
Perhalophenyl–Phosphide: A Couple Needed to Stabilize Phosphide–Gold Complexes

 , ,
, ,
Abstract
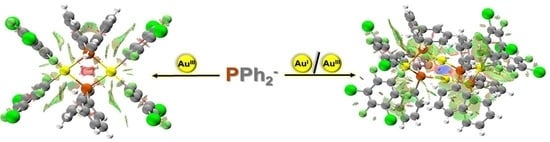
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
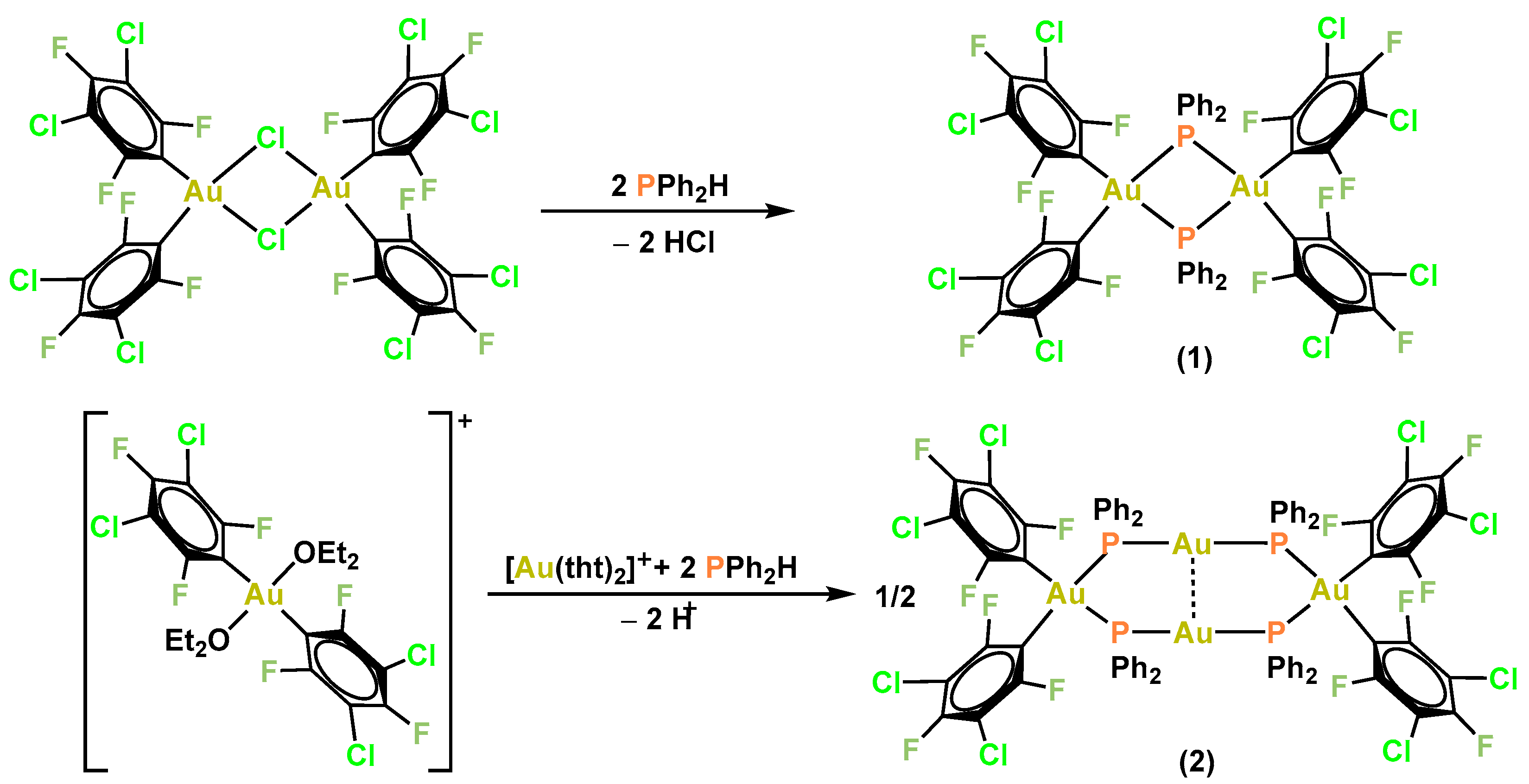
2.1. Synthesis and Characterization of the Complexes
2.1.1. Synthesis and Spectroscopic Characterization
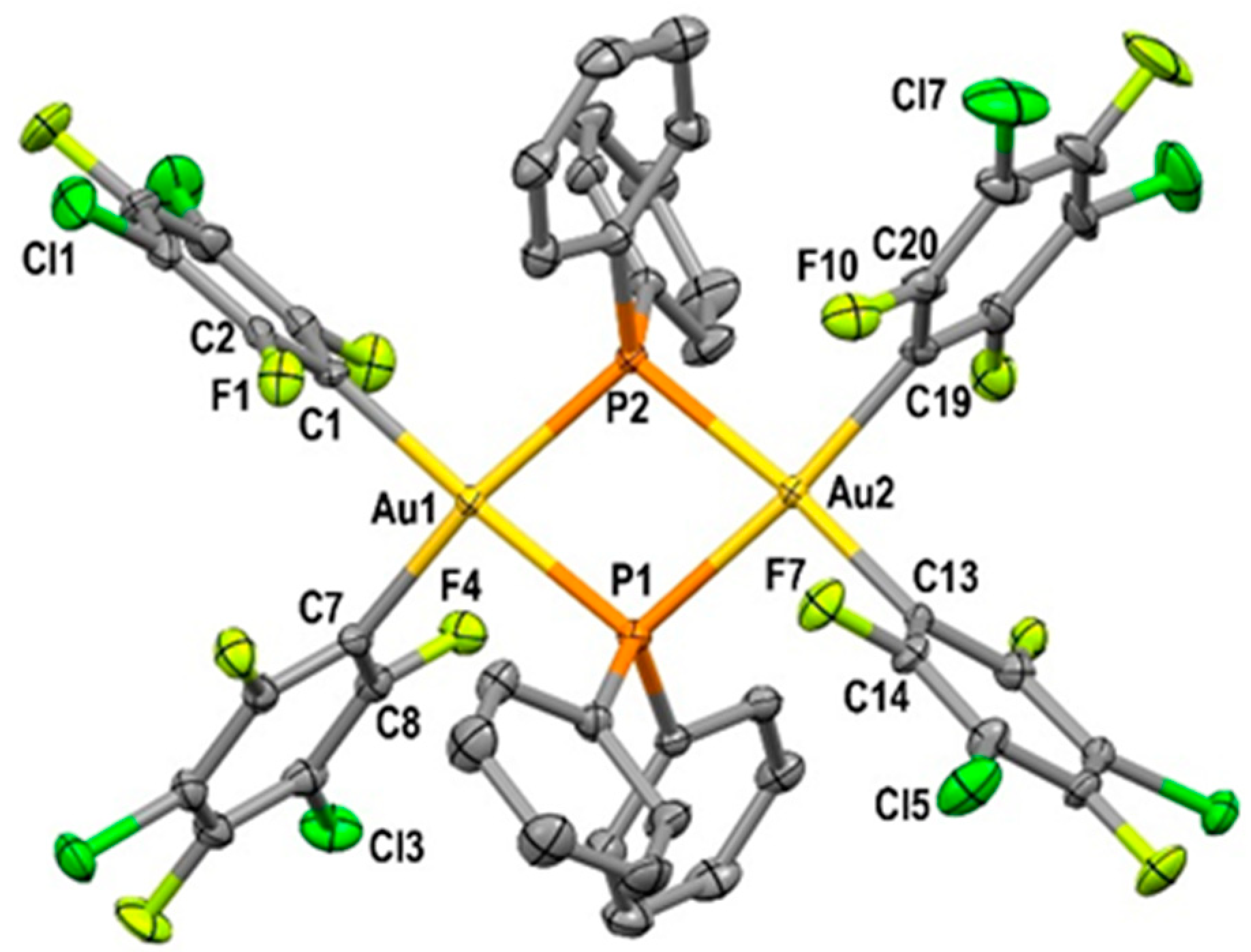
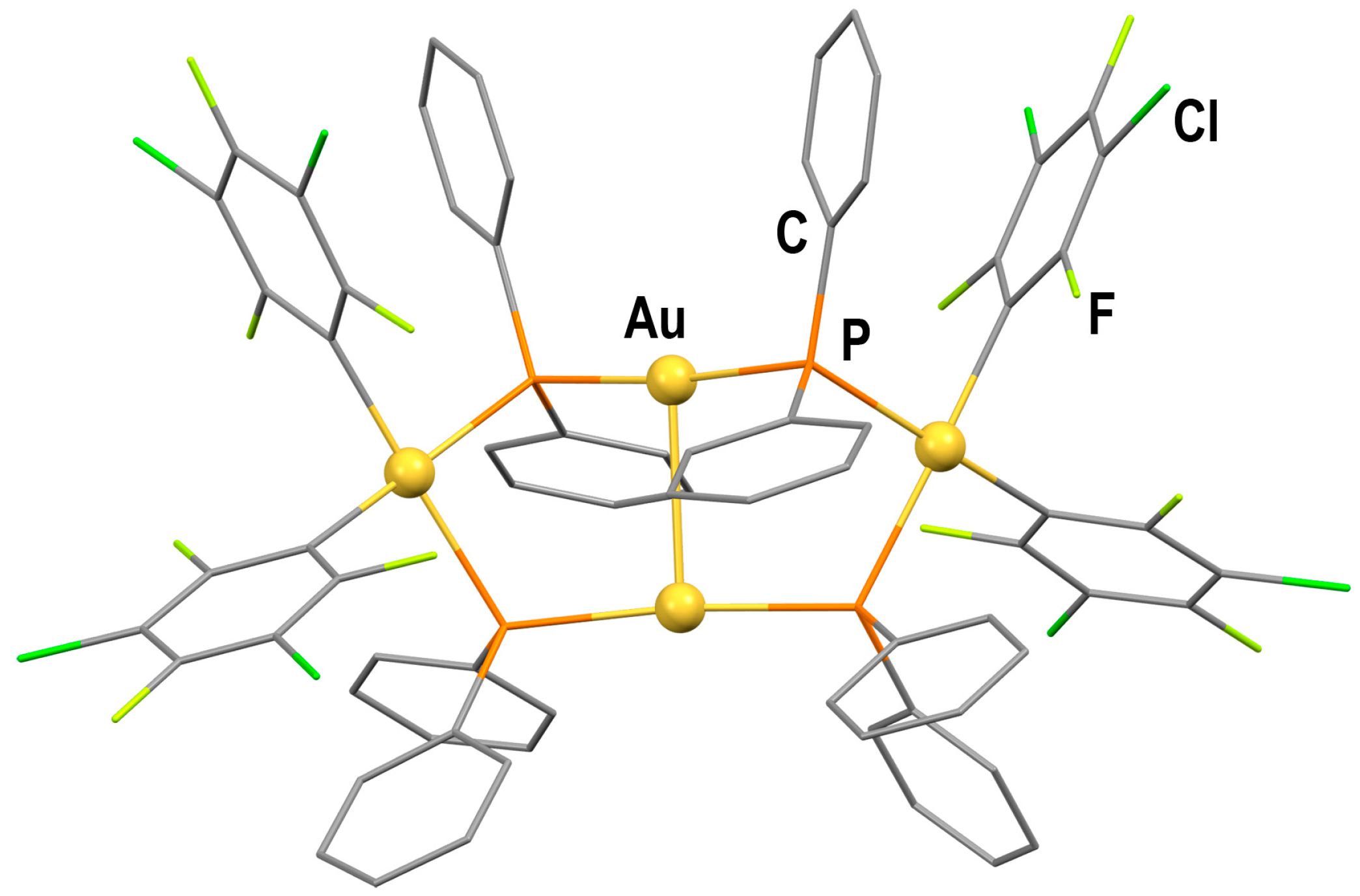
2.1.2. Crystal Structures
2.2. Computational Studies
3. Materials and Methods
3.1. General
3.2. Materials and Physical Measurements
3.3. Synthesis
3.3.1. Synthesis of [{Au(C6Cl2F3)2}2(µ-PPh2)2] (1)
3.3.2. Synthesis of [{Au(C6Cl2F3)2(µ-PPh2)2Au}2] (2)
3.4. Crystallography
3.5. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dyson, D.B.; Parish, R.V.; McAuliffe, C.A.; Pritchard, R.G.; Fields, R.; Beagley, B. Gold(I) Complexes Derived from Secondary Phosphines: [{Au(µ-PR2)}n], [(AuBr)2(µ-PPh2)]–, [AuX(PHR2)], and [{Au(PHR2)N}]+. Crystal Structure of [AuBr(PHPh2)]. J. Chem. Soc. Dalton Trans. 1989, 5, 907–914. [Google Scholar] [CrossRef]
- Vicente, J.; Chicote, M.T.; Jones, P.G. (Diphenylphosphine)-, (Diphenylphosphido)-, and (Diphenylphosphinito)Gold(I) Complexes. Crystal Structure of [(PPh3)2N][Au{P(O)Ph2}2]. Inorg. Chem. 1993, 32, 4960–4964. [Google Scholar] [CrossRef]
- Puddephatt, R.J.; Thompson, P.J. Some Reactions of Methylplatinum and Methylgold Compounds with Phenylselenol, Diphenylphosphine, Diphenylarsine, N-Bromosuccinimide and 2-Nitrophenylsulphenyl Chloride. J. Organomet. Chem. 1976, 117, 395–403. [Google Scholar] [CrossRef]
- Li, X.-S.; Mo, J.; Zhang, S.-M.; Yuan, L.; Liu, J.-H. (μ-Diphenylphosphanido-Κ2P:P′)bis [2,2′-(pyridine-2,6-diyl)diphenyl-Κ3C1,N,C1′)Gold(III)] Perchlorate Acetonitrile Solvate. Acta Crystallogr. Sect. E Struct. Rep. Online 2008, 64, m1126–m1127. [Google Scholar] [CrossRef]
- Blanco, M.C.; Fernández, E.J.; López-de-Luzuriaga, J.M.; Olmos, M.E.; Crespo, O.; Gimeno, M.C.; Laguna, A.; Jones, P.G. Heteropolynuclear Phosphide Complexes: Phosphorus as Unique Atom Bridging Coinage Metal Centres. Chem. Eur. J. 2000, 6, 4116–4123. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.C.; Fernández, E.J.; Fischer, A.K.; Jones, P.G.; Laguna, A.; Olmos, M.E.; Villacampa, M.D. NBu4[{Au(C6F5)3}2(m-PPh2)]: A Gold(III) Phosphide with a Single Bridging the Metallic Centers. Inorg. Chem. Commun. 2000, 3, 163–165. [Google Scholar] [CrossRef]
- Coconubo-Guio, L.; López-de-Luzuriaga, J.M.; Moreno, S.; Olmos, M.E. Synthesis and Structural Characterization of Phosphide Gold(III)/Gold(I) Complexes and Their Thallium(III) and Gold(III) Precursors. Molecules 2023, 28, 447. [Google Scholar] [CrossRef] [PubMed]
- Weber, L.; Lassahn, U.; Stammler, H.-G.; Neumann, B.; Karaghiosoff, K. Synthesis and Structure of the Decanuclear Gold(I) Cluster Cation [Au8(AuCl)2{μ3-P(TBu)}2{μ-P(TBu)=C(NMe2)2}6]4+ with Bridging Phosphaalkene and Phosphanediide Ligands. Eur. J. Inorg. Chem. 2002, 2002, 3272–3277. [Google Scholar] [CrossRef]
- Azizpoor Fard, M.; Rabiee Kenaree, A.; Boyle, P.D.; Ragogna, P.J.; Gilroy, J.B.; Corrigan, J.F. Coinage Metal Coordination Chemistry of Stable Primary, Secondary and Tertiary Ferrocenylethyl-Based Phosphines. Dalton Trans. 2016, 45, 2868–2880. [Google Scholar] [CrossRef] [PubMed]
- Paderina, A.V.; Koshevoy, I.O.; Grachova, E.V. Keep It Tight: A Crucial Role of Bridging Phosphine Ligands in the Design and Optical Properties of Multinuclear Coinage Metal Complexes. Dalton Trans. 2021, 50, 6003–6033. [Google Scholar] [CrossRef] [PubMed]
- Fernández, E.; Laguna, A.; Olmos, M.E. Perfluoroarylgold Complexes. Coord. Chem. Rev. 2008, 252, 1630–1667. [Google Scholar] [CrossRef]
- Mastrorilli, P.; Fortuño, C. Platinum and Palladium Promoted Couplings of Bridging PPh2–with Terminally Bonded Ligands. Inorganica Chim. Acta 2020, 513, 119947. [Google Scholar] [CrossRef]
- Ara, I.; Forniés, J.; Ibáñez, S.; Mastrorilli, P.; Todisco, S.; Gallo, V. Polynuclear Platinum Phosphanido/Phosphinito Complexes: Formation of P–O and P–O–P Bonds through Reductive Coupling Processes. Dalton Trans. 2016, 2156–2171. [Google Scholar] [CrossRef] [PubMed]
- Álvarez, S. A cartography of the van der Waals territories. Dalton Trans. 2013, 42, 8617–8636. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.Z.; Zhou, Z.H.; Xie, Z.X.; Robertson, B.E. A comparative study of crystallographic van der Waals radii. Z. Krist. Mater. 2014, 229, 517–523. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Electronic Structure Calculations on Workstation Computers: The Program System Turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Vangala, V.R.; Nangia, A.; Lynch, V.M. Interplay of Phenyl–Perfluorophenyl Stacking, C–H⋯F, C–F⋯π and F⋯F Interactions in Some Crystalline Aromatic Azines. Chem. Commun. 2002, 12, 1304–1305. [Google Scholar] [CrossRef]
- Sonoda, Y.; Goto, M.; Tsuzuki, S.; Tamaoki, N. Fluorinated Diphenylpolyenes: Crystal Structures and Emission Properties. J. Phys. Chem. A 2007, 111, 13441–13451. [Google Scholar] [CrossRef]
- Echeverría, R.; López-de-Luzuriaga, J.M.; Monge, M.; Moreno, S.; Olmos, M.E.; Rodríguez-Castillo, M. Lead Encapsulation by a Golden Clamp through Multiple Electrostatic, Metallophilic, Hydrogen Bonding and Weak Interactions. Chem. Commun. 2018, 54, 295–298. [Google Scholar] [CrossRef]
- Laguna, A.; Laguna, M.; Jiménez, J.; Fumanal, A.J. 2,4,6-Trifluorophenyl Gold(I) and Gold(III) Complexes. J. Organomet. Chem. 1990, 396, 121–128. [Google Scholar] [CrossRef]
- Uson, R.; Laguna, A.; Garcia, J.; Laguna, M. Anionic Perfluorophenyl Complexes of Gold(I) and Gold(III). Inorganica Chim. Acta 1979, 37, 201–207. [Google Scholar] [CrossRef]
- Usón, R.; Laguna, A.; Laguna, M.; Abad, M. Synthesis and Reactions of Di-μ-Halo- or -Pseudohalotetrakis(Pentafluorophenyl)Digold(III). J. Organomet. Chem. 1983, 249, 437–443. [Google Scholar] [CrossRef]
- Usón, R.; Laguna, A.; Navarro, A.; Parish, R.V.; Moore, L.S. Synthesis and Reactivity of Perchlorate Bis(Tetrahydrothiophen)Gold(I). 197Au Mössbauer Spectra of Three-Coordinate Gold(I) Complexes. Inorganica Chim. Acta 1986, 112, 205–208. [Google Scholar] [CrossRef]
- Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. The Anatomy of a Comprehensive Constrained, Restrained Refinement Program for the Modern Computing Environment—Olex2 Dissected. Acta Crystallogr. A Found. Adv. 2015, 71, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Gaussian 16, Revision C.01. Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. (Eds.) Gaussian, Inc.: Wallingford, CT, USA, 2016.
- GaussView, Version 6. Dennington, R.; Keith, T.A.; Millam, J.M. (Eds.) Semichem Inc.: Shawnee Mission, KS, USA, 2016.


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coconubo-Guio, L.; Rodríguez-Castillo, M.; Moreno, S.; Monge, M.; Olmos, M.E.; López-de-Luzuriaga, J.M. Perhalophenyl–Phosphide: A Couple Needed to Stabilize Phosphide–Gold Complexes. Inorganics 2024, 12, 78. https://doi.org/10.3390/inorganics12030078
Coconubo-Guio L, Rodríguez-Castillo M, Moreno S, Monge M, Olmos ME, López-de-Luzuriaga JM. Perhalophenyl–Phosphide: A Couple Needed to Stabilize Phosphide–Gold Complexes. Inorganics. 2024; 12(3):78. https://doi.org/10.3390/inorganics12030078
Chicago/Turabian StyleCoconubo-Guio, Laura, María Rodríguez-Castillo, Sonia Moreno, Miguel Monge, M. Elena Olmos, and José M. López-de-Luzuriaga. 2024. "Perhalophenyl–Phosphide: A Couple Needed to Stabilize Phosphide–Gold Complexes" Inorganics 12, no. 3: 78. https://doi.org/10.3390/inorganics12030078