
Structure Transformation among Deca-, Dodeca- and Tridecavanadates and Their Properties for Thioanisole Oxidation

Abstract
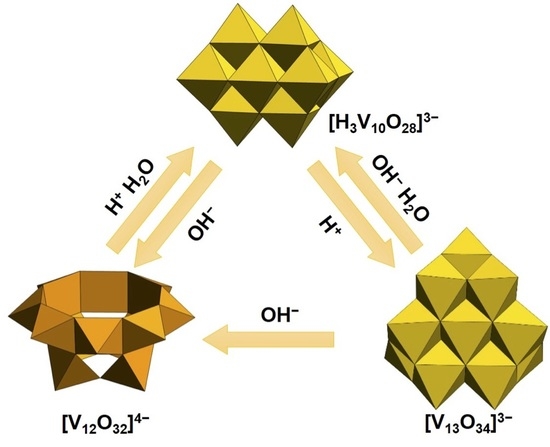
:
1. Introduction

2. Results and Discussion
2.1. Structure Transformation among Deca-, Dodeca- and Tridecavanadates
2.1.1. Transformation between Deca- and Dodecavanadates

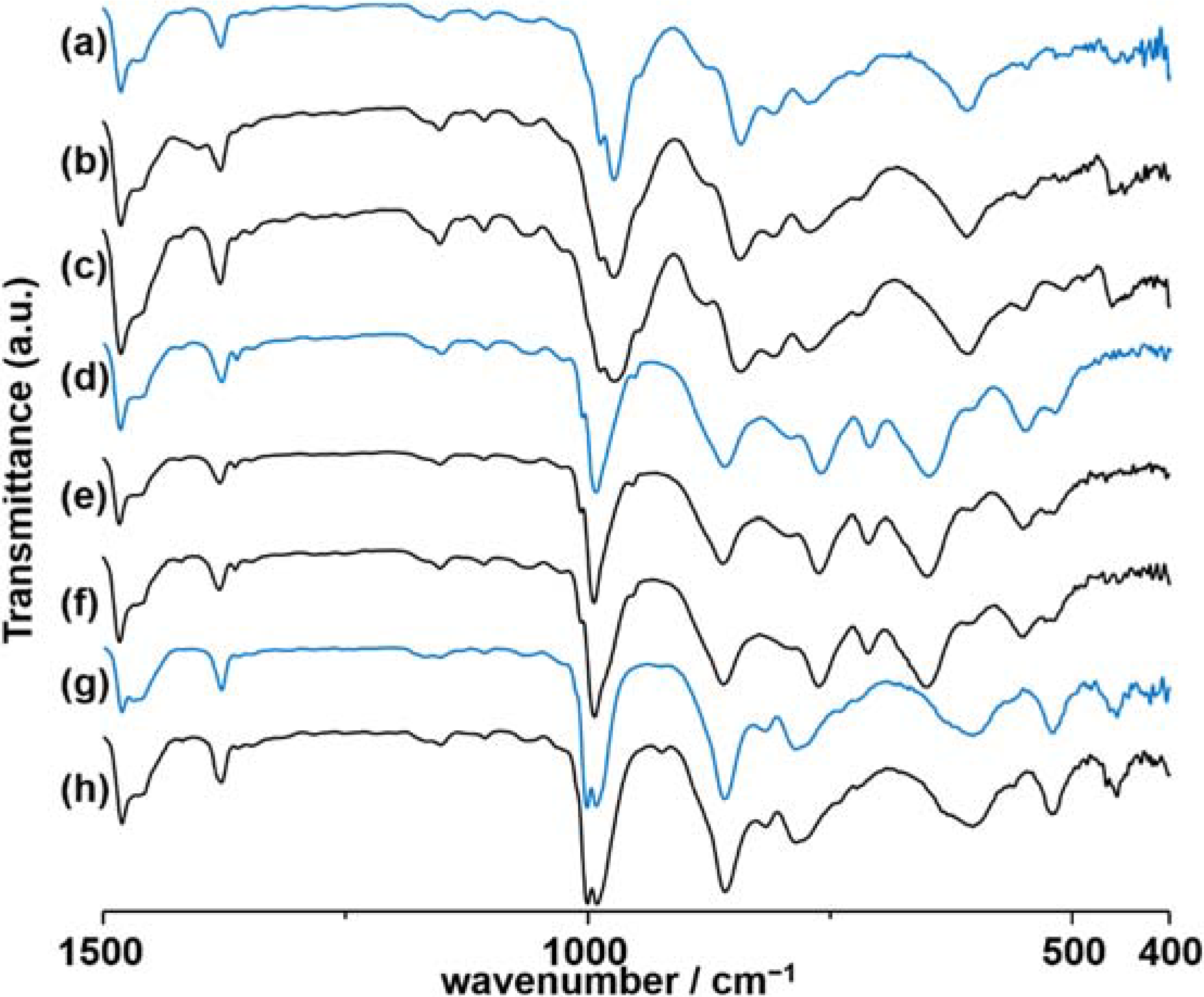
2.1.2. Transformation between Deca- and Tridecavanadates
2.1.3. Transformation between Dodeca- and Tridecavanadates

2.2. Oxidation of Thioanisole

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
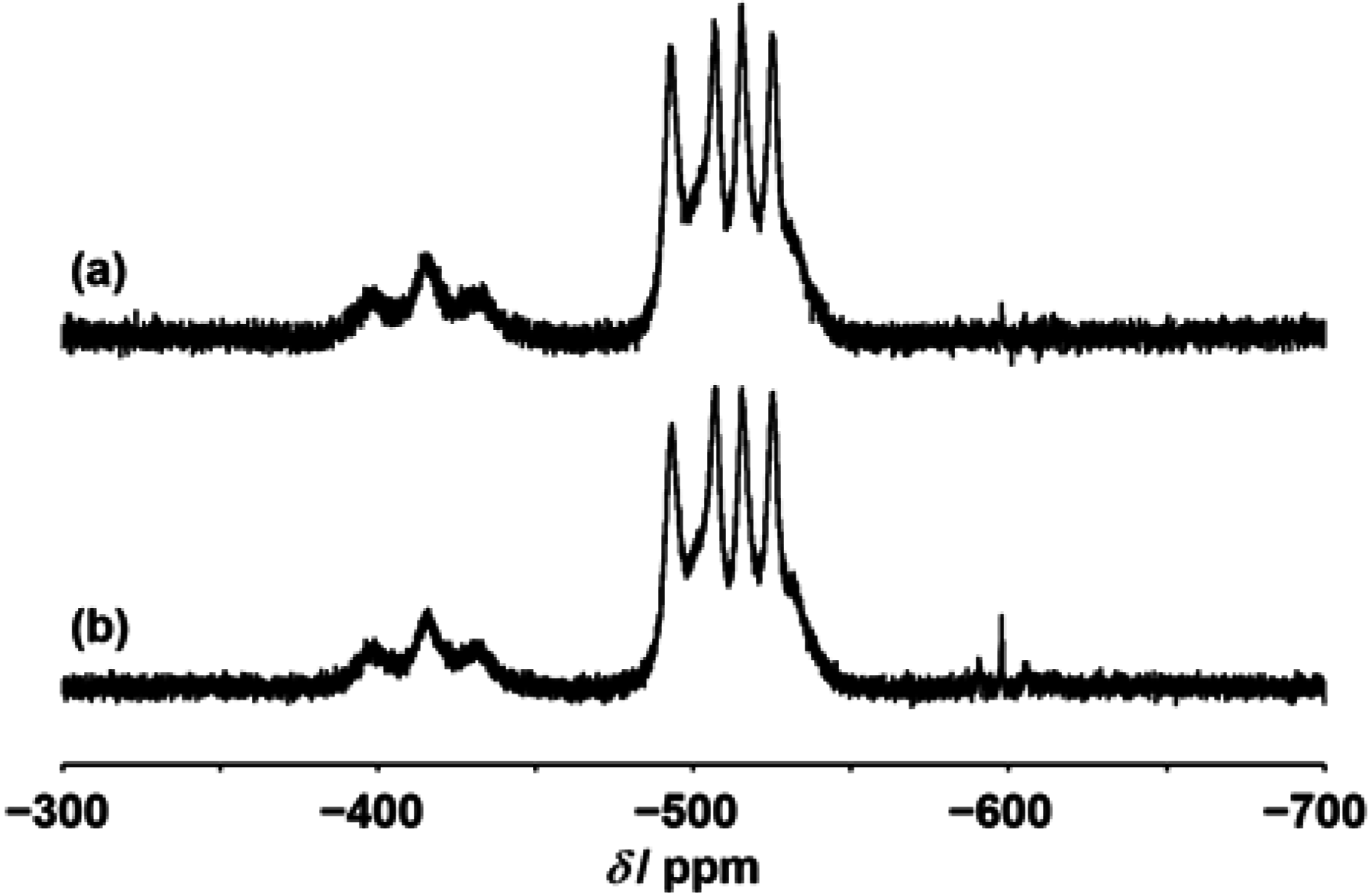{kind=link}
{kind=link}
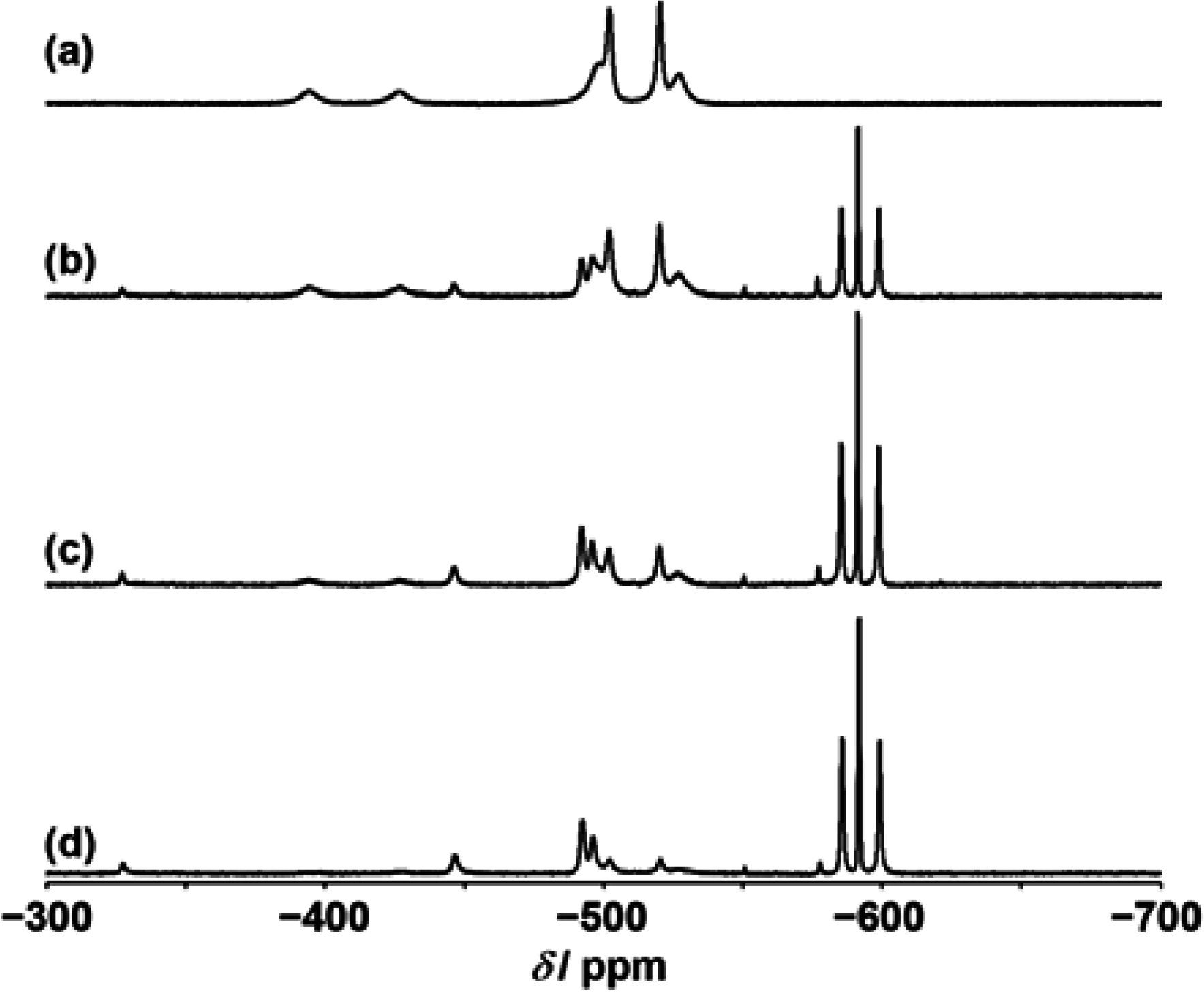{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
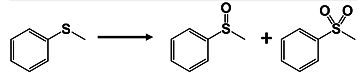 | |||
|---|---|---|---|
| Entry | Vanadium Species (μmol) | Total Yield (%) | Ratio of Sulfoxide:Sulfone |
| 1 | {(n-C4H9)4N}3[V13O34] (7.7) | 91 | 93:7 |
| 2 | {(n-C4H9)4N}4[V12O32] (8.4) | 79 | 79:21 |
| 3 | {(n-C4H9)4N}3[H3V10O28] (10) | 33 | 94:6 |
| 4 b | V2O5 (20) | 67 | 97:3 |
| 5 | - | 3 | - |
3. Experimental Section
3.1. Chemicals and Instruments
3.2. Transformation of {(n-C4H9)4N}3[H3V10O28] to {(n-C4H9)4N}4[V12O32]
3.3. Transformation of {(n-C4H9)4N}3[H3V10O28] to {(n-C4H9)4N}3[V13O34]
3.4. Transformation of {(n-C4H9)4N}4[V12O32] to {(n-C4H9)4N}3[H3V10O28]
3.5. Transformation of {(n-C4H9)4N}3[V13O34] to {(n-C4H9)4N}3[H3V10O28]
3.6. Transformation of {(n-C4H9)4N}3[V13O34] to {(n-C4H9)4N}4[V12O32]
3.7. Oxidation of Thioanisole
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix







References
- Hill, C.L. Thematic issue on polyoxometalates. Chem. Rev. 1998, 98, 1–390. [Google Scholar] [CrossRef] [PubMed]
- Kozhevnikov, I.V. Catalysis by Polyoxometalates; John Wiley & Sons, Ltd.: Chichester, UK, 2002. [Google Scholar]
- Cronin, L.; Müller, A. Thematic issue on polyoxometalates. Chem. Soc. Rev. 2012, 41, 7325–7648. [Google Scholar]
- Khan, M.I. Novel Extended Solids Composed of Transition Metal Oxide Clusters. J. Solid Stat. Chem. 2000, 152, 105–112. [Google Scholar] [CrossRef]
- Baes, C.F., Jr.; Mesmer, R.E. The Hydrolysis of Cations; Wiley & Sons: New York, NY, USA, 1976. [Google Scholar]
- Hamilton, E.E.; Fanwick, P.E.; Wilker, J.J. The Elusive Vanadate (V3O9)3–: Isolation, Crystal Structure, and Nonaqueous Solution Behavior. J. Am. Chem. Soc. 2002, 124, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Román, P.; José, A.S.; Luque, A.; Gutiérrez-Zorrilla, J.M. Observation of a Novel Cyclic Tetrametavanadate Anion Isolated from Aqueous Solution. Inorg. Chem. 1993, 32, 775–776. [Google Scholar] [CrossRef]
- Day, V.W.; Klemperer, W.G.; Yaghi, O.M. A New Structure Type in Polyoxoanion Chemistry: Synthesis and Structure of the V5O143− Anion. J. Am. Chem. Soc. 1989, 111, 4518–4519. [Google Scholar] [CrossRef]
- Okaya, K.; Kobayashi, T.; Koyama, Y.; Hayashi, Y.; Isobe, K. Formation of VV Lacunary Polyoxovanadates and Interconversion Reactions of Dodecavanadate Species. Eur. J. Inorg. Chem. 2009, 2009, 5156–5163. [Google Scholar] [CrossRef]
- Day, V.W.; Klemperer, W.G.; Yaghi, O.M. Synthesis and Characterization of a Soluble Oxide Inclusion Complex, [CH3CN⊂(V12O324−)]. J. Am. Chem. Soc. 1989, 111, 5959–5961. [Google Scholar] [CrossRef]
- Kastner, K.; Margraf, J.T.; Clark, T.; Streb, C. A Molecular Placeholder Strategy to Access a Family of Transition-Metal-Functionalized Vanadium Oxide Clusters. Chem. Eur. J. 2014, 20, 12269–12273. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Kuwajima, S.; Kurata, T.; Hayashi, Y. Structural Conversion from Bowl- to Ball-Type Polyoxovanadates: Synthesis of a Spherical Tetradecavanadate through a Chloride-Incorporated Bowl-Type Dodecavanadate. Inorg. Chim. Acta 2014, 420, 69–74. [Google Scholar] [CrossRef]
- Omri, I.; Graia, M.; Mhiri, T. Synthesis, Crystal Structure, Vibrational and Optical Properties of (Hdea)4(V7O19F)∙0.42H2O, an Original (V7O19F)4− Cluster Oxyfluoride. J. Clust. Sci. 2015, 26, 815–825. [Google Scholar] [CrossRef]
- Kikukawa, Y.; Yokoyama, T.; Kashio, S.; Hayashi, Y. Synthesis and Characterization of Fluoride-Incorporated Polyoxovanadates. J. Inorg. Biochem. 2015, 147, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zubieta, J. Synthesis and Structural Characterization of a Polyoxovanadate Coordination Complex with a Hexametalate Core: [(n-C4H9)4N]2[V6O13{O2NC(CH2O)3}2]. Inorg. Chem. 1990, 29, 1456–1458. [Google Scholar] [CrossRef]
- Domae, K.; Uchimura, D.; Koyama, Y.; Inami, S.; Hayashi, Y.; Isobe, K.; Kameda, H.; Shimoda, T. Synthesis of a Bowl-Type Dodecavanadate by the Coupling Reaction of Alkoxohexavanadate and Discovery of a Chiral Octadecavanadate. Pure Appl. Chem. 2009, 81, 1323–1330. [Google Scholar] [CrossRef]
- Day, V.W.; Klemperer, W.G.; Maltbie, D.J. Where Are the Protons in H3V10O283−? J. Am. Chem. Soc. 1987, 109, 2991–3002. [Google Scholar] [CrossRef]
- Nakamura, S.; Ozeki, T. Hydrogen-bonded Aggregates of Protonated Decavanadate Anions in Their Tetraalkylammonium Salts. J. Chem. Soc. Dalton Trans. 2001. [Google Scholar] [CrossRef]
- Hou, D.; Hagen, K.S.; Hill, C.L. Tridecavanadate, [V13O34]3−, a New High-Potential Isopolyvanadate. J. Am. Chem. Soc. 1992, 114, 5864–5866. [Google Scholar] [CrossRef]
- Klemperer, W.G.; Marquart, T.A.; Yaghi, O.M. Shape-Selective Binding of Nitriles to the Inorganic Cavitand, V12O324−. Mater. Chem. Phys. 1991, 29, 97–104. [Google Scholar] [CrossRef]
- Kawanami, N.; Ozeki, T.; Yagasaki, A. NO− Anion Trapped in a Molecular Oxide Bowl. J. Am. Chem. Soc. 2000, 122, 1239–1240. [Google Scholar] [CrossRef]
- Kurata, T.; Hayashi, Y.; Isobe, K. Synthesis and Characterization of Chloride-Incorporated Dodecavanadate from Dicopper Complex of Macrocyclic Octadecavanadate. Chem. Lett. 2010, 39, 708–709. [Google Scholar] [CrossRef]
- Rohmer, M.-M.; Devémy, J.; Wiest, R.; Bénard, M. Ab initio Modeling of the Endohedral Reactivity of Polyoxometallates: 1. Host-Guest Interactions in [RCN⊂(V12O32)4−] (R = H, CH3, C6H5). J. Am. Chem. Soc. 1996, 118, 13007–13014. [Google Scholar]
- Kurata, T.; Hayashi, Y.; Uehara, A.; Isobe, K. Synthesis of a Reduced Tridecavanadate Dimer Linked by Eight Hydrogen Bonds. Chem. Lett. 2003, 32, 1040–1041. [Google Scholar] [CrossRef]
- Li, F.; Long, D.-L.; Cameron, J.M.; Miras, H.N.; Pradeep, C.P.; Xu, L.; Cronin, L. Cation Induced Structural Transformation and Mass Spectrometric Observation of the Missing Dodecavanadomanganate(IV). Dalton Trans. 2012, 41, 9859–9862. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Tan, J.M.; Besson, C.; Geletti, Y.V.; Musaev, D.G.; Kuznetsov, A.E.; Luo, Z.; Hardcastle, K.I.; Hill, C.L. A Fast Soluble Carbon-Free Molecular Water Oxidation Catalyst Based on Abundant Metals. Science 2010, 328, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Kikukawa, Y.; Yamaguchi, K.; Mizuno, N. Zinc(II) Containing γ-Keggin Sandwich-Type Silicotungstate: Synthesis in Organic Media and Oxidation Catalysis. Angew. Chem. Int. Ed. 2010, 49, 6096–6100. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.; Lan, Y.; Bassil, B.S.; Xiang, Y.; Suchopar, A.; Powell, A.K.; Kortz, U. Hexadecacobalt(II)-Containing Polyoxometalate-Based Single-Molecule Magnet. Angew. Chem. Int. Ed. 2011, 50, 4708–4711. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Sato, R.; Mizuno, N. Reversible Switching of Single-Molecule Magnet Behaviors by Transformation of Dinuclear Dysprosium Cores in Polyoxometalates. Chem. Sci. 2013, 4, 596–600. [Google Scholar] [CrossRef]
- Wagner, G.W. Two-Dimensional 17O–51V Heteronuclear Shift Correlation NMR Spectroscopy of the 17O-Enriched Inclusion Complex [CH3CN⊂(V12O324−)]. Relationship of Cross-Peak Intensity to Bond Order. Inorg. Chem. 1991, 30, 1960–1962. [Google Scholar] [CrossRef]
- Hayashi, Y.; Fukuyama, K.; Takatera, T.; Uehara, A. Synthesis and Structure of a New Reduced Isopolyvanadate, [V17O42]4−. Chem. Lett. 2000, 29, 770–771. [Google Scholar] [CrossRef]
- Koyama, Y.; Hayashi, Y.; Isobe, K. Self-Assembled All-Inorganic Chiral Polyoxovanadate: Spontaneous Resolution of Nitrate-Incorporated Octadecavanadate. Chem. Lett. 2008, 37, 578–579. [Google Scholar] [CrossRef]
- Carreño, M.C. Applications of Sulfoxides to Asymmetric Synthesis of Biologically Active Compounds. Chem. Rev. 1995, 95, 1717–1760. [Google Scholar] [CrossRef]
- Zannikos, F.; Lois, E.; Stournas, S. Desulfurization of Petroleum Fractions by Oxidation and Solvent Extraction. Fuel Process. Tech. 1995, 42, 35–45. [Google Scholar] [CrossRef]
- Csányi, L.J.; Jáky, K.; Dombi, G.; Evanics, F.; Dezsö, G.; Kóta, Z. Onium-Decavanadate Ion-Pair Complexes as Catalysts in the Oxidation of Hydrocarbons by O2. J. Mol. Catal. A 2003, 195, 101–111. [Google Scholar] [CrossRef]
- Coletti, A.; Whiteoak, C.J.; Conte, V.; Kleij, A.W. Vanadium Catalyzed Synthesis of Cyclic Organic Carbonates. ChemCatChem 2012, 4, 1190–1196. [Google Scholar] [CrossRef] [Green Version]
- Adam, W.; Malisch, W.; Roschmann, K.J.; Saha-Möller, C.R.; Schenk, W.A. Catalytic Oxidations by Peroxy, Peroxo and Oxo Metal Complexes: An Interdisciplinary Account with a Personal View. J. Organomet. Chem. 2002, 661, 3–16. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kikukawa, Y.; Ogihara, K.; Hayashi, Y. Structure Transformation among Deca-, Dodeca- and Tridecavanadates and Their Properties for Thioanisole Oxidation. Inorganics 2015, 3, 295-308. https://doi.org/10.3390/inorganics3020295
Kikukawa Y, Ogihara K, Hayashi Y. Structure Transformation among Deca-, Dodeca- and Tridecavanadates and Their Properties for Thioanisole Oxidation. Inorganics. 2015; 3(2):295-308. https://doi.org/10.3390/inorganics3020295
Chicago/Turabian StyleKikukawa, Yuji, Kazuhiro Ogihara, and Yoshihito Hayashi. 2015. "Structure Transformation among Deca-, Dodeca- and Tridecavanadates and Their Properties for Thioanisole Oxidation" Inorganics 3, no. 2: 295-308. https://doi.org/10.3390/inorganics3020295