
Molecular Switching through Chalcogen-Bond-Induced Isomerization of Binuclear (Diaminocarbene)PdII Complexes †

Institute of Chemistry, Saint Petersburg State University, 26 Universitetskii Prospect, Petergof, St. Petersburg 198504, Russia
*
Author to whom correspondence should be addressed.
†
In commemoration of the 300th anniversary of Saint Petersburg State University’s founding.
Inorganics 2023, 11(6), 255; https://doi.org/10.3390/inorganics11060255
Submission received: 25 May 2023
/
Revised: 5 June 2023
/
Accepted: 7 June 2023
/
Published: 9 June 2023
(This article belongs to the Special Issue Non-covalent Interactions in Coordination Chemistry)
Abstract
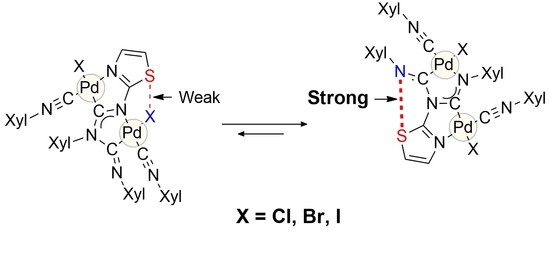
:Binuclear diaminocarbene complexes, which form as a regioisomer mixture in the reaction between isocyanide–palladium(II) complex cis-[PdCl2(CNXyl)2] and 1,3-thiazol-2-amine, are able to exchange an anionic chloride ligand with other halides, such as Br or I. This process also affords binuclear complexes as mixtures of kinetically and thermodynamically controlled regioisomers. In CDCl3 solutions, we observed interconversion of kinetically and thermodynamically controlled regioisomers. The results of the DFT calculations revealed that in CHCl3 solution, each pair of the isomers exhibited two different types of chalcogen bonding such as S···X or S···N; the presence of CBs for two complexes in the solid state was also proven through X-ray crystallographic study. Based on the combined experimental and theoretical data, it could be concluded that thermodynamic favorability for the formation of thermodynamically controlled regioisomers increases in the Cl < Br ≈ I row and correlate well with the energy difference between S···N and S···X (X = Cl, Br, I) chalcogen bonds in kinetically and thermodynamically controlled products. This means that it is possible to change the structure of metallocycles in binuclear diaminocarbene complexes by simply replacing one halide ligand with another.
1. Introduction
Chalcogen bonding, or a chalcogen bond (CB), is one of the varieties of σ-hole non-covalent interactions [1,2]. This interaction consists of bonding the region of low electron density lying on the axis of a covalent bond between an electron withdrawing group (EWG) and an element outside of this bond (σ-hole), and the region of increased electron density [3]. In the case of the chalcogen bond, sulfur acts as this element. The region of increased electron density is most often due to the proximity to an element that has a lone electron pair. Such interactions are denoted σ-lp.
Recently, this phenomenon has been the subject of much research in the fields of polymer science [4,5,6], drug development [7,8], biochemistry [9,10,11,12], crystal engineering [13,14,15,16,17], and supramolecular chemistry [18,19,20]. The reason for this is that chalcogen bonds (as well as other non-covalent interactions) determine many practically significant physicochemical properties of chalcogen-containing compounds: conformational stabilization [21], molecular assembly [22,23,24], protein folding [25,26], molecular recognition [9,27], and even reactivity [28,29]. Of course, the heavy chalcogen atoms—namely selenium and tellurium– are better CB donors than sulfur due to their higher polarizability [30,31]. However, the much more common sulfur-containing compounds play a much more important role in applied research. This causes an ever-increasing interest in the study of CBs, including S-centers.
The best known types of CB involving S atoms are S···N, S···O, and S···S interactions, which are often found both in biomolecules [10,32] and in organic [33,34,35] and coordination compounds [36,37,38]. Most of the examples described relate to the stabilization by CB of a certain conformation in intermediates or in starting compounds. These are papers on stabilization of the Pummerer reaction intermediate by inter- and intramolecular S···O contacts [39]; the use of chiral derivatives of isothiourea as organocatalysts, forming intramolecular S···O CB in reaction intermediates [40,41,42]; photochromic cyclization of 2,3-dithiazolylbenzothiophene [43]; assistance in intramolecular redox cyclization of S-containing diazene [44]; and bond-switching rearrangement of substituted isothiazoles and isothiadiazoles [45] to form, in both cases, intramolecular S···N-CB in the starting compounds. Other reports reveal the effect of CB on the reaction center, namely, hydrodeboronation of 5-thiazolylboronic acid [46], facilitation of S–S bond cleavage in benzo-1,2-dithiolan-3-one-1-oxides by formation of S···O contacts [47], and the Mann–Pope reaction [48], which are due to intramolecular S···O interactions. So far, CBs of sulfur atoms with halogens have been less studied, although the interest in such interactions is undeniable. For example, in the previously mentioned work on benzo-1,2-dithiolan-3-on-1-oxides, S–S bond cleavage can also be facilitated by the interaction of S···Cl.
We have previously shown [36] that the cause of a chemical reaction can be not only CB itself, but also the difference between two CBs. Binuclear diaminocarbene complexes formed upon treatment of cis-[PdCl2(CNXyl)2] (Xyl = 2,6-Me2C6H3) with 1,3-thiazol-2-amines exist as a mixture of regioisomers (Scheme 1), one of which is the product of the kinetic control (isomer I), and the other is the thermodynamic one (isomer II). In CHCl3 solutions, isomer I is reversibly converted to isomer II. According to theoretical calculations, the reason for this transformation is that chalcogen bond S···N in II is 1.8–2.3 kcal/mol more stable than chalcogen bond S···Cl in I.
In this work, we have shown that the equilibrium in this system can be further shifted by a change of the halide ligand at the PdII-center. The latter can be caused by a change of the energy difference between two types of S···Hal and S···N intramolecular chalcogen bonds.
2. Results and Discussions
2.1. Synthesis of Binuclear Diaminocarbene Complexes
As has been previously reported [36], the reaction of cis-[PdCl2(CNXyl)2] (1) with 1,3- thiazol-2-amine (2) in a molar ratio 1:1.5 in CH2Cl2 at RT affords a mixture of two binuclear aminocarbene palladium(II) complexes bearing chloride anion ligands (Scheme 1). These products are the kinetically controlled regioisomer 3a and the thermodynamically controlled regioisomer 3b.
The treatment of the reaction mixture containing the chloride complexes 3a and 3b with two-fold excess of AgOTf in CH2Cl2 at RT, followed by the addition of KBr or KI, affords a mixture of two new binuclear aminocarbene products for each case, corresponding to kinetically (4a or 5a) and thermodynamically (4b or 5b) controlled regioisomers (Scheme 2). The mixture is enriched with the thermodynamically controlled regioisomer (Scheme 3).
2.2. The Regioisomerization
As the reaction proceeds, a mixture of both isomers (a and b) is formed. However, over time, the mixture is enriched with the thermodynamically controlled regioisomer (Scheme 3).
The isomerization of the kinetically to the thermodynamically controlled regioisomers was monitored via 1H NMR in CDCl3 (Table 1). The isomerization of 3a was slow. At 45 °C, the equilibrium was reached after 10 days (Figure 1). In contrast to the chlorine-containing complexes, the isomerization of 4a (Figure 2) and 5a (Figure 3) proceeded much faster; at 45 °C, it took 30 h.
The observed regioisomerization can proceed only with cleavage of the Pd−N and C−N bonds of the aminocarbene fragment. The kinetical study of the regioisomerization of the similar binuclear diaminocarbene complexes showed that it is a first-order reaction; that is, it occurs intramolecularly [37]. It was concluded that the rate-determining step of the isomerization is breaking the carbon–nitrogen bond in the carbene fragment of the binuclear complex.
Data on the isomeric ratios and the equilibrium constants are presented in Table 1. It can be seen from the obtained data that the bromide and iodide complexes are relatively quickly converted into the almost pure regioisomers, b. This means that it is impossible to isolate regioisomers, a, from the initially formed mixtures. To obtain the pure complexes 4a and 5a, we applied the following method. The replacement of the halogen was carried out not for mixture 3a/b, but for the pure isomer 3a (Scheme 4). Since the reaction was carried out at RT for a short time (10 min), regiomerization did not have time to occur, and we were able to isolate the pure complexes 4a and 5a.
Such a dependence of the structure of binuclear complexes on the nature of the halogen makes it possible to obtain the desired specific regioisomer; that is, by changing the halide ligand, we can switch the mutual arrangement of atoms in the bicyclic structure.
2.3. Characterization of the Regioisomers 4a,b–5a,b
Complexes 4–5a,b were obtained as yellow solids and characterized by elemental (C, H, N) analysis (as the regioisomer mixtures), high-resolution ESI+–MS (as the regioisomer mixtures), FTIR, and 1D (1H, 13C{1H}) and 2D (1H,1H-COSY, 1H,1H-NOESY, 1H,13C-HSQC, 1H,13C-HMBC) NMR spectroscopies. The assignments of the 1H and 13C signals were performed with 1H,1H-COSY, 1H,1HNOESY, 1H,13C-HSQC, and 1H,13C-HMBC NMR methods. For all isomer mixtures, the HRESI+ mass spectra displayed a fragmentation pattern corresponding to [M − Hal]+ with the characteristic isotopic distribution.
The FTIR spectra for all complexes exhibited one strong and broad ν(C≡N) band in the range 2210–2190 cm−1 from isocyanide ligands.
The 1H NMR spectra of the complexes displayed a set of overlapping and individual signals in the δ range 6.10–7.25 ppm corresponding to the twelve aromatic C–H protons of the xylyl groups and signals of the thiaazaheterocyclic fragment in the δ range 8.25–6.80 ppm. Signals of the Me groups were observed in the δ range 2.45–2.00 ppm.
The 13C NMR spectra of the complexes exhibited two signals of carbons in the NCN fragments in the δ range 150–195 ppm. These resonances belong to the typical range specific to Pd–Ccarbene (δC = 160–200 ppm) in acyclic diaminocarbenes [36,49,50].
In addition, the XRD data for complexes 4b and 5b were obtained. The plots of the structures 4b and 5b with the highlighted intramolecular chalcogen bonds are presented in Figure 4. Selected bond lengths and angles are given in Table 2. The metal centers have a slightly distorted square planar structure, and the halogen and nitrogen atoms bonded to the metal center are in the cis position to each other. In the NCN fragments bonded to the metal atom, the length of one of the CN bonds is in the range 1.407–1.444 Å; that is, it is close in length to the simple C–N bond (1.451(1) Å) [51]. The other has a length of 1.259–1.290 Å, which corresponds to the length of the double C=N bond (1.260(1) Å) [51]. All bond lengths and angles are similar to those for the previously reported thiazole-2-amine-based analogues [36].
Checking of the XRD data indicates the existence of chalcogen bonding S···N in the bromide and iodide complexes 4b and 5b in solid state. Indeed, the distances between the S and N atoms in 4b and 5b (2.708(2) Å and 2.704(3), respectively) are shorter than their van der Waals radii sum (viz., 3.35 Å according to Bondi’s definition) [52] and very close to the same contact in the previously reported structure of the thiazole-2-amin-based binuclear aminocarbene chloride complex 3b (2.672 Å).
2.4. Analysis of the Intramolecular Chalcogen Bonding
We performed quantum chemical calculations and carried out topological analysis of the electron density distribution (aka AIM analysis) [53] to confirm the existence of these non-covalent interactions and quantitatively estimate their energies. Note that we already used such an approach in studies of these non-covalent contacts for the previously reported thiazole-2-amin-based binuclear aminocarbene chloride complexes 3a and 3b [36]. The results of the current quantum chemical study are given in Table 3, and the Laplacian of electron density distribution ∇2ρ(r) contour line diagram, together with surfaces of zero-flux and bond paths for 4b, are illustrated in Figure 5. The results of the AIM analysis reveal that the S···N chalcogen bonds in 4b and 5b are slightly weaker than in the chloride complex 3b [36] in the solid state (4.9–5.3 kcal/mol vs. 5.1−6.0 kcal/mol).
To estimate the effect of these non-covalent interactions on the chemical equilibrium of the 4–5a,b complexes in the chloroform solution, we carried out geometry optimization of these two isomeric pairs using DFT calculations followed by additional AIM analysis, as well as theoretical analysis of relative stability for these isomer pairs in the chloroform solution. In all cases, AIM analysis indicated the presence of chalcogen bonds S···X (X = Br, I) for 4–5a and S···N for 4–5b in the optimized equilibrium model structures. Analysis of energy differences between the S···X (X = Cl, Br, I) and S···N intramolecular chalcogen bonds in the complex pairs 4–5a/4–5b indicated that in all cases the S···N interaction was stronger than for S···X (X = Cl, Br, I) (Table 3). Note that elongation of S···X contacts in the optimized equilibrium model structures in the row Cl<Br<I correlates well with increasing of atomic radii in the same order (viz. Cl<Br<I) (Table 3). It was experimentally observed that the change of chloride ligand on bromide and iodide led to a shift of equilibrium towards complexes 3–5b during the isomerization of complexes 3–5a/3–5b, and this observation can also be supported by theoretical analysis of the relative stability of these isomer pairs in the chloroform solution; in accordance with the conducted quantum chemical calculations, the relative stability of b complexes with an S···N chalcogen bond also increased in the row Cl < Br ≈ I (Table 4).
3. Materials and Methods
3.1. General
Solvents, PdCl2, AgOTf, heterocycle 2, and xylyl isocyanide were obtained from commercial sources and used as received. Complex cis-[PdCl2(CNXyl)2] was synthesized via the procedure outlined in the literature [56]. Elemental analyses were carried out on a Euro EA 3028 HT CHNSO analyzer. ESI mass-spectra were recorded on a Bruker micrOTOF (Bruker, Billerica, MA, USA) spectrometer with CH2Cl2/MeOH mixture as a solvent. The analyses were carried out in positive ion mode (m/z range of 50–3000). The most intensive peak in the isotopic pattern was reported. IR spectra were recorded on Shimadzu FTIR 8400S (Shimadzu, Kyoto, Japan) instrument in KBr pellets (4000–400 cm−1, resolution 2 cm−1). The 1D NMR spectra were recorded on a Bruker Avance 400 (Bruker Corporation, Billerica, MA, USA) spectrometer; 2D NMR correlation experiments were recorded on a Bruker Avance II+ 500 MHz (UltraShield Magnet) (Bruker Corporation, Billerica, MA, USA) spectrometer. All NMR spectra were recorded in CDCl3. X-ray diffraction data were collected at a single crystal diffractometer (Agilent Technologies, Santa Clara, CA, USA) (Oxford Diffraction) “Xcalibur” using Mo-Kα (λ = 0.71073 nm) radiation.
3.2. Synthesis of the Complexes
Synthesis of 3a. The solid isocyanide complex 1 (0.2 mmol) and heterocycle 2 (0.3 mmol) were placed in a 10 mL flat-bottom flask and filled with 5 mL of CH2Cl2. The resulting suspension was stirred at room temperature for 2 days. As the reaction proceeded, the reaction mixture turned yellow and aminothiazole hydrochloride precipitate formed. The solution was filtered from the precipitate and dried under reduced pressure. The resulting mixture contained complexes 3a and 3b. The summary yield was 82%. For the further synthesis of complexes 4b and 5b, the obtained mixture of complexes 3a and 3b was used further without separation. The pure complex 3a was synthesized as per the literature [36].
Synthesis of 4a and 5a. Pure complex 3a (0.03 mmol) and silver triflate (0.06 mmol) were added to a 10 mL flat-bottom flask and filled with 2 mL of CH2Cl2. The resulting suspension was put into a room temperature ultrasonic bath for 10 min. As the reaction proceeded, a yellow solution of complex 6a and white precipitate of AgCl formed. The solution was filtered from the precipitate and then 1 mmol KBr (in the case of 4a) or KI (in the case of 5a) was added. The resulting suspension was stirred at room temperature for 10 min. After that, the solution was filtered from the precipitate and dried under reduced pressure.
Synthesis of 4a/b (mixture) and 5a/b (mixture). A mixture of complexes 3a and 3b (0.03 mmol) and silver triflate (0.06 mmol) was added to a 10 mL flat-bottom flask and filled with 2 mL of CHCl3. The resulting suspension was put into a room temperature ultrasonic bath for 10 min. As the reaction proceeded, a yellow solution of mixture of complexes 6a and 6b and white precipitate of AgCl formed. The solution was filtered from the precipitate and then 1 mmol KBr (in the case of 4a,b) or KI (in the case of 5a,b) was added. The resulting suspension was stirred at room temperature for 10 min. After that, the solution was filtered from the precipitate and dried under reduced pressure.
Synthesis of 4b and 5b. The resulting mixture from the previous stage was dissolved in 2 mL of CHCl3 and heated for 2 d at 45 °C. After that, the solution was dried under reduced pressure.
3.3. Characterisation of the Complexes
3a/b mixtures. Anal. calcd for C39H38Cl2N6Pd2S·0.2CDCl3: C, 50.59; H, 4.16; N, 9.03, found: C, 50.19; H, 4.12; N, 9.43. HRESI+ MS: calcd for C39H38ClN6Pd2S+ 871.0641, found m/z 871.0611 [M – Cl]+. IR (KBr, selected bands, cm−1): ν(C–H) 2920 (w), 2850 (w), ν(C≡N) 2193 (s), ν(C=N) 1634 (s), 1554 (s), δ(C–H) 772 (m).

3a. Yield 68%. NMR spectra fully corresponding with the literature data [36]. 1H NMR (δ, ppm, J/Hz): 2.04 (s, 6H, CH3, Xyl-C), 2.21 (s, 6H, CH3, Xyl-D), 2.26 (s, 6H, CH3, Xyl-A), 2.42 (s, 6H, CH3, Xyl-B), 6.13 (t, 1H, H4, Xyl-C, J = 7.6), 6.43 (t, 1H, H4, Xyl-B, J = 7.6), 6.63 (d, 2H, H3,5, Xyl-C, J = 7.6), 6.82–6.88 (m, 2H, H3,5, Xyl-B and 1H, H5, thiazole), 6.93–6.96 (m, 2H, H3,5, Xyl-D), 7.02–7.05 (m, 2H, H3,5, Xyl-A), 7.10 (t, 1H, H4, Xyl-D, J = 7.6), 7.16–7.21 (m, 1H, H4, Xyl-A), 7.86 (d, 1H, H4, thiazole, J = 4.0). 13C{1H} NMR (δ, ppm): 18.40 (2C, CH3, Xyl-D), 18.46 (2C, CH3, Xyl-A), 19.42 (2C, CH3, Xyl-B), 19.84 (2C, CH3, Xyl-C), 112.70 (1C, C5, thiazole), 123.71 (1C, C4, Xyl-C), 126.43 (1C, C1, Xyl-A), 126.80 (2C, C2,6, Xyl-C), 127.34 (2C, C3,5, Xyl-D), 127.54 (2C, C3,5, Xyl-C), 127.72 (2C, C3,5, Xyl-A), 127.74 (2C, C3,5, Xyl-B), 128.95 (1C, C4, Xyl-B), 129.48 (1C, C4, Xyl-D), 129.60 (1C, C4, Xyl-A), 134.15 (4C, C2,6, Xyl-A and Xyl-D), 134.13 (1C, C4, thiazole), 136.29 (2C, C2,6, Xyl-B), 142.51 (1C, C1, Xyl-B), 149.71 (1C, C1, Xyl-C), 164.16 (1C, C2, carbene), 180.26 (1C, C2, thiazole), 194.67 (1C, C1, carbene). The signals of the isocyanide quaternary atoms and C1 Xyl-D were not found even at high acquisition.

3b. NMR spectra fully corresponding with the literature data [36]. 1H NMR (δ, ppm, J/Hz): 2.21 (s, 6H, CH3, Xyl-D), 2.27 (s, 6H, CH3, Xyl-C), 2.28 (s, 6H, CH3, Xyl-A), 2.42 (s, 6H, CH3, Xyl-B), 6.23 (t, 1H, H4, Xyl-C, J = 7.5), 6.33 (t, 1H, H4, Xyl-B, J = 7.6), 6.78 (d, 2H, H3,5, Xyl-C, J = 7.5), 6.83 (d, 2H, H3,5, Xyl-B, J = 7.6), 6.93–6.96 (m, 3H, H3,5, Xyl-D and 1H, H5, thiazole), 7.03 (d, 2H, H3,5, Xyl-A, J = 7.6), 7.11 (t, 1H, H4, Xyl-D, J = 7.6), 7.19 (t, 1H, H4, Xyl-A, J = 7.6), 8.07 (d, 1H, H4, thiazole, J = 4.0). 13C{1H} NMR (δ, ppm): 18.43 (2C, CH3, Xyl-D), 18.50 (2C, CH3, Xyl-A), 19.09 (2C, CH3, Xyl-B), 19.42 (2C, CH3, Xyl-C), 113.56 (1C, C5, thiazole), 124.01 (1C, C4, Xyl-C), 127.34 (2C, C3,5, Xyl-D), 127.38 (2C, C3,5, Xyl-B), 127.57 (1C, C4, Xyl-B), 127.61 (2C, C3,5, Xyl-A), 127.69 (2C, C3,5, Xyl-C), 127.97 (2C, C2,6, Xyl-C), 128.95 (1C, C4, Xyl-D), 129.48 (1C, C4, Xyl-A), 132.63 (2C, C2,6, Xyl-B), 134.13 (2C, C2,6, Xyl-A), 134.18 (1C, C4, thiazole), 134.36 (2C, C2,6, Xyl-D), 148.16 (1C, C1, Xyl-B), 148.40 (1C, C1, Xyl-C), 160.23 (1C, C2, carbene), 161.76 (1C, C2, thiazole), 179.22 (1C, C1, carbene). The signals of the isocyanide quaternary atoms and C1 Xyl-A and Xyl-D were not found even at high acquisition.
4a/b mixtures. Anal. calcd for C39H38Br2N6Pd2S: C, 47.05; H, 3.85; N, 8.44, found: C, 46.70; H, 3.76; N, 8.92. HRESI+ MS: calcd for C39H38BrN6Pd2S+ 915.0124, found m/z 915.0136 [M – Br]+.

4a. Yield 90% (27 mg). IR (KBr, selected bands, cm−1): ν(C–H) 2921 (w), 2920 (w), 2853 (w), ν(C≡N) 2206 (s), ν(C=N) 1648 (s), δ(C–H) 771 (m), 638 (s). 1H NMR (δ, ppm, J/Hz): 2.02 (s, 6H, CH3, Xyl-D), 2.24 (s, 6H, CH3, Xyl-C), 2.29 (s, 6H, CH3, Xyl-A), 2.45 (s, 6H, CH3, Xyl-B), 6.18 (t, 1H, H4, Xyl-C, J = 7.6), 6.48 (t, 1H, H4, Xyl-B, J = 7.6), 6.66 (d, 2H, H3,5, Xyl-C, J = 7.6), 6.90 (d, 2H, H3,5, Xyl-B, J = 7.6), 6.96 (d, 2H, H3,5, Xyl-D, J = 7.6), 7.01 (d, 1H, H5, thiazole, J = 4.0), 7.06 (d, 2H, H3,5, Xyl-A), 7.13 (t, 1H, H4, Xyl-D, J = 7.6), 7.23 (m, 1H, H4, Xyl-A), 7.47 (s, 1H, H4, thiazole, J = 4.0). 13C{1H} NMR (δ, ppm): 18.13 (2C, CH3, Xyl-A), 18.21 (2C, CH3, Xyl-D), 19.10 (2C, CH3, Xyl-C), 19.37 (2C, CH3, Xyl-B), 114.59 (1C, C5, thiazole), 118.07 (1C, C1, Xyl-D), 120.61 (1C, C1, Xyl-A), 124.43 (1C, C4, Xyl-C), 126.24 (2C, C2,6, Xyl-C), 127.48 (2C, C3,5, Xyl-D), 127.79 (2C, C3,5, Xyl-C), 127.90 (2C, C3,5, Xyl-A), 128.12 (2C, C3,5, Xyl-B), 129.64 (1C, C4, Xyl-D), 130.36 (1C, C4, Xyl-A), 130.40 (1C, C4, Xyl-B), 134.38 (1C, C4, thiazole), 134.73 (2C, C2,6, Xyl-A), 134.83 (2C, C2,6, Xyl-D), 136.16 (2C, C2,6, Xyl-B), 141.64 (1C, C1, Xyl-B), 147.91 (1C, C1, Xyl-C), 150.89 (1C, C2, carbene), 176.70 (1C, C2, thiazole), 185.68 (1C, C1, carbene). The signals of the isocyanide quaternary atoms were not found even at high acquisition.

4b. Yield 86% (26 mg). IR (KBr, selected bands, cm−1): ν(C–H) 2977 (w), 2920 (w), 2851 (w), ν(C≡N) 2202 (s), ν(C=N) 1627 (s), 1556 (s), δ(C–H) 773 (m), 747 (m). 1H NMR (δ, ppm, J/Hz): 2.25 (s, 12H, CH3, Xyl-C and Xyl-D), 2.28 (s, 6H, CH3, Xyl-A), 2.44 (s, 6H, CH3, Xyl-B), 6.31 (t, 1H, H4, Xyl-C, J = 7.7), 6.40 (t, 1H, H4, Xyl-B, J = 7.6), 6.80–6.84 (m, 4H, H3,5, Xyl-B and H3,5, Xyl-C), 6.95 (d, 2H, H3,5, Xyl-D, J = 7.8), 7.04–7.06 (m, 3H, 1H, H5, thiazole, and H3,5, Xyl-A), 7.13 (t, 1H, H4, Xyl-D, J = 7.8), 7.22 (t, 1H, H4, Xyl-A, J = 7.7), 7.85 (s, 1H, H4, thiazole). 13C{1H} NMR (δ, ppm): 18.14 (2C, CH3, Xyl-A), 18.25 (2C, CH3, Xyl-D), 19.39 (2C, CH3, Xyl-C), 19.54 (2C, CH3, Xyl-B), 115.27 (1C, C5, thiazole), 117.63 (1C, C1, Xyl-A), 120.80 (1C, C1, Xyl-D), 124.97 (1C, C4, Xyl-C), 126.29 (1C, CNXyl-D), 127.50 (2C, C3,5, Xyl-D), 127.70 (2C, C2,6, Xyl-C), 127.84 (2C, C3,5, Xyl-A), 127.97 (2C, C3,5, Xyl-B), 128.03 (2C, C3,5, Xyl-C), 128.35 (1C, C4, Xyl-B), 129.80 (1C, C4, Xyl-D), 130.35 (1C, C4, Xyl-A), 132.59 (2C, C2,6, Xyl-B), 133.93 (1C, C4, thiazole), 134.77 (2C, C2,6, Xyl-D), 135.00 (2C, C2,6, Xyl-A), 145.41 (1C, C1, Xyl-B), 147.10 (1C, C1, Xyl-C), 148.25 (1C, C2, carbene), 161.29 (1C, C2, thiazole), 168.88 (1C, C1, carbene). The signal of the isocyanide quaternary atom CNXyl-A was not found even at high acquisition.
5a/b mixtures. Anal. calcd for C39H38I2N6Pd2S·0.1CHCl3: C, 42.64; H, 3.49; N, 7.63, found: C, 43.03; H, 3.42; N, 7.12. HRESI+ MS: calcd for C39H38IN6Pd2S+ 962.9988, found m/z 962.9960 [M – I]+.

5a. Yield 92% (30 mg). IR (KBr, selected bands, cm−1): ν(C–H) 2922 (w), 2852 (w), 2851 (w), ν(C≡N) 2203 (s), ν(C=N) 1650 (m), δ(C–H) 773 (m), 638 (s). 1H NMR (δ, ppm, J/Hz): 2.02 (s, 6H, CH3, Xyl-D), 2.25 (s, 6H, CH3, Xyl-C), 2.29 (s, 6H, CH3, Xyl-A), 2.45 (s, 6H, CH3, Xyl-B), 6.18 (t, 1H, H4, Xyl-C, J = 7.6), 6.48 (t, 1H, H4, Xyl-B, J = 7.6), 6.66 (d, 2H, H3,5, Xyl-C, J = 7.6), 6.91 (d, 2H, H3,5, Xyl-B, J = 7.6), 6.96 (d, 2H, H3,5, Xyl-D, J = 7.6), 7.01 (d, 1H, H5, thiazole, J = 4.0), 7.06 (d, 2H, H3,5, Xyl-A, J = 7.6), 7.13 (t, 1H, H4, Xyl-D, J = 7.6), 7.23 (t, 1H, H4, Xyl-A, J = 7.6), 7.47 (d, 1H, H4, thiazole, J = 4.0). 13C{1H} NMR (δ, ppm): 18.12 (2C, CH3, Xyl-D), 18.21 (2C, CH3, Xyl-A), 19.09 (2C, CH3, Xyl-B), 19.37 (2C, CH3, Xyl-C), 114.56 (1C, C5, thiazole), 118.07 (1C, C1, Xyl-D), 120.61 (1C, C1, Xyl-A), 124.43 (1C, C4, Xyl-C), 126.23 (2C, C2,6, Xyl-C), 127.47 (2C, C3,5, Xyl-D), 127.78 (2C, C3,5, Xyl-C), 127.89 (2C, C3,5, Xyl-A), 128.11 (2C, C3,5, Xyl-B), 129.63 (1C, C4, Xyl-D), 130.35 (1C, C4, Xyl-A), 130.38 (1C, C4, Xyl-B), 134.40 (1C, C4, thiazole), 134.71 (2C, C2,6, Xyl-A), 134.82 (2C, C2,6, Xyl-d), 136.15 (2C, C2,6, Xyl-B), 141.65 (1C, C1, Xyl-B), 147.90 (1C, C1, Xyl-C), 150.87 (1C, C2, carbene), 176.72 (1C, C2, thiazole), 185.76 (1C, C1, carbene). The signals of the isocyanide quaternary atoms were not found even at high acquisition.

5b. Yield 88% (29 mg). IR (KBr, selected bands, cm−1): ν(C–H) 2975 (w), 2920 (w), 2850 (w), ν(C≡N) 2201 (s), ν(C=N) 1629 (s), 1555 (s), ν(C–H) 771 (m). 1H NMR (δ, ppm, J/Hz): 2.02 (s, 12H, CH3, Xyl-D and Xyl-C), 2.29 (s, 6H, CH3, Xyl-A), 2.45 (s, 6H, CH3, Xyl-B), 6.28 (t, 1H, H4, Xyl-C, J = 7.6), 6.38 (t, 1H, H4, Xyl-B, J = 7.6), 6.81 (d, 2H, H3,5, Xyl-C, J = 7.6), 6.84 (d, 2H, H3,5, Xyl-B, J = 7.6), 6.95 (m, 2H, H3,5, Xyl-D), 7.04–7.08 (m, 3H, 2H3,5, Xyl-A and H5, thiazole), 7.13 (t, 1H, H4, Xyl-D, J = 7.6), 7.22 (t, 1H, H4, Xyl-A, J = 7.7), 7.78 (s, 1H, H4, thiazole). 13C{1H} NMR (δ, ppm): 18.17 (2C, CH3, Xyl-A), 18.24 (2C, CH3, Xyl-D), 19.38 (2C, CH3, Xyl-C), 19.51 (2C, CH3, Xyl-B), 115.09 (1C, C5, thiazole), 117.62 (1C, C1, Xyl-A), 120.79 (1C, C1, Xyl-D), 124.92 (1C, C4, Xyl-C), 126.16 (1C, CNXyl-D), 127.47 (2C, C3,5, Xyl-D), 127.68 (2C, C2,6, Xyl-C), 127.79 (2C, C3,5, Xyl-A), 127.94 (2C, C3,5, Xyl-B), 128.11 (2C, C3,5, Xyl-C), 128.55 (1C, C4, Xyl-B), 129.74 (1C, C4, Xyl-D), 130.23 (1C, C4, Xyl-A), 132.48 (2C, C2,6, Xyl-B), 133.98 (1C, C4, thiazole), 134.67 (2C, C2,6, Xyl-D), 134.98 (2C, C2,6, Xyl-A), 145.34 (1C, C1, Xyl-B), 147.14 (1C, C1, Xyl-C), 148.67 (1C, C2, carbene), 161.32 (1C, C1, carbene). The signals of the isocyanide quaternary atom CNXyl-A and C2 thiazole were not found even at high acquisition.
3.4. Data for the Intermediate Complexes 6a and 6b
6a. HRESI+ MS: calcd for C40H38F3N6O3Pd2S2+ 985.0468, found m/z 985.0444 [M – OTf]+. 1H NMR (δ, ppm, J/Hz): 2.02 (s, 6H, CH3, Xyl-D), 2.25 (s, 6H, CH3, Xyl-C), 2.29 (s, 6H, CH3, Xyl-A), 2.45 (s, 6H, CH3, Xyl-B), 6.18 (t, 1H, H4, Xyl-C, J = 7.6), 6.49 (t, 1H, H4, Xyl-B, J = 7.6), 6.66 (d, 2H, H3,5, Xyl-C, J = 7.6), 6.90 (d, 2H, H3,5, Xyl-B, J = 7.6), 6.96 (m, 2H, H3,5, Xyl-D), 7.04–7.08 (d, 1H, H5, thiazole, J = 4.1), 7.04–7.08 (d, 2H, H3,5, Xyl-A, J = 7.7), 7.13 (t, 1H, H4, Xyl-D, J = 7.6), 7.24 (m, 1H, H4, Xyl-A), 7.48 (d, 1H, H4, thiazole, J = 4.1).

6b. HRESI+ MS: calcd for C40H38F3N6O3Pd2S2+ 985.0468, found m/z 985.0444 [M – OTf]+. 1H NMR (δ, ppm, J/Hz): 2.25 (s, 12H, CH3, Xyl-D and Xyl-C), 2.29 (s, 6H, CH3, Xyl-A), 2.45 (s, 6H, CH3, Xyl-B), 6.29 (t, 1H, H4, Xyl-C, J = 7.6), 6.49 (t, 1H, H4, Xyl-B, J = 7.6), 6.81 (d, 2H, H3,5, Xyl-C, J = 7.6), 6.83 (d, 2H, H3,5, Xyl-B, J = 7.6), 6.96 (d, 2H, H3,5, Xyl-D, J = 7.6), 7.05 (d, 2H, H3,5, Xyl-A, J = 7.7), 7.09 (d, 1H, H5, thiazole, J = 4.1), 7.14 (t, 1H, H4, Xyl-D, J = 7.6), 7.23 (t, 1H, H4, Xyl-A J = 7.7), 7.72 (d, 1H, H4, thiazole, J = 4.1).

3.5. X-ray Diffraction
Single crystal X-ray diffraction experiments for all crystals were carried out using an Oxford Diffraction “Xcalibur” diffractometer with monochromated MoKα radiation. The crystals were kept at 100 K throughout data collection. Initial data processing was carried out using the CrysAlisPro (Agilent Technologies, ver. 1.171.36.32) data reduction program. The structures were solved using the Superflip [57] structure solution program and refined by means using the ShelXL [58] structure refinement program incorporated in the Olex2 [59] program package. All crystallographic data for this paper can be obtained free of charge via the Cambridge Crystallographic Database (CCDC 2263951, 2263952).
3.6. Computational Details
The quantum chemical computations for model compounds were performed utilizing the DFT-functional M06 [60] (it was specifically developed to describe weak dispersion forces and non-covalent interactions) using Gaussian-09 [61] software. The experimentally obtained XRD geometries were used as initial points. The quantum chemical computations were carried out using the quasi-relativistic Stuttgart effective core potentials that considered 28 or 46 core electrons and the corresponding basis sets [62] for the Pd or I atoms, respectively, and the 6-31 + G* basis sets for other atoms. The continuum solvation model SMD [63], with chloroform as the solvent, was used to take into account the solvent effects. The Hessian matrices were analytically calculated for the optimized model structures (no symmetry restrictions were applied in all cases) in order to prove the location of true minima on the potential energy surface (no imaginary frequencies were found in all cases), and to estimate the thermodynamic parameters at 25 °C and 1 atm. The AIM analysis [53] was carried out using Multiwfn 3.3.4 software [64]. The atomic coordinates for optimized equilibrium model structures are presented in the Supplementary Materials, Table S2.
4. Conclusions
Binuclear diaminocarbene complexes, which form as the regioisomer mixture 3a/b in the reaction of cis-[PdCl2(CNXyl)2] with 1,3-thiazol-2-amine, are able to exchange an anionic chloride ligand with other halides, such as Br or I. The exchange occurs upon the sequential addition of first AgOTf and then KBr or KI to a mixture of complex 3. This process also affords binuclear complexes 4a/b or 5a/b as mixtures of kinetically (a) and thermodynamically (b) controlled regioisomers. All these species were characterized by elemental analysis, MS, and NMR in CDCl3 solutions. The solid-state structures of two complexes—the thermodynamically controlled regioisomers 4b and 5b—were elucidated with X-ray diffraction.
In CDCl3 solutions, we observed interconversion of kinetically (a) and thermodynamically (b) controlled regioisomers affording equilibrium mixtures of the isomers. Compared to the previously described chloride complex 3, this equilibrium for the bromide complex 4 and for the iodide complex 5 is, firstly, established much faster, and secondly, shifted to a much greater extent towards the thermodynamically controlled isomer. The results of the quantum chemical computations together with AIM analysis showed that in a chloroform solution, each pair of the isomers exhibited two different types of chalcogen bonding, such as S···X (4a and 5a) and S···N (4b and 5b); the presence of CBs for 4b and 5b in the solid state was experimentally proven by XRD analysis as well.
Based on the combined experimental and theoretical data, it can be concluded that thermodynamic favorability for the formation of thermodynamically controlled regioisomers increases in the row Cl<Br≈I and correlates well with the energy difference between S···N and S···X (where X = Cl, Br, or I) chalcogen bonds in kinetically and thermodynamically controlled products. This means that it is possible to change the structure of metallocycles in binuclear diaminocarbene complexes by simply replacing one halide ligand with another.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/inorganics11060255/s1, Figures S1–S8: NMR 1H, and 13C spectra of complexes 4–5a,b; Tables S1 and S2: Crystal data and structure refinement for complexes 4b, 5b and cartesian atomic coordinates of optimized equilibrium model structures; Crystallographic information files (CIF) and checkCIF report files for complexes 4b, 5b.
Author Contributions
Conceptualization, V.P.B.; methodology, V.P.B.; investigation, R.A.P., A.S.N. and V.V.S.; writing—original draft preparation, A.S.N. and R.A.P.; writing—review and editing, V.P.B.; visualization, V.V.S.; supervision, V.P.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Russian Science Foundation, grant number 19-13-00008.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
Physicochemical studies were performed at the Center for Magnetic Resonance, the Center for X-ray Diffraction Studies, and the Center for Chemical Analysis and Materials Research of Saint Petersburg State University.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Wang, W.; Ji, B.; Zhang, Y. Chalcogen Bond: A Sister Noncovalent Bond to Halogen Bond. J. Phys. Chem. A 2009, 113, 8132–8135. [Google Scholar] [CrossRef] [PubMed]
- Aakeroy, C.B.; Bryce, D.L.; Desiraju, G.R.; Frontera, A.; Legon, A.C.; Nicotra, F.; Rissanen, K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the Chalcogen Bond (IUPAC Recommendations 2019). Pure Appl. Chem. 2019, 91, 1889–1892. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Politzer, P. Expansion of the σ-Hole Concept. J. Mol. Model. 2009, 15, 723–729. [Google Scholar] [CrossRef]
- Jackson, N.E.; Savoie, B.M.; Kohlstedt, K.L.; Olvera de la Cruz, M.; Schatz, G.C.; Chen, L.X.; Ratner, M.A. Controlling Conformations of Conjugated Polymers and Small Molecules: The Role of Nonbonding Interactions. J. Am. Chem. Soc. 2013, 135, 10475–10483. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Gong, Z.; Yan, Q. Chalcogen-Bonding Supramolecular Polymers. J. Org. Chem. 2020, 85, 8397–8404. [Google Scholar] [CrossRef]
- Zeng, R.; Gong, Z.; Chen, L.; Yan, Q. Solution Self-Assembly of Chalcogen-Bonding Polymer Partners. ACS Macro. Lett. 2020, 9, 1102–1107. [Google Scholar] [CrossRef]
- Beno, B.R.; Yeung, K.-S.; Bartberger, M.D.; Pennington, L.D.; Meanwell, N.A. A Survey of the Role of Noncovalent Sulfur Interactions in Drug Design. J. Med. Chem. 2015, 58, 4383–4438. [Google Scholar] [CrossRef]
- Reid, R.C.; Yau, M.-K.; Singh, R.; Lim, J.; Fairlie, D.P. Stereoelectronic Effects Dictate Molecular Conformation and Biological Function of Heterocyclic Amides. J. Am. Chem. Soc. 2014, 136, 11914–11917. [Google Scholar] [CrossRef]
- Fick, R.J.; Kroner, G.M.; Nepal, B.; Magnani, R.; Horowitz, S.; Houtz, R.L.; Scheiner, S.; Trievel, R.C. Sulfur–Oxygen Chalcogen Bonding Mediates AdoMet Recognition in the Lysine Methyltransferase SET7/9. ACS Chem. Biol. 2016, 11, 748–754. [Google Scholar] [CrossRef]
- Iwaoka, M.; Babe, N. Mining and Structural Characterization of S···X Chalcogen Bonds in Protein Database. Phosphorus Sulfur Silicon Relat. Elem. 2015, 190, 1257–1264. [Google Scholar] [CrossRef]
- Carugo, O.; Resnati, G.; Metrangolo, P. Chalcogen Bonds Involving Selenium in Protein Structures. ACS Chem. Biol. 2021, 16, 1622–1627. [Google Scholar] [CrossRef] [PubMed]
- Iwaoka, M. Chalcogen-Containing Protein and Nucleic Acid Derivatives—Synthesis and Applications. In Chalcogen Chemistry: Fundamentals and Applications; The Royal Society of Chemistry: London, UK, 2023; pp. 625–647. [Google Scholar]
- Suresh, K.; Minkov, V.S.; Namila, K.K.; Derevyannikova, E.; Losev, E.; Nangia, A.; Boldyreva, E.V. Novel Synthons in Sulfamethizole Cocrystals: Structure–Property Relations and Solubility. Cryst. Growth Des. 2015, 15, 3498–3510. [Google Scholar] [CrossRef]
- Kumar, V.; Triglav, M.; Morin, V.M.; Bryce, D.L. Predictability of Chalcogen-Bond-Driven Crystal Engineering: An X-Ray Diffraction and Selenium-77 Solid-State NMR Investigation of Benzylic Selenocyanate Cocrystals. ACS Org. Inorg. Au 2022, 2, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, W. Pseudo-Bifurcated Chalcogen Bond in Crystal Engineering. Crystals 2018, 8, 163. [Google Scholar] [CrossRef] [Green Version]
- Peloquin, A.J.; McMillen, C.D.; Iacono, S.T.; Pennington, W.T. Crystal Engineering Using Polyiodide Halogen and Chalcogen Bonding to Isolate the Phenothiazinium Radical Cation and Its Rare Dimer, 10-(3-Phenothiazinylidene)Phenothiazinium. Chem. Eur. J. 2021, 27, 8398–8405. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Kumar, V.; Bradshaw, M.J.Z.; Bryce, D.L. Chalcogen-Bonded Cocrystals of Substituted Pyridine N-Oxides and Chalcogenodiazoles: An X-ray Diffraction and Solid-State NMR Investigation. Cryst. Growth Des. 2020, 20, 7910–7920. [Google Scholar] [CrossRef]
- Cozzolino, A.F.; Elder, P.J.W.; Lee, L.M.; Vargas-Baca, I. The Role of the Lewis Acid−base Properties in the Supramolecular Association of 1,2,5-Chalcogenadiazoles. Can. J. Chem. 2013, 91, 338–347. [Google Scholar] [CrossRef]
- Mikherdov, A.; Novikov, A.; Kinzhalov, M.; Zolotarev, A.; Boyarskiy, V. Intra-/Intermolecular Bifurcated Chalcogen Bonding in Crystal Structure of Thiazole/Thiadiazole Derived Binuclear (Diaminocarbene)PdII Complexes. Crystals 2018, 8, 112. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.Y.C.; Marques, I.; Thompson, A.L.; Christensen, K.E.; Félix, V.; Beer, P.D. Chalcogen Bonding Macrocycles and [2]Rotaxanes for Anion Recognition. J. Am. Chem. Soc. 2017, 139, 3122–3133. [Google Scholar] [CrossRef]
- Wojtkowiak, K.; Michalczyk, M.; Zierkiewicz, W.; Jezierska, A.; Panek, J.J. Chalcogen Bond as a Factor Stabilizing Ligand Conformation in the Binding Pocket of Carbonic Anhydrase IX Receptor Mimic. Int. J. Mol. Sci. 2022, 23, 13701. [Google Scholar] [CrossRef]
- Alfuth, J.; Zadykowicz, B.; Wicher, B.; Kazimierczuk, K.; Połoński, T.; Olszewska, T. Cooperativity of Halogen- and Chalcogen-Bonding Interactions in the Self-Assembly of 4-Iodoethynyl- and 4,7-Bis(Iodoethynyl)Benzo-2,1,3-Chalcogenadiazoles: Crystal Structures, Hirshfeld Surface Analyses, and Crystal Lattice Energy Calculations. Cryst. Growth Des. 2022, 22, 1299–1311. [Google Scholar] [CrossRef]
- Mullin, W.J.; Sharber, S.A.; Thomas, S.W. Optimizing the Self-assembly of Conjugated Polymers and Small Molecules through Structurally Programmed Non-covalent Control. J. Polym. Sci. 2021, 59, 1643–1663. [Google Scholar] [CrossRef]
- Rahman, F.-U.; Tzeli, D.; Petsalakis, I.D.; Theodorakopoulos, G.; Ballester, P.; Rebek, J.; Yu, Y. Chalcogen Bonding and Hydrophobic Effects Force Molecules into Small Spaces. J. Am. Chem. Soc. 2020, 142, 5876–5883. [Google Scholar] [CrossRef]
- Iwaoka, M.; Isozumi, N. Hypervalent Nonbonded Interactions of a Divalent Sulfur Atom. Implications in Protein Architecture and the Functions. Molecules 2012, 17, 7266–7283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piña, M.d.l.N.; Frontera, A.; Bauza, A. Charge Assisted S/Se Chalcogen Bonds in SAM Riboswitches: A Combined PDB and Ab Initio Study. ACS Chem. Biol. 2021, 16, 1701–1708. [Google Scholar] [CrossRef]
- Pizzi, A.; Daolio, A.; Beccaria, R.; Demitri, N.; Viani, F.; Resnati, G. Chalcogen Bonding (ChB) as a Robust Supramolecular Recognition Motif of Benzisothiazolinone Antibacterials. Chem. Eur. J. 2023, 2023, e202300571. [Google Scholar] [CrossRef]
- Shukla, R.; Dhaka, A.; Aubert, E.; Vijayakumar-Syamala, V.; Jeannin, O.; Fourmigué, M.; Espinosa, E. Understanding Reactivity and Assembly of Dichalcogenides: Structural, Electrostatic Potential, and Topological Analyses of 3H -1,2-Benzodithiol-3-One and Selenium Analogs. Cryst. Growth Des. 2020, 20, 7704–7725. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, M.; Deng, R.; Zheng, P.; Chi, Y.R. Chalcogen Bond-Guided Conformational Isomerization Enables Catalytic Dynamic Kinetic Resolution of Sulfoxides. Nat. Commun. 2022, 13, 4793. [Google Scholar] [CrossRef]
- Gatti, C.; Dessì, A.; Dallocchio, R.; Mamane, V.; Cossu, S.; Weiss, R.; Pale, P.; Aubert, E.; Peluso, P. Factors Impacting σ- and π-Hole Regions as Revealed by the Electrostatic Potential and Its Source Function Reconstruction: The Case of 4,4′-Bipyridine Derivatives. Molecules 2020, 25, 4409. [Google Scholar] [CrossRef]
- Bauzá, A.; Quiñonero, D.; Deyà, P.M.; Frontera, A. Halogen Bonding Versuschalcogen and Pnicogen Bonding: A Combined Cambridge Structural Database and Theoretical Study. CrystEngComm 2013, 15, 3137–3144. [Google Scholar] [CrossRef]
- Carugo, O.I. Chalcogen Bonds Formed by Protein Sulfur Atoms in Proteins. A Survey of High-Resolution Structures Deposited in the Protein Data Bank. J. Biomol. Struct. Dyn. 2022, 91, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bai, M.; Thomas, S.P.; Kottokkaran, R.; Nayak, S.K.; Ramamurthy, P.C.; Guru Row, T.N. A Donor–Acceptor–Donor Structured Organic Conductor with S···S Chalcogen Bonding. Cryst. Growth Des. 2014, 14, 459–466. [Google Scholar] [CrossRef]
- Yasuda, T.; Shimizu, T.; Liu, F.; Ungar, G.; Kato, T. Electro-Functional Octupolar π-Conjugated Columnar Liquid Crystals. J. Am. Chem. Soc. 2011, 133, 13437–13444. [Google Scholar] [CrossRef]
- Kříž, K.; Fanfrlík, J.; Lepšík, M. Chalcogen Bonding in Protein−Ligand Complexes: PDB Survey and Quantum Mechanical Calculations. ChemPhysChem 2018, 19, 2540–2548. [Google Scholar] [CrossRef] [PubMed]
- Mikherdov, A.S.; Kinzhalov, M.A.; Novikov, A.S.; Boyarskiy, V.P.; Boyarskaya, I.A.; Dar’in, D.V.; Starova, G.L.; Kukushkin, V.Y. Difference in Energy between Two Distinct Types of Chalcogen Bonds Drives Regioisomerization of Binuclear (Diaminocarbene)PdII Complexes. J. Am. Chem. Soc. 2016, 138, 14129–14137. [Google Scholar] [CrossRef] [PubMed]
- Mikherdov, A.S.; Popov, R.A.; Kinzhalov, M.A.; Haukka, M.; Polukeev, V.A.; Boyarskiy, V.P.; Roodt, A. Reaction Mechanism of Regioisomerization in Binuclear (Diaminocarbene)PdII Complexes. Inorg. Chim. Acta 2021, 514, 120012. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Gurbanov, A.V.; Aliyeva, V.A.; Guedes da Silva, M.F.C.; Resnati, G.; Pombeiro, A.J.L. Chalcogen Bonding in Coordination Chemistry. Coord. Chem. Rev. 2022, 464, 214556. [Google Scholar] [CrossRef]
- Nagao, Y.; Miyamoto, S.; Miyamoto, M.; Takeshige, H.; Hayashi, K.; Sano, S.; Shiro, M.; Yamaguchi, K.; Sei, Y. Highly Stereoselective Asymmetric Pummerer Reactions That Incorporate Intermolecular and Intramolecular Nonbonded S···O Interactions. J. Am. Chem. Soc. 2006, 128, 9722–9729. [Google Scholar] [CrossRef]
- Fukata, Y.; Asano, K.; Matsubara, S. Facile Net Cycloaddition Approach to Optically Active 1,5-Benzothiazepines. J. Am. Chem. Soc. 2015, 137, 5320–5323. [Google Scholar] [CrossRef]
- Abdelhamid, Y.; Kasten, K.; Dunne, J.; Hartley, W.C.; Young, C.M.; Cordes, D.B.; Slawin, A.M.Z.; Ng, S.; Smith, A.D. Isothiourea-Catalyzed [2 + 2] Cycloaddition of C(1)-Ammonium Enolates and N-Alkyl Isatins. Org. Lett. 2022, 24, 5444–5449. [Google Scholar] [CrossRef]
- Zhang, S.; Hartley, W.C.; Greenhalgh, M.D.; Ng, S.; Slawin, A.M.Z.; Smith, A.D. Isothiourea-Catalyzed Synthesis of Pyrrole- and Indole-Functionalized Tetrasubstituted Pyridines. ChemCatChem 2020, 12, 4522–4525. [Google Scholar] [CrossRef]
- Fukumoto, S.; Nakashima, T.; Kawai, T. Photon-Quantitative Reaction of a Dithiazolylarylene in Solution. Angew. Chem. Int. Ed. 2011, 50, 1565–1568. [Google Scholar] [CrossRef]
- Chandrasekhar, V.; Chivers, T.; Ellis, L.; Krouse, I.; Parvez, M.; Vargas-Baca, I. Intramolecular Redox Cyclization upon Oxidation of a Sulfur(II)-Containing Diazene: X-ray Structures of (Ar = 4-CH3C6H4) and MeSO2N(4-CH3C6H4)CN=NC(C6H4CH3-4)NSO2Me. Can. J. Chem. 1997, 75, 1188–1194. [Google Scholar] [CrossRef] [Green Version]
- Akiba, K.; Kobayashi, T.; Arai, S. Chemistry of Hypervalent Sulfur. 9. Bond Switch with Participation of .Pi.-Bonded SIV in a Thiadiazole Ring System. J. Am. Chem. Soc. 1979, 101, 5857–5858. [Google Scholar] [CrossRef]
- Cox, P.A.; Leach, A.G.; Campbell, A.D.; Lloyd-Jones, G.C. Protodeboronation of Heteroaromatic, Vinyl, and Cyclopropyl Boronic Acids: PH–Rate Profiles, Autocatalysis, and Disproportionation. J. Am. Chem. Soc. 2016, 138, 9145–9157. [Google Scholar] [CrossRef] [Green Version]
- Sawwan, N.; Brzostowska, E.M.; Greer, A. Substituent Effects on the Reactivity of Benzo-1,2-Dithiolan-3-One 1-Oxides and Their Possible Application to the Synthesis of DNA-Targeting Drugs. J. Org. Chem. 2005, 70, 6968–6971. [Google Scholar] [CrossRef]
- Ruff, F.; Kapovits, I.; Rábai, J.; Kucsman, Á. Neighbouring Group Participation in Reactions of Sulphides with Chloramine-t. Tetrahedron 1978, 34, 2767–2774. [Google Scholar] [CrossRef]
- Kinzhalov, M.A.; Timofeeva, S.A.; Luzyanin, K.V.; Boyarskiy, V.P.; Yakimanskiy, A.A.; Haukka, M.; Kukushkin, V.Y. Palladium(II)-Mediated Addition of Benzenediamines to Isocyanides: Generation of Three Types of Diaminocarbene Ligands Depending on the Isomeric Structure of the Nucleophile. Organometallics 2016, 35, 218–228. [Google Scholar] [CrossRef] [Green Version]
- Mikhaylov, V.N.; Sorokoumov, V.N.; Korvinson, K.A.; Novikov, A.S.; Balova, I.A. Synthesis and Simple Immobilization of Palladium(II) Acyclic Diaminocarbene Complexes on Polystyrene Support as Efficient Catalysts for Sonogashira and Suzuki–Miyaura Cross-Coupling. Organometallics 2016, 35, 1684–1697. [Google Scholar] [CrossRef]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of Bond Lengths Determined by X-Ray and Neutron-Diffraction. Part 1. Bond Lengths in Organic-Compounds. J. Chem. Soc. Perkin Trans. 2 1987, 12, S1–S19. [Google Scholar] [CrossRef]
- Alvarez, S. A Cartography of the van Der Waals Territories. Dalton Trans. 2013, 42, 8617. [Google Scholar] [CrossRef] [Green Version]
- Bader, R.F.W. A Quantum Theory of Molecular Structure and Its Applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen Bond Strengths Revealed by Topological Analyses of Experimentally Observed Electron Densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Vener, M.V.; Egorova, A.N.; Churakov, A.V.; Tsirelson, V.G. Intermolecular Hydrogen Bond Energies in Crystals Evaluated Using Electron Density Properties: DFT Computations with Periodic Boundary Conditions. J. Comput. Chem. 2012, 33, 2303–2309. [Google Scholar] [CrossRef]
- Kinzhalov, M.A.; Kashina, M.V.; Mikherdov, A.S.; Mozheeva, E.A.; Novikov, A.S.; Smirnov, A.S.; Ivanov, D.M.; Kryukova, M.A.; Ivanov, A.Y.; Smirnov, S.N.; et al. Dramatically Enhanced Solubility of Halide-Containing Organometallic Species in Diiodomethane: The Role of Solvent⋅⋅⋅Complex Halogen Bonding. Angew. Chem. Int. Ed. 2018, 57, 12785–12789. [Google Scholar] [CrossRef] [PubMed]
- Palatinus, L.; Prathapa, S.J.; van Smaalen, S. EDMA: A Computer Program for Topological Analysis of Discrete Electron Densities. J. Appl. Crystallogr. 2012, 45, 575–580. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta. Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-Adjustedab Initio Pseudopotentials for the Second and Third Row Transition Elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
The reactions of cis-[PdCl2(XylNC)2] with thiazol-2-amines.

Scheme 2.
Obtaining aminocarbene complexes with different halogens.

Scheme 3.
Regioisomerization of the obtained aminocarbene complexes.

Figure 1.
Monitoring of 3a→3b isomerization at 45 °C (CDCl3, 1H NMR data). Blue circles correspond to signals of Xyl-C and Xyl-B aromatic protons (H4) for complex 3a. Red circles correspond to signals of Xyl-C and Xyl-B aromatic protons (H4) for complex 3b.
Figure 1.
Monitoring of 3a→3b isomerization at 45 °C (CDCl3, 1H NMR data). Blue circles correspond to signals of Xyl-C and Xyl-B aromatic protons (H4) for complex 3a. Red circles correspond to signals of Xyl-C and Xyl-B aromatic protons (H4) for complex 3b.

Figure 2.
Monitoring of 4a→4b isomerization at 45 °C (CDCl3, 1H NMR data). Blue circles correspond to signals of Xyl-C and Xyl-B aromatic protons (H4) for complex 4a. Red circles correspond to signals of Xyl-C and Xyl-B aromatic protons (H4) for complex 4b.
Figure 2.
Monitoring of 4a→4b isomerization at 45 °C (CDCl3, 1H NMR data). Blue circles correspond to signals of Xyl-C and Xyl-B aromatic protons (H4) for complex 4a. Red circles correspond to signals of Xyl-C and Xyl-B aromatic protons (H4) for complex 4b.

Figure 3.
Monitoring of 5a→5b isomerization at 45 °C (CDCl3, 1H NMR data). Blue circles correspond to signals of Xyl-C and Xyl-B aromatic protons (H4) for complex 5a. Red circles correspond to signals of Xyl-C and Xyl-B aromatic protons (H4) for complex 5b.
Figure 3.
Monitoring of 5a→5b isomerization at 45 °C (CDCl3, 1H NMR data). Blue circles correspond to signals of Xyl-C and Xyl-B aromatic protons (H4) for complex 5a. Red circles correspond to signals of Xyl-C and Xyl-B aromatic protons (H4) for complex 5b.

Scheme 4.
Obtaining pure kinetically controlled regioisomers 4a and 5a.

Figure 4.
Views of XRD structures of 4b (left) and 5b (right). Thermal ellipsoids are drawn at the 50% probability level. Yellow stripped lines indicate S···N intramolecular chalcogen bonds.
Figure 4.
Views of XRD structures of 4b (left) and 5b (right). Thermal ellipsoids are drawn at the 50% probability level. Yellow stripped lines indicate S···N intramolecular chalcogen bonds.

Figure 5.
Laplacian of electron density distribution ∇2ρ(r) contour line diagram together with surfaces of zero-flux and bond paths for 4b. Nuclear critical points (3, −3) are indicated as pale brown dots, bond critical points (3, −1) are indicated as blue dots, ring critical points (3, +1) are indicated as orange dots, and length units are given in angstroms.
Figure 5.
Laplacian of electron density distribution ∇2ρ(r) contour line diagram together with surfaces of zero-flux and bond paths for 4b. Nuclear critical points (3, −3) are indicated as pale brown dots, bond critical points (3, −1) are indicated as blue dots, ring critical points (3, +1) are indicated as orange dots, and length units are given in angstroms.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Ratios and constants based on 1H NMR monitoring at 45 °C.
| Mixture | Equilibrium Ratio | Equilibrium Constant | –∆G(CHCl3), kcal/mol |
|---|---|---|---|
| 3a/3b [36] | 13/87 | 6.7 | 1.2 |
| 4a/4b | 4/96 | 24 | 2.0 |
| 5a/5b | 4/96 | 24 | 2.0 |
Table 2.
Selected bond lengths (Å) and angles (o) for compounds 4b, 5b.
| Complex | 4b | 5b |
|---|---|---|
| d(Pd1–Ccarbene) | 2.014(2) | 2.034(3) |
| d(N2–Ccarbene) | 1.407(3) | 1.409(4) |
| d(N3–Ccarbene) | 1.286(3) | 1.290(4) |
| d(Pd2–Ccarbene2) | 1.986(2) | 2.007(3) |
| d(N2–Ccarbene2) | 1.438(3) | 1.444(4) |
| d(N4–Ccarbene2) | 1.265(3) | 1.259(4) |
| ∠(N1–Pd1–Br1) | 94.94(6) | |
| ∠(N3–Pd2–Br2) | 97.05(6) | |
| ∠(N1–Pd1–I1) | 95.95(8) | |
| ∠(N3–Pd2–I2) | 97.73(7) |
Table 3.
Electron density [ρ(r)], Laplacian of electron density [∇2ρ(r)], energy density [Hb], potential energy density [V(r)], and Lagrangian kinetic energy [G(r)] (in Hartree) at the bond-critical points (3, −1), associated with chalcogen bonding S···X (X = Cl, Br, I) and S···N in 3–5a and 3–5b in solid state and chloroform solution, contact length [l] (in angstroms), and estimated energies for these contacts [Eint] (in kcal/mol).
Table 3.
Electron density [ρ(r)], Laplacian of electron density [∇2ρ(r)], energy density [Hb], potential energy density [V(r)], and Lagrangian kinetic energy [G(r)] (in Hartree) at the bond-critical points (3, −1), associated with chalcogen bonding S···X (X = Cl, Br, I) and S···N in 3–5a and 3–5b in solid state and chloroform solution, contact length [l] (in angstroms), and estimated energies for these contacts [Eint] (in kcal/mol).
| Compound | Contact | ρ(r) | ∇2ρ(r) | Hb | V(r) | G(r) | Eint a | Eint b | l |
|---|---|---|---|---|---|---|---|---|---|
| Solid-state structures | |||||||||
| 3b c | S···N | 0.026 | 0.080 | 0.001 | −0.019 | 0.019 | 6.0 | 5.1 | 2.67 |
| 4b | S···N | 0.024 | 0.076 | 0.001 | −0.017 | 0.018 | 5.3 | 4.9 | 2.71 |
| 5b | S···N | 0.024 | 0.076 | 0.001 | −0.017 | 0.018 | 5.3 | 4.9 | 2.70 |
| Optimized structures (CHCl3) | |||||||||
| 3a c | S···Cl | 0.015 | 0.051 | 0.002 | −0.009 | 0.011 | 3.0 | 2.8 | 3.14 |
| 3b c | S···N | 0.024 | 0.071 | 0.001 | −0.016 | 0.017 | 5.0 | 4.6 | 2.73 |
| 4a | S···Br | 0.016 | 0.049 | 0.002 | −0.009 | 0.011 | 2.8 | 3.0 | 3.22 |
| 4b | S···N | 0.024 | 0.073 | 0.001 | −0.017 | 0.018 | 5.3 | 4.9 | 2.72 |
| 5a | S···I | 0.011 | 0.031 | 0.001 | −0.005 | 0.006 | 1.6 | 1.6 | 3.58 |
| 5b | S···N | 0.025 | 0.074 | 0.001 | −0.017 | 0.018 | 5.3 | 4.9 | 2.71 |
Table 4.
Values of experimental and theoretically calculated –∆G318 for isomerization of complexes 4–5.
Table 4.
Values of experimental and theoretically calculated –∆G318 for isomerization of complexes 4–5.
| System | −∆Gexp | −∆Gcalcd |
|---|---|---|
| 3a –> 3b * | 1.2 | 3.2 |
| 4a –> 4b | 2.0 | 5.4 |
| 5a –> 5b | 2.0 | 5.8 |
* Data taken from Ref. [36].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Popov, R.A.; Novikov, A.S.; Suslonov, V.V.; Boyarskiy, V.P. Molecular Switching through Chalcogen-Bond-Induced Isomerization of Binuclear (Diaminocarbene)PdII Complexes. Inorganics 2023, 11, 255. https://doi.org/10.3390/inorganics11060255
AMA Style
Popov RA, Novikov AS, Suslonov VV, Boyarskiy VP. Molecular Switching through Chalcogen-Bond-Induced Isomerization of Binuclear (Diaminocarbene)PdII Complexes. Inorganics. 2023; 11(6):255. https://doi.org/10.3390/inorganics11060255
Chicago/Turabian StylePopov, Roman A., Alexander S. Novikov, Vitalii V. Suslonov, and Vadim P. Boyarskiy. 2023. "Molecular Switching through Chalcogen-Bond-Induced Isomerization of Binuclear (Diaminocarbene)PdII Complexes" Inorganics 11, no. 6: 255. https://doi.org/10.3390/inorganics11060255
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.