
Lu-Lu Bond in Lu2@C60 Metallofullerenes
1
Engineering Research Center of Light Stabilizers for Polymer Materials, School of Materials Science and Chemical Engineering, Xi’an Technological University, Xi’an 710021, China
2
School of Materials Science and Engineering, Hefei University of Technology, Hefei 230009, China
3
State Key Laboratory of Materials Processing and Die & Mould Technology, School of Materials Science and Engineering, Huazhong University of Science and Technology, 1037 Luoyu Road, Wuhan 430074, China
*
Authors to whom correspondence should be addressed.
Inorganics 2023, 11(7), 277; https://doi.org/10.3390/inorganics11070277
Submission received: 31 May 2023
/
Revised: 22 June 2023
/
Accepted: 26 June 2023
/
Published: 28 June 2023
(This article belongs to the Special Issue Research on Metallofullerenes)
Abstract
:This study on Lu2@C60 isomers provides insights into the metal–metal bond through the confinement effect of fullerene cages. Density functional theory calculations were used to study the nature of the Lu-Lu bond in two stable endohedral metallofullerenes (EMFs), Lu2@C2v_C60 and Lu2@Ih_C60, both with negative endohedral energy. These two isomers are geometrically connected through a simple Stone–Wales (SW) transformation. The electronic configuration of (Lu2)4+@C604− was also confirmed, leading to the formation of a two-center two-electron (2c–2e) Lu-Lu σ single bond. By comparing the Lu-Lu bonds in Lu2@C60 with those in acknowledged Lu2@C2n, the smaller C60 fullerene compressed the geometry of Lu2 resulting in a much shorter Lu-Lu bond length. However, the Lu-Lu bond strength is slightly weaker in Lu2@C60 than that in large fullerenes, as the Lu-Lu bond in C60 is likely a p-p σ bond with an above the 40% contribution of p orbital and a strong metal–cage interaction. Additionally, the vis-NIR spectra of Lu2@C2v_C60 and Lu2@Ih_C60 were simulated, which could provide valuable information for future experimental studies on Lu-based EMFs.
1. Introduction
Metal–metal bonds have been a subject of significant research interest in past decades, which show important roles in catalysis and biology [1,2]. Metal–metal bonds provide a large perturbation in electronic structure and the unique properties of these dinuclear fragments can be harnessed in a broad range of applications [3]. Since the synthesis and characterization of Re2Cl82− [4], there is growing evidence for the formation of metal–metal bonds under specific conditions [5,6], and growing research interest in understanding the nature of the metal–metal bonds. Generally, the transition metals could form multinuclear complexes with direct metal–metal interactions, but a complex coordination environment makes it difficult to understand the nature of metal–metal interaction [7].
Luckily, endohedral dimetallofullerenes (di-EMFs), i.e., fullerenes with two metal atoms trapped inside are considered as the ideal model to study the metal–metal bond [8]. In 2014, di-EMF Sc2@C1(4059)-C66 was determined as the geometry of Sc2C66 molecule by the single-crystal X-ray diffraction (XRD) [9]. La2@C94 are also proposed with experimental and theoretical methods [10], and later, theoretical study revealed that the La–La bond plays a key role in the stability of dimetallofullerenes [La2@C2n]− (2n = 92–96) [11]. Recently, Chen et al. have studied Sc-Y σ2 bond in ScY@C3v(8)-C82 [12].
Additionally, the previous reports clarified strong confinement effects of fullerene size on the metal-metal bond in EMFs [7,13,14,15]. For example, Poblet et al. theoretically reveal a U-U triple bond in U2@C60 with the effective bond order of 2.52, singly occupied molecular orbitals (MOs) with metal–metal bond characters in U2@C80, non-negligible U···U interaction in U2@C78, but the metal–metal interaction almost disappears in U2@C104 [8]. In 2017, di-EMFs Lu2@C2n (2n = 82, 84, and 86) were synthesized and characterized with the single-crystal XRD method, which gives the crystallographic evidence of direct Lu-Lu bond between two divalent lutetium ions inside fullerenes [16]. Later, additional Lu2C2n (2n = 76–90) molecules were reported with a wide range cage size, and the successful isolation and unambiguous crystallographic assignment of a series of lutetium-containing EMFs, Lu2C2n (2n = 76, 78, 80, 84, 86, 88, and 90), reveal an unrecognized decisive effect of the cage size on the configuration of the encapsulated clusters [17]. Following theoretical studies reveal a stable two-center two-electron (2c–2e) Lu-Lu σ bond in Lu2@C84 and Lu2@C86 [18,19]. However, experimental and theoretical study focused on the Lu-containing di-EMFs with medium- or large-size fullerene cages and Lu-Lu bond in small fullerene cages is still unclear.
Herein, Lu-Lu bond is studied in the smaller C60 fullerene cage with the highest yield in the fullerene family by using density functional theory calculations and bonding analysis. The aim is to evaluate the confinement effect of fullerene size on the Lu-Lu bond. Two isomers, C2v_C60 and Ih_C60, which have been previously verified as the stable host cage of C60 fullerene to encage inner moieties, were selected as they have well-established thermodynamic stability and were used to encapsulate the Lu2 dimer. The bonding features of the Lu-Lu in the C60 were evaluated via frontier molecular orbital (FMO), natural bond orbital (NBO), and bond orders analyses. Additionally, the visible-near-infrared (vis-NIR) spectra of Lu2@C2v_C60 and Lu2@Ih_C60 were simulated to study their electronic features and gain a better understanding of the Lu-Lu bonds both in theory and experiment.
2. Results and Discussion
2.1. Stability and Geometries of Lu2@C60
The good stability of fullerenes C2v_C60 and Ih_C60, connected through a single-step Stone-Wales transformation, has been previously reported [20], and they have been characterized as suitable isomers of C60 fullerenes to encage metal atoms or clusters [8,21]. In this work, we focus on both Lu2@C2v_C60 and Lu2@Ih_C60 to generally elucidate the confinement effect of the fullerene on metal–metal bond.
Figure 1 shows the optimized geometries of Lu2@C2v_C60 and Lu2@Ih_C60. The lowest frequencies for Lu2@C2v_C60 and Lu2@Ih_C60 are calculated to be 36 and 45 cm−1, respectively, suggesting their possible existence, as shown in Figures S1 and S2. The single-point energy calculations showed that the Lu2@C2v_C60 possessed energy of 5.3 kcal·mol−1 higher than the energy of Lu2@Ih_C60, of which results are confirmed by the hybrid functional PBE0 (PBE0/6-311G(d,p)~SDD//PBE0/6-31G(d)~CEP-4G) showing Lu2@C2v_C60 with energy of 0.7 kcal·mol−1 higher than the energy of Lu2@Ih_C60.
In order to evaluate the effect of temperature, the Boltzmann distribution has been calculated for Lu2@C2v_C60 and Lu2@Ih_C60. The results indicate that the Boltzmann distribution of Lu2@Ih_C60 is about 97%, 84%, 75%, 70%, 66%, and 63% at 500, 1000, 1500, 2000, 2500, and 3000 K, respectively, which is higher than Lu2@C2v_C60 with the Boltzmann distribution values of about 4%, 16%, 25%, 30%, 34%, and 37% at 500, 1000, 1500, 2000, 2500, and 3000 K, respectively. The energy gaps between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are 0.19 and 0.35 eV for Lu2@Ih_C60 and Lu2@C2v_C60, respectively, which are larger than the DFT-computed HOMO-LUMO gaps of Lu2@Cs(17,490)_C76 and Lu2@C2v(19,138)_C76 [22].
The formation energy for Lu2@C2v_C60 and Lu2@Ih_C60 has been calculated by basis set superposition error (BSSE) correction with a counterpoise calculation on the PBE/6-311G(d,p)~SDD considering the Grimme’s dispersion with the original D3 damping function. The formation energy for Lu2@C2v_C60 and Lu2@Ih_C60 are −179.7 and −156.4 kcal·mol−1, indicating the possible synthesis and isolation in experiment. On the other hand, the much negative formation energy indicates their good stability. The similar stability for Lu2@Ih_C60 and Lu2@C2v_C60 might be explained by the geometrical connection with only the single-step Stone–Wales transformation, and this case is similar to the hollow cage of C2v_C60 and Ih_C60 isomers.
The Lu-Lu bond length in Lu2@Ih_C60 and Lu2@C2v_C60 are 3.04 and 3.14 Å, respectively, which are much smaller than the previously reported Lu-Lu bond length (~3.50 Å) in Lu2@C2n (2n ≥ 76) [17,18,19,22]. This suggests that there is a confinement effect of fullerenes on the Lu-Lu bond length. The bond length of Sc-Sc and Y-Y in C82 are 3.201 and 3.695 Å, respectively, and bond length of La-La in C80 is 3.826 Å, indicating the presence of a metal–metal single bond, which has been verified in both theory and experiment [22]. Based on the geometry, further study into the confinement effect of fullerenes on the Lu-Lu bond is warranted.
2.2. Electronic Structures of Lu2@C60
The electronic structure of Lu2@C60 was further investigated through NBO calculations. The results in Table 1 show that there is charge transfer from inner Lu2 dimer to the C60 fullerenes, and in combination with the Lu-Lu σ bond in C60, the 6s orbitals of Lu atoms lose four electrons in Lu2@C60. Therefore, the electronic configurations of both Lu2@Ih_C60 and Lu2@C2v_C60 can be expressed as (Lu2)4+@(C60)4−.
In addition, the location of fractional charge on 6p, 6d, and 7p orbitals of Lu atoms implies the occurrence of back-donation, similar to that observed in other EMFs [23,24,25,26,27,28,29,30,31,32,33,34,35,36,37]. This is further supported by the presence of electrostatic interaction between the Lu2 dimer and fullerene cages, as depicted in Figure 2. These observations are consistent with the ionic model of EMFs [13,38,39].
Furthermore, the FMOs have been mapped as shown in Figure 3, in which the C60 fullerenes based on single-point calculations are obtained from the optimized Lu2@Ih_C60 and Lu2@C2v_C60. The highest occupied molecular orbitals (HOMOs) of Lu2@C60 are contributed by the Lu2 dimer, presenting the Lu-Lu bond. It is clear that the LUMO and LUMO + 1 of the C60 fullerenes become the HOMO-1 and HOMO-2 of the Lu2@C60 indicating the four-electron transfer from inner moiety to fullerene cage, and this result is in line with the natural bond order analysis again confirming the electronic configuration of (Lu2)4+@(C60)4−. This electronic configuration has been verified in the previous report on Lu2@C2n (2n ≥ 76) [17,18,19,22]. Additionally, the t1u LUMO has been split after encapsulation of Lu2 dimer. As shown in Figure 3, after encapsulation of Lu2 dimer in Ih-C60, its energy level of LUMO + 1 and LUMO is equal, but the energy level of LUMO + 2 is −4.22 eV. This is derived from the distortion of C60 fullerene. The symmetry of Ih-C60 and C2v-C60 have reduced to D3d and C1, respectively, after encapsulation, and the calculated distortion energies of Ih-C60 and C2v-C60 are 35.4 and 29.6 kcal·mol−1, respectively, on the theoretical level of PBE/6-311G(d,p)~SDD.
2.3. Bonding Features of Lu2@C60
Figure 3 illustrates the presence of a clearly localized bonding molecular orbital between two Lu atoms in Lu2@C60, which has been reported in previous studies [16,17,18,19]. The energy level of the bonding molecular orbitals is −4.66 and −4.54 eV for Lu2@Ih_C60 and Lu2@C2v_C60, respectively. However, due to the confinement effect of the smaller fullerene cage size, such as C60, further investigation is needed to determine the strength of the Lu-Lu bond. The LUMOs are shown in Figure S3, which display both a metal–metal antibonding orbital and a π antibonding orbital.
To study the nature of the Lu-Lu bond in small fullerene sizes, we calculated the Mayer bond order (MBO) and Wiberg bond order (WBO) and the results are presented in Table 2. Despite the much shorter bond length of Lu-Lu in fullerene C60, the MBO for Lu-Lu bond in C60 is smaller than that in larger fullerenes. For example, in Lu2@C2n (2n = 84 and 86), the MBO is a little larger than 1 for Lu-Lu bond with a larger bond length [18,19]. The WBOs for Lu-Lu bond in C60 are also smaller than 1. To gain further insights into this abnormal phenomenon, the hybrid compositions of M-M bond in Lu2@C60 were studied with NBO calculations. As shown in Table 2, the p orbital contributes more than 40% to the Lu-Lu bond, while the rest of the contributions come from the s and d atomic orbitals. Thus, the Lu-Lu σ bond in C60 is likely a p-p σ bond. Previous studies have shown that the Lu-Lu bond is mainly contributed by s orbitals [18,19], in which the metal–metal bond is more like a s-s σ bond. Generally, the overlap of s orbitals is more effective than that of p orbitals, meaning that the s-s σ bond is much stronger than the p-p σ bond. On the other hand, the interaction between Lu2 dimer and fullerene cage C60 is stronger with MBO values of 4.19 and 4.82 for Lu2@Ih_C60 and Lu2@C2v_C60, respectively.
In Lu2@Ih_C60 and Lu2@C2v_C60, the Lu-Lu σ bond is mainly derived from p orbitals, for which the bond strength is related to the bond length. The Lu-Lu bond in Ih_C60 with higher values of bond orders (0.99 for MBO and 0.73 for WBO, Table 2) show stronger bond strength than that in C2v_C60, because the shorter bond length in Ih_C60 than that in C2v_C60.
2.4. Simulated Spectra of Lu2@C60
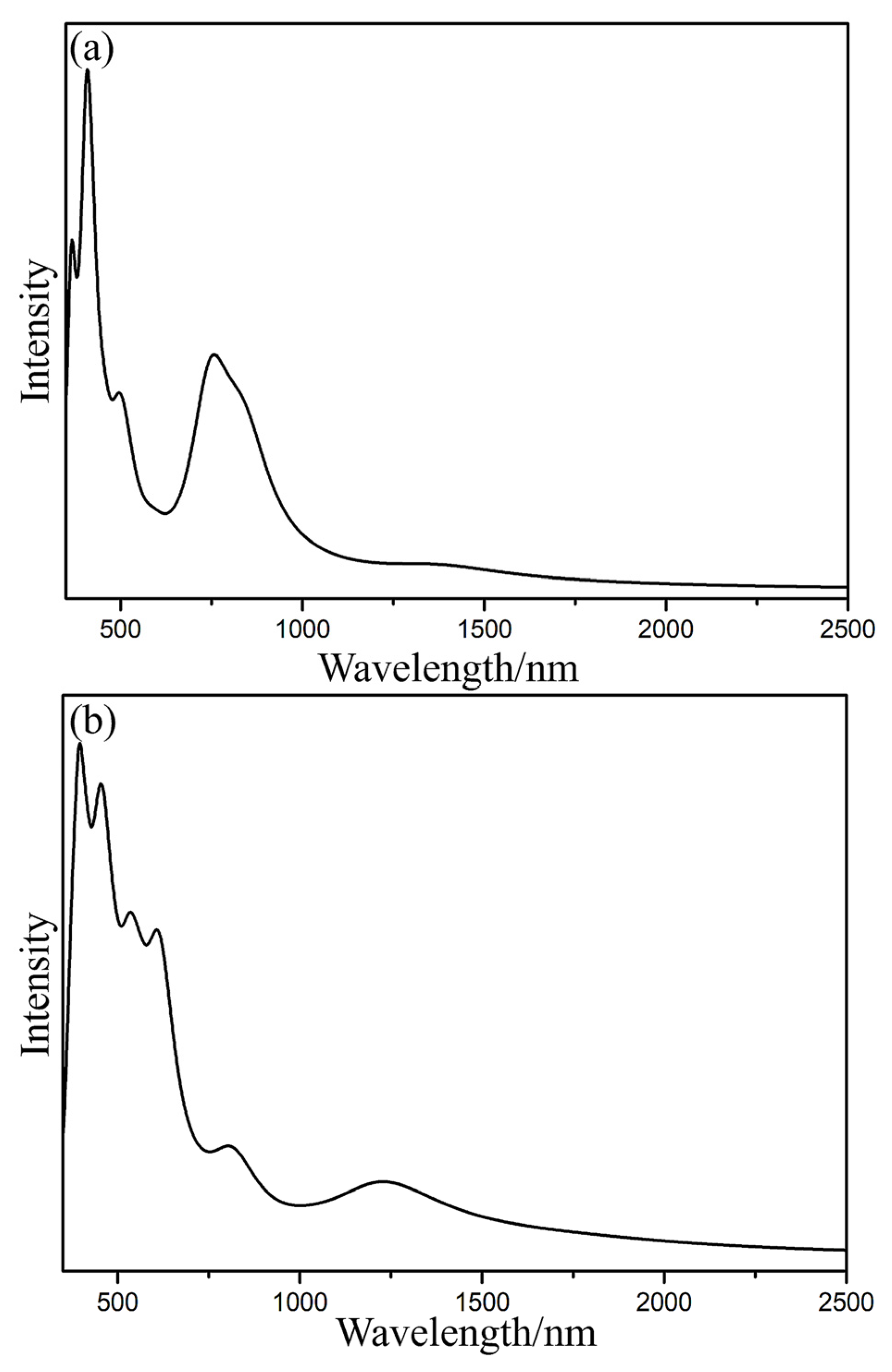
Figure 4 illustrates that the excitation energies of the first excited state of Lu2@Ih_C60 and Lu2@C2v_C60 are 0.49 eV (2542 nm) and 0.47 eV (2637 nm), respectively, which are smaller than the first excitation energy of approximately 0.90 eV (1384 nm) for Lu2@C86 in both theory and experiment [16,19]. The excitation form S0 to S1 is primarily derived from the transition of electrons from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) of Lu2@C60, and the energy required for this transition is 0.78 e− and 0.61 e− for Lu2@Ih_C60 and Lu2@C2v_C60, respectively. As demonstrated in Figure 3 and Figure S3, the HOMO corresponds to the metal–metal bonding orbital and the LUMO corresponds to the metal–metal antibonding orbital and the π antibonding orbital in Lu2@C60, which is potential reason why their first excitation energy is low. The much lower first excitation energy and the ease of electron transition suggest the poor photochemical stability of Lu2@C60.
In addition, although the absorption peaks of Lu2@Ih_C60 and Lu2@C2v_C60 isomers have similar shapes (Figure 4), there is a clear difference in their absorption strength, which can be useful for distinguishing them in the future experiments. Specifically, Lu2@Ih_C60 exhibits a much stronger absorption peak at around 750 nm compared to Lu2@C2v_C60. The absorption band at 200–700 nm for Lu2@C2v_C60 is slightly wider than that of Lu2@Ih_C60. Furthermore, the IR spectra of Lu2@Ih_C60 and Lu2@C2v_C60 were simulated (Figure S4) to evaluate their vibration models. These characterized absorption peaks in the vis-NIR and IR spectra can be beneficial for future characterization of Lu2@C60 in experiments.
3. Computational Methods
The optimizations of Lu2@C2v_C60 and Lu2@Ih_C60 were carried out on the PBE/6-31G(d)~CEP-4G theoretical level [40,41], in which the basis function 6-31G(d) was used for the carbon atom and CEP-4G containing pseudopotential was used for the lutetium atom with valance electronic configuration of 4f145d16s2. The frequencies were calculated on the same theoretical level for the optimized geometries of Lu2@C60, and all of them are free from imaginary frequency meaning the existence of local minima point for Lu2@C2v_C60 and Lu2@Ih_C60. The PBE has been previously proved to be the suitable functional for the lutetium-based EMFs [18,19,42,43]. To obtain accurate energy and frontier molecular orbitals (FMOs), a single-point calculation was performed on the PBE/6-311G(d,p)~SDD theoretical level, in which a larger basis set 6-311G(d,p) [40] with polarization functions and SDD with effective core pseudopotential were used for the carbon and lutetium atoms, respectively. In order to confirm the calculated results on PBE, the hybrid functional PBE0 (PBE0/6-311G(d,p)~SDD//PBE0/6-31G(d)~CEP-4G) calculations were also carried out for both Lu2@C2v_C60 and Lu2@Ih_C60. In order to evaluate the effect of temperature, the Boltzmann distribution has been calculated for Lu2@C2v_C60 and Lu2@Ih_C60. The formation energy for Lu2@C2v_C60 and Lu2@Ih_C60 was calculated based on basis set superposition errors via counterpoise corrections on the theoretical level of PBE/6-311G(d,p)~SDD. NBO [44] analyses were also conducted on the PBE/6-311G(d,p)~SDD theoretical level. The vis-NIR spectra were simulated on the PBE/6-31G(d)~CEP-4G theoretical level. All the above calculations were carried out by Gaussian16 software package [45]. In addition, Mayer bond order (MBO) [46] together with localized molecular orbitals (LMOs) [47,48] analyses for Lu2@C60 were performed with the Multiwfn program [49].
4. Conclusions
Based on the Lu2@Ih_C60 and Lu2@C2v_C60 which are related by a single-step Stone–Wales transformation, we provide insight into the confinement effects of fullerene on the metal–metal bonding. Although, in the Lu2@C60, there is a much shorter Lu-Lu bond length, its bond strength is a little weaker than the Lu-Lu bond in large fullerenes, because the Lu-Lu σ bond in C60 is likely a p-p σ bond with the contribution p orbital above 40% and a strong metal–cage interaction. Clearly, the confinement effects of fullerene play important roles in the geometry of the inner cluster, especially the bond length in the present work, and the electronic effect is more important for the bonding nature. Furthermore, the electronic configurations of (Lu2)4+@C604− were confirmed. Additionally, the vis-NIR spectra of Lu2@C2v_C60 and Lu2@Ih_C60 were simulated, which could give some valuable information for the future experimental study on Lu-based EMFs.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/inorganics11070277/s1, Figure S1: Several selected displacement vectors of Lu2@Ih_C60 with vibration frequencies, including the lowest vibration one; Figure S2: Several selected displacement vectors of Lu2@C2v_C60 with vibration frequencies, including the lowest vibration one; Figure S3: Lowest unoccupied molecular orbitals (LUMO) with localization of Lu2@Ih_C60 (a) and Lu2@C2v_C60 (b) with isovalue of 0.03 a.u. for the surface; Figure S4: IR vibration spectra for Lu2@Ih_C60 (a) and Lu2@C2v_C60 (b). The broadening function is Gaussian function and full width at half maximum is 12 cm−1; coordinates for optimized geometries: Lu2@Ih_C60 and Lu2@C2v_C60.
Author Contributions
Conceptualization, X.L. and W.S.; methodology, Y.Z.; software, Y.Z.; validation, X.L., W.S., W.C. and Y.Z.; formal analysis, Y.Z. and W.S.; investigation, Y.Z.; data curation, Y.Z.; writing—original draft preparation, Y.Z. and W.S.; writing—review and editing, W.C. and X.L.; visualization, Y.Z.; supervision, X.L.; project administration, X.L.; funding acquisition, X.L. and W.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (21925104, 22001084, and 92261204).
Data Availability Statement
Data are available on request from the authors.
Acknowledgments
We thank the Analytical and Testing Center at the Hefei University of Technology and Huazhong University of Science and Technology for all related measurements, and national supercomputing center in Xi’an.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rodriguez, J.A.; Goodman, D.W. The nature of the metal-metal bond in bimetallic surfaces. Science 1992, 257, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, P.A. Metal–metal bonds in biology. J. Inorg. Biochem. 2012, 106, 172–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powers, I.G.; Uyeda, C. Metal–metal bonds in catalysis. ACS Catal. 2017, 7, 936–958. [Google Scholar] [CrossRef]
- Cotton, F.A. Strong homonuclear metal-metal bonds. Acc. Chem. Res. 1969, 2, 240–247. [Google Scholar] [CrossRef]
- Foroutan-Nejad, C.; Vicha, J.; Marek, R.; Patzschke, M.; Straka, M. Unwilling U-U bonding in U2@C80: Cage-driven metal-metal bonds in di-uranium fullerenes. Phys. Chem. Chem. Phys. 2015, 17, 24182–24192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, J.; Morales-Martínez, R.; Zhang, J.; Wang, Y.; Yao, Y.R.; Pei, C.; Rodríguez-Fortea, A.; Wang, S.; Echegoyen, L.; de Graaf, C.; et al. Characterization of a strong covalent Th3+-Th3+ bond inside an Ih(7)-C80 fullerene cage. Nat. Commun. 2021, 12, 2372. [Google Scholar] [CrossRef]
- Li, M.; Zhao, R.; Dang, J.; Zhao, X. Theoretical study on the stabilities, electronic structures, and reaction and formation mechanisms of fullerenes and endohedral metallofullerenes. Coordin. Chem. Rev. 2022, 471, 214762. [Google Scholar] [CrossRef]
- Moreno-Vicente, A.; Roselló, Y.; Chen, N.; Echegoyen, L.; Dunk, P.W.; Rodríguez-Fortea, A.; de Graaf, C.; Poblet, J.M. Are U–U Bonds Inside Fullerenes Really Unwilling Bonds? J. Am. Chem. Soc. 2023, 145, 6710–6718. [Google Scholar] [CrossRef]
- Yamada, M.; Kurihara, H.; Suzuki, M.; Guo, J.D.; Waelchli, M.; Olmstead, M.M.; Balch, A.L.; Nagase, S.; Maeda, Y.; Hasegawa, T. Sc2@C66 revisited: An endohedral fullerene with scandium ions nestled within two unsaturated linear triquinanes. J. Am. Chem. Soc. 2014, 136, 7611–7614. [Google Scholar] [CrossRef]
- Zhao, S.; Zhao, P.; Cai, W.; Bao, L.; Chen, M.; Xie, Y.; Zhao, X.; Lu, X. Stabilization of Giant Fullerenes C2(41)-C90, D3(85)-C92, C1(132)-C94, C2(157)-C96, and C1(175)-C98 by Encapsulation of a Large La2C2 Cluster: The Importance of Cluster–Cage Matching. J. Am. Chem. Soc. 2017, 139, 4724–4728. [Google Scholar] [CrossRef]
- Li, Q.-Z.; Zheng, J.-J.; He, L.; Nagase, S.; Zhao, X. La–La bonded dimetallofullerenes [La2@C2n]−: Species for stabilizing C2n (2n = 92–96) besides La2C2@C2n. Phys. Chem. Chem. Phys. 2018, 20, 14671–14678. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Roselló, Y.; Yan, Y.; Yao, Y.R.; Fan, X.; Poblet, J.M.; Rodríguez-Fortea, A.; Chen, N. ScY@C3v(8)-C82: Metal-Metal σ2 Bond in Mixed Rare-Earth Di-metallofullerenes. Chin. J. Chem. 2023; early view. [Google Scholar] [CrossRef]
- Popov, A.A.; Yang, S.; Dunsch, L. Endohedral fullerenes. Chem. Rev. 2013, 113, 5989–6113. [Google Scholar] [CrossRef]
- Lu, X.; Akasaka, T.; Nagase, S. Chemistry of endohedral metallofullerenes: The role of metals. Chem. Commun. 2011, 47, 5942–5957. [Google Scholar] [CrossRef]
- Shen, W.; Bao, L.; Lu, X. Endohedral Metallofullerenes: An Ideal Platform of Sub-Nano Chemistry. Chin. J. Chem. 2022, 40, 275–284. [Google Scholar] [CrossRef]
- Shen, W.; Bao, L.; Wu, Y.; Pan, C.; Zhao, S.; Fang, H.; Xie, Y.; Jin, P.; Peng, P.; Li, F.-F. Lu2@C2n (2n = 82, 84, 86): Crystallographic evidence of direct Lu–Lu bonding between two divalent lutetium ions inside fullerene cages. J. Am. Chem. Soc. 2017, 139, 9979–9984. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Bao, L.; Hu, S.; Yang, L.; Jin, P.; Xie, Y.; Akasaka, T.; Lu, X. Crystallographic characterization of Lu2C2n (2n = 76–90): Cluster selection by cage size. Chem. Sci. 2019, 10, 829–836. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Dang, H.; Zhao, Y.; Gu, Y.-X.; Li, M.; Li, Q.-Z.; Zhao, X. Theoretical Investigations of Lu2C84: Unexpected Impact of Metal Electronic Configuration toward the Metal–Metal σ-Bond in Fullerene. Inorg. Chem. 2020, 59, 10113–10122. [Google Scholar] [CrossRef]
- Zhang, K.; Zheng, H.; Li, M.; Li, Q.-Z.; Zhao, Y.; Zhao, X. Significant roles of a particularly stable two-center two-electron Lu–Lu σ bond in Lu2@C86: Electronic structure of Lu and radius of Lu2+. Inorg. Chem. 2021, 60, 2425–2436. [Google Scholar] [CrossRef]
- Stone, A.J.; Wales, D.J. Theoretical studies of icosahedral C60 and some related species. Chem. Phys. Lett. 1986, 128, 501–503. [Google Scholar] [CrossRef]
- Wang, Y.; Tomanek, D.; Bertsch, G.F.; Ruoff, R.S. Stability of C60 fullerite intercalation compounds. Phys. Rev. B 1993, 47, 6711–6720. [Google Scholar] [CrossRef]
- Popov, A.A.; Avdoshenko, S.M.; Pendás, A.M.; Dunsch, L. Bonding between strongly repulsive metal atoms: An oxymoron made real in a confined space of endohedral metallofullerenes. Chem. Commun. 2012, 48, 8031–8050. [Google Scholar] [CrossRef]
- Fu, W.; Zhang, J.; Fuhrer, T.; Champion, H.; Furukawa, K.; Kato, T.; Mahaney, J.E.; Burke, B.G.; Williams, K.A.; Walker, K.; et al. Gd2@C79N: Isolation, characterization, and monoadduct formation of a very stable heterofullerene with a magnetic spin state of S = 15/2. J. Am. Chem. Soc. 2011, 133, 9741–9750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Morales-Martinez, R.; Zhuang, J.; Yao, Y.R.; Li, X.; Poblet, J.M.; Rodriguez-Fortea, A.; Chen, N. Th@D5h(6)-C80: A highly symmetric fullerene cage stabilized by a single metal ion. Chem. Commun. 2021, 57, 6624–6627. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Abella, L.; Yang, W.; Yao, Y.R.; Liu, X.; Zhuang, J.; Li, X.; Echegoyen, L.; Autschbach, J.; Chen, N. UCN@Cs(6)-C82: An Encapsulated Triangular UCN Cluster with Ambiguous U Oxidation State [U(III) versus U(I)]. J. Am. Chem. Soc. 2021, 143, 16226–16234. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Yu, Y.; Slanina, Z.; Wang, F.; Lian, Y.; Uhlik, F.; Feng, L. Ho2C2 Cluster with Flexible Configurations inside a Large C2(61)-C92 Cage. Inorg. Chem. 2022, 61, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, L.; Liu, B.; Wang, D.; Zhao, Y.; Gao, X. Theoretical study on the ground state structure of uranofullerene U@C82. J. Phys. Chem. A 2012, 116, 11651–11655. [Google Scholar] [CrossRef]
- Mulet-Gas, M.; Abella, L.; Dunk, P.W.; Rodríguez-Fortea, A.; Kroto, H.W.; Poblet, J.M. Small endohedral metallofullerenes: Exploration of the structure and growth mechanism in the Ti@C2n (2n = 26–50) family. Chem. Sci. 2015, 6, 675–686. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Gu, X.; Jin, P. Overlooked Effects of La-4f Orbitals in Endohedral Metallofullerenes. Inorg. Chem. 2022, 61, 5891–5902. [Google Scholar] [CrossRef]
- Cai, W.; Alvarado, J.; Metta-Magana, A.; Chen, N.; Echegoyen, L. Interconversions between Uranium Mono-metallofullerenes: Mechanistic Implications and Role of Asymmetric Cages. J. Am. Chem. Soc. 2020, 142, 13112–13119. [Google Scholar] [CrossRef]
- Shen, W.; Hu, Z.; Yu, P.; Wei, Z.; Jin, P.; Shi, Z.; Lu, X. An experimental and theoretical study of LuNC@C76,82 revealing a cage-cluster selection rule. Inorg. Chem. Front. 2020, 7, 4563–4571. [Google Scholar] [CrossRef]
- Bao, L.; Li, Y.; Yu, P.; Shen, W.; Jin, P.; Lu, X. Preferential Formation of Mono-Metallofullerenes Governed by the Encapsulation Energy of the Metal Elements: A Case Study on Eu@C2n (2n = 74–84) Revealing a General Rule. Angew. Chem. Int. Ed. 2020, 59, 5259–5262. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Peng, P.; Lu, X. Bonding inside and outside Fullerene Cages. Acc. Chem. Res. 2018, 51, 810–815. [Google Scholar] [CrossRef]
- Xu, D.; Jiang, Y.; Wang, Y.; Zhou, T.; Shi, Z.; Omachi, H.; Shinohara, H.; Sun, B.; Wang, Z. Turning on the Near-Infrared Photoluminescence of Erbium Metallofullerenes by Covalent Modification. Inorg. Chem. 2019, 58, 14325–14330. [Google Scholar] [CrossRef]
- Guan, R.; Chen, Z.-C.; Huang, J.; Tian, H.-R.; Xin, J.; Ying, S.-W.; Chen, M.; Zhang, Q.; Li, Q.; Xie, S.-Y. Self-driven carbon atom implantation into fullerene embedding metal–carbon cluster. Proc. Natl. Acad. Sci. USA 2022, 119, e22025631192022. [Google Scholar] [CrossRef] [PubMed]
- Xiang, W.; Jiang, X.; Yao, Y.-R.; Xin, J.; Jin, H.; Guan, R.; Zhang, Q.; Chen, M.; Xie, S.-Y.; Popov, A.A. Monometallic Endohedral Azafullerene. J. Am. Chem. Soc. 2022, 144, 21587–21595. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhao, Y.; Yuan, K.; Zhao, R.; Zhao, P.; Zhao, X. Insight into the thermodynamically preferred V3N@Ih(31924)-C80 and acknowledged VxSc3-xN@Ih(31924)-C80 (x = 0, 1 and 2). Carbon 2018, 132, 312–322. [Google Scholar] [CrossRef]
- Zhao, Y.-X.; Yuan, K.; Li, M.-Y.; Ehara, M.; Zhao, X. In-Depth Theoretical Probe into Novel Mixed-Metal Uranium-Based Endohedral Clusterfullerenes Sc2UX@Ih(31924)-C80 (X = C, N). Inorg. Chem. 2019, 58, 10769–10777. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Torres, A.; Esper, R.; Dunk, P.W.; Morales-Martínez, R.; Rodríguez-Fortea, A.; Echegoyen, L.; Poblet, J.M. Small Cage Uranofullerenes: 27 Years after Their First Observation. Helv. Chim. Acta 2019, 102, e1900046. [Google Scholar] [CrossRef]
- Raghavachari, K.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. 20. Basis set for correlated wave-functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Samoylova, N.A.; Avdoshenko, S.M.; Krylov, D.S.; Thompson, H.R.; Kirkhorn, A.C.; Rosenkranz, M.; Schiemenz, S.; Ziegs, F.; Wolter, A.U.; Yang, S. Confining the spin between two metal atoms within the carbon cage: Redox-active metal–metal bonds in dimetallofullerenes and their stable cation radicals. Nanoscale 2017, 9, 7977–7990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popov, A.A. Redox-active metal–metal bonds between lanthanides in dimetallofullerenes. Curr. Opin. Electrochem. 2018, 8, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Natural bond orbital methods. WIREs Comput. Mol. Sci. 2012, 2, 1–42. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Bridgeman, A.J.; Cavigliasso, G.; Ireland, L.R.; Rothery, J. The Mayer bond order as a tool in inorganic chemistry. J. Chem. Soc. Dalton Trans. 2001, 1, 2095–2108. [Google Scholar] [CrossRef]
- Popov, A.A.; Dunsch, L. Bonding in endohedral metallofullerenes as studied by quantum theory of atoms in molecules. Chem.—Eur. J. 2009, 15, 9707–9729. [Google Scholar] [CrossRef]
- Foster, J.; Boys, S. Canonical configurational interaction procedure. Rev. Modern Phys. 1960, 32, 300. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
Figure 1.
Geometries of optimized Lu2@Ih_C60 (a) and Lu2@C2v_C60 (b) on the PBE/6-31G(d)~CEP-4G, including the relative energy (ΔE) on the theoretical level of PBE/6-311G(d,p)~SDD with single-point calculations.
Figure 1.
Geometries of optimized Lu2@Ih_C60 (a) and Lu2@C2v_C60 (b) on the PBE/6-31G(d)~CEP-4G, including the relative energy (ΔE) on the theoretical level of PBE/6-311G(d,p)~SDD with single-point calculations.

Figure 2.
NPA charges for Lu2@Ih_C60 (a) and Lu2@C2v_C60 (b) on the PBE/6-311G(d,p)~SDD, in which the atoms are colored by corresponding NPA charges.
Figure 2.
NPA charges for Lu2@Ih_C60 (a) and Lu2@C2v_C60 (b) on the PBE/6-311G(d,p)~SDD, in which the atoms are colored by corresponding NPA charges.

Figure 3.
FMOs for stable C2v_C60, Ih_C60, Lu2@Ih_C60, and Lu2@C2v_C60 molecules with isovalue of 0.03 a.u. for the surface with the PBE functional, 6-311G(d,p) basis set for carbon atoms, and SDD basis set for Lu atoms. The highest occupied molecular orbitals (HOMO) represented the Lu-Lu bond in C60. The blue numbers represent the energies of orbitals.
Figure 3.
FMOs for stable C2v_C60, Ih_C60, Lu2@Ih_C60, and Lu2@C2v_C60 molecules with isovalue of 0.03 a.u. for the surface with the PBE functional, 6-311G(d,p) basis set for carbon atoms, and SDD basis set for Lu atoms. The highest occupied molecular orbitals (HOMO) represented the Lu-Lu bond in C60. The blue numbers represent the energies of orbitals.
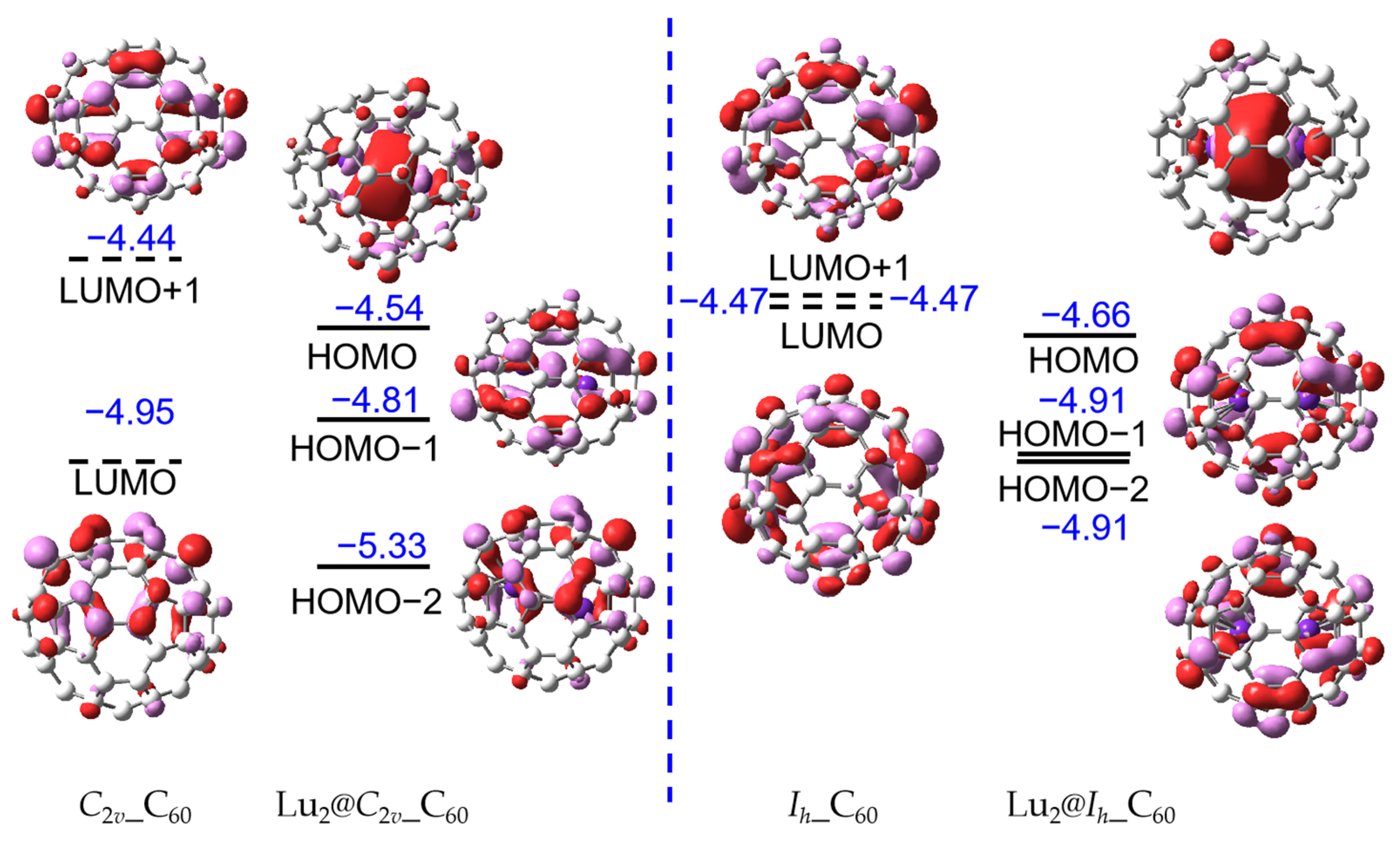
Figure 4.
Simulated vis-NIR spectra of the Lu2@Ih_C60 (a) and Lu2@C2v_C60 (b) isomers. The broadening function is Lorentzian and full width at half maximum is 0.30 eV.
Figure 4.
Simulated vis-NIR spectra of the Lu2@Ih_C60 (a) and Lu2@C2v_C60 (b) isomers. The broadening function is Lorentzian and full width at half maximum is 0.30 eV.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Natural population analysis (NPA) on Lu2@Ih_C60 and Lu2@C2v_C60, and the NPA charges (e) for metal atoms on the PBE/6-311G(d,p)~SDD.
Table 1.
Natural population analysis (NPA) on Lu2@Ih_C60 and Lu2@C2v_C60, and the NPA charges (e) for metal atoms on the PBE/6-311G(d,p)~SDD.
| Molecules | Atoms | Populations | NPA Charges |
|---|---|---|---|
| Lu2@Ih_C60 | Lu1 | 5d0.376s0.296p0.486d0.887p0.29 | 0.78 |
| Lu2 | 5d0.366s0.376p0.466d0.857p0.28 | 0.78 | |
| Lu2@C2v_C60 | Lu1 | 5d0.366s0.306p0.416d0.977p0.39 | 0.65 |
| Lu2 | 5d0.346s0.306p0.326d0.897p0.40 | 0.83 |
Table 2.
Mayer bond order (MBO) and Wiberg bond order (WBO) of Lu-Lu bond, and hybrid compositions of M-M bond in the Lu2@C60 at the theoretical level of PBE/6-311G(d,p)~SDD.
Table 2.
Mayer bond order (MBO) and Wiberg bond order (WBO) of Lu-Lu bond, and hybrid compositions of M-M bond in the Lu2@C60 at the theoretical level of PBE/6-311G(d,p)~SDD.
| Molecules | MBO | WBO | Atoms | Hybrid Composition |
|---|---|---|---|---|
| Lu2@Ih_C60 | 0.99 | 0.73 | Lu1 | s(26%)p(46%)d(28%) |
| Lu2 | s(36%)p(43%)d(21%) | |||
| Lu2@C2v_C60 | 0.84 | 0.67 | Lu1 | s(26%)p(44%)d(30%) |
| Lu2 | s(28%)p(45%)d(27%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhao, Y.; Shen, W.; Chen, W.; Lu, X. Lu-Lu Bond in Lu2@C60 Metallofullerenes. Inorganics 2023, 11, 277. https://doi.org/10.3390/inorganics11070277
AMA Style
Zhao Y, Shen W, Chen W, Lu X. Lu-Lu Bond in Lu2@C60 Metallofullerenes. Inorganics. 2023; 11(7):277. https://doi.org/10.3390/inorganics11070277
Chicago/Turabian StyleZhao, Yaoxiao, Wangqiang Shen, Weixing Chen, and Xing Lu. 2023. "Lu-Lu Bond in Lu2@C60 Metallofullerenes" Inorganics 11, no. 7: 277. https://doi.org/10.3390/inorganics11070277
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.