


Probing the Electronic Structure of Dinuclear Carbon-Rich Complexes Containing an Octa-3,5-diene-1,7-diyndiyl Bridging Ligand


School of Molecular Sciences, University of Western Australia, 35 Stirling Highway, Crawley 6009, Australia
*
Author to whom correspondence should be addressed.
Inorganics 2024, 12(1), 20; https://doi.org/10.3390/inorganics12010020
Submission received: 14 November 2023
/
Revised: 14 December 2023
/
Accepted: 14 December 2023
/
Published: 1 January 2024
(This article belongs to the Special Issue 10th Anniversary of Inorganics: Organometallic Chemistry)
Abstract
:One electron oxidation of the monometallic alkenylacetylide complexes [Ru{C≡CC(R)=CH2}(dppe)Cp*] (1) and [Ru{C≡CC(R)=CH2}Cl(dppe)2] (2) (R = Ph (a); R = 4-MeS-C6H4 (b)) generates in each case a dinuclear bis(allenylidene) complex [{Ru}2{μ-C=C=C(R)–CH2–H2C–(R)C=C=C}][PF6]2 ({Ru} = Ru(dppe)Cp* ([3a,b][PF6]2); {Ru} = RuCl(dppe)2 ([4a,b][PF6]2), containing an unsaturated ethane bridge between both allenylidene moieties. Deprotonation of this ethane bridge results in the formation of the previously reported octa-3,5-diene-1,7-diyndiyl-bridged bimetallic species [{Ru}2{μ-C≡CC(R)=CH–HC=(R)CC≡C}] ({Ru} = Ru(dppe)Cp* (5a,b); {Ru} = RuCl(dppe)2 (6a,b). The isolation of these complexes illustrates a general synthetic route to these conjugated bimetallic species from monomeric alkenylacetylide precursors. Electrochemical and spectroelectrochemical investigations evince the ready formation of the representative redox series [5a]n+, and TD-DFT calculations performed on optimised structures featuring the simplified {Ru(dmpe)Cp} coordination sphere [{Ru(dmpe)Cp}2{μ-C≡CC(Ph)=HC–CH(Ph)CC≡C}]n+ ([5a†]n+) (n = 0, 1, 2) reveal significant delocalisation of the unpaired charge in the formally mixed-valent species (n = 1), consistent with Class III assignment within the Robin–Day classification scheme.
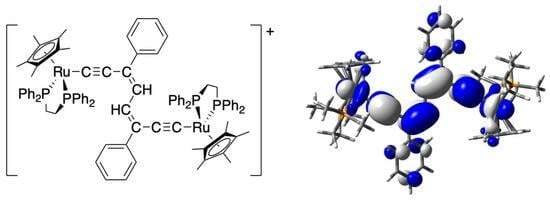
1. Introduction
Mixed-valent hetero- and homo-bimetallic complexes containing π-conjugated all-carbon and carbon-rich bridging ligands of the general form [{LxM}(μ–Bridge){MLx}] have drawn sustained interest as model systems with which to explore intramolecular electron and charge-transfer phenomena [1,2,3]. The orbital overlaps along the metal–bridge–metal assemblies are critical to the intramolecular electron transfer properties of the complex [4,5]; consequently, the chemical composition and structure of both the metal-ancillary ligand end-capping fragments, {MLx}, and the bridging ligand afford a significant degree of control over these metal–bridge coupling interactions [6,7,8]. This electronic control by chemical design permits facile manipulation of the degree of (de)localisation along the molecular scaffold, facilitating the application of mixed-valent species as sensors, electrochromes or components for molecular electronics [9,10,11,12,13]. However, the relative orientation of the metal end-caps with respect to each other and features of low axial symmetry incorporated in the bridge can induce dynamic aspects in unconstrained systems that necessitate careful interpretation of data collected from measurements of the entire population with respect to any single conformation or structure [14,15,16,17,18].
Against this background, polyyndiyl complexes of general form [{LxM}{μ-(C≡C)n}{MLx}] can be identified as useful ‘all-carbon’ bridged systems and precursors through which to further explore chemical structure: electronic-property relationships and electron-transfer phenomena [19,20,21,22,23,24]. Within the metal capping fragments, {MLx}, the energy and symmetry of the frontier metal d-orbitals can be manipulated through choice of the metal [23,25,26,27,28,29] and ancillary ligands [30,31], which in turn mediates the extent of d-orbital overlap with the π-conjugated frontier orbitals of the polyyndiyl bridge. Electrochemical measurements of polyyndiyl complexes [{LxM}{μ-(C≡C)n}{MLx}] typically reveal a decreasing separation of the first and second oxidation processes with increasing n, a phenomenon best attributed to the increasing spin delocalisation onto the carbon chain in the case of complexes of the heavier 4d and 5d metals [24,32,33], and increasingly localised metal-based redox processes for the lighter metal examples with more contracted and higher energy 3d fragment orbitals [8,20,27].
As a consequence of the increasing carbon character in the frontier orbitals of the heavier metal complexes, spectroscopic studies of the redox products derived from these all-carbon-bridged species [{LxM}{μ-(C≡C)n}{MLx}] are generally limited to the n = 1, 2 and 3 examples, the latter being only possible in the case of complexes bearing sterically demanding ancillary ligands [8]. This recalls the use of large end-groups to stabilise very long polyynes employed by, for example, Walton [34,35] and Tykwinski [36]. For smaller ancillary ligand sets, dimerisation of the radicals derived from the hexatriyndiyl-bridged complexes has been reported from spectroelectrochemical (SEC) and preparative scale chemical oxidation of these complexes, hindering further experimental measurements [37].
Metal polyendiyl complexes [{LxM}{μ-(CH=CH)n}{MLx}] are also known [38,39], and although spectroscopic studies of the redox products derived from these complexes are rare [40], a number of electrochemical studies have hinted at the efficacy of charge transport through the polyene chain [41,42,43]. The bridging structures themselves are intriguing as the functionalisation these sp2 centres, a feature not possible with polyyndiyl ligands composed of sp carbon centres, which would allow introduction of electron-donating or -withdrawing groups to stabilise charges along the carbon-rich backbone [44].
In seeking to develop longer bridging ligands with increased chemical stability in the mixed-valent state, attention has been largely drawn to ‘carbon-rich’ derivatives of these metal-alkynyl structures. As a result, there are numerous examples of bimetallic complexes and mixed-valence systems derived from bridging ligands featuring metal-alkynyl and -vinyl fragments and a host of interpolated arylene fragments [1,20,45,46]. The studies of such compounds, augmented by advances in (TD-)DFT methods, have been critical to unravelling further details of the electronic structure and nature of mixed-valence compounds and complexes [5,47,48,49,50,51] and also the fundamentals of the electron transfer reaction in both the ground and excited states [52,53,54]. However, whilst these compounds have allowed significant advances to be made in both the synthesis and subsequent analysis of these mixed-valent derivatives bridged by all-carbon or carbon-rich ligands, the inclusion of aromatic spacing groups introduces a degree of conformational uncertainty and significantly alters the underlying electronic structures [15,16,17,55,56].
Resting between these structure types are less well-explored families of complexes featuring hybrid bridging ligands containing both alkynyl/ethynyl and vinyl/eneyl motifs [57,58,59,60,61,62]. We have recently reported the synthesis of a range of bimetallic ruthenium complexes containing an octa-3,5-diene-1,7-diyl bridging ligand [{Ru(dppe)Cp*}2{μ-C≡CC(R)=CH–HC=(R)C≡CC}] (5), formed via in situ oxidation of [Ru{C≡CC(R)=CH2}(dppe)Cp*] [63] and sequential dimerisation and deprotonation [64]. Containing a highly conjugated carbon-rich bridging ligand, readily modified by aryl side groups, R, complexes 5 present as a structural motif, complementary to the polyyndiyl, polyendiyl, diethynylarylene or divinylarylene complexes discussed above, through which to study electronic structure and electron-transfer processes in mixed-valence complexes (Figure 1).
Here, we report further investigation of the electronic properties of these coupled products, alongside a modified stoichiometric oxidative coupling procedure that provides a more rational synthesis of these compounds (Scheme 1). For the series [{Ru(dppe)Cp*}2{μ-C≡CC(R)=HC–CH(R)CC≡C}], UV-Vis-NIR measurements supported by TD-DFT calculations performed on the model complexes [{Ru(dmpe)Cp}2(μ-C≡CC(Ph)=HC–CH(Ph)CC≡C}]n+ ([5a†]n+, n = 0, 1, 2), with analysis of the natural transition orbitals (NTOs) of the associated transitions, allow formal assignment of [5]+ as a strongly delocalised, Robin and Day Class III, mixed-valent species.
2. Results and Discussion
2.1. Synthesis
Solutions of alkenylacetylide complexes [Ru{C≡CC(R)=CH2}(dppe)Cp*] are known to readily dimerise following aerial oxidation, resulting in the initial formation of ethane-bridged bis(allenylidene) complexes [Ru(dppe)Cp*}2{=C=C=C(R)-CH2-H2C-(R)C=C=C=}][PF6]2 [3][PF6]2. In turn, complexes [3][PF6]2 readily deprotonate to give bimetallic complexes [{Ru(dppe)Cp*}2{C≡CC(R)=HC-CH=(R)CC≡C}] (5) containing octa-3,5-diene-1,7-diyl bridging ligands (Scheme 1) [64]. In an effort to expand the scope of this reaction, the alkenylacetylide complexes [Ru{C≡CC(R)=CH2}(dppe)Cp*] (1a, R = Ph; 1b, R = C6H4SMe-4) and [Ru{C≡CC(R)=CH2}Cl(dppe)2] (2a, R = Ph; 2b, R = C6H4SMe-4) were each treated with one equivalent of [FeCp]2PF6 to promote 1-electron oxidation (noting the relatively low first oxidation potential of 1a (−0.14 V vs. ferrocene/ferrocenium)) [64] and allowed to stir in solution for 2 h to promote dimerisation. Work-up afforded the ethane-bridged bis(allenylidene) complexes [3a,b][PF6]2 and [4a,b][PF6]2 (Scheme 1), characterised by the intense ν(C=C=C) bands near 1920 cm−1, as well as resonances in the 1H NMR spectrum near δ 1.9 ppm arising from the protons of the ethane bridge. Deprotonation of [3a,b][PF6]2 and [4a,b][PF6]2 by treatment with t-BuOK in THF gave the bis(acetylide) complexes 5a,b and 6a,b (Scheme 1) characterised by ν(C≡C) bands near 2030 cm−1, with other spectroscopic characteristics in agreement with those previously reported for similar compounds [64]. Related chemistry from ethane-bridged bis(carbyne) complexes leading to bis(vinylidene) and ultimately butadiyndiyl complexes on rhenium scaffolds is also noted as providing precedence for such transformations of carbon-rich scaffolds [65].
The bimetallic complexes [4][PF6]2 and 6 featuring the {RuCl(dppe)2} coordination sphere were poorly soluble in most common solvents, limiting the extent to which they could be characterised, especially via 13C{1H} NMR spectroscopy. This limited solubility also hampered efforts at obtaining pure samples, and as such [4a][PF6]2 could only be obtained as part of a crude mixture. Furthermore, attempts to deprotonate this complex within this mixture gave a solid from which spectroscopic evidence supported the formation of 6a, but which could not be satisfactorily purified. However, oxidation of 2a with two or three equivalents [FeCp]2PF6, followed by treatment of the reactions solution with t-BuOK, permitted isolation of the more soluble and hence more readily purified monocationic and dicationic complexes [6a]PF6 (30% isolated yield) and [6a][PF6]2 (19% isolated yield), respectively (Scheme 2). In this modified one-pot, two-step procedure, any amount of [4a]2+ formed from the initial oxidation of 2a is deprotonated in situ upon the addition of a base to give 6a (as part of a complex mixture), which is then further oxidised with the remaining equivalent(s) of ferrocenium.
Single crystals of [6a]PF6 were grown from the slow diffusion of MeOH into a CH2Cl2 solution of the complex and these were characterised by X-ray diffraction (Figure 2, Table 1). An inversion centre in the mid-point of the central C4–C4A bond renders each half of the cation identical, necessitating caution in any discussion of mixed-valence characteristics based on the structural data. Nevertheless, the Ru1–C1 [1.958(4) Å], C1–C2 [1.217(6) Å], C2–C3 [1.400(6) Å] and C3–C4 [1.401(6) Å] bond lengths show modestly reduced bond length alternation comparted with the bridging ligands in complexes 5 [64], consistent with involvement of the bridging ligand in the redox process [66]. These observations are also consistent with the conclusions drawn from electrochemical and spectroelectrochemical studies of 5a, and related species, which undergo two sequential one-electron oxidations to give the delocalised ‘mixed-valent’ complex [5a]PF6 and the crystallographically characterised bis(allenylidene) [5a][PF6]2 [64].
Beyond these solution-based chemical oxidation reactions, aerial oxidation of the alkene unit occurred over the course of several weeks when solid samples of 2a and 2b were stored under ambient conditions, resulting in the formation of the alkynylketone complexes 2aox and 2box in a manner similar to that observed previously for related samples (Scheme 3) [63,67]. Both 2aox and 2box were structurally characterised by single crystal X-ray diffraction (Figure 3, Table 2) in addition to the conventional spectroscopic methods. These carbonyl species were differentiated from their alkenylacetylide precursors by a characteristically low-energy shift of the ν(C≡C) band at ca. 2000 cm−1, reflecting the conjugation of the alkynyl and carbonyl moieties, the absence of geminal proton signals in the 1H NMR spectra, as well as observation of the appropriate cationic ion envelope by ESI(+)-MS. The formation of these complexes necessitated the storage of solid samples of 1 and 2 under an inert atmosphere.
2.2. Electronic Structure Calculations and Spectroscopy
To better rationalise the course of the reactions leading from complexes 1 and 2 to 5 and 6, and the sequence of redox reactions that characterise the bimetallic compounds, (TD-)DFT calculations (BLYP35/COSMO(CH2Cl2)//Ru(LANL2DZ)/all other atoms 6-31G**) were performed on the representative model structures [Ru{C≡CC(Ph)=CH2}(dmpe)Cp] (1a†) and [{Ru(dmpe)Cp}2(μ-C≡CC(Ph)=HC–CH(Ph)CC≡C}] (5a†), featuring the simplified {Ru(dmpe)Cp} coordination sphere (Figure 4), and their oxidation products [1a†]+ and [5a†]n+ (n = 1, 2) (dmpe = bis(dimethylphosphino)ethane). As has been demonstrated by Kaupp and others elsewhere [49,68], the BLYP35 global hybrid functional, used in concert with a COSMO solvent model, provides a balance between accuracy and computational efficiency in accurately describing localised vs. delocalised mixed-valence complexes. The optimised geometries determined in this manner, and which were verified as true minima through the absence of imaginary frequencies, are in good agreement with the formal valence bond descriptions of these species.
The use of simplified ancillary ligand sets in model calculations of this type has been shown to provide results in good agreement with larger models and experiment [69]. Here, the close agreement of the calculated ν(C≡C) frequencies (after application of a scaling factor 0.93) [70] for 1a† (2043 cm−1) and 5a† (2024 cm−1) with those observed for 1a (2057 cm−1) [63] and 5a (2036 cm−1) [64] gives further confidence in the validity of these models.
The HOMO of 1a† has appreciable contribution from the buta-3-ene-ynyl ligand (46%), which includes significant CδH2 character (15%). This observations helps rationalise the heightened nucleophilicity and propensity for this site to enter into reactions with electrophiles (Figure 5a) [63]. The removal of one electron from 1a† affords the radical cation [1a†]+, with analysis of the frontier orbital composition revealing that both the α- and β-HOSO have significant character at CδH2 (25 and 14% respectively), providing a rationale for the selective Cδ–Cδ coupling observed following oxidation of 1, and, by analogy, 2 (Figure 5a).
Turning to the bimetallic model 5a†, the bond lengths and angles in the DFT-optimised geometry (Table 3) and the nodal pattern of the HOMO (Figure 6) are consistent with the description of the carbon-rich bridging ligand as an octa-3,5-diene-1,7-diyndiyl fragment (Scheme 1). The 10-atom RuC8Ru (Ru–C≡C–C(Ph)=CH–CH=C(Ph)–C≡C–Ru) chain features heavily in the frontier orbitals with a significant contribution from the carbon atoms of the diene-diyndiyl bridge (HOMO, Ru/C10/Ru 10/63/10%; LUMO, Ru/C10/Ru 2/58/2%), which is reminiscent of the frontier orbital composition of polyyndiyl [{Cp*(dppe)Ru}{μ-(C≡C)n}{Ru(dppe)Cp*}] complexes [14,27,71,72,73].
Cyclic voltammetry measurements have revealed that complex 5a undergoes two sequential one-electron oxidation processes ( = −0.62 V; = −0.37 V vs. ferrocene/ferrocenium), generating [5a]+ and [5a]2+ (crystallographically characterised as the bis-PF6 salt) [64]. These results, together with isolation of [6a]n+ (n = 0, 1, 2) described here, point to the utility of the octa-3,5-diene-1,7-diyndiyl ligand as a bridging structure in bimetallic complexes, complementary in structure to octa-1,3,5,7-tetrayn-1,7-diyl but better able to support stable oxidation products. The DFT-optimised geometries of [5a†]+ and [5a†]2+ (Table 3) reveal a progressive evolution of the valence bond description of the bridging ligand towards more cumulated structures for the higher oxidation states. Thus, for [5a†]n+, the Ru(1)–C(1), C(2)–C(3) and C(4)–C(4′) bond lengths progressively decrease with increasing n, whilst the C(1)≡C(2) and C(3)=C(4) bond lengths increase. This structural progression is associated with a decrease in the calculated ν(C≡C) wavenumbers (ν(C≡C)/cm−1: 5a† 2024; [5a†]+ 1904; [5a†]2+ 1899), which is in agreement with experimental data from spectroelectrochemically generated samples (ν(C≡C)/cm−1: 5a 2034; [5a]+ 1939; [5a]2+ 1920) [64]. Overall, the tendency for these octa-1,3,5,7-tetrayn-1,7-diyl-bridged bimetallic ruthenium complexes to evolve towards more structures featuring more cumulated bridging ligand structures on oxidation is a consequence of the substantial ligand character of the oxidation process, which in turn parallels the behaviour of polyyndiyl complexes of the 4d [27,37,72,74] and 5d [75,76,77] metals.
The UV-Vis-NIR spectrum of the deep red complex 5a contains an intense absorption at ca. 19,417 cm−1, with several further high energy absorptions forming a large shoulder arising past 30,000 cm−1 (Figure 7). Similar visible absorption bands are also observed for other complexes of general form [{Ru(dppe)Cp*}2{C≡CC(R)=HC–CH=(R)CC≡C}] (R = C6H4-SMe (5b), C5H4N (5c), C6H4-NO2 (5d)), with the energy being sensitive to the electronic nature of the R group [64]. This observation clearly indicates that the conjugated bridging ligand plays a role in these transitions, and these optical features can therefore in turn provide an experimental reporter on changes to the electronic structure on a change in the redox state. As with the IR data, UV-Vis-NIR spectra of [5a]n+ (n = 0, 1, 2) were collected via spectroelectrochemical methods), and data analysed with the aid of TD-DFT calculations carried out on [5a†]n+ (n = 0, 1, 2) (Figure 7, Figure 8, Figure 9, Figure 10, Figure 11 and Figure 12). In each case, analysis of the transitions revealed contributions from numerous molecular orbitals. To simplify the description of these spectra, interpretations were made on the basis of natural transition orbital (NTO) plots, generated for each associated ground (particle) to excited (hole) state transition.
For 5a, the intense absorption observed at ca. 19,417 cm−1 (515 nm) in CH2Cl2/0.1 M NBu4PF6 corresponds to a calculated transition in 5a† at 20,952 cm−1 (f = 0.9687) (Figure 7), and for which the NTO analysis (Figure 8) reveals significant transfer of electron density from the metals to ligand, allowing assignment as a metal-to-ligand charge-transfer (MLCT) band. This assignment is consistent with both the substantial solvatochromic behaviour of this transition in 5a, and the significant shift to lower energy that tracks with the increasing electron-withdrawing character of the bridging ligand alkenyl substituents in complexes of general form [{Ru(dppe)Cp*}2{C≡CC(R)=HC–CH=(R)CC≡C}] (R = Ph, 5a; C6H4-SMe, 5b; C5H4N, 5c; C6H4NO2, 5d) (Table 4).
Less intense transitions are calculated for 5a† at ca. 25,407 cm−1 (f = 0.0082) and 25,488 cm−1 (f = 0.0453) with the associated particle and hole states illustrating significant ligand-to-metal charge-transfer (LMCT) character. A transition of moderate intensity at ca. 33,576 cm−1 (f = 0.3080) has substantial intra-ligand π–π* character, accompanied by some charge transfer back to the metal centres.
The spectroelectrochemically generated UV-Vis-NIR spectrum of [5a]+ contains three primary absorption band envelopes, although TD-DFT calculations indicate several mixed-transitions are responsible for the observed features (Figure 9). The consideration of all particle and hole wavefunctions shows the accumulation of spin density along the bridging ligand, supporting a strongly delocalised structure for the cationic species [5a]+ (Figure 10). The relatively intense, low-energy band envelope observed at 6242 cm−1 in [5a]+ corresponds to a calculated transition in [5a†]+ at 8646 cm−1 (Figure 9). This transition takes place between β-spin orbitals that are extensively delocalised over the 10-atom RuC8Ru (Ru–C≡C–C(Ph)=CH–CH=C(Ph)–C≡C–Ru) chain and has significant π–π* character. This band may therefore be considered the ‘intervalence charge transfer’ (IVCT) band associated with the strongly delocalised (Class III) mixed-valent radical cation [5a]+ that is perhaps better described as a charge resonance transition [78,79,80]. The intense absorption band envelopes between 12,000 and 14,000 cm−1 in [5a]+ correspond to a calculated transition in [5a†]+ at 14,056 cm−1 and arise from transitions in the α-spin manifold with a greater degree of metal-to-ligand charge-transfer (MLCT) character. Less intense transitions calculated at ca. 23,973 cm−1 and 25,265 cm−1 with mixed metal-to-ligand and intra-ligand charge-transfer character fit well to the heavily overlapped transitions that form the unresolved features through the visible region of the experimental spectrum (Figure 9 and Figure 10).
The UV-Vis-NIR spectrum of the spectroelectrochemically generated dication [5a][PF6]2 contains a single intense absorption at 14,245 cm−1, with many overlapped, poorly resolved absorption bands at higher energy (Figure 11). The TD-DFT calculated spectrum reveals an additional band with a low-energy shoulder at ca. 30,000 cm−1. The intense absorption at 14,245 cm−1 observed for [5a]2+ correlates well with the transition at 14,876 cm−1 calculated via TD-DFT methods for [5a†]2+ (Figure 11). The NTO analysis supports the description of this transition as an MLCT band. A less intense transition calculated at 26,675 cm−1 shows intra-ligand charge-transfer character from the pendant phenyl substituents to the octa-diene-diyldiyl fragment, whilst a second MLCT-like transition at 29,844 cm−1 also contributes to absorption in the visible region (Figure 12).
Overall, the primary feature of the UV-Vis-NIR spectra of [5a]n+ (n = 0, 1, 2) appears to be intense MLCT bands at 19,417 (5a), 12,000–14,000 ([5a]+) and 14,245 ([5a]2+), whilst [5a]+ contains an additional IVCT or charge resonance band at ca. 6200 cm−1. Additional transitions show significant accumulation of spin density to the bridging ligand and phenyl substituents in particular, supporting a strongly delocalised electronic structure.
3. Experimental Details
All reactions were performed under an atmosphere of nitrogen employing standard Schlenk techniques. Unless stated otherwise, no particular care was taken to exclude air upon work-up of reaction products. Solvents were dried by literature methods or by an Innovative Technologies Solvent Purification System and sparged with nitrogen before use. For chromatography, silica gel was used as received and alumina (basic) was oven dried (100 °C) overnight before use. The compounds [Ru{C≡CC(R)=CH2}(dppe)Cp*] (R = Ph (1a), 4-MeS-C6H4 (1b)) [63], [Ru{C≡CC(R)=CH2}Cl(dppe)2] (R = Ph (2a), 4-MeS-C6H4 (2b)) [63], [{Ru(dppe)Cp*}2{μ-C=C=C(Ph)–H2C–CH2–C(Ph)=C=C}][PF6]2 ([3a][PF6]2) [64] and [FeCp2]PF6 [81] were all prepared either in accordance with, or with slight refinements to, existing literature procedures. All other chemicals were purchased and used as received.
The various 1H, 13C{1H} and 31P{1H} spectra were recorded on Bruker 400 MHz (1H: 399.86 MHz, 13C: 100.6 MHz, 31P: 161.9 MHz), Bruker 500 MHz (1H: 500.10 MHz, 13C: 125.8 MHz, 31P: 202.4) and Bruker 600 MHz (1H: 600.10 MHz, 13C: 150.9 MHz, 31P: 242.9) spectrometers at room temperature. Chemical shifts are all reported relative to the residual solvent peaks. Unless stated otherwise 31P{1H}, chemical shifts are reported relative to the NMR spectrometer lock signal. For all NMR spectra, multiplets are reported according to their closest first order approximation. For all NMR assignments, Ho, Hm and Hp refer to the ortho, meta and para protons of the phenyl rings of the ancillary phosphine ligands respectively, whilst Ci, Co, Cm and Cp, similarly refer to the ipso, ortho, meta and para carbons of these same phenyl rings. IR spectra were recorded on an Agilent Cary 630 FTIR Spectrometer using ATR or in transmission mode from solutions between CaF2 plates. Mass spectra were obtained from a Waters Liquid Chromatograph Premier Mass Spectrometer using positive-mode electrospray ionisation (ESI(+)) or atmospheric pressure chemical ionisation (APCI(+)). Samples were prepared in MeCN, EtOAc or MeOH and inserted by direct injection via the on-board injector.
Single crystals of 2aox, 2box and [6a]PF6 were used as supplied and mounted on a XtaLAB Synergy, Single source at home/near, HyPix diffractometer. All crystals were kept at a steady T = 100.0 K during data collection. Data were collected using Cu Kα radiation (λ = 1.54184 Å). The structures were solved, and the space group determined by the ShelXT 2018/2 structure solution program using dual methods and refined by full matrix least-squares minimisation on F2 using version 2018/3 of XL [82,83], while using Olex2 1.5 [84,85] or WinGX 2018.3 [86] as graphical interfaces. For all structures, all non-hydrogen atoms were refined anisotropically. Hydrogen atom positions were calculated geometrically and refined using the riding model. Disordered phenyl groups in 2box were refined as rigid fragments, with thermal similarity constraints, while the PF6 group in [6a]PF6 was refined with 1,2 similarity restraints on the P-F bonds due to disorder of the F-atoms in the equatorial plane. Crystallographic data for the structures reported in this paper have been deposited at the Cambridge Crystallographic Data Centre (2307001-2307003).
DFT calculations were carried out using the Gaussian09 suite of programs (revision A.02) [87], and the results analysed with the aid of GaussView5.0 and GassSum3.0 [88]. All calculations were carried out with the BLYP35 functional [5], with the LANL2DZ basis set for Ru [89,90,91] and 6-31G** for all other atoms [92,93], and a COOSMO(CH2Cl2) solvent model [94,95]. All optimised structures were confirmed as true minima through the absence of imaginary frequencies. Reported values of vibrational frequencies have been scaled by a factor of 0.93) [70], with descriptions of the optical spectra performed within the framework of natural transition orbital analyses [96].
Cyclic voltammetry measurements were performed in CH2Cl2 solutions containing 0.1 M NBu4PF6-supporting electrolyte using a platinum working electrode and a PalmSens Emstat2+ or Emstat3+ potentiostat. Potential measurements were referenced against an internal ferrocene/ferrocenium (FeCp2/[FeCp2]+) couple reference.
Spectroelectrochemistry was conducted in an OTTLE cell of Hartl design [97] using CH2Cl2 solutions containing 0.1 M NBu4PF6-supporting electrolyte. Spectra were recorded on an Agilent Technologies Cary 660 FTIR. Electrolysis in the cell was performed using a PalmSens Emstat3+ potentiostat at a scan rate of 0.0025 V s−1.
3.1. Synthesis of [{Ru(dppe)Cp*}2{μ-C=C=C(MeS-4-C6H4)–H2C–CH2–C(MeS-4-C6H4)=C=C}][PF6]2 ([3b][PF6]2)

A solution of 1b (0.16 g, 0.20 mmol) in CH2Cl2 (5 mL) was cooled to −78 °C after which it was treated with [FeCp2]PF6 (0.061 g, 0.18 mmol) and stirred for 2 h. The solvent volume was reduced, and the concentrated solution filtered into stirred hexanes, giving the title product as a purple powder (0.160 g, 0.099 mmol, 98%).
1H NMR (CDCl3 400 MHz) δ/ppm: 7.51–7.50 (m, 2H, H6/7); 7.47–7.45 (m, 4H, Ho, dppe); 7.36–7.28 (m, 8H, Ho + Hp, dppe); 7.17–7.12 (m, 4H, Hm, dppe); 2.82 (m, 2H, dppe); 2.62 (m, 2H, dppe); 2.50 (s, 3H, SMe); 1.93 (s, 2H, H4); 1.59 (s, 15H, Me of Cp*). 13C{1H} NMR (CDCl3 100.6 MHz) δ/ppm: 287.8–287.5 (m, C1); 198.4 (s, C2); 156.0 (s, C3); 146.9 (s, C8); 139.1 (s, C5); 134.7–134.2 (m, 2 × Ci, dppe); 132.7–132.6 (app t., Co, dppe); 132.5–132.4 (app t., Co, dppe); 131.4 (s, Cp, dppe); 131.3 (s, Cp, dppe); 128.9–128.8 (app t., Cm, dppe); 128.7–128.6 (app t., Cm, dppe); 127.5 (s, C6/7); 125.7 (s, C6/7); 102.3 (s, Cp*); 40.2 (s, C4); 30.5–30.0 (m, dppe); 14.7 (s, SMe); 10.2 (s, Cp*). 31P{1H} NMR (CDCl3 161.9 MHz) δ/ppm: 78.76 (s, dppe); −144.81 (sept, PF6). IR ATR ν/cm−1: 1917 ν(C=C=C); 834 ν(PF6−). ESI(+)-MS m/z: Calculated for [M]2+ ([C94H96P4S2Ru2]2+) = 807.1912. Observed: 807.1918 [M]2+.
3.2. Synthesis of [{RuCl(dppe)2}2{μ-C=C=C(MeS-4-C6H4)–H2C–CH2–C(MeS-4-C6H4)=C=C}][PF6]2 ([4b][PF6]2)

A solution of 2b (0.097 g, 0.088 mmol) in CH2Cl2 (5 mL) was cooled to −78 °C after which it was treated with [FeCp2]PF6 (0.031 g, 0.094 mmol) and stirred for 2 h. The solvent volume was reduced and the concentrated solution filtered into stirred hexanes, giving the title product as a dark purple powder (0.095 g, 0.043 mmol, 97%). Extremely poor solubility prevented informative 13C{1H} measurements from being performed.
1H NMR (CD2Cl2 500 MHz) δ/ppm: 7.32–6.99 (m, 40H, dppe); 6.71 (m, 2H, H6/7); 6.49 (m, 2H, H6/7); 3.16 (m, 4H, dppe); 2.78 (m, 4H, dppe); 2.50 (s, 3H, SMe); 1.92 (s, 2H, H4). 31P{1H} NMR (CD2Cl2 161.9 MHz) δ/ppm: 40.41 (s, dppe); −144.81 (sept, PF6). IR ATR ν/cm−1: 1915 ν(C=C=C); 835 ν(PF6−). ESI(+)-MS m/z: Calculated for [M]2+ ([C126H114Cl2P8S2Ru2]2+) = 1106.1858. Observed: 1106.1789 [M]2+.
3.3. Synthesis of [{Ru(dppe)Cp*}2{μ-C≡CC(R)=HC–CH=C(R)C≡C}] (R = Ph (5a), MeS-4-C6H4 (5b)

A solution of [3a][PF6]2 or [3b][PF6]2 (ca. 0.06 mmol) in THF (5 mL) was treated with t-BuOK (2.5 equiv., ca. 0.17 mmol) and stirred for 2 h. Removal of solvent under reduced pressure was followed by passage of a CH2Cl2 extract through a basic alumina plug. The solid obtained after taking the eluted fraction to dryness was triturated with hexanes gave the title products as red-orange powders in near-quantitative yield.
5a: data were in accord with previous reports of an authentic sample [64].
5b: 1H NMR (CDCl3 400 MHz) δ/ppm: 7.71–7.70 (m, 4H, Ho, dppe); 7.34–7.15 (m, 16H, Ho + Hp + Hm, dppe); 6.94 (s, 1H, H4); 6.77 (app d., JHH = 8.0 Hz, 2H, H6/7); 6.64 (app d., JHH = 8.0 Hz, 2H, H6/7); 2.76 (m, 2H, dppe); 2.39 (s, 3H, SMe); 2.09 (m, 2H, dppe); 1.57 (s, 15H, Me of Cp*). 13C{1H} NMR (CDCl3, 100.6 MHz) δ/ppm: 133.6–133.5 (m, Co, dppe); 133.3–133.2 (m, Co, dppe); 129.1 (s, Cp, dppe); 129.0 (s, Cp, dppe); 127.6 –127.4 (m, 2 × Cm, dppe); 93.0 (s, Cp*); 30.5 (m, dppe); 10.4 (s, Me of Cp*).* Limited solubility prevented observation of C1, C2, C3 and C4 13C{1H} resonances. 31P{1H} NMR (CDCl3 161.9 MHz) δ/ppm: 81.95 (s, dppe). IR ATR ν/cm−1: 2027 ν(C≡C). ESI(+)-MS m/z: Calculated for [M]+ ([C94H94P4S2Ru2]+) = 1614.3834. Observed: 1614.3862 [M]+.
3.4. Synthesis of [{RuCl(dppe)2}2{μ-C≡CC(MeS-4-C6H4)=HC–CH=C(MeS-4-C6H4)C≡C}] (6b)

A solution of [4b][PF6]2 (0.097 g, 0.039 mmol) in THF (5 mL) was treated with t-BuOK (0.011 g, 0.098 mmol) and stirred for 2 h. Removal of solvent under reduced pressure, followed by passage through a basic alumina plug and trituration of the solid obtained from the eluted fraction with hexanes gave the title product as a red-brown powder (0.044 g, 0.020 mmol, 51%).
1H NMR (CDCl3 400 MHz) δ/ppm: 7.71 (m, 8H, Ho, dppe); 7.19–7.11 (m, 8H, Ho, dppe); 7.10–7.00 (m, 8H, Hp, dppe); 6.97–6.90 (m, 21H, H6 + H7 + H4 + Hm, dppe); 2.76 (m, 4H, dppe); 2.58 (m, 4H, dppe); 2.44 (s, 3H, SMe). 13C{1H} NMR (CDCl3 100.6 MHz) δ/ppm: 136.7–136.0 (m, 4 × Ci); 135.0–134.9 (m, Co, dppe); 134.6 (); 134.3–134.2 (m, Co, dppe); 133.9 (); 129.3 (s, Cp, dppe); 128.6 (s, Cp, dppe); 127.5–127.4 (m, Cm, dppe); 127.2 (s, C6/7); 127.0–126.9 (m, Cm, dppe); 126.3 (s, C6/7); 30.8–30.6 (m, dppe); 22.8 (s, SMe). Poor solubility prevented assignment of several 13C{1H} NMR resonances. 31P{1H} NMR (CDCl3 161.9 MHz) δ/ppm: 49.74 (s, dppe). IR ATR ν/cm−1: 2035 ν(C≡C). ESI(+)-MS m/z: Calculated for [M–Cl− + MeCN]+ ([C128H115ClNP8S2Ru2]+) = 2216.4147. Observed: 2216.4173 [M–Cl− + MeCN]+.
3.5. Synthesis of [{RuCl(dppe)2}2{μ-C≡CC(Ph)=HC–CH=C(Ph)C≡C}]PF6 ([6a]PF6)

A solution of 2a (0.098 g, 0.092 mmol) in CH2Cl2 (10 mL) was cooled to −78 °C and then treated with [FeCp2]PF6 (0.061 g, 0.18 mmol) and stirred for 2 h, after which the solution was allowed to reach room temperature before t-BuOK (2.2 equiv.) was added. The solvent volume was reduced, and the concentrated solution filtered into stirred hexanes, giving the title product as a green-brown powder (0.062 g, 0.027 mmol, 30%). Single crystals suitable for X-ray diffraction were obtained by layering a CH2Cl2 solution of [6a]PF6 with MeOH. Low solubility and the inherent paramagnetism of the sample prevented a satisfactory 13C{1H} NMR spectrum from being obtained.
1H NMR (CDCl3 500 MHz) δ/ppm: 8.14–6.93 (m, 45H, dppe + Ph); 6.44 (s, 1H, H4); 2.68 (m, 4H, dppe); 2.58 (m, 4H, dppe). 31P{1H} NMR (CDCl3 202.4 MHz) δ/ppm: 78.76 (s, dppe); −144.81 (sept, PF6). IR ATR ν/cm−1: 1918 ν(C=C=C); 833 ν(PF6−). ESI(+)-MS m/z: Calculated for [M–H − 2 × Cl− + MeOH + MeCN]+ ([C127H114NOP8Ru2]2+) = 2120.4888. Observed: 2120.3842 [M–H − 2 × Cl− + MeOH + MeCN]+.
3.6. Preparation of [{RuCl(dppe)2}2{μ-C=C=C(Ph)–HC=CH–C(Ph)=C=C}][PF6]2 ([6a][PF6]2)

A solution of 2a (0.102 g, 0.096 mmol) in CH2Cl2 (10 mL) was cooled to −78 °C and then treated with [FeCp2]PF6 (0.091 g, 0.27 mmol) and stirred for 2 h, after which the solution was allowed to reach room temperature before t-BuOK (2.2 equiv.) was added. The solvent was reduced and the concentrated solution filtered into stirred hexanes, giving the title product as a blue powder (0.044 g, 0.018 mmol, 19%).
1H NMR (CDCl3 600 MHz) δ/ppm: 7.54–7.03 (m, 45H, dppe + Ph); 6.30 (s, 1H, H4); 3.01 (m, 4H, dppe); 2.89 (m, 4H, dppe). 13C{1H} NMR (CDCl3 150.9 MHz) δ/ppm: 214.3 (s, C2); 161.2 (s, C3); 136.6–135.3 (m, Ci, dppe); 134.2 (m, Co, dppe); 133.9 (s, Ph); 133.2 (m, Co, dppe); 132.5 (s, Ph); 131.5 (s, Cp, dppe); 131.2 (s, Cp, dppe); 128.8 (m, Cm, dppe); 128.7 (s, Ph); 128.2 (s, Cm, dppe); 126.9 (s, Ph); 29.3–29.0 (m, dppe). C1 was not observed due to poor solubility. 31P{1H} NMR (CDCl3 242.9 MHz) δ/ppm: 40.42 (s, dppe); −147.94 (sept, PF6). IR ATR ν/cm−1: 1899 ν(C=C=C); 831 ν(PF6−). ESI(+)-MS m/z: Calculated for [M]2+ ([C124H110Cl2P8Ru2]2+) = 1060.1981. Observed: 1060.1921 [M]2+.
3.7. Preparation of trans-[Ru{C≡CC(=O)Ph}Cl(dppe)2] (2aox)

This compound was obtained from aerial oxidation of a solid sample of 2a stored under ambient conditions, purified by preparative TLC (acetone/hexanes 3:7) and obtained as a yellow solid. Single crystals suitable for X-ray diffraction were grown by layering a CH2Cl2 solution of 2aox with MeOH.
1H NMR (CDCl3 400 MHz) δ/ppm: 7.40–7.38 (m, 8H, Ho, dppe); 7.29–7.24 (m, 8H, Ho, dppe); 7.22–7.21 (m, 6H, Hp + Ph); 7.07–7.03 (m, 12H, Hp + Hm, dppe); 6.98–6.94 (m, 2H, Ph); 6.89–6.85 (m, 8H, Hm, dppe); 2.92 (m, 4H, dppe); 2.81 (m, 4H, dppe). 13C{1H} NMR (CDCl3 100.6 MHz) δ/ppm: 173.5 (s, C3); 139.1 (s, C4); 135.9–135.4 (m, 2 × Ci, dppe); 134.6–134.5 (m, Co, dppe); 134.0–133.9 (m, Co, dppe); 130.7 (s, CPh); 129.4 (s, CPh); 129.3 (s, Cp, dppe); 127.6–127.5 (m, Cm, dppe); 127.4 (s, CPh); 127.3–127.2 (m, Cm, dppe); 120.8 (s, C2); 30.8–30.3 (m, dppe). 31P{1H} NMR (CDCl3 161.9 MHz) δ/ppm: 47.09 (s, dppe). IR ATR ν/cm−1: 2000 ν(C≡C); 1705 ν(C=O). ESI(+)-MS m/z: Calculated for [M–Cl− + MeCN]+ ([C63H56NOP4Ru]+) = 1068.2356. Observed: 1068.2341 [M–Cl− + MeCN]+.
3.8. Preparation of trans-[Ru{C≡CC(=O)-4-MeS-C6H4}Cl(dppe)2] (2box)

This compound was obtained from aerial oxidation of a solid sample of 2b stored under ambient conditions, purified by preparative TLC (acetone/hexanes 3:7) and obtained as a yellow solid. Single crystals suitable for X-ray diffraction were grown by layering a CH2Cl2 solution of 2box with MeOH.
1H NMR (CDCl3 400 MHz) δ/ppm: 7.41–7.39 (m, 8H, Ho, dppe); 7.24–7.20 (m, 8H, Ho, dppe); 7.09 (app d., JHH = 8.5 Hz, 2H, H5/6); 7.05–7.02 (m, 16H, Hp + Hm, dppe); 6.88 (app t, JHH = 7.6 Hz, 8H, Hm, dppe); 6.76 (app d, JHH = 8.5 Hz, 2H, H5/6); 2.92 (m, 4H, dppe); 2.79 (m, 4H, dppe); 2.45 (s, 3H, SMe). 13C{1H} NMR (CDCl3 100.6 MHz) δ/ppm: 195.2 (s, C3); 142.1 (s, C4); 136.0–135.4 (m, 2 × Ci, dppe); 134.6 (m, Co, dppe); 133.9 (m, Co, dppe); 129.7 (s, C5/6); 129.3 (s, Cp, dppe); 129.2 (s, Cp, dppe); 127.6 (m, Cm, dppe); 127.3 (m, Cm, dppe); 126.6 (s, C2); 124.3 (s, C5/6); 29.9 (m, dppe); 15.3 (s, SMe). 31P{1H} NMR (CDCl3 161.9 MHz) δ/ppm: 46.92 (s, dppe). IR ATR ν/cm−1: 1998 ν(C≡C); 1704 ν(C=O). ESI(+)-MS m/z: Calculated for [M–Cl− + MeCN]+ ([C64H58NOP4SRu]+) = 1114.2233. Observed: 1114.2227 [M–Cl− + MeCN]+.
4. Conclusions
The dinuclear octa-3,5-diene-1,7-diyndiyl-bridged complexes [{Ru}2{μ-C≡CC(R)=CH–HC=(R)CC≡C}] ({Ru} = Ru(dppe)Cp* (5a,b); {Ru} = RuCl(dppe)2 (6a,b)) have been prepared via the one-electron oxidation of alkenylacetylide complexes [Ru{C≡CC(R)=CH2}(dppe)Cp*] (1) and [Ru{C≡CC(R)=CH2}Cl(dppe)2] (2) (R = Ph (a); R = 4-MeS-C6H4 (b)) and deprotonation of the resultant ethane-bridged dinuclear bis(allenylidene) complexes [{Ru}2{μ-C=C=C(R)–CH2–H2C–(R)C=C=C}][PF6]2 ({Ru} = Ru(dppe)Cp* ([3a,b][PF6]2); {Ru} = RuCl(dppe)2 ([4a,b][PF6]2), formed by Cδ–Cδ’ homo-dimerisation of [1]+ or [2]+, respectively. The chemical reactivity profiles and redox/electrochemical response of 1 (and by inference 2) and 5 (and by inference 6), coupled with spectroscopic and (TD-)DFT calculations, support a number of important conclusions, which were explored with the more soluble {Ru(dppe)Cp*}-based complexes 1 and 5 and the computational model systems [Ru{C≡CC(Ph)=CH2}(dmpe)Cp] (1a†) and [{Ru(dmpe)Cp}2(μ-C≡CC(Ph)=HC–CH(Ph)CC≡C}] (5a†). Oxidation of the alkenylacetylide complex 1 and 2 results in the accumulation of significant radical character at Cδ, rationalising the formation of the new C-C bond in [3][PF6]2 and [4][PF6]2. After deprotonation, the resulting octa-3,5-diene-1,7-diyndiyl are found to undergo a sequence of two, facile one-electron oxidation steps to give the redox series [5]n+ and [6]n+ (n = 0, 1, 2). The greater stability of the one- and two-electron oxidation products permit a range of more detailed investigations than are possible with similar octa-1,3,5,7-tetrayndiyl complexes. A combination of crystallographic characterisation, spectroelectrochemical study and (TD-)DFT calculations indicate a significant degree of charge delocalisation and ligand redox non-innocence for the dinuclear complexes [5]n+ (and hence [6]n+). The formally mixed-valence one-electron oxidised species are assigned as Robin–Day Class III or fully delocalised species.
Author Contributions
Conceptualization, P.J.L. and M.R.H.; methodology, M.R.H., S.A.M. and P.J.L.; formal analysis, M.R.H., S.A.M. and P.J.L.; investigation, M.R.H.; resources, S.A.M. and P.J.L.; data curation, M.R.H., S.A.M. and P.J.L.; writing—original draft preparation, M.R.H.; writing—review and editing, M.R.H., S.A.M. and P.J.L.; supervision, P.J.L.; project administration, P.J.L.; funding acquisition, M.R.H., S.A.M. and P.J.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the University of Western Australia through a Dean’s Scholarship for Excellence to M.R.H and the Australian Research Council through a Future Fellowship to S.A.M. (FT200100243).
Data Availability Statement
The data presented in this study are available in the article.
Acknowledgments
M.R.H. held a Dean’s Excellence in Science Ph.D. Scholarship from the Faculty of Science, University of Western Australia. S.A.M. thanks the Australian Research Council (ARC) for a Future Fellowship (FT200100243). The authors gratefully acknowledge the facilities and the scientific and technical assistance of Microscopy Australian at the Centre for Microscopy, Characterisation Analysis, the University of Western Australia, a facility funded by the University, State, and Commonwealth Governments.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Aguirre-Etcheverry, P.; O’Hare, D. Electronic Communication through Unsaturated Hydrocarbon Bridges in Homobimetallic Organometallic Complexes. Chem. Rev. 2010, 110, 4839–4864. [Google Scholar] [CrossRef] [PubMed]
- Launay, J.P. Long-distance intervalence electron transfer. Chem. Soc. Rev. 2001, 30, 386–397. [Google Scholar] [CrossRef]
- Launay, J.P. Mixed-Valent Compounds and their Properties—Recent Developments. Eur. J. Inorg. Chem. 2020, 2020, 329–341. [Google Scholar] [CrossRef]
- Launay, J.P. An orbital approach of electron transfer in multisite systems. Implications for carbon-rich spacers. Polyhedron 2015, 86, 151–166. [Google Scholar] [CrossRef]
- Renz, M.; Theilacker, K.; Lambert, C.; Kaupp, M. A Reliable Quantum-Chemical Protocol for the Characterization of Organic Mixed-Valence Compounds. J. Am. Chem. Soc. 2009, 131, 16292–16302. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, E.C.; Brown, N.J.; Edge, R.; Helliwell, M.; Roberts, H.N.; Tuna, F.; Beeby, A.; Collison, D.; Low, P.J.; Whiteley, M.W. Orbital Symmetry Control of Electronic Coupling in a Symmetrical, All-Carbon-Bridged “Mixed Valence” Compound: Synthesis, Spectroscopy, and Electronic Structure of [{Mo(dppe)(η-C7H7)}2(μ-C4)]n+ (n = 0, 1, or 2). Organometallics 2012, 31, 157–169. [Google Scholar] [CrossRef]
- Pieslinger, G.E.; Aramburu-Troselj, B.M.; Cadranel, A.; Baraldo, L.M. Influence of the Electronic Configuration in the Properties of d6-d5 Mixed-Valence Complexes. Inorg. Chem. 2014, 53, 8221–8229. [Google Scholar] [CrossRef] [PubMed]
- Gückel, S.; Safari, P.; Ghazvini, S.M.B.H.; Hall, M.R.; Gluyas, J.B.G.; Kaupp, M.; Low, P.J. Iron Versus Ruthenium: Evidence for the Distinct Differences in the Electronic Structures of Hexa-1,3,5-triyn-1,6-diyl-bridged Complexes [Cp*(dppe)M}{μ-(C≡C)3}{M(dppe)Cp*}]+ (M = Fe, Ru). Organometallics 2021, 40, 346–357. [Google Scholar] [CrossRef]
- Launay, J.P. Electron transfer in molecular binuclear complexes and relation with electron transport through nanojunctions. Coord. Chem. Rev. 2013, 257, 1544–1554. [Google Scholar] [CrossRef]
- Zhang, J.; Ouyang, J.; Ye, Y.X.; Li, Z.; Lin, Q.; Chen, T.; Zhang, Z.; Xiang, S.C. Mixed-Valence Cobalt(II/III) Metal–Organic Framework for Ammonia Sensing with Naked-Eye Color Switching. ACS Appl. Mater. Interfaces 2018, 10, 27465–27471. [Google Scholar] [CrossRef]
- Corrente, G.A.; Cospito, S.; Capodilupo, A.L.; Beneduci, A. Mixed-Valence Compounds as a New Route for Electrochromic Devices with High Coloration Efficiency in the Whole Vis-NIR Region. Appl. Sci. 2020, 10, 8372. [Google Scholar] [CrossRef]
- Ma, X.; Pang, C.; Li, S.; Li, J.; Wang, M.; Xiong, Y.; Su, L.; Luo, J.; Xu, Z.; Lin, L. Biomimetic Synthesis of Ultrafine Mixed-Valence Metal–Organic Framework Nanowires and Their Application in Electrochemiluminescence Sensing. ACS Appl. Mater. Interfaces 2021, 13, 41987–41996. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.; Long, G.J.; Rebbouh, L.; Grandjean, F.; Beatty, A.M.; Fehlner, T.P. Properties of a Mixed-Valence (FeII)2(FeIII)2 Square Cell for Utilization in the Quantum Cellular Automata Paradigm for Molecular Electronics. J. Am. Chem. Soc. 2005, 127, 17819–17831. [Google Scholar] [CrossRef] [PubMed]
- Gluyas, J.B.G.; Gückel, S.; Kaupp, M.; Low, P.J. Rational Control of Conformational Distributions and Mixed-Valence Characteristics in Diruthenium Complexes. Chem. Eur. J. 2016, 22, 16138–16146. [Google Scholar] [CrossRef] [PubMed]
- Parthey, M.; Gluyas, J.B.G.; Fox, M.A.; Low, P.J.; Kaupp, M. Mixed-Valence Ruthenium Complexes Rotating through a Conformational Robin-Day Continuum. Chem. Eur. J. 2014, 20, 6895–6908. [Google Scholar] [CrossRef] [PubMed]
- Costuas, K.; Cador, O.; Justaud, F.; Le Stang, S.; Paul, F.; Monari, A.; Evangelisti, S.; Toupet, L.; Lapinte, C.; Halet, J.F. 3,5-Bis(ethynyl)pyridine and 2,6-Bis(ethynyl)pyridine Spanning Two Fe(Cp*)(dppe) Units: Role of the Nitrogen Atom on the Electronic and Magnetic Couplings. Inorg. Chem. 2011, 50, 12601–12622. [Google Scholar] [CrossRef]
- Fitzgerald, E.C.; Ladjarafi, A.; Brown, N.J.; Collison, D.; Costuas, K.; Edge, R.; Halet, J.F.; Justaud, F.; Low, P.J.; Meghezzi, H.; et al. Spectroscopic Evidence for Redox Isomerism in the 1,4-Diethynylbenzene-Bridged Heterobimetallic Cation [{Fe(dppe)Cp*}(μ-C≡CC6H4C≡C){Mo(dppe)(η-C7H7)}]PF6. Organometallics 2011, 30, 4180–4195. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, M.X.; Sun, C.F.; Xu, M.; Hartl, F.; Yin, J.; Yu, G.A.; Rao, L.; Liu, S.H. Diruthenium Complexes with Bridging Diethynyl Polyaromatic Ligands: Synthesis, Spectroelectrochemistry, and Theoretical Calculations. Organometallics 2015, 34, 3967–3978. [Google Scholar] [CrossRef]
- Bruce, M.; Low, P. Transition metal complexes containing all-carbon ligands. Adv. Organomet. Chem. 2004, 50, 179–444. [Google Scholar]
- Halet, J.F.; Lapinte, C. Charge delocalization vs localization in carbon-rich iron mixed-valence complexes: A subtle interplay between the carbon spacer and the (dppe)Cp*Fe organometallic electrophore. Coord. Chem. Rev. 2013, 257, 1584–1613. [Google Scholar] [CrossRef]
- Zheng, Q.L.; Schneider, J.F.; Amini, H.; Hampel, F.; Gladysz, J.A. Wire like diplatinum, triplatinum, and tetraplatinum complexes featuring X[PtC≡CC≡CC≡CC≡C]mPtX segments; iterative syntheses and functionalization for measurements of single molecule properties. Dalton Trans. 2019, 48, 5800–5816. [Google Scholar] [CrossRef] [PubMed]
- Ballmann, S.; Hieringer, W.; Secker, D.; Zheng, Q.L.; Gladysz, J.A.; Gorling, A.; Weber, H.B. Molecular Wires in Single-Molecule Junctions: Charge Transport and Vibrational Excitations. ChemPhysChem 2010, 11, 2256–2260. [Google Scholar] [CrossRef] [PubMed]
- Szafert, S.; Paul, F.; Meyer, W.E.; Gladysz, J.A.; Lapinte, C. Synthesis and reactivity of new heterodinuclear iron/rhenium Cx complexes of the formula (η5-C5Me5)Re(NO)(PPh3)(C≡C)n(η2-dppe)Fe(η5-C5Me5) (n = 3, 4): Redox properties and a dicobalt hexacarbonyl adduct. C. R. Chim. 2008, 11, 693–701. [Google Scholar] [CrossRef]
- Meyer, W.E.; Amoroso, A.J.; Horn, C.R.; Jaeger, M.; Gladysz, J.A. Synthesis and Oxidation of Dirhenium C4, C6, and C8 Complexes of the Formula (η5-C5Me5)Re(NO)(PR3)(C≡C)n(R3P)(ON)Re(η5-C5Me5) (R = 4-C6H4R’, c-C6H11): In Search of Dications and Radical Cations with Enhanced Stabilities. Organometallics 2001, 20, 1115–1127. [Google Scholar] [CrossRef]
- Gendron, F.; Burgun, A.; Skelton, B.W.; White, A.H.; Roisnel, T.; Bruce, M.I.; Halet, J.F.; Lapinte, C.; Costuas, K. Iron and Ruthenium sigma-Polyynyls of the General Formula [{M(dppe)Cp*}-(C≡C)n-R]0/+ (M = Fe, Ru): An Experimental and Theoretical Investigation. Organometallics 2012, 31, 6796–6811. [Google Scholar] [CrossRef]
- Bruce, M.I.; Costuas, K.; Davin, T.; Ellis, B.G.; Halet, J.F.; Lapinte, C.; Low, P.J.; Smith, M.E.; Skelton, B.W.; Toupet, L.; et al. Iron versus ruthenium: Dramatic changes in electronic structure result from replacement of one Fe by Ru in [{Cp*(dppe)Fe}-CC-CC-{Fe(dppe)Cp*}]n+) (n = 0, 1, 2). Organometallics 2005, 24, 3864–3881. [Google Scholar] [CrossRef]
- Gückel, S.; Gluyas, J.B.G.; El-Tarhuni, S.; Sobolev, A.N.; Whiteley, M.W.; Halet, J.-F.; Lapinte, C.; Kaupp, M.; Low, P.J. Iron versus Ruthenium: Clarifying the Electronic Differences between Prototypical Mixed-Valence Organometallic Butadiyndiyl Bridged Molecular Wires. Organometallics 2018, 37, 1432–1445. [Google Scholar] [CrossRef]
- Jiao, H.J.; Costuas, K.; Gladysz, J.A.; Halet, J.F.; Guillemot, M.; Toupet, L.; Paul, F.; Lapinte, C. Bonding and electronic structure in consanguineous and conjugal iron and rhenium sp carbon chain complexes [MC4M’]n+: Computational analyses of the effect of the metal. J. Am. Chem. Soc. 2003, 125, 9511–9522. [Google Scholar] [CrossRef]
- Paul, F.; Meyer, W.E.; Toupet, L.; Jiao, H.J.; Gladysz, J.A.; Lapinte, C. A “conjugal” consanguineous family of butadiynediyl-derived complexes: Synthesis and electronic ground states of neutral, radical cationic, and dicationic iron/rhenium C4 species. J. Am. Chem. Soc. 2000, 122, 9405–9414. [Google Scholar] [CrossRef]
- Roberts, H.N.; Brown, N.J.; Edge, R.; Fitzgerald, E.C.; Ta, Y.T.; Collison, D.; Low, P.J.; Whiteley, M.W. Synthesis, Redox Chemistry, and Electronic Structure of the Butadiynyl and Hexatriynyl Complexes [Mo{(C≡C)nC≡CR}(L2) (η-C7H7)]z+ (n = 1, 2; z = 0, 1; R = SiMe3, H.; L2 = 2,2′-bipyridine, Ph2PCH2CH2PPh2). Organometallics 2012, 31, 6322–6335. [Google Scholar] [CrossRef]
- Brown, N.J.; Collison, D.; Edge, R.; Fitzgerald, E.C.; Low, P.J.; Helliwell, M.; Ta, Y.T.; Whiteley, M.W. Metal-stabilised diynyl radicals: Structure and reactivity of [Mo(C≡C-C≡CSiMe3)L2(η-C7H7)]•+ (L2 = 2,2′-bipyridine or dppe). Chem. Commun. 2010, 46, 2253–2255. [Google Scholar] [CrossRef] [PubMed]
- Zhuravlev, F.; Gladysz, J.A. Electronic structure and chain-length effects in diplatimun polyynediyl complexes trans, trans-[(X)(R3P)2Pt(C≡C)nPt(PR3)2(X)]: A computational investigation. Chem. Eur. J. 2004, 10, 6510–6522. [Google Scholar] [CrossRef] [PubMed]
- Bruce, M.I.; Cole, M.L.; Ellis, B.G.; Gaudio, M.; Nicholson, B.K.; Parker, C.R.; Skelton, B.W.; White, A.H. The series of carbon-chain complexes {Ru(dppe)Cp*}2{μ-(C≡C)x} (x = 4–8, 11): Synthesis, structures, properties and some reactions. Polyhedron 2015, 86, 43–56. [Google Scholar] [CrossRef]
- Johnson, T.R.; Walton, D.R.M. Silylation as a Protective Method in Acetylene Chemistry—Polyyne Chain Extensions Using Reagents, Et3Si(C≡C)mH (m = 1, 2, 4) in Mixed Oxidative Couplings. Tetrahedron 1972, 28, 5221–5236. [Google Scholar] [CrossRef]
- Eastmond, R.; Walton, D.R.M.; Johnson, T.R. Silylation as a Protective Method for Terminal Alkynes in Oxidative Couplings—General Synthesis of Parent Polyynes H(C≡C)nH (n = 4–10, 12). Tetrahedron 1972, 28, 4601–4616. [Google Scholar] [CrossRef]
- Chalifoux, W.A.; Tykwinski, R.R. Synthesis of polyynes to model the sp-carbon allotrope carbyne. Nat. Chem. 2010, 2, 967–971. [Google Scholar] [CrossRef] [PubMed]
- Burgun, A.; Gendron, F.; Schauer, P.A.; Skelton, B.W.; Low, P.J.; Costuas, K.; Halet, J.F.; Bruce, M.I.; Lapinte, C. Straightforward Access to Tetrametallic Complexes with a Square Array by Oxidative Dimerization of Organometallic Wires. Organometallics 2013, 32, 5015–5025. [Google Scholar] [CrossRef]
- Xia, H.P.; Jia, G.C. C5H5-bridged dimeric ruthenium complexes. Organometallics 1997, 16, 1–4. [Google Scholar] [CrossRef]
- Xia, H.P.; Yeung, R.C.Y.; Jia, G.C. Synthesis of symmetrical C5H5-bridged dimeric ruthenium complexes. Organometallics 1997, 16, 3557–3560. [Google Scholar] [CrossRef]
- Ribou, A.C.; Launay, J.P.; Sachtleben, M.L.; Li, H.; Spangler, C.W. Intervalence electron transfer in mixed valence diferrocenylpolyenes. Decay law of the metal-metal coupling with distance. Inorg. Chem. 1996, 35, 3735–3740. [Google Scholar] [CrossRef]
- Liu, S.H.; Chen, Y.H.; Wan, K.L.; Wen, T.B.; Zhou, Z.Y.; Lo, M.F.; Williams, I.D.; Jia, G.C. Synthesis and characterization of linear (CH)8-bridged bimetallic ruthenium complexes. Organometallics 2002, 21, 4984–4992. [Google Scholar] [CrossRef]
- Liu, S.H.; Xia, H.P.; Wen, T.B.; Zhou, Z.Y.; Jia, G.C. Synthesis and characterization of bimetallic ruthenium complexes with (CH)6 and related bridges. Organometallics 2003, 22, 737–743. [Google Scholar] [CrossRef]
- Yuan, P.; Liu, S.H.; Xiong, W.C.; Yin, J.; Yu, G.A.; Sung, H.Y.; Williams, I.D.; Jia, G.C. Synthesis and characterization of (CH=CH)3-bridged heterobimetallic ferrocene-ruthenium complexes. Organometallics 2005, 24, 1452–1457. [Google Scholar] [CrossRef]
- Sahnoune, H.; Mahias, V.; Halet, J.F.; Lapinte, C. 1,4-Dimethoxybutadienediyl-Bridged Diiron Compounds in Three Oxidation States: Evaluation of Delocalization Effects. Organometallics 2019, 38, 2724–2737. [Google Scholar] [CrossRef]
- Low, P.J. Twists and turns: Studies of the complexes and properties of bimetallic complexes featuring phenylene ethynylene and related bridging ligands. Coord. Chem. Rev. 2013, 257, 1507–1532. [Google Scholar] [CrossRef]
- Costuas, K.; Rigaut, S. Polynuclear carbon-rich organometallic complexes: Clarification of the role of the bridging ligand in the redox properties. Dalton Trans. 2011, 40, 5643–5658. [Google Scholar] [CrossRef] [PubMed]
- Haasler, M.; Maier, T.M.; Grotjahn, R.; Gückel, S.; Arbuznikov, A.V.; Kaupp, M. A Local Hybrid Functional with Wide Applicability and Good Balance between (De)Localization and Left–Right Correlation. J. Chem. Theory Comput. 2020, 16, 5645–5657. [Google Scholar] [CrossRef]
- Kaupp, M.; Karton, A.; Bischoff, F.A. [Al2O4]−, a Benchmark Gas-Phase Class II Mixed-Valence Radical Anion for the Evaluation of Quantum-Chemical Methods. J. Chem. Theory Comput. 2016, 12, 3796–3806. [Google Scholar] [CrossRef]
- Parthey, M.; Kaupp, M. Quantum-chemical insights into mixed-valence systems: Within and beyond the Robin-Day scheme. Chem. Soc. Rev. 2014, 43, 5067–5088. [Google Scholar] [CrossRef]
- Renz, M.; Kess, M.; Diedenhofen, M.; Klamt, A.; Kaupp, M. Reliable Quantum Chemical Prediction of the Localized/Delocalized Character of Organic Mixed-Valence Radical Anions. From Continuum Solvent Models to Direct-COSMO-RS. J. Chem. Theory Comput. 2012, 8, 4189–4203. [Google Scholar] [CrossRef]
- Renz, M.; Kaupp, M. Predicting the Localized/Delocalized Character of Mixed-Valence Diquinone Radical Anions. Toward the Right Answer for the Right Reason. J. Phys. Chem. A 2012, 116, 10629–10637. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Wierzbicki, I.; Cotic, A.; Cadranel, A. Photoinduced Intervalence Charge Transfers: Spectroscopic Tools to Study Fundamental Phenomena and Applications. ChemPhysChem 2022, 23, e202200384. [Google Scholar] [CrossRef] [PubMed]
- Pieslinger, G.E.; Ramirez-Wierzbicki, I.; Cadranel, A. The Excited-State Creutz-Taube Ion. Angew. Chem. Int. Ed. 2022, 61, e202211747. [Google Scholar] [CrossRef] [PubMed]
- Glover, S.D.; Goeltz, J.C.; Lear, B.J.; Kubiak, C.P. Inter- or intramolecular electron transfer between triruthenium clusters: We’ll cross that bridge when we come to it. Coord. Chem. Rev. 2010, 254, 331–345. [Google Scholar] [CrossRef]
- Safari, P.; Gückel, S.; Gluyas, J.B.G.; Moggach, S.A.; Kaupp, M.; Low, P.J. The Use of Bridging Ligand Substituents to Bias the Population of Localized and Delocalized Mixed-Valence Conformers in Solution. Chem. Eur. J. 2022, 28, e202200926. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.P.; Grotjahn, R.; Naher, M.; Ghazvini, S.; Mazzucato, D.M.; Korb, M.; Moggach, S.A.; Lambert, C.; Kaupp, M.; Low, P.J. Quantum Interference in Mixed-Valence Complexes: Tuning Electronic Coupling Through Substituent Effects. Angew. Chem. Int. Ed. 2022, 61, e202211000. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, M.J.; Frogley, B.J.; Hill, A.F.; Sharma, M.; Smith, M.K.; Ward, J.S. Hydrogenating an organometallic carbon chain: Buten-yn-diyl (CH=CHC≡C) as a missing link. Dalton Trans. 2019, 48, 16534–16554. [Google Scholar] [CrossRef]
- Wuttke, E.; Pevny, F.; Hervault, Y.M.; Norel, L.; Drescher, M.; Winter, R.F.; Rigaut, S. Fully Delocalized (Ethynyl)(vinyl)phenylene Bridged Triruthenium Complexes in up to Five Different Oxidation States. Inorg. Chem. 2012, 51, 1902–1915. [Google Scholar] [CrossRef]
- Olivier, C.; Costuas, K.; Choua, S.; Maurel, V.; Turek, P.; Saillard, J.Y.; Touchard, D.; Rigaut, S. “Chain-Like” Trimetallic Ruthenium Complexes with C7 Carbon-Rich Bridges: Experimental and Theoretical Investigations of Electronic Communication Tuning in Five Distinct Oxidation States. J. Am. Chem. Soc. 2010, 132, 5638–5651. [Google Scholar] [CrossRef]
- Vacher, A.; Benameur, A.; Ndiaye, C.M.; Touchard, D.; Rigaut, S. Linked C-7 Carbon-Rich Bridges: A New Dimension for Ruthenium Redox-Active Organometallics. Organometallics 2009, 28, 6096–6100. [Google Scholar] [CrossRef]
- Rigaut, S.; Olivier, C.; Costuas, K.; Choua, S.; Fadhel, O.; Massue, J.; Turek, P.; Saillard, J.Y.; Dixneuf, P.H.; Touchard, D. C7 and C9 carbon-rich bridges in diruthenium systems: Synthesis, spectroscopic, and theoretical investigations of different oxidation states. J. Am. Chem. Soc. 2006, 128, 5859–5876. [Google Scholar] [CrossRef] [PubMed]
- Rigaut, S.; Perruchon, J.; Guesmi, S.; Fave, C.; Touchard, D.; Dixneuf, P.H. Carbon-rich ruthenium complexes containing Bis(allenylidene) and mixed alkynyl-allenylidene bridges. Eur. J. Inorg. Chem. 2005, 2005, 447–460. [Google Scholar] [CrossRef]
- Hall, M.R.; Korb, M.; Moggach, S.A.; Low, P.J. Further Chemistry of Ruthenium Alkenyl Acetylide Complexes: Routes to Allenylidene Complexes via a Series of Electrophilic Addition Reactions. Organometallics 2020, 39, 2838–2853. [Google Scholar] [CrossRef]
- Hall, M.R.; Korb, M.; Moggach, S.A.; Low, P.J. Oxidative Coupling of Ruthenium Alkenyl Acetylide Complexes as a Route to Dinuclear Complexes Featuring Carbon-Rich Bridging Ligands. Organometallics 2022, 41, 2958–2973. [Google Scholar] [CrossRef]
- Li, Y.; Blacque, O.; Fox, T.; Luber, S.; Polit, W.; Winter, R.F.; Venkatesan, K.; Berke, H. Electronic communication in phosphine substituted bridged dirhenium complexes—Clarifying ambiguities raised by the redox non-innocence of the C4H2- and C4-bridges. Dalton Trans. 2016, 45, 5783–5799. [Google Scholar] [CrossRef] [PubMed]
- Bruce, M.I.; Ellis, B.G.; Low, P.J.; Skelton, B.W.; White, A.H. Syntheses, structures, and spectro-electrochemistry of {Cp*(PP)Ru}C≡CC≡C{Ru(PP)Cp*} (PP = dppm, dppe) and their mono- and dications. Organometallics 2003, 22, 3184–3198. [Google Scholar] [CrossRef]
- Hall, M.R.; Moggach, S.A.; Low, P.J. Syntheses and Structures of trans-bis(Alkenylacetylide) Ruthenium Complexes. Chem. Asian, J. 2021, 16, 3385–3403. [Google Scholar] [CrossRef]
- Kaupp, M.; Renz, M.; Parthey, M.; Stolte, M.; Wurthner, F.; Lambert, C. Computational and spectroscopic studies of organic mixed-valence compounds: Where is the charge? Phys. Chem. Chem. Phys. 2011, 13, 16973–16986. [Google Scholar] [CrossRef]
- Ladjarafi, A.; Costuas, K.; Meghezzi, H.; Halet, J.F. Electronic structure of modelized vs. real carbon-chain containing organometallic dinuclear complexes: Similarities and differences. J. Mol. Model. 2015, 21, 71. [Google Scholar] [CrossRef]
- Scott, A.P.; Radom, L. Harmonic vibrational frequencies: An evaluation of Hartree-Fock, Moller-Plesset, quadratic configuration interaction, density functional theory, and semiempirical scale factors. J. Phys. Chem. 1996, 100, 16502–16513. [Google Scholar] [CrossRef]
- Bruce, M.I.; Costuas, K.; Ellis, B.G.; Halet, J.F.; Low, P.J.; Moubaraki, B.; Murray, K.S.; Ouddai, N.; Perkins, G.J.; Skelton, B.W.; et al. Redox-active complexes containing group 8 metal centers linked by C2 bridges. Organometallics 2007, 26, 3735–3745. [Google Scholar] [CrossRef]
- Bruce, M.I.; Low, P.J.; Costuas, K.; Halet, J.F.; Best, S.P.; Heath, G.A. Oxidation chemistry of metal-bonded C4 chains: A combined chemical, spectroelectrochemical, and computational study. J. Am. Chem. Soc. 2000, 122, 1949–1962. [Google Scholar] [CrossRef]
- Parthey, M.; Gluyas, J.B.G.; Schauer, P.A.; Yufit, D.S.; Howard, J.A.K.; Kaupp, M.; Low, P.J. Refining the Interpretation of Near-Infrared Band Shapes in a Polyynediyl Molecular Wire. Chem. Eur. J. 2013, 19, 9780–9784. [Google Scholar] [CrossRef] [PubMed]
- Bruce, M.I.; Hall, B.C.; Low, P.J.; Smith, M.E.; Skelton, B.W.; White, A.H. Heterometallic complexes containing C4 chains. X-ray structures of {Cp(OC)3W}C≡CC≡C{Ir(CO)(PPh3)2(O2)} and cis-Pt{C≡C[W(CO)3Cp]}2(PEt3)2. Inorg Chim Acta 2000, 300, 633–644. [Google Scholar] [CrossRef]
- Brady, M.; Weng, W.Q.; Zhou, Y.L.; Seyler, J.W.; Amoroso, A.J.; Arif, A.M.; Bohme, M.; Frenking, G.; Gladysz, J.A. Consanguineous families of coordinated carbon: A ReC4Re assembly that is isolable in three oxidation states, including crystallographically characterized ReC≡CC≡CRe and Re+=C=C=C=C=Re+ adducts and a radical cation in which charge is delocalized between rhenium termini. J. Am. Chem. Soc. 1997, 119, 775–788. [Google Scholar]
- Weng, W.G.; Bartik, T.; Gladysz, J.A. Towards One-Dimensional Carbon Wires Connecting Single Metal Centers—A Cumulenic C5 Chain That Mediates Charge-Transfer between Rhenium and Manganese Termini. Angew. Chem. Int. Ed. 1994, 33, 2199–2202. [Google Scholar] [CrossRef]
- Seyler, J.W.; Weng, W.Q.; Zhou, Y.L.; Gladysz, J.A. An Isolable Organometallic Cation-Radical in Which a C4 Chain Conducts Charge between 2 Chiral and Configurationally Stable Rhenium Termini. Organometallics 1993, 12, 3802–3804. [Google Scholar] [CrossRef]
- Lear, B.J.; Chisholm, M.H. Oxalate Bridged MM (MM = Mo2, MoW, and W2) Quadruply Bonded Complexes as Test Beds for Current Mixed Valence Theory: Looking beyond the Intervalence Charge Transfer Transition. Inorg. Chem. 2009, 48, 10954–10971. [Google Scholar] [CrossRef]
- Szeghalmi, A.V.; Erdmann, M.; Engel, V.; Schmitt, M.; Amthor, S.; Kriegisch, V.; Noll, G.; Stahl, R.; Lambert, C.; Leusser, D.; et al. How delocalized is N,N,N′,N′-tetraphenylphianylenediamine radical cation? An experimental and theoretical study on the electronic and molecular structure. J. Am. Chem. Soc. 2004, 126, 7834–7845. [Google Scholar] [CrossRef]
- Badger, B.; Brocklehurst, B. Absorption spectra of dimer cations. Part 4—Theoretical considerations and dimer structure. Trans. Faraday Soc. 1970, 66, 2939–2947. [Google Scholar] [CrossRef]
- Connelly, N.G.; Geiger, W.E. Chemical redox agents for organometallic chemistry. Chem. Rev. 1996, 96, 877–910. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Puschmann, H.; Dolomanov, O. Olex(2): A Comprehensive Molecular Graphics Tool for Small-Molecule Structures. Acta Crystallogr. A 2006, 62, S246. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision, A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- O’Boyle, N.M.; Tenderholt, A.L.; Langner, K.M. cclib: A library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef]
- Wadt, W.R.; Hay, P.J. Abinitio Effective Core Potentials for Molecular Calculations—Potentials for Main Group Elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Abinitio Effective Core Potentials for Molecular Calculations—Potentials for K to Au Including the Outermost Core Orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Abinitio Effective Core Potentials for Molecular Calculations—Potentials for the Transition-Metal Atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Petersson, G.A.; Allaham, M.A. A Complete Basis Set Model Chemistry. 2. Open-Shell Systems and the Total Energies of the 1st-Row Atoms. J. Chem. Phys. 1991, 94, 6081–6090. [Google Scholar] [CrossRef]
- Petersson, G.A.; Bennett, A.; Tensfeldt, T.G.; Allaham, M.A.; Shirley, W.A.; Mantzaris, J. A Complete Basis Set Model Chemistry.1. The Total Energies of Closed-Shell Atoms and Hydrides of the 1st-Row Elements. J. Chem. Phys. 1988, 89, 2193–2218. [Google Scholar] [CrossRef]
- Klamt, A. The COSMO and COSMO-RS solvation models. Wires Comput. Mol. Sci. 2011, 1, 699–709. [Google Scholar] [CrossRef]
- Klamt, A. Calculation of UV/Vis spectra in solution. J. Phys. Chem. 1996, 100, 3349–3353. [Google Scholar] [CrossRef]
- Martin, R.L. Natural transition orbitals. J. Chem. Phys. 2003, 118, 4775–4777. [Google Scholar] [CrossRef]
- Krejcik, M.; Danek, M.; Hartl, F. Simple Construction of an Infrared Optically Transparent Thin-Layer Electrochemical-Cell—Applications to the Redox Reactions of Ferrocene, Mn2(CO)10 and Mn(CO)3(3,5-Di-Tert-Butyl-Catecholate)−. J. Electroanal. Chem. 1991, 317, 179–187. [Google Scholar] [CrossRef]
Figure 1.
Compounds discussed in this study.

Scheme 1.
Formation of ethane-bridged dinuclear bis(allenylidene) complexes [3,4]2+ and subsequent octa-3,5-diene-1,7-diyndiyl-bridged bimetallic species 5,6. a Previously reported [64]. b Generated as a component of a crude product mixture that could not be purified.
Scheme 1.
Formation of ethane-bridged dinuclear bis(allenylidene) complexes [3,4]2+ and subsequent octa-3,5-diene-1,7-diyndiyl-bridged bimetallic species 5,6. a Previously reported [64]. b Generated as a component of a crude product mixture that could not be purified.

Scheme 2.
A schematic showing the synthesis of [6a][PF6]n (n = 1, 2) via [4a][PF6]2; note that the additional equivalents of [FeCp2]PF6 added at the commencement of the reaction drive the oxidation of 6a to [6a][PF6]n (n = 1, 2).
Scheme 2.
A schematic showing the synthesis of [6a][PF6]n (n = 1, 2) via [4a][PF6]2; note that the additional equivalents of [FeCp2]PF6 added at the commencement of the reaction drive the oxidation of 6a to [6a][PF6]n (n = 1, 2).

Figure 2.
Molecular structure of [6a]PF6 showing the atom labelling scheme. Selected hydrogen atoms are omitted and dppe ligands drawn in wireframe for clarity. Thermal ellipsoids are shown at the 50% level.
Figure 2.
Molecular structure of [6a]PF6 showing the atom labelling scheme. Selected hydrogen atoms are omitted and dppe ligands drawn in wireframe for clarity. Thermal ellipsoids are shown at the 50% level.

Scheme 3.
Alkynylketone complexes 2aox and 2box generated from aerial oxidation of alkenylacetylide complexes 2a and 2b.
Scheme 3.
Alkynylketone complexes 2aox and 2box generated from aerial oxidation of alkenylacetylide complexes 2a and 2b.

Figure 3.
Molecular structures of (a) 2aox and (b) 2box (right) showing the atom labelling scheme. Hydrogen atoms are omitted and dppe ligands drawn in wireframe for clarity. Thermal ellipsoids are shown at the 50% level.
Figure 3.
Molecular structures of (a) 2aox and (b) 2box (right) showing the atom labelling scheme. Hydrogen atoms are omitted and dppe ligands drawn in wireframe for clarity. Thermal ellipsoids are shown at the 50% level.

Figure 4.
The computational model compounds 1a† and 5a†, and the associated atom labelling scheme.

Figure 5.
Plots (contours at ± 0.02 (e/bohr3)1/2) of the frontier molecular (spin) orbitals of [1a†]n+ ((a) n = 0; (b) n = 1), and spin density plot of [1a†]+ (c).
Figure 5.
Plots (contours at ± 0.02 (e/bohr3)1/2) of the frontier molecular (spin) orbitals of [1a†]n+ ((a) n = 0; (b) n = 1), and spin density plot of [1a†]+ (c).

Figure 6.
Plots (contours at ± 0.02 (e/bohr3)1/2) of the frontier molecular (spin) orbitals of [5a†]n+ ((a) n = 0; (b) n = 1; (c) n = 2), and spin density plot of [5a†]+ (d).
Figure 6.
Plots (contours at ± 0.02 (e/bohr3)1/2) of the frontier molecular (spin) orbitals of [5a†]n+ ((a) n = 0; (b) n = 1; (c) n = 2), and spin density plot of [5a†]+ (d).

Figure 7.
TD-DFT calculated UV-Vis-NIR spectrum of 5a and associated transitions, with authentic SEC-UV-Vis-NIR generated spectrum overlay (CH2Cl2/0.1 M NBu4PF6).
Figure 7.
TD-DFT calculated UV-Vis-NIR spectrum of 5a and associated transitions, with authentic SEC-UV-Vis-NIR generated spectrum overlay (CH2Cl2/0.1 M NBu4PF6).

Figure 8.
Mapped isosurfaces showing particle and hole wavefunctions for NTO pairs for selected excited states of 5a† (oscillator strength given in italic font).
Figure 8.
Mapped isosurfaces showing particle and hole wavefunctions for NTO pairs for selected excited states of 5a† (oscillator strength given in italic font).

Figure 9.
The TD-DFT calculated UV-Vis-NIR spectrum of [5a†]+ and associated transitions, overlaid on the authentic spectroelectrochemically generated UV-Vis-NIR spectrum of [5a]+ (CH2Cl2/0.1 M NBu4PF6).
Figure 9.
The TD-DFT calculated UV-Vis-NIR spectrum of [5a†]+ and associated transitions, overlaid on the authentic spectroelectrochemically generated UV-Vis-NIR spectrum of [5a]+ (CH2Cl2/0.1 M NBu4PF6).

Figure 10.
Mapped isosurfaces showing particle and hole wavefunctions for NTO pairs for selected excited states of [5a]+ (oscillator strength given in italic font).
Figure 10.
Mapped isosurfaces showing particle and hole wavefunctions for NTO pairs for selected excited states of [5a]+ (oscillator strength given in italic font).

Figure 11.
The TD-DFT calculated UV-Vis-NIR spectrum of [5a†]2+ and associated transitions, overlaid on the authentic spectroelectrochemically generated UV-Vis-NIR spectrum of [5a]2+ (CH2Cl2/0.1 M NBu4PF6).
Figure 11.
The TD-DFT calculated UV-Vis-NIR spectrum of [5a†]2+ and associated transitions, overlaid on the authentic spectroelectrochemically generated UV-Vis-NIR spectrum of [5a]2+ (CH2Cl2/0.1 M NBu4PF6).

Figure 12.
Mapped isosurfaces showing particle and hole wavefunctions for NTO pairs for selected excited states of [5a][PF6]2 (oscillator strength given in italic font).
Figure 12.
Mapped isosurfaces showing particle and hole wavefunctions for NTO pairs for selected excited states of [5a][PF6]2 (oscillator strength given in italic font).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Selected bond lengths (Å) and angles (°) from the crystallographically determined structure of [6a]PF6.
Table 1.
Selected bond lengths (Å) and angles (°) from the crystallographically determined structure of [6a]PF6.
| [6a]PF6 | |
|---|---|
| Ru1-P1 | 2.3729 (11) |
| Ru1-P2 | 2.3946 (11) |
| Ru1-P3 | 2.3803 (11) |
| Ru1-P4 | 2.3934 (11) |
| Ru1-Cl1 | 2.4591 (11) |
| Ru1-C1 | 1.958 (4) |
| C1-C2 | 1.217 (6) |
| C2-C3 | 1.400 (6) |
| C3-C4 | 1.401 (6) |
| C4-C4A | 1.403 (9) |
| C3-C5 | 1.490 (6) |
| Ru1-C1-C2 | 175.1 (4) |
| C1-C2-C3 | 175.4 (4) |
| C2-C3-C4 | 121.1 (4) |
| C3-C4-C4A | 124.3 (5) |
| C4-C3-C5 | 119.4 (4) |
Table 2.
Selected bond lengths (Å) and angles (°) from the crystallographically determined structures of 2aox and 2box.
Table 2.
Selected bond lengths (Å) and angles (°) from the crystallographically determined structures of 2aox and 2box.
| 2aox | 2box | |
|---|---|---|
| Ru1-P1 | 2.3884(5) | 2.3944(15) |
| Ru1-P2 | 2.3979(5) | 2.3529(14) |
| Ru1-P3 | 2.3407(5) | 2.3860(15) |
| Ru1-P4 | 2.3612(5) | 2.3856(15) |
| Ru1-Cl1 | 2.5067(5) | 2.5053(15) |
| Ru1-C1 | 1.986(2) | 1.972(6) |
| C1-C2 | 1.196(3) | 1.218(9) |
| C2-C3 | 1.428(3) | 1.424(9) |
| C3-C4 | 1.495(3) | 1.497(9) |
| O1-C3 | 1.245(3) | 1.240(8) |
| Ru1-C1-C2 | 175.58(17) | 178.2(5) |
| C1-C2-C3 | 167.6(2) | 166.0(7) |
| C2-C3-C4 | 119.45(18) | 119.3(6) |
Table 3.
Selected bond lengths (Å) and angles (°) from the optimised structures of [1a†]n+ (n = 0, 1) and [5a†]n+ (n = 1, 2) (labelling as per Figure 4).
Table 3.
Selected bond lengths (Å) and angles (°) from the optimised structures of [1a†]n+ (n = 0, 1) and [5a†]n+ (n = 1, 2) (labelling as per Figure 4).
| 1a† | [1a†]+ | 5a† | [5a†]+ | [5a†]2+ | |
|---|---|---|---|---|---|
| Ru1-P1 | 2.3137 | 2.3572 | 2.3226/2.3136 | 2.3260/2.3253 | 2.3446/2.3434 |
| Ru1-P2 | 2.3084 | 2.3393 | 2.3071/2.3075 | 2.3166/2.3176 | 2.3292/2.3287 |
| Ru1-C1 | 2.0293 | 1.9633 | 2.0278/2.0286 | 1.9661/1.9687 | 1.9114/1.9117 |
| C1-C2 | 1.2283 | 1.2372 | 1.2301/2.0286 | 1.2437/1.2433 | 1.2591/1.2587 |
| C2-C3 | 1.4307 | 1.4195 | 1.4250/1.4251 | 1.3884/1.3891 | 1.3565/1.3571 |
| C3-C4 | 1.3481 | 1.3546 | 1.3699/1.3702 | 1.4069/1.4065 | 1.4539/1.4538 |
| C4-C4′ | 1.4289 | 1.3895 | 1.3532 | ||
| Ru1-C1-C2 | 176.18 | 175.04 | 176.71/175.84 | 175.64/176.61 | 175.77/175.31 |
| C1-C2-C3 | 178.99 | 179.17 | 178.77/178.59 | 177.33/178.87 | 178.22/177.15 |
| C2-C3-C4 | 121.91 | 119.23 | 122.46/122.20 | 121.74/121.81 | 120.65/120.55 |
| C3-C4-C4′ | 125.01/122.20 | 124.83/124.90 | 124.41/124.23 |
Table 4.
Absorption maxima (λ) for 5a–d and [5a]n+ (n = 1, 2)/nm.
| n-Hexane | Cyclohexane | THF | CH2Cl2 | PhCN | |
|---|---|---|---|---|---|
| 5a | 476 | 496 | 511 | 513 | 520 |
| 5b | 490 | 489 | 521 | 533 | 533 |
| 5c | 516 | 525 | 555 | 562 | 549 |
| 5d | 630 | 620 | 685 | 707 | 720 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hall, M.R.; Moggach, S.A.; Low, P.J. Probing the Electronic Structure of Dinuclear Carbon-Rich Complexes Containing an Octa-3,5-diene-1,7-diyndiyl Bridging Ligand. Inorganics 2024, 12, 20. https://doi.org/10.3390/inorganics12010020
AMA Style
Hall MR, Moggach SA, Low PJ. Probing the Electronic Structure of Dinuclear Carbon-Rich Complexes Containing an Octa-3,5-diene-1,7-diyndiyl Bridging Ligand. Inorganics. 2024; 12(1):20. https://doi.org/10.3390/inorganics12010020
Chicago/Turabian StyleHall, Michael R., Stephen A. Moggach, and Paul J. Low. 2024. "Probing the Electronic Structure of Dinuclear Carbon-Rich Complexes Containing an Octa-3,5-diene-1,7-diyndiyl Bridging Ligand" Inorganics 12, no. 1: 20. https://doi.org/10.3390/inorganics12010020
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.