. Phosphorus Sulfur Silicon Relat. Elem. 1998, 134, 487–492. [Google Scholar] [CrossRef]
- Aliev, Z.G.; Shurov, S.N.; Nekrasov, D.D.; Podvintsev, I.B.; Atovmyan, L.O. Character of enolization of the β-dicarbonyl fragment in α,γ-dioxocarboxylic acids. Crystal and molecular structure of benzoyl- and cinnamoylpyruvic acids. J. Struct. Chem. 2000, 41, 1041–1045. [Google Scholar] [CrossRef]
- Berclas, T.; Bernardinelli, G.; Geoffroy, M.; Rao, G.; Tancic, Z. EPR/ENDOR study of an X-irradiated single crystal of 1-triphenylphosphoranylidene-2-propanone: The role of hydrogen bonds in the trapping of radiogenic radicals. Radiation Phys. Chem. 1999, 56, 539–545. [Google Scholar] [CrossRef]
- Aitken, R.A.; Cleghorn, L.P.; Leitch, R.M.; Morrill, L.C.; Slawin, A.M.Z. Unexpected rearrangement leading to formation of a 1,3-bis(triphenylphosphonio)prop-1-en-3-idyl carboxylate. Eur. J. Org. Chem. 2010, 2010, 3211–3214. [Google Scholar] [CrossRef]
- Aitken, R.A.; Karodia, N. Flash vacuum pyrolysis of stabilized phosphorus ylides, 9. Preparation and pyrolysis of β,γ-dioxo ylides, β,β’,γ,γ’-tetraoxo ylides and hexaoxo bis(ylides). Liebigs Ann./Recueil 1997, 1997, 779–783. [Google Scholar] [CrossRef]
- Brömme, E.; Claisen, L. Ueber die Einwirkung des Oxaläthers auf Acetophenon. Ber. Dtsch. Chem. Ges. 1888, 21, 1131–1135. [Google Scholar] [CrossRef]
- Ramirez, F.; Dershowitz, S. Phosphinemethylenes II. Triphenylphosphineacylmethylenes. J. Org. Chem. 1957, 22, 41–45. [Google Scholar] [CrossRef]
- Aitken, R.A.; Cadogan, J.I.G.; Gosney, I. Convenient preparation of unsymmetrically substituted benzils by permanganate oxidation of β-oxo phosphorus ylides. Phosphorus Sulfur Silicon Relat. Elem. 1995, 101, 281–286. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELXL. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Structures of Ph3PO 1 and vinylogous oxo-stabilised ylides 2.

Figure 2.
Previously reported intramolecular hydrogen-bonded structures.

Scheme 1.
Discovery of hydrogen bonded adduct 9.

Figure 3.
Molecular structure of 9 with anisotropic displacement ellipsoids drawn at the 50% probability level and the numbering system used.
Figure 3.
Molecular structure of 9 with anisotropic displacement ellipsoids drawn at the 50% probability level and the numbering system used.

Figure 4.
Hydrogen bonding interactions in the crystal structure of 9.

Figure 5.
Ylide—carboxylic acid adducts formed.

Figure 6.
Molecular structure of 11 with anisotropic displacement ellipsoids drawn at the 50% probability level and the numbering system used.
Figure 6.
Molecular structure of 11 with anisotropic displacement ellipsoids drawn at the 50% probability level and the numbering system used.

Figure 7.
Hydrogen bonding interactions in the crystal structure of 11.

Figure 8.
Bond lengths (Å) in triphenylphosphoranylideneacetone and its adducts as determined by X-ray diffraction.
Figure 8.
Bond lengths (Å) in triphenylphosphoranylideneacetone and its adducts as determined by X-ray diffraction.

Figure 9.
Formation of adducts with other donor types.

Figure 10.
Molecular structure of 21 with anisotropic displacement ellipsoids drawn at the 50% probability level and the numbering system used.
Figure 10.
Molecular structure of 21 with anisotropic displacement ellipsoids drawn at the 50% probability level and the numbering system used.

Figure 11.
Hydrogen bonding interactions in the crystal structure of 21 and schematic representation.
Figure 11.
Hydrogen bonding interactions in the crystal structure of 21 and schematic representation.

Scheme 2.
Synthesis of bis(ylide) 26.

Figure 12.
Molecular structure of 27 with anisotropic displacement ellipsoids drawn at the 50% probability level and the numbering system used for non-C,H atoms.
Figure 12.
Molecular structure of 27 with anisotropic displacement ellipsoids drawn at the 50% probability level and the numbering system used for non-C,H atoms.

Figure 13.
Hydrogen bonding interactions in the crystal structure of 27 and schematic representation.
Figure 13.
Hydrogen bonding interactions in the crystal structure of 27 and schematic representation.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Hydrogen bonding parameters for ylide adducts (Å, °).
| Compound | D–H...A | D–H | H...A | D...A | D–H...A |
|---|---|---|---|---|---|
| 9 | O(7)–H(7)...O(3) | 0.98(8) | 1.62(9) | 2.490(9) | 145(6) |
| 9 | O(9)–H(9)...O(11) | 0.98(6) | 1.59(6) | 2.501(8) | 153(6) |
| 9 | O(47)–H(47)...O(43) | 0.98(4) | 1.51(2) | 2.471(8) | 165(7) |
| 9 | O(49)–H(49)...O(51) | 0.98(9) | 1.58(8) | 2.507(9) | 156(8) |
| 11 | O(6)–H(6)...O(3) | 0.93(5) | 1.53(5) | 2.501(6) | 173(6) |
| 11 | O(36)–H(36)...O(33) | 0.98(5) | 1.53(5) | 2.506(6) | 171(6) |
| 21 | N(5)–H(5A)...O(3) | 0.86(3) | 2.04(3) | 2.889(3) | 169(2) |
| 21 | N(5)–H(5B)...O(6) | 0.94(3) | 1.97(3) | 2.901(4) | 173(3) |
| 27 | O(102)–H(10B)...O(2) | 0.980(4) | 1.639(3) | 2.555(5) | 153.83(19) |
| 27 | O(108)–H(10M)...O(52) | 0.980(3) | 1.803(3) | 2.783(4) | 177.8(2) |
Table 2.
Changes observed in key 13C NMR shifts upon adduct formation (ppm).
| Adduct | CO2H (δ) | P=C (δ) | ||||
|---|---|---|---|---|---|---|
| Adduct | Free Acid | Δδ | Adduct | Free Ylide | Δδ | |
| 10 | 170.2 | 172.6 | −2.4 | 58.8 | 57.0 | +1.8 |
| 11 | 169.6 | 172.6 | −3.0 | 56.3 | 51.5 | +4.8 |
| 12 | 169.7 | 172.6 | −2.9 | 75.1 | 71.0 | +4.1 |
| 13 | 174.6 | 178.2 | −3.6 | 56.1 | 51.5 | +4.6 |
| 14 | 175.2 | 179.0 | −3.8 | 56.0 | 51.5 | +4.5 |
| 15 | 177.0 | 180.5 | −3.5 | 55.4 | 51.5 | +3.9 |
| 16 | 174.5 | 178.2 | −3.7 | 52.7 | 50.6 | +2.1 |
| 17 | 174.9 | 179.0 | −4.1 | 52.5 | 50.6 | +1.9 |
| 18 | 177.7 | 180.5 | −2.8 | 53.5 | 50.6 | +2.9 |
Table 3.
Solvent effect on the key 13C NMR shifts upon adduct formation for 11 (ppm).
| Solvent | CO2H (δ) | P=C (δ) | ||||
|---|---|---|---|---|---|---|
| Adduct | Free Acid | Δδ | Adduct | Free Ylide | Δδ | |
| C6D6 | 168.8 | 168.9 | −0.1 | 55.9 | 50.2 | +5.7 |
| CDCl3 | 169.6 | 172.6 | −3.0 | 56.3 | 51.5 | +4.8 |
| CD3COCD3 | 168.1 | 168.0 | +0.1 | 54.4 | 51.7 | +2.7 |
| CD3OD | 170.7 | 169.8 | +0.9 | 54.1 | 51.8 | +2.3 |
| CD3SOCD3 | 167.4 | 167.3 | +0.1 | 50.2 | 49.7 | +0.5 |
Table 4.
Formation of adducts with other types of donor in CDCl3.
| Adduct | Donor | δC Adduct | δC Free Ylide | Δδ |
|---|---|---|---|---|
| 19 | MeCO2H | 56.5 | 51.5 | +5.0 |
| 20 | PhCH2OH | 58.0 | 51.5 | +6.5 |
| 21 | PhCONH2 | 56.2 | 51.5 | +4.7 |
| 22 | PhCSNH2 | 54.5 | 51.5 | +3.0 |
| 23 | PhSO2H | 57.2 | 51.5 | +5.7 |
| 24 | Ph2P(=O)OH | 59.1 | 51.5 | +7.6 |
| - | PhCH2NH2 | 51.8 | 51.5 | +0.3 |
| - | PhSO3H | - | 51.5 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Aitken, R.A.; Cleghorn, L.P.; Dawson, G.; Gray, I.P.; Lashtabeg, A.; Slawin, A.M.Z. Oxo-Stabilised Phosphonium Ylides as Hydrogen Bond Acceptors. Inorganics 2023, 11, 50. https://doi.org/10.3390/inorganics11020050
AMA Style
Aitken RA, Cleghorn LP, Dawson G, Gray IP, Lashtabeg A, Slawin AMZ. Oxo-Stabilised Phosphonium Ylides as Hydrogen Bond Acceptors. Inorganics. 2023; 11(2):50. https://doi.org/10.3390/inorganics11020050
Chicago/Turabian StyleAitken, R. Alan, Lee P. Cleghorn, Graham Dawson, Ian P. Gray, Anna Lashtabeg, and Alexandra M. Z. Slawin. 2023. "Oxo-Stabilised Phosphonium Ylides as Hydrogen Bond Acceptors" Inorganics 11, no. 2: 50. https://doi.org/10.3390/inorganics11020050
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.