
In Situ Synthesis of Hexadentate Cyclometalated Ir(III) Complexes as Photocatalysts for the Oxidation of Sulfides into Sulfoxides in Water
1
MOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Chemistry, Sun Yat-sen University, Guangzhou 510275, China
2
School of Chemistry and Materials Science, Guangdong University of Education, Guangzhou 510303, China
*
Authors to whom correspondence should be addressed.
Inorganics 2024, 12(3), 73; https://doi.org/10.3390/inorganics12030073
Submission received: 9 February 2024
/
Revised: 21 February 2024
/
Accepted: 26 February 2024
/
Published: 28 February 2024
(This article belongs to the Section Organometallic Chemistry)
Abstract
:The aerobic photooxidation of sulfides into sulfoxides in eco-friendly solvents, notably water, at room temperature, represents a significant interest in the domain of synthetic chemistry. This study introduces four highly stable hexadentate Ir(III) complexes: [Ir(fpqen)](PF6) (1), [Ir(btqen)](PF6) (2), [Ir(bmpqen)](PF6) (3), and [Ir(bnqen](PF6) (4) (where bfpqen is N,N′-bis(2-(4-fluorophenyl)quinolin-8-yl)ethane-1,2-diamine, btqen is N,N′-bis(2-(4-tolyl)quinolin-8-yl)ethane-1,2-diamine, bmpqen is N,N′-bis(2-(4-methoxyphenyl)quinolin-8-yl)ethane-1,2-diamine, and bnqen is N,N′-bis(2-naphthylquinolin-8-yl)ethane-1,2-diamine). These complexes were synthesized utilizing an in situ inter-ligand C-N cross-coupling photoreaction of the precursors [Ir(L)2(en)](PF6) (L is 2-(4-fluorophenyl)quinoline, (2-(4-tolyl)quinoline, 2-(4-methoxyphenyl)quinoline or 2-naphthylquinoline, and en is 1,2-diamine) under benign conditions. This methodology furnishes a valuable and complementary approach for the in situ generation of multidentate complexes through a post-coordination inter-ligand-coupling strategy under mild conditions. Moreover, these hexadentate Ir(III) complexes exhibit pronounced catalytic activity and chemo-selectivity toward the aerobic photooxidations of sulfides into sulfoxides in aqueous media at room temperature, offering a new avenue for the sustainable synthesis of sulfoxides.
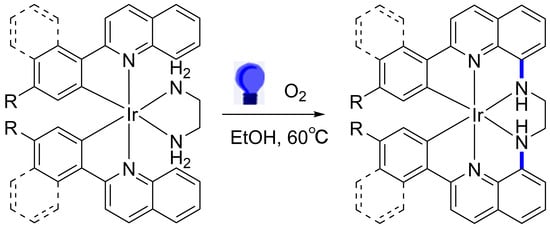
1. Introduction
Cyclometalated Ir(III) complexes have emerged as pivotal components in optoelectronic devices, photocatalysts, luminescent sensors for metal ions, and biological labels. They provide exceptional phosphorescence quantum yield, substantial Stoke shifts, and formidable chemical and thermal photostability [1,2,3]. Moreover, the employment of multidentate ligands has significantly augmented the phosphorescence properties of Ir(III) complexes by reinforcing metal-ligand bond strength and facilitating thermally accessible metal-centered d-d transitions [4,5]. A prominent synthesis strategy for multidentate ligands involves the conjugation of two bidentate ligands through a bridging entity. Notably, Lee et al. reported an enhancement in phosphorescence efficiency upon integrating ortho-carboranes at the 5 positions of Ir(III) ppy complexes (where ppy is 2-phenylpyridine) [6]. In a remarkable instance, an Ir(III) complex endowed with a C,C,C,C-tetradentate ligand, bridging two bis(1-phenylimidazolium) ligands via a butylene bridge, showcased blue-green emissions with a quantum yield of 0.96 [7]. Nonetheless, the synthesis of multidentate ligand entails a complex and extended procedure. Recently, in situ synthesis in the presence of a metal ion template has gained considerable attention [8,9,10], where ligands, upon coordination to the metal ion, undergo activation, facilitating mutual cross-coupling [11,12,13]. This coordination-induced pre-arrangement of ligands around the metal center benefits the subsequent coupling reaction in terms of regio- and stereoselectivity [14,15], thus presenting in situ synthesis as a viable route for crafting new multidentate complexes post-coordination.
Sulfoxide compounds hold critical importance in organic synthesis and the pharmaceutical industry [16,17]. The pursuit of green synthetic routes for sulfoxides has intensified, with aerobic photooxidation of sulfides to sulfoxides employing visible light and oxygen emerging as a particularly eco-friendly methodology [18,19]. However, the sustainability and efficiency of this process are hampered by the photodegradation of photosensitizers. Thus, the development of a durable and recyclable photosensitizer is crucial. Moreover, although water is an ideal solvent for green chemistry, catalytic oxidation of sulfides into sulfoxides in aqueous media faces challenges due to poor catalyst solubility and sulfide reactivity issues [20,21,22,23].
Building on our group’s previous work, we have established a robust protocol for the amination of coordinated 2-phenylquinoline (pq) complexes at the C8 position via in situ inter-ligand C-N cross-coupling in the presence of O2 under visible light irradiation [24,25]. Mechanistic studies suggest that metal aminyl radicals are instrumental in new C-N bond formation [26,27], paving the way for the in situ synthesis of new multidentate complexes and inspiring the development of new stable Ir(III) photosensitizers under mild conditions. In this paper, we report the synthesis of four new hexadentate cyclometalated Ir(III) complexes [Ir(bfpqen)](PF6) (1), [Ir(btqen)](PF6) (2), [Ir(bmpqen)](PF6) (3), and [Ir(bnqen)](PF6) (4) (where bfpqen is N,N′-bis(2-(4-fluorophenyl)quinolin-8-yl)ethane-1,2-diamine, btqen is N,N′-bis(2-(4-tolyl)quinolin-8-yl)ethane-1,2-diamine, bmpqen is N,N′-bis(2-(4-methoxyphenyl)quinolin-8-yl)ethane-1,2-diamine, and bnqen is N,N′-bis(2-naphthylquinolin-8-yl)ethane-1,2-diamine) through in situ inter-ligand C-N cross-coupling under benign conditions. This method provides a straightforward and complementary approach for the in situ generation of hexadentate complexes via a post-coordination interligand-coupling strategy. Additionally, we explored the selective photooxidations of sulfides into sulfoxides using these complexes as photosensitizers under blue-light irradiation in an O2 atmosphere at room temperature in aqueous media.
2. Results and Discussion
2.1. Synthesis of Hexadentate Ir(III) Complexes
The synthesis pathway of the hexadentate Ir(III) complexes is summarized in Scheme 1. Initially, the 2-phenylquinoline (pq) ligands were prepared through the Suzuki-Miyaura cross-coupling reaction, employing phenylboronic acid and 2-bromoquinoline as reactants in the presence of Pd catalysts. This step was followed by the formation of dimeric Ir(III) complexes, achieving by reacting the pq ligands with IrCl3 in 2-methoxyethanol solution at 120 °C for 24 h, adhering to a previously published protocol [28,29]. The precursors [Ir(fpq)2(en)](PF6) (1a) (fpq is 2-(4-fluorophenyl)quinoline and en is 1,2-diamine), [Ir(tq)2(en)](PF6) (2a) (tq is 2-(4-tolyl)quinoline), [Ir(mpq)2(en)](PF6) (3a) (mpq is 2-(4-methoxyphenyl)quinoline), and [Ir(nq)2(en)](PF6) (4a) (nq is 2-naphthylquinoline) were synthesized from these dimeric complexes upon the introduction of the diamine ligand under mild conditions, yielding good to excellent yields.
The structural and compositional integrity of these complexes was thoroughly confirmed through elemental analysis, NMR, and high-resolution mass spectrometry (HRMS) spectrometry. Detailed spectroscopic data are presented in the Supporting Information. The 1H NMR spectra showcased distinct resonance peaks associated with the coordinated en, specifically between 3.8~4.1 ppm and 2.52~2.7 ppm (attributed to the NH2 group), alongside another set of peaks between 2.4~2.57 ppm and 1.7~1.98 ppm (assigned to the CH2 group). Furthermore, the 13C NMR spectra revealed resonance peaks at 43.45, 43.41, 43.45, and 43.50 ppm for the complexes 1a, 2a, 3a, and 4a, respectively, which is consistent with the spectral characteristics previously reported for the [Ir(pq)2(en)](PF6) complex [26].
To elucidate the structural configuration, single-crystal X-ray diffraction analyses were performed on 1a and 3a, with detailed crystallographic data. Figure 1 illustrates the molecular structures, where each Ir(III) ion is coordinated by an en ligand alongside two pq ligands, culminating in a distorted octahedral geometry. A notable observation is the elongation of the Ir-N bond lengths within the en ligand, ranging from 2.217(4) Å to 2.240(4) Å, in contrast to the more compact Ir-N bond lengths observed in the pq units, which span from 2.078(3) Å to 2.097(3) Å. This elongation is attributed to the trans influence exerted by the Ir-C bonds. These structural findings align closely with those previously reported for the [Ir(pq)2(en)](PF6) complex [26].
The photocatalytic reactions of the diamine Ir(III) complexes were carried out employing our established in situ inter-ligand C-N cross-coupling protocol under visible light irradiation [24,25], as delineated in Scheme 1. Complex 1a was utilized as a benchmark to refine the photocatalytic conditions and monitor spectral transformations. Upon irradiating an ethanol solution of 1a with visible light at 60 °C for 85 h in an oxygen environment, complete consumption of the starting material 1a was observed, leading to the formation of a new C-N coupling hexadentate product 1, isolated in an impressive yield of 92% post-purification. HRMS of product 1 exhibited a molecular ion peak at m/z 693.14117, congruent with the expected isotope pattern for C32H22F2IrN4 (m/z = 693.14363, [M-PF6]+), signifying a reduction of four hydrogen atoms relative to 1a (m/z = 697.17149, [M-PF6]+), thus indicating the formation of two new C-N bonds during the photoreaction. Further spectral analysis revealed significant shifts in the 1H NMR spectra as shown in Figure 2. Notably, the characteristic resonance of H8 proton of the fpq ligand at 7.77 ppm in 1a vanished in 1. Additionally, resonances attributed to the NH2 (originally at 3.99 and 2.66 ppm) and CH2 (2.50 and 1.90 ppm) groups in the en ligand of 1a shifted to 6.61 ppm and 3.31 and 3.02 ppm in 1, respectively. The 13C NMR spectra of 1 showed a notable peak shift from 43.45 ppm in 1a to 53.94 ppm in 1, underscoring the transformation of the en ligand (detailed spectroscopic data available in the Supporting Information). These findings collectively underscore the in situ C-N cross-coupling of the en ligand with the fpq ligands under visible light irradiation, culminating in the formation of the hexadentate ligand bfpqen. Notably, this method represents the inaugural instance of employing in situ C-N cross-coupling for the synthesis of hexadentate cyclometalated complexes. Following this protocol, complexes 2, 3, and 4 were synthesized from substrates 2a, 3a, and 4a, respectively, under identical conditions, yielding satisfactory results of 88%, 92%, and 85%, respectively. The structural and compositional veracity of these complexes was rigorously confirmed by elemental analysis, HRMS, and NMR spectroscopy, as documented in the Supporting Information. This strategy delineates a practical and methodical protocol for the generation of hexadentate cyclometalated Ir(III) complexes via in situ post-coordinated inter-ligand C-N cross-coupling reactions under benign conditions.
The molecular structure of the photoreaction product 3 was definitively characterized through X-ray diffraction analysis, with crystallographic details. The complex was determined to crystallize in the P21/n space group. As depicted in Figure 3, the Ir(III) ion in 3 is coordinated by a hexadentate ligand comprising four nitrogen atoms and two carbon atoms, arranging in a distorted octahedral geometry. This arrangement confirms the formation of two new C-N bonds, C2-N3 (1.469(7) Å) and N4-C17 (1.480(7) Å), through inter-ligand C-N coupling between mpq and en ligands, successfully synthesizing the hexadentate ligand bmpqen. A comparative analysis of the structural parameters between 3 and its precursor 3a highlighted notable changes in the coordination geometry. Specifically, the angles N3-Ir1-N4 and N1-Ir1-N2 in 3 expanded to 79.63(18)° and 177.65(19)°, respectively, from the 77.45(13)° and 173.98(12)° observed in 3a. These modifications in bond angles reflect the structural adjustments ensuing from the formation of the hexadentate ligand bmpqen, with other geometric parameters remaining largely consistent.
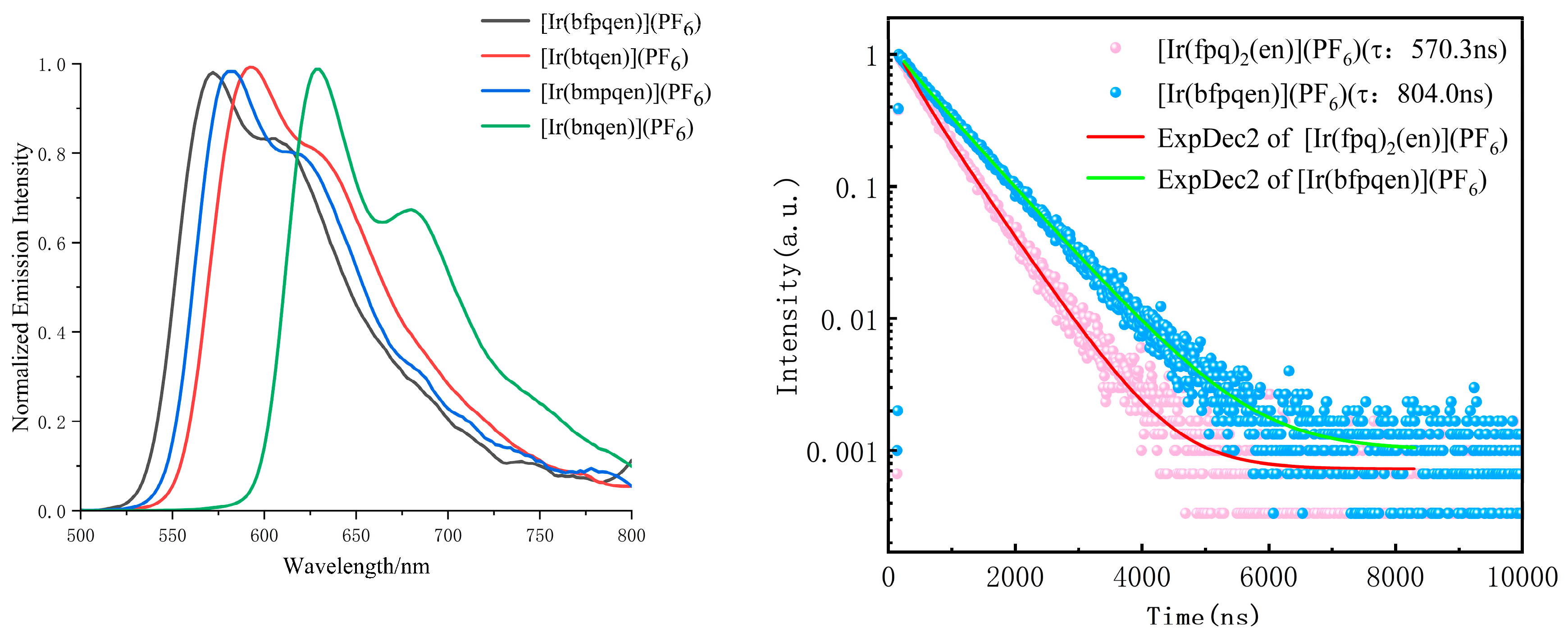
The absorption spectra of the cyclometalated Ir(III) complexes 1, 2, 3, and 4 were investigated in MeOH solutions, corroborating with prior studies on analogous Ir(III)–pq complexes [30]. Detailed emission spectra, lifetimes, and quantum yields of these complexes were also determined in a MeOH solution at room temperature (see the Supporting Information). Figure 4 delineates that complex 2 exhibits an orange-red emission, with a principal emission peak at approximately 592 nm, accompanied by a shoulder at 634 nm. This emission profile mirrors that of the [Ir(bpqen)](PF6) complex (bpqen is N,N′-bis(2-phenylquinolin-8-yl)ethane-1,2-diamine), attributable to a 3LC (π–π*) and 3MLCT transitions [31,32], underscoring that the methyl substitution does not alter the maximum emission wavelength. Contrastingly, the introduction of electron-withdrawing fluorine and electron-donating methoxy groups resulted in blue shifts of the emission maximum to 572 and 582 nm, respectively, indicative of the strong electronic effect of these substituents. Conversely, incorporation of the naphthyl group, in lieu of a phenyl ring, significantly red-shifted the emission peak to 630 nm, with an additional shoulder at 680 nm. This shift is attributed to the conjugate effect of the naphthyl group, enhancing π electrons delocalization. These results indicate that changing the substituent in phenyl is an important strategy to regulate the fluorescence and photoelectric properties of molecules. A notable increase in photoluminescence lifetimes was observed for the hexadentate complexes, with values of 798 ns for 1, 513 ns for 2, 476 ns for 3, and 420 ns for 4, significantly surpassing those of their precursors (575 ns, 269 ns, 327 ns, and 362 ns, respectively) in argon-saturated methanol. This enhancement is likely due to the increased rigidity conferred by the hexadentate coordination sphere. Additionally, the photoluminescence quantum yields of the complexes 1, 2, 3, and 4 were measured at 9.4%, 5.0%, 8.1%, and 4.6%, respectively, employing an absolute measurement method with an integrating sphere in argon-saturated methanol at room temperature.
2.2. Photocatalysts for the Oxidation of Sulfides into Sulfoxides
Leveraging the robust stability of hexadentate cyclometalated Ir(III) complexes, we explored their efficacy as photosensitizers in the photooxidation of sulfide into sulfoxide. Methyl phenyl sulfide (5a) served as the model substrate for optimizing the reaction conditions, which included an O2 atmosphere, water as the solvent, and blue-light irradiation at ambient temperature over 14 h. The photocatalytic reactions proceeded with remarkable efficiency, yielding methyl phenyl sulfoxide (6a) in excellent yields of 97%, 92%, and 100% for complexes 1, 3, and 4 as catalysts (1 mol%), respectively (see entries 1–3 in Table 1). Notably, the photooxidation reactions exhibited high chemo-selectivity without the detection of by-products. For comparative analysis, complex 4a, under identical conditions, produced 6a with a yield of 79% (entry 4 in Table 1), underscoring the superior stability and reactivity of the hexadentate structure. To elucidate the underlying photooxidation mechanism, control experiments with 5a and catalyst 4 were conducted, confirming the indispensability of the photocatalyst, light, and oxygen for successful sulfide to sulfoxide conversion (entries 5–7 in Table 1). The involvement of reactive oxygen species (ROS) was investigated to determine the dominant oxidative intermediates. The addition of 2 equiv of 1,4-diazabicyclo [2.2.2]octane (DABCO), a known 1O2 scavenger, significantly reduced the yield to 24%, implicating 1O2 as the primary ROS. Conversely, the presence of benzoquinone (BQ), isopropyl alcohol (iPrOH), and 1,4-dimethoxybenzene (DMB), scavengers for O2•–, •OH, and sulfide radical cation, respectively, had minimal effects on the yield, reinforcing the critical role of 1O2 in the oxidation process. Based on these findings, we propose a photooxidation mechanism wherein the Ir(III) complex is photoexcited to a high-energy singlet state (1Ir(III)*), which then undergoes intersystem crossing to a triplet state (3Ir(III)*). The 3Ir(III)* then facilitates the energy transfer to ground-state O2, generating 1O2, which reacts with sulfide to form a per-sulfoxide intermediate. This intermediate further reacts with a sulfide molecule to yield two molecules of sulfoxide [33,34,35].
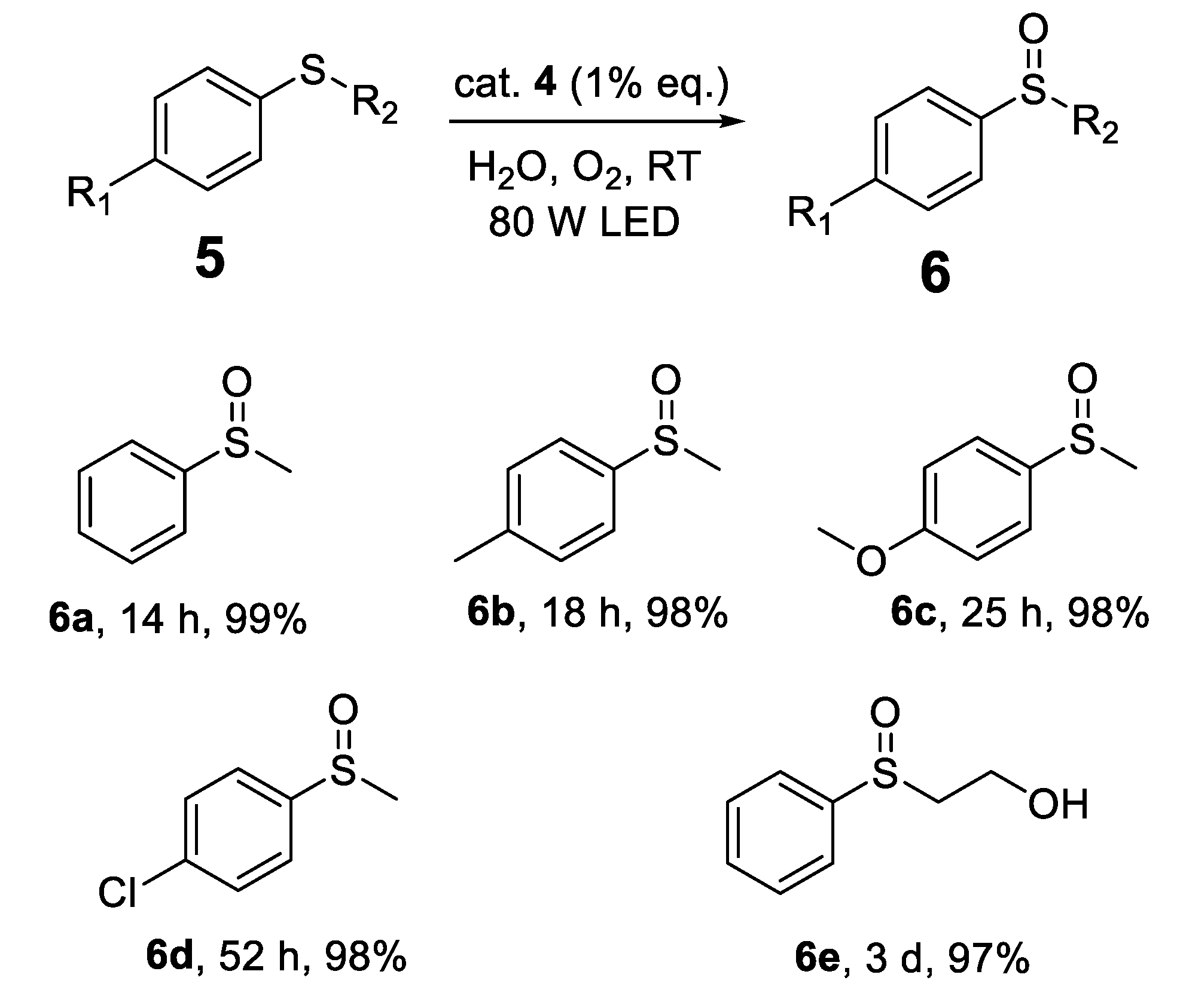
Armed with optimized reaction conditions, we extended our investigation to assess the protocol’s versatility in synthesizing sulfoxides from various sulfides in water under aerobic conditions and blue-light irradiation. Employing para-methylphenyl and para-methoxyphenyl sulfides as substrates yielded the sulfoxides 6b and 6c with excellent efficiencies of 98% in 18 h and 25 h, respectively (Scheme 2). Conversely, introducing an electron-withdrawing chloro group at the para position markedly extended the reaction time to 52 h, albeit still achieving a high yield of 98% for sulfoxide 6d. This observation highlights the impact of electron-withdrawing groups on retarding the oxidation process, suggesting a nuanced interaction between the sulfide substrate’s electronic properties and the photo-oxidative reactivity. Furthermore, the protocol demonstrated significant versatility in accommodating steric demands. For instance, replacing methyl sulfide with 2-(phenylthio)ethanol led to the formation of sulfoxide 6e with a yield of 97% over a period of 3 days. This experiment not only confirms the method’s robustness across different sulfide classes but also its potential for synthesizing a wide array of sulfoxide compounds through an environmentally benign pathway.
3. Materials and Methods
Unless specified otherwise, all reagents were procured from commercial sources and utilized as received without further purification. The ligands 2-(4-fluorophenyl) quinoline (fpq), 2-(p-tolyl) quinoline (tq), 2-(4-methoxyphenyl) quinoline (mpq), and 2-(naphthyl-1) quinoline (nq) were prepared using the Suzuki reaction. [36,37,38]. The complexes [Ir(nyq)2Cl]2, [Ir(ptq)2Cl]2, [Ir(mpq)2Cl]2, and [Ir(fpq)2Cl]2 were synthesized according to the literature [28,29]. Photocatalytic experiments employed a blue light-emitting diode (LED) UV lamp (λ = 450–470 nm, 10 W, Shenzhen Nuoguan Technology Co., shenzhen, China.) as the light source. For chromatographic separations, silica gel (300–400 mesh) was used. Electrospray ionization mass spectrometry (ESI-MS) analyses were conducted on a Thermo LCQ DECA XP mass spectrometer, while HRMS data were acquired using a Thermo Fisher Scientific Q Exactive mass spectrometer (Schwerte, Germany). Elemental (C, H and N) analysis was performed on the Elementar Vario EL analyzer (Schwerte, Germany). Using TMS as the internal standard, 1H and 13C NMR spectra were recorded with Bruker AV 400 and Bruker AV 600 spectrometers (Fällanden, Switzerland). Absorption spectra of the complexes were measured using a Perkin Elmer Lambda 950 UV-VIS spectrophotometer (Beaconsfiel, UK) in MeOH solution at room temperature. Steady-state photoluminescence spectra were recorded on an Edinburgh Instruments FLS 980 Photoluminescence Spectrometer(Edinburgh, UK). Fluorescence lifetimes were ascertained using an IBH 5000F coaxial nanosecond flash lamp (Atlanta, GA, USA). Fluorescence Quantum yields were quantified employing the Edinburgh Instruments FLS 1000 Quantum Yield Integrating Sphere Measurement System with PMT980 Detector.
3.1. Single-Crystal X-ray Crystallography
The crystal structures of complexes [Ir(fpq)2(en)](PF6) (1a), [Ir(mpq)2(en)](PF6) (3a), and [Ir(bmpqen)](PF6) (3) were determined using an Agilent SuperNova, Dual, Cu at home/near, AtlasS2 four-circle diffractometer, equipped with mirror-monochromated Cu Kα radiation (λ = 1.54184 Å). Absorption corrections were applied to all datasets using the multi-scan technique to account for absorption effects. The initial structural solutions were obtained through direct methods using olex2.solve [39], with subsequent refinements performed via iterative cycles of least-squares refinement on F2, complemented by difference Fourier synthesis [40]. Anisotropic refinement was applied to all non-hydrogen atoms. Hydrogen atoms were treated using a riding model, positioned at geometrically idealized locations with fixed isotropic displacement parameters. To address the presence of disordered solvent molecules within the crystal lattice, the Solvent Mask feature in Olex2 (an implementation of the BYPASS method, also known as SQUEEZE) [41] was employed. This approach allowed for the refinement of the disordered solvent molecules without explicitly modeling their positions, ensuring accurate structural parameters for the complexes. Detailed crystallographic information, data collection, and refinement statistics for each complex are presented in Table 2.
3.2. Synthesis and Characterization of Complexes
General procedure for the synthesis of Ir(III)-diamine complexes. To a stirred solution of the corresponding cyclometalated Ir(III) complex ([Ir(nyq)2Cl]2, [Ir(ptq)2Cl]2, [Ir(mpq)2Cl]2, or [Ir(fpq)2Cl]2; 0.05 mmol) in 10 mL of methanol, ethylenediamine (0.12 mmol) was added. The reaction mixture was then stirred at 50 °C under a nitrogen atmosphere for 8 h. Following this, the solvent was removed under reduced pressure via vacuum distillation. To the resultant residue, 30 mL of dichloromethane was added. The organic layer was subsequently washed with saturated KPF6 solution (3 × 20 mL) to ensure the complete conversion of the chloride to the PF6 counterion. The organic phase was then dried over anhydrous Na2SO4 and filtered to remove the drying agent. Finally, the solvent was evaporated under reduced pressure to afford the desired Ir(III)-diamine complex as the target product.
For 1a. Yield, 96%. Anal. Calcd. for C32H26F8IrN4P: C 45.66, H 3.11, N 6.66. Found: C 45.53, H 3.24, N 6.52. HRMS: Calcd. m/z = 697.17493 [M − PF6]+; Found: 697.17149 [M − PF6]+. 1H NMR (600 MHz, CD3CN): δ 8.53 (d, J = 8.8 Hz, 1H), 8.26 (d, J = 8.7 Hz, 1H), 8.10 (dd, J = 6.2, 3.4 Hz, 1H), 8.03 (dd, J = 8.7, 5.6 Hz, 1H), 7.79–7.75 (m, 1H), 7.71 (dd, J = 6.6, 3.2 Hz, 2H), 6.82–6.85 (m, 1H), 6.29 (dd, J = 9.9, 2.6 Hz, 1H), 3.99 (d, J = 12.8 Hz, 1H), 2.72–2.60 (m, 1H), 2.50–2.52(m, 1H), 1.93–1.84 (m, 1H). 13C NMR (151 MHz, CD3CN): δ 170.57, 163.14 (d, J = 252.17 Hz), 153.36 (d, J = 7.55 Hz), 148.04, 143.24, 140.26, 132.00, 129.62, 128.71 (d, J = 10.57 Hz), 128.37, 126.89, 124.34, 120.71 (d, J = 16.61 Hz), 117.82, 109.36 (d, J = 24.16 Hz), 43.45.
For 2a. Yield, 88%. Anal. Calcd. for C34H32F6IrN4P: C 48.98, H 3.87, N 6.72. Found: C 49.00, H 3.92, N 6.42. HRMS: Calcd. m/z = 689.22507 [M − PF6]+; Found: 689.22528 [M− PF6]+. 1H NMR (400 MHz, CD3CN): δ 8.47 (d, J = 8.8 Hz, 1H), 8.25 (d, J = 8.8 Hz, 1H), 8.09–8.03 (m, 1H), 7.86 (d, J = 8.0 Hz, 1H), 7.80–7.74 (m, 1H), 7.70–7.62 (m, 2H), 6.88 (dd, J = 8.1, 1.7 Hz, 1H), 6.48 (s, 1H), 3.84 (d, J = 12.3 Hz, 1H), 2.61–2.42 (m, 2H), 2.19 (s, 3H), 1.91–1.82 (m, 1H). 13C NMR (151 MHz, CD3CN): δ 171.50, 150.75, 148.28, 144.02, 140.00, 139.57, 135.98, 131.58, 129.38, 128.17, 126.46, 126.38, 124.81, 124.54, 123.05, 43.41, 20.45.
For 3a. Yield, 92%. Anal. Calcd. for C34H32F6IrN4O2P: C 47.17, H 3.73, N 6.47. Found: C 47.23, H 3.64, N 6.51. HRMS: Calcd. m/z = 721.21490 [M − PF6]+; Found: 721.21290 [M− PF6]+. 1H NMR (600 MHz, CD3CN): δ 8.39 (d, J = 8.8 Hz, 1H), 8.14 (d, J = 8.9 Hz, 1H), 8.00 (dd, J = 7.2, 2.3 Hz, 1H), 7.88 (d, J = 8.7 Hz, 1H), 7.74 (d, J = 8.3 Hz, 1H), 7.59–7.64 (m, 2H), 6.62 (dd, J = 8.7, 2.6 Hz, 1H), 6.06 (d, J = 2.5 Hz, 1H), 3.88 (d, J = 12.8 Hz, 1H), 3.34 (s, 3H), 2.61 (s, 1H), 2.52–2.45 (m, 1H), 1.27 (s, 1H). 13C NMR (151 MHz, CD3CN) δ 171.21, 160.70, 152.79, 148.12, 139.57, 139.43, 131.47, 129.39, 128.29, 127.87, 126.19, 124.48, 119.70, 117.49, 108.30, 54.37, 43.45.
For 4a. Yield, 85%. Anal. Calcd. for C40H32F6IrN4P: C 53.03, H 3.56, N 6.18. Found: C 52.88, H 3.35, N 6.02. HRMS: Calcd. m/z = 761.22507 [M − PF6]+; Found: 761.22183 [M− PF6]+. 1H NMR (400 MHz, CD3CN): δ 8.85 (d, J = 9.0 Hz, 1H), 8.78 (d, J = 8.7 Hz, 1H), 8.57 (d, J = 9.1 Hz, 1H), 8.11 (dd, J = 8.1, 1.6 Hz, 1H), 7.79–7.72 (m, 2H), 7.70–7.63 (m, 2H), 7.59 (m, 1H), 7.44 (t, J = 7.4 Hz, 1H), 7.15 (d, J = 8.4 Hz, 1H), 6.83 (d, J = 8.4 Hz, 1H), 3.95 (d, J = 12.0 Hz, 1H), 2.60–2.65 (m, 1H), 2.47–2.50 (m, 1H), 1.88–1.81 (m, 1H). 13C NMR (151 MHz, CD3CN): δ 172.39, 158.94, 148.71, 140.01, 139.24, 134.12, 132.27, 131.75, 131.47, 129.72, 129.31, 129.29, 127.35, 127.20, 126.52, 124.20, 123.48, 121.75, 121.47, 43.50.
General procedure for the synthesis of hexadentate Ir(III) complexes. The synthesis commenced with the addition of an Ir(III)-diamine complex (0.05 mmol) to a reaction mixture consisting of 100 mL ethanol under an oxygen atmosphere facilitated by an oxygen balloon. This mixture was subjected to stirring at an appropriate rate and irradiated with a 10 W blue light LED lamp at a temperature of 60 °C. The progress of the reaction was monitored via 1H NMR spectroscopy to ensure completeness. Upon completion, the ethanol solvent was removed under reduced pressure through vacuum distillation. The resulting crude product underwent purification via silica gel column chromatography, employing a gradient elution with a dichloromethane/methanol mixture, varying the composition from 100/0.5 to 100/5 (v/v), to isolate the carbon-nitrogen coupling product.
For 1. Yield, 92% for 85 h. Anal. Calcd. C32H22F8IrN4P: C 45.88, H 2.65, N 6.69. Found: C 45.63, H 2.86, N 6.74. HRMS: Calcd. m/z = 693.14363 [M − PF6]+; Found: 693.14117 [M− PF6]+. 1H NMR (600 MHz, CD3CN): δ 8.52 (d, J = 8.9 Hz, 1H), 8.25 (d, J = 8.9 Hz, 1H), 8.12 (d, J = 8.2 Hz, 1H), 8.00 (dd, J = 8.6, 5.7 Hz, 1H), 7.88 (d, J = 7.5 Hz, 1H), 7.78 (t, J = 7.9 Hz, 1H), 6.71–6.74 (m, 1H), 6.62 (s, 1H), 5.59 (dd, J = 9.4, 2.6 Hz, 1H), 3.31 (d, J = 10.3 Hz, 1H), 3.05–3.00 (m, 1H). 13C NMR (151 MHz, CD3CN): δ 164.21, 162.57(d, J = 13.59 Hz), 151.12(d, J = 6.04 Hz), 145.94, 143.31, 142.69, 135.97, 129.74, 128.76, 128.68(d, J = 10.57 Hz), 128.25, 128.17, 118.47,117.46, 108.64(d, J = 22.65 Hz), 53.94.
For 2. Yield, 80% for 88 h. Anal. Calcd. C34H28F6IrN4P: C 49.21, H 3.40, N 6.75. Found: C 49.03, H 3.55, N 6.78. HRMS: Calcd. m/z = 685.19377 [M − PF6]+; Found: 685.19434 [M − PF6]+. 1H NMR (400 MHz, CD3CN): δ 8.46 (d, J = 9.0 Hz, 1H), 8.22 (d, J = 9.0 Hz, 1H), 8.10 (d, J = 8.1 Hz, 1H), 7.84 (dd, J = 12.8, 7.7 Hz, 2H), 7.74 (t, J = 7.9 Hz, 1H), 6.78 (d, J = 7.9 Hz, 1H), 6.45 (s, 1H), 5.78 (s, 1H), 3.29 (d, J = 10.0 Hz, 1H), 3.04–2.97 (m, 1H), 2.18 (s, 3H). 13C NMR (151 MHz, CD3CN): δ 163.62, 147.92, 146.10, 143.51, 143.37, 140.45, 135.26, 132.22, 129.44, 128.41, 128.14, 128.01, 126.53, 122.55, 118.25, 53.77, 20.60.
For 3. Yield, 90% for 92 h. Anal. Calcd. C34H28F6IrN4O2P: C 47.39, H 3.27, N 6.50. Found: C 47.53, H 3.35, N 6.38. HRMS: Calcd. m/z = 717.18360 [M− PF6]+; Found: 717.18372 [M − PF6]+. 1H NMR (400 MHz, CD3CN): δ 8.42 (d, J = 9.0 Hz, 1H), 8.15 (d, J = 9.0 Hz, 1H), 8.06 (d, J = 8.1 Hz, 1H), 7.88 (d, J = 8.6 Hz, 1H), 7.83 (d, J = 7.5 Hz, 1H), 7.71 (t, J = 7.9 Hz, 1H), 6.55 (dd, J = 8.6, 2.6 Hz, 1H), 6.47 (s, 1H), 5.34 (d, J = 2.6 Hz, 1H), 3.48 (s, 3H), 3.31 (d, J = 10.1 Hz, 1H), 3.05–2.99 (m, 1H). 13C NMR (151 MHz, CD3CN): δ 163.17, 161.07, 150.08, 146.14, 143.17, 138.98, 135.22, 129.42, 128.40, 128.04, 127.99, 127.73, 118.07, 117.01, 106.20, 54.37, 53.84.
For 4. Yield, 70% for 96 h. Anal. Calcd. C40H28F6IrN4P: C 53.27, H 3.13, N 6.21. Found: C 53.11, H 3.25, N 6.38. HRMS: Calcd. m/z = 757.19377 [M− PF6]+; Found: 757.19014 [M − PF6]+. 1H NMR (400 MHz, CD3CN): δ 9.00 (d, J = 9.3 Hz, 1H), 8.87 (d, J = 8.8 Hz, 1H), 8.61 (d, J = 9.3 Hz, 1H), 8.17 (d, J = 8.0 Hz, 1H), 7.85 (d, J = 7.5 Hz, 1H), 7.77 (t, J = 7.8 Hz, 1H), 7.68 (d, J = 8.1 Hz, 1H), 7.63 (m, 1H), 7.38 (m, 1H), 7.14 (d, J = 8.3 Hz, 1H), 6.60 (s, 1H), 6.10 (d, J = 8.3 Hz, 1H), 3.35 (d, J = 10.1 Hz, 1H), 3.14–3.06 (m, 1H). 13C NMR (151 MHz, CD3CN): δ 164.56, 157.40, 146.74, 143.05, 138.77, 135.45, 133.28, 130.90, 130.34, 130.22, 129.91, 129.82, 129.70, 128.61, 127.89, 127.47, 123.18, 122.67, 121.32, 53.62.
3.3. Procedure for Photooxidation of Sulfide into Sulfoxide
In a typical procedure, the sulfide substrate (0.1 mmol) and photocatalyst (1 mol %) were combined in 8 mL of distilled water in a 10 mL quartz reaction tube, sealed with an O2 balloon to maintain an oxygen-rich environment. The assembled reaction setup was then exposed to irradiation from an 80 W blue LED lamp, operating at room temperature for a duration determined by preliminary studies or until completion as indicated by analytical methods. Post-irradiation, the reaction mixture was subjected to extraction with CH2Cl2 in three portions (3 × 6 mL) to recover the reaction products from the aqueous phase. The combined CH2Cl2 extracts were then sequentially washed with water and brine to remove any remaining water-soluble impurities and salts. The organic layer was dried over anhydrous Na2SO4 to remove residual water, followed by the removal of CH2Cl2 under reduced pressure. The crude product was further purified through silica gel column chromatography, employing a hexane:ethyl acetate (10:1, v/v) mixture as the elution solvent to afford the desired sulfoxide product.
4. Conclusions
We have devised a novel, mild, and efficient methodology for the synthesis of hexadentate cyclometalated Ir(III) complexes through an in situ inter-ligand C-N cross-coupling photoreaction, leveraging quinoline and diamine in the presence of an Ir(III) ion template. This approach introduces a versatile and complementary strategy for the in situ fabrication of multidentate complexes via post-coordination inter-ligand coupling under benign conditions. Furthermore, our study reveals that these newly synthesized hexadentate cyclometalated Ir(III) complexes exhibit remarkable photocatalytic efficiency and chemo-selectivity in facilitating the aerobic oxidation of sulfides to sulfoxides in aqueous media at ambient temperature. These findings contribute significantly to the development of innovative strategies for the design and application of multidentate cyclometalated Ir(III) complexes as potent photocatalysts, marking a significant advancement in the field of sustainable and green chemistry.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/inorganics12030073/s1, Figure S1: 1H-NMR spectra of fpq in CDCl3; Figure S2: 1H-NMR spectra of [Ir(fpq)2Cl]2 in DMSO-d6; Figure S3. 13C-NMR spectra of [Ir(fpq)2Cl]2 in DMSO-d6; Figure S4. 1H-NMR spectra of [Ir(fpq)2(en)](PF6) (1a) in CD3CN; Figure S5. 13C-NMR spectra of [Ir(fpq)2(en)](PF6) (1a) in CD3CN; Figure S6. HRMS of [Ir(fpq)2(en)](PF6) (up) and calculated for [Ir(fpq)2(en)]+ (down); Figure S7. 1H-NMR spectra of [Ir(bfpqen)](PF6) (1) in CD3CN; Figure S8. 13C-NMR spectra of [Ir(bfpqen)](PF6) (1) in CD3CN; Figure S9. HRMS of [Ir(bfpqen)](PF6) (up) and calculated for [Ir(bfpqen)]+ (down); Figure S10. 1H-NMR spectra of tq in CDCl3; Figure S11. 1H-NMR spectra of [Ir(tq)2(en)](PF6) (2a) in CD3CN; Figure S12. 13C-NMR spectra of [Ir(tq)2(en)](PF6) (2a) in CD3CN; Figure S13. HRMS of [Ir(tq)2(en)](PF6) (up) and calculated for [Ir(tq)2(en)]+ (down); Figure S14. 1H-NMR spectra of [Ir(btqen)](PF6) (2) in CD3CN; Figure S15. 13C-NMR spectra of [Ir(btqen)](PF6) (2) in CD3CN; Figure S16. HRMS of [Ir(btqen)](PF6) (up) and calculated for [Ir(btqen)]+ (down); Figure S17. 1H-NMR spectra of mpq in CDCl3; Figure S18. 1H-NMR spectra of [Ir(mpq)2Cl]2 in DMSO-d6; Figure S19. 13C-NMR spectra of [Ir(mpq)2Cl]2 in DMSO-d6; Figure S20. 1H-NMR spectra of [Ir(mpq)2(en)](PF6) (3a) in CD3CN; Figure S21. 13C-NMR spectra of [Ir(mpq)2(en)](PF6) (3a) in CD3CN; Figure S22. HRMS of [Ir(mpq)2(en)](PF6) (up) and calculated for [Ir(mpq)2(en)]+ (down); Figure S23. 1H-NMR spectra of [Ir(bmpqen)](PF6) (3) in CD3CN; Figure S24. 13C-NMR spectra of [Ir(bmpqen)](PF6) (3) in CD3CN; Figure S25. HRMS of [Ir(bmpqen)](PF6) (up) and calculated for [Ir(bmpqen)]+ (down); Figure S26. 1H-NMR spectra of nq in CDCl3; Figure S27. 113C-NMR spectra of nq in CDCl3; Figure S28. 1H-NMR spectra of [Ir(nq)2Cl]2 in DMSO-d6; Figure S29. 13C-NMR spectra of [Ir(nq)2Cl]2 in DMSO-d6; Figure S30. 1H-NMR spectra of [Ir(nq)2(en)](PF6) (4a) in CD3CN; Figure S31. 13C-NMR spectra of [Ir(nq)2(en)](PF6) in CD3CN; Figure S32. HRMS of [Ir(nq)2(en)](PF6) (up) and calculated for [Ir(nq)2(en)]+ (down); Figure S33. 1H-NMR spectra of [Ir(bnqen)](PF6) (4) in CD3CN; Figure S34. 13C-NMR spectra of [Ir(bnqen)](PF6) (4) in CD3CN; Figure S35. HRMS of [Ir(bnqen)](PF6) (up) and calculated for [Ir(bnqen)]+ (down); Figure S36. Single-wavelength decay traces for 1a (left) and 1 (right) in Ar-saturated MeOH at room temperature; Figure S37. Single-wavelength decay traces for 2a (left) and 2 (right) in Ar-saturated MeOH at room temperature; Figure S38. Single-wavelength decay traces for 3a (left) and 3 (right) in Ar-saturated MeOH at room temperature; Figure S39. Single-wavelength decay traces for 4a (left) and 4 (right) in Ar-saturated MeOH at room temperature; Figure S40. 1H-NMR spectra of methyl phenyl sulfoxide (6a) in CDCl3; Figure S41. 1H-NMR spectra of methyl 4-methylphenyl sulfoxide (6b) in CDCl3; Figure S42. 1H-NMR spectra of methyl 4-methoxyphenyl sulfoxide (6c) in CDCl3; Figure S43. 1H-NMR spectra of methyl 4-chloro-phenyl sulfoxide (6d) in CDCl3; Figure S44. 1H-NMR spectrum of 2-(phenylsulfinyl)ethan-1-ol (6e) in CDCl3.
Author Contributions
Conceptualization, B.-H.Y. and J.-Y.F.; methodology, J.-Y.F.; formal analysis, J.-Y.F.; investigation, J.-Y.F.; data curation, J.-Y.F.; writing—original draft preparation, B.-H.Y.; writing—review and editing, S.-Y.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Natural Science Foundation of China (Grant no. 21971266). Science and Technology Research Project of Guangzhou (Grant no. 202102021122).
Data Availability Statement
The data presented in this study are available in this article and Supplementary Materials.
Acknowledgments
We thank L. Jiang at SYSU for his help in the resolution of the crystal structures.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- You, Y.; Nam, W. Photofunctional triplet excited states of cyclometalated Ir(iii) complexes: Beyond electroluminescence. Chem. Soc. Rev. 2012, 41, 7061–7084. [Google Scholar] [CrossRef]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef]
- Lee, L.C.-C.; Lo, K.K.-W. Luminescent and Photofunctional Transition Metal Complexes: From Molecular Design to Diagnostic and Therapeutic Applications. J. Am. Chem. Soc. 2022, 144, 14420–14440. [Google Scholar] [CrossRef]
- Gorczyński, A.; Harrowfield, J.M.; Patroniak, V.; Stefankiewicz, A.R. Quaterpyridines as Scaffolds for Functional Metallosupramolecular Materials. Chem. Rev. 2016, 116, 14620–14674. [Google Scholar] [CrossRef]
- Chi, Y.; Chang, T.-K.; Ganesan, P.; Rajakannu, P. Emissive bis-tridentate Ir(III) metal complexes: Tactics, photophysics and applications. Coord. Chem. Rev. 2017, 346, 91–100. [Google Scholar] [CrossRef]
- Lee, Y.H.; Park, J.; Lee, J.; Lee, S.U.; Lee, M.H. Impact of Restricted Rotation of o-Carborane on Phosphorescence Efficiency. J. Am. Chem. Soc. 2015, 137, 8018–8021. [Google Scholar] [CrossRef]
- Esteruelas, M.A.; Lόpez, A.M.; Oñate, E.; San-Torcuato, A.; Tsai, J.-Y.; Xia, C. Preparation of Phosphorescent Iridium(III) Complexes with a Dianionic C,C,C,C-Tetradentate Ligand. Inorg. Chem. 2018, 57, 3720–3730. [Google Scholar] [CrossRef]
- Zhang, X.-M. Hydro(solvo)thermal in situ ligand syntheses. Coord. Chem. Rev. 2005, 249, 1201–1219. [Google Scholar] [CrossRef]
- Chen, X.-M.; Tong, M.-L. Solvothermal in Situ Metal/Ligand Reactions: A New Bridge between Coordination Chemistry and Organic Synthetic Chemistry. Acc. Chem. Res. 2007, 40, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Villafañe, F. ReI(CO)3 complexes with diimine ligands synthesized in situ. Coord. Chem. Rev. 2017, 339, 128–137. [Google Scholar] [CrossRef]
- Liu, C.-M.; Gao, S.; Kou, H.-Z. Dehydrogenative coupling of phenanthroline under hydrothermal conditions: Crystal structure of a novel layered vanadate complex constructed of 4,8,10-net sheets: [(2,2′-biphen)Co]V3O8. 5. Chem. Commun. 2001, 17, 1670–1671. [Google Scholar] [CrossRef]
- Viguri, M.E.; Huertos, M.A.; Pérez, J.; Riera, L.; Ara, I. Re-Mediated C-C Coupling of Pyridines and Imidazoles. J. Am. Chem. Soc. 2012, 134, 20326–20329. [Google Scholar] [CrossRef]
- Omori, H.; Hiroto, S.; Takeda, Y.; Fliegl, H.; Minakata, S.; Shinokubo, H. Ni(II) 10-Phosphacorrole: A Porphyrin Analogue Containing Phosphorus at the Meso Position. J. Am. Chem. Soc. 2019, 141, 4800–4805. [Google Scholar] [CrossRef]
- Vallavoju, N.; Sivaguru, J. Supramolecular photocatalysis: Combining confinement and non-covalent interactions to control light initiated reactions. Chem. Soc. Rev. 2014, 43, 4084–4101. [Google Scholar] [CrossRef]
- Espinal-Viguri, M.; Fombona, S.; Álvarez, D.; Díaz, J.; Menéndez, M.I.; Lόpez, R.; Pérez, J.; Riera, L. Regiochemistry Control by Bipyridine Substituents in the Deprotonation of ReI and MoII N-Alkylimidazole Complexes. Chem. Eur. J. 2019, 25, 9253–9265. [Google Scholar] [CrossRef]
- Sipos, G.; Drinkel, E.E.; Dorta, R. The Emergence of Sulfoxides as Efficient Ligands in Transition Metal Catalysis. Chem. Soc. Rev. 2015, 44, 3834–3860. [Google Scholar] [CrossRef] [PubMed]
- Nishiguchi, S.; Izumi, T.; Kouno, T.; Sukegawa, J.; Ilies, L.; Nakamura, E. Synthesis of Esomeprazole and Related Proton Pump Inhibitors through Iron-Catalyzed Enantioselective Sulfoxidation. ACS Catal. 2018, 8, 9738–9743. [Google Scholar] [CrossRef]
- Kinen, C.O.; Rossi, L.I.; de Rossi, R.H. The Development of an Environmentally Benign Sulfide Oxidation Procedure and Its Assessment by Green Chemistry Metrics. Green Chem. 2009, 11, 223–228. [Google Scholar] [CrossRef]
- Zhang, P.F.; Wang, Y.; Li, H.; Antonietti, M. Metal-Free Oxidation of Sulfides by Carbon Nitride with Visible Light Illumination at Room Temperature. Green Chem. 2012, 14, 1904–1908. [Google Scholar] [CrossRef]
- Miller, B.L.; Williams, T.D.; Schöneich, C. Mechanism of Sulfoxide Formation through Reaction of Sulfur Radical Cation Complexes with Superoxide or Hydroxide Ion in Oxygenated Aqueous Solution. J. Am. Chem. Soc. 1996, 118, 11014–11025. [Google Scholar] [CrossRef]
- Xing, C.; Deng, J.; Tan, R.; Gao, M.; Hao, P.; Yin, D.; Yin, D. Cooperative Chiral Salen TiIV Catalyst Supported on Ionic Liquid-Functionalized Graphene Oxide Accelerates Asymmetric Sulfoxidation in Water. Catal. Sci. Technol. 2017, 7, 5944–5952. [Google Scholar] [CrossRef]
- Heydari-turkmani, A.; Zakavi, S. The First Solid State Porphyrin-Weak Acid Molecular Complex: A Novel Metal Free, Nanosized and Porous Photocatalyst for Large Scale Aerobic Oxidations in Water. J. Catal. 2018, 364, 394–405. [Google Scholar] [CrossRef]
- Wei, L.-Q.; Ye, B.-H. Cyclometalated Ir–Zr Metal–Organic Frameworks as Recyclable Visible-Light Photocatalysts for Sulfide Oxidation into Sulfoxide in Water. ACS Appl. Mater. Interfaces 2019, 11, 41448–41457. [Google Scholar] [CrossRef]
- Peng, H.-L.; Li, Y.; Chen, X.-Y.; Li, L.-P.; Ke, Z.; Ye, B.-H. Visible-Light-Induced Amination of Quinoline at the C8 Position via a Postcoordinated Interligand-Coupling Strategy under Mild Conditions. Inorg. Chem. 2021, 60, 908–918. [Google Scholar] [CrossRef] [PubMed]
- Xiong, M.-F.; Peng, H.-L.; Zhang, X.-P.; Ye, B.-H. Discrepancy between Proline and Homoproline in Chiral Recognition and Diastereomeric Photoreactivity with Iridium(III) Complexes. Inorg. Chem. 2021, 60, 5423–5431. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-K.; Li, L.-P.; Zhou, H.-Y.; Xiong, M.-F.; Fan, J.-Y.; Ye, B.-H. Switching the Photoreactions of Ir(III) Diamine Complexes between C-N Coupling and Dehydrogenation under Visible Light Irradiation. Inorg. Chem. 2022, 61, 20834–20847. [Google Scholar] [CrossRef] [PubMed]
- Xiong, M.-F.; Liu, G.-F.; Ye, B.-H. Solvent-Induced Umpolung Reaction from Dioxygenation to C-S Coupling in Bis(2-phenylquinoline) Iridium(III) Thiolate Complexes. Inorg. Chem. 2023, 62, 11654–11664. [Google Scholar] [CrossRef]
- Matsuo, N. Benzo[h]quinolin-10-yl-N Iridium(III) Complexes. Bull. Chem. Soc. Jpn. 1974, 47, 767–768. [Google Scholar]
- Yao, S.-Y.; Ou, Y.L.; Ye, B.-H. Asymmetric Synthesis of Enantiomerically Pure Mono- and Binuclear Bis(cyclometalated) Iridium(III) Complexes. Inorg. Chem. 2016, 55, 6018–6026. [Google Scholar] [CrossRef]
- Zhang, K.Y.; Li, S.P.-Y.; Zhu, N.; Or, I.W.-S.; Cheung, M.S.-H.; Lam, Y.-W.; Lo, K.K.-W. Structure, Photophysical and Electrochemical Properties, Biomolecular Interactions, and Intracellular Uptake of Luminescent Cyclometalated Iridium(III) Dipyridoquinoxaline Complexes. Inorg. Chem. 2010, 49, 2530–2540. [Google Scholar] [CrossRef] [PubMed]
- Lamansky, S.; Djurovich, P.; Murphy, D.; Abdel-Razzaq, F.; Lee, H.-E.; Adachi, C.; Burrows, P.E.; Forrest, S.R.; Thompson, M.E. Highly Phosphorescent Bis-Cyclometalated Iridium Complexes: Synthesis, Photophysical Characterization, and Use in Organic Light Emitting Diodes. J. Am. Chem. Soc. 2001, 123, 4304–4312. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Liu, R.; Qiu, Y.; Duan, L. High-performance yellow and orange-emitting diodes based on novel sublimable cationic iridium(III) complexes by ligand control. J. Mater. Chem. C 2018, 6, 5630–5638. [Google Scholar] [CrossRef]
- Bonesi, S.M.; Manet, I.; Freccero, M.; Fagnoni, M.; Albini, A. Photosensitized Oxidation of Sulfides: Discriminating between the Singlet-Oxygen Mechanism and Electron Transfer Involving Superoxide Anion or Molecular Oxygen. Chem. Eur. J. 2006, 12, 4844–4857. [Google Scholar] [CrossRef] [PubMed]
- Baptista, M.S.; Cadet, J.; Di Mascio, P.; Ghogare, A.A.; Greer, A.; Hamblin, M.R.; Lorente, C.; Nunez, S.C.; Ribeiro, M.S.; Thomas, A.H.; et al. Type I and Type II Photosensitized Oxidation Reactions: Guidelines and Mechanistic Pathways. Photochem. Photobiol. 2017, 93, 912–919. [Google Scholar] [CrossRef]
- Baciocchi, E.; Giacco, T.D.; Elisei, F.; Gerini, M.F.; Guerra, M.; Lapi, A.; Liberali, P. Electron Transfer and Singlet Oxygen Mechanisms in the Photooxygenation of Dibutyl Sulfide and Thioanisole in MeCN Sensitized by N-Methylquinolinium Tetrafluoborate and 9,10-Dicyanoanthracene. The Probable Involvement of a Thiadioxirane Intermediate in Electron Transfer Photooxygenations. J. Am. Chem. Soc. 2003, 125, 16444–16454. [Google Scholar] [PubMed]
- Chen, Y.; Zhu, C.; Guo, Z.; Liu, W.; Yang, X. Asymmetric Synthesis of Hydroquinolines with α,α-Disubstitution through Organocatalyzed Kinetic Resolution. Angew. Chem. Int. Ed. 2021, 60, 5268–5272. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Liu, W.; Zhao, F.; Chen, Y.; Tao, H.; He, Y.-P.; Yang, X. Kinetic Resolution of 2,2-Disubstituted Dihydroquinolines through Chiral Phosphoric Acid-Catalyzed C6-Selective Asymmetric Halogenations. Organ. Lett. 2021, 23, 4104–4108. [Google Scholar] [CrossRef] [PubMed]
- Mitra, R.; Zhu, H.; Grimme, S.; Niemeyer, J. Functional Mechanically Interlocked Molecules: Asymmetric Organocatalysis with a Catenated Bifunctional Brønsted Acid. Angew. Chem. Int. Ed. 2017, 56, 11456–11459. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Diagram of the synthesis of hexadentate Ir(III) complexes.
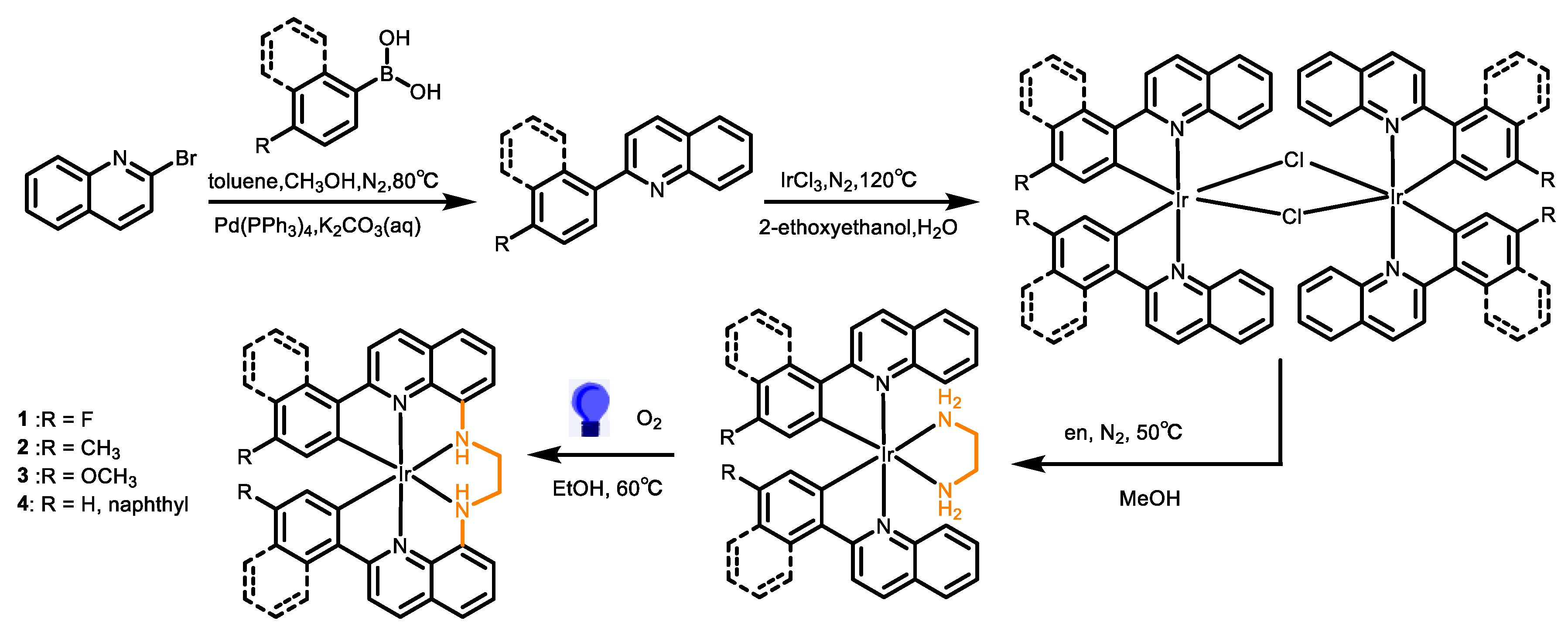
Figure 1.
Crystal structures of 1a (left) and 3a (right) with 50% probability ellipsoids. H atoms and anion are omitted for clarity. Selected bond lengths (Å) and angles (°) for 1a and 3a (in parentheses): Ir1-N1 = 2.082(3) (2.087(3)), Ir1-N2 = 2.097(3) (2.078(3)), Ir1-N3 = 2.240(4) (2.220(3)), Ir1-N4 = 2.217(4) (2.236(3)), Ir1-C11 = 2.010(4) (2.002(4)), Ir1-C26 = 1.995(5) (1.994(4)), N4-Ir1-N3 = 76.65(15) (77.45(13)), N1-Ir1-N2 = 170.87(15) (173.98(12)).
Figure 1.
Crystal structures of 1a (left) and 3a (right) with 50% probability ellipsoids. H atoms and anion are omitted for clarity. Selected bond lengths (Å) and angles (°) for 1a and 3a (in parentheses): Ir1-N1 = 2.082(3) (2.087(3)), Ir1-N2 = 2.097(3) (2.078(3)), Ir1-N3 = 2.240(4) (2.220(3)), Ir1-N4 = 2.217(4) (2.236(3)), Ir1-C11 = 2.010(4) (2.002(4)), Ir1-C26 = 1.995(5) (1.994(4)), N4-Ir1-N3 = 76.65(15) (77.45(13)), N1-Ir1-N2 = 170.87(15) (173.98(12)).

Figure 2.
1H-NMR spectra of 1a and 1 in CD3CN.
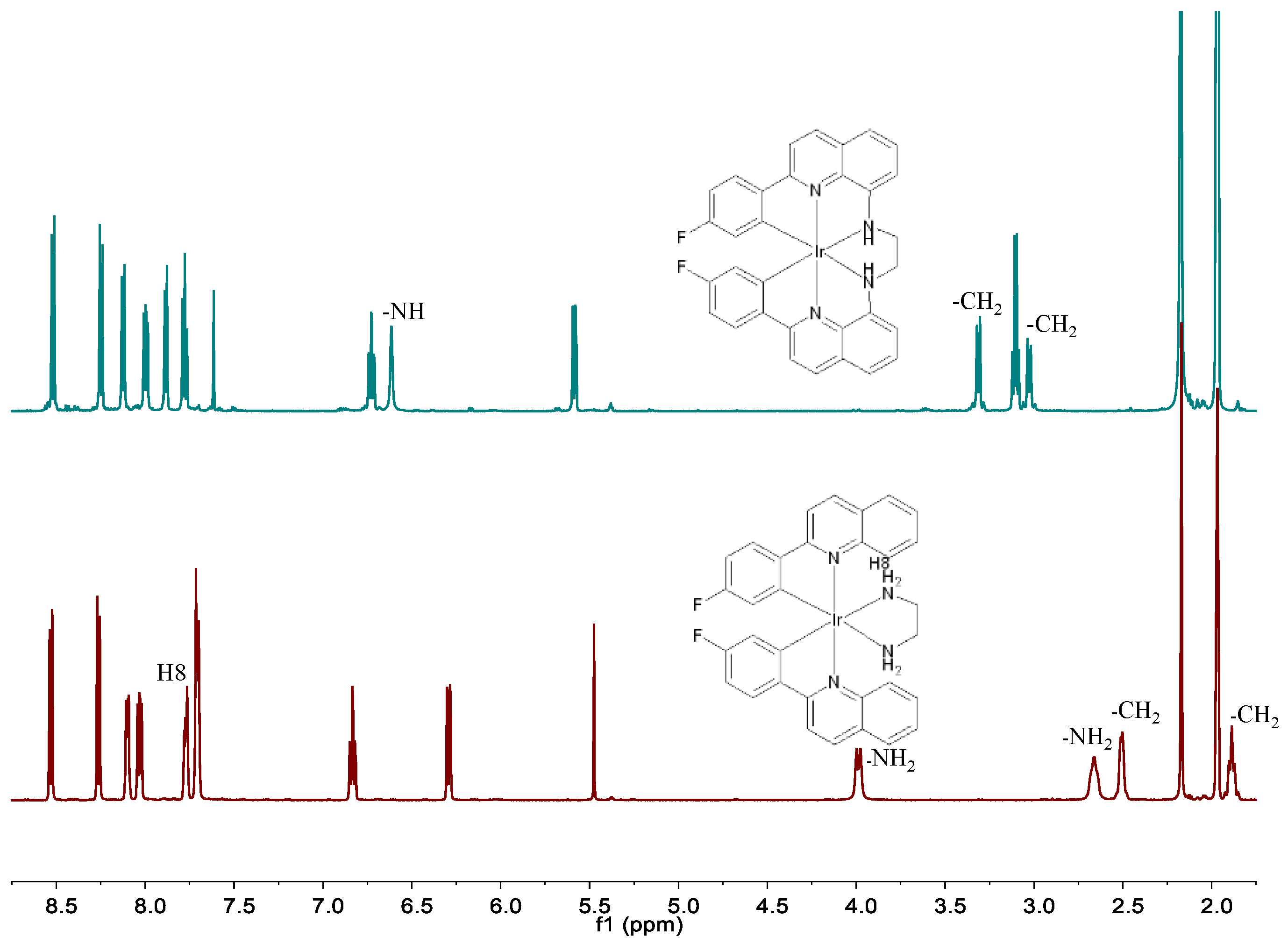
Figure 3.
Crystal structure of 3 with 50% probability ellipsoids. H atoms and anion are omitted for clarity. Selected bond lengths (Å) and angles (°): Ir1-N1 = 2.000(4), Ir1-N2 = 1.999(5), Ir1-N3 = 2.240(5), Ir1-N4 = 2.238(5), Ir1-C11 = 2.016(5), Ir1-C26 = 2.025(5), N3-C2 = 1.469(7), N4-C17= 1.480(7), N3-C33 = 1.503(8), N4-C34 = 1.492(7); N4-Ir1-N3 = 79.63(18), N1-Ir1-N2 = 177.65(19).
Figure 3.
Crystal structure of 3 with 50% probability ellipsoids. H atoms and anion are omitted for clarity. Selected bond lengths (Å) and angles (°): Ir1-N1 = 2.000(4), Ir1-N2 = 1.999(5), Ir1-N3 = 2.240(5), Ir1-N4 = 2.238(5), Ir1-C11 = 2.016(5), Ir1-C26 = 2.025(5), N3-C2 = 1.469(7), N4-C17= 1.480(7), N3-C33 = 1.503(8), N4-C34 = 1.492(7); N4-Ir1-N3 = 79.63(18), N1-Ir1-N2 = 177.65(19).

Figure 4.
Normalized emission spectra of 1, 2, 3, and 4 in air-saturated MeOH solution (10−5 M) at room temperature (left). Single-wavelength decay traces for 1 and 1a in Ar-saturated MeOH at room temperature (right).
Figure 4.
Normalized emission spectra of 1, 2, 3, and 4 in air-saturated MeOH solution (10−5 M) at room temperature (left). Single-wavelength decay traces for 1 and 1a in Ar-saturated MeOH at room temperature (right).
Scheme 2.
Photooxidation of Sulfides (Isolated yield).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Photooxidation of Sulfides a.
 | |||
|---|---|---|---|
| Entry | Catalyst | DFSC b | Yield (%) c |
| 1 | 1 | - | 97 |
| 2 | 3 | - | 92 |
| 3 | 4 | - | 100 |
| 4 | 4a | - | 79 |
| 5 | 4 | N2 | 0 |
| 6 | 4 | dark | 0 |
| 7 | - | - | 0 |
| 8 | 4 | 2 eq. DABCO | 24 |
| 9 | 4 | 2 eq. BQ | 81 |
| 10 | 4 | 2 eq. DMB | 90 |
| 11 | 4 | 2 eq. iPrOH | 98 |
a Reaction conditions: 5a (0.1 mmol) and catalyst (1 mol %) in 8 mL H2O with an O2 balloon under an 80 W blue LED at RT for 14 h. b DFSC is the deviation from standard conditions. c Determined by 1H NMR. DABCO is diazabicyclo[2.2.2]octane; BQ is benzoquinone; DMB is 1,4-dimethoxybenzene.
Table 2.
Crystallographic data for Ir(III) complexes.
| Complex | 1a | 3a | 3 |
|---|---|---|---|
| Molecular formula | C32H26F8IrN4P | C34H32F6IrN4O2P | C34H28F6IrN4O2P |
| Temp/K | 150 | 150 | 150 |
| Mr | 841.74 | 865.80 | 861.77 |
| Crystal system | triclinic | triclinic | monoclinic |
| Space group | P-1 | P-1 | P21/n |
| a/Å | 8.3615(3) | 10.6791(3) | 10.58160(10) |
| b/Å | 10.6980(5) | 12.5175(4) | 18.5399(2) |
| c/Å | 19.2560(7) | 13.8170(4) | 17.3118(2) |
| α/° | 76.136(4) | 81.202(2) | 90 |
| β/° | 84.487(3) | 84.247(2) | 90.3740(10) |
| γ/° | 77.048(3) | 82.229(2) | 90 |
| V/Å3 | 1628.11(12) | 1802.52(9) | 3396.19(6) |
| Z | 2 | 2 | 4 |
| Dc (g cm−3) | 1.717 | 1.595 | 1.685 |
| Μ (mm−1) | 9.072 | 8.171 | 8.674 |
| aR1[I > 2σ(I)] | 0.0365 | 0.0329 | 0.0392 |
| bωR2 (all data) | 0.0930 | 0.0807 | 0.1009 |
| GOF | 1.055 | 1.039 | 1.062 |
| CCDC No. | 2291984 | 2291986 | 2291985 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fan, J.-Y.; Yao, S.-Y.; Ye, B.-H. In Situ Synthesis of Hexadentate Cyclometalated Ir(III) Complexes as Photocatalysts for the Oxidation of Sulfides into Sulfoxides in Water. Inorganics 2024, 12, 73. https://doi.org/10.3390/inorganics12030073
AMA Style
Fan J-Y, Yao S-Y, Ye B-H. In Situ Synthesis of Hexadentate Cyclometalated Ir(III) Complexes as Photocatalysts for the Oxidation of Sulfides into Sulfoxides in Water. Inorganics. 2024; 12(3):73. https://doi.org/10.3390/inorganics12030073
Chicago/Turabian StyleFan, Jing-Yan, Su-Yang Yao, and Bao-Hui Ye. 2024. "In Situ Synthesis of Hexadentate Cyclometalated Ir(III) Complexes as Photocatalysts for the Oxidation of Sulfides into Sulfoxides in Water" Inorganics 12, no. 3: 73. https://doi.org/10.3390/inorganics12030073
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.