
Computational Studies on the Selective Polymerization of Lactide Catalyzed by Bifunctional Yttrium NHC Catalyst


State Key Laboratory of Fine Chemicals, School of Chemical Engineering, Dalian University of Technology, Dalian 116024, China
*
Author to whom correspondence should be addressed.
Inorganics 2017, 5(3), 46; https://doi.org/10.3390/inorganics5030046
Submission received: 20 June 2017
/
Revised: 13 July 2017
/
Accepted: 13 July 2017
/
Published: 20 July 2017
(This article belongs to the Special Issue N-Heterocyclic Carbene Metal Complexes: From Design to Applications)
Abstract
:A theoretical investigation of the ring-opening polymerization (ROP) mechanism of rac-lactide (LA) with an yttrium complex featuring a N-heterocyclic carbine (NHC) tethered moiety is reported. It was found that the carbonyl of lactide is attacked by N(SiMe3)2 group rather than NHC species at the chain initiation step. The polymerization selectivity was further investigated via two consecutive insertions of lactide monomer molecules. The insertion of the second monomer in different assembly modes indicated that the steric interactions between the last enchained monomer unit and the incoming monomer together with the repulsion between the incoming monomer and the ligand framework are the primary factors determining the stereoselectivity. The interaction energy between the monomer and the metal center could also play an important role in the stereocontrol.
1. Introduction
Polylactide (PLA) is a biodegradable and biocompatible material derived from biorenewable feedstock, and it is considered an excellent commercial alternative for conventional petroleum-based materials. It is of great interest for various biomedical as well as ecological applications, including drug-delivery systems and tissue engineering [1,2,3,4,5,6,7,8]. PLA has attracted much attention from researchers in both industry and academia. The most efficient method for the synthesis of PLA is ring-opening polymerization (ROP) of the six-membered cyclic ester lactide with a variety of metal-based complexes [4,9,10,11]. Organocatalysts also have been employed to more precisely control the synthesis of ring-opening polymerization [12]. Among the metal-based catalysts, rare-earth metal complexes show excellent performance for the ROP of lactides, due to their qualities such as living behaviours, controlled molar weights, and very narrow molar weight distributions [13,14,15,16]. Many different lithium complexes have been identified as excellent catalysts for the ROP of lactides in alcohol [17]. Particularly, divalent metal catalysts bearing a monoanionic auxiliary skeleton are advantageous due to their low cost, and group 3 metal complexes are also exquisite initiators in the synthesis of PLAs by the ROP of rac-lactide [18,19]. The explicit group 4 metal complexes bearing polydentate ligands have also been proved to be active initiators for the synthesis of PLA [20]. As compared to the other metal-based catalysts for the synthesis of PLAs by ROP, titanium complexes are not high in terms of activity and stereoselectivity, but their low toxicity is an attractive property for the synthesis of PLAs [21]. Regardless of the high cost, Ga(III) and In(III) precursors have captivated an increasing interest for ROP catalysis and are considered to be potentially effective initiators with biocompatible metal centers [22].
As for organocatalysts, the application of N-heterocyclic carbene (NHC) and its derivatives to ROP has attracted considerable attention. It can efficiently promote the polymerization of lactide and related monomers under mild conditions [23,24,25,26]. These organocatalysts often exhibit higher functional group tolerance [27] than metal-based complexes, and inherently work under milder conditions. Inspired by above features, bifunctional catalysts formed by combining metal and organic catalysis, which may result in a combination of their respective advantages, have been demonstrated to be a very powerful strategy in organic synthesis; these dual systems have attracted considerable interest in the last few years [28,29,30,31,32]. To exploit the potential of NHC–ZnR2 complexes, different approaches have been followed in which the synthesis and characterization of NHC–ZnR2 are carried out, and these complexes have then been used in ROP [33]. Controlled ROP of lactide was carried out by a dual system Zn(C6F5)2 combined with an amine or phosphine as organic bases [34]. In this context, Bourissou and co-workers reported a dual catalytic system combining an original cationic zinc complex with a tertiary amine, which was shown to promote efficiently and in a controlled manner the ROP of lactide under mild conditions [31]. In addition, Guillaume and Carpentier used analogous discrete cationic zinc and magnesium complexes for dual organic/organometallic-catalyzed ROP of trimethylene carbonate. The zinc cationic compounds are highly active catalysts for the ROP of TMC in the presence of an exogenous alcohol and addition of a small amount of tertiary amine [32]. However, in this content, bifunctional rare earth metal catalysts had been rarely explored for polymer synthesis.
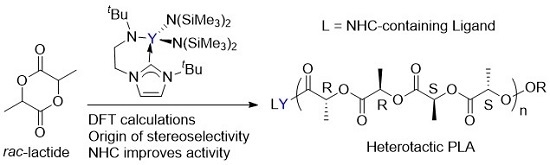
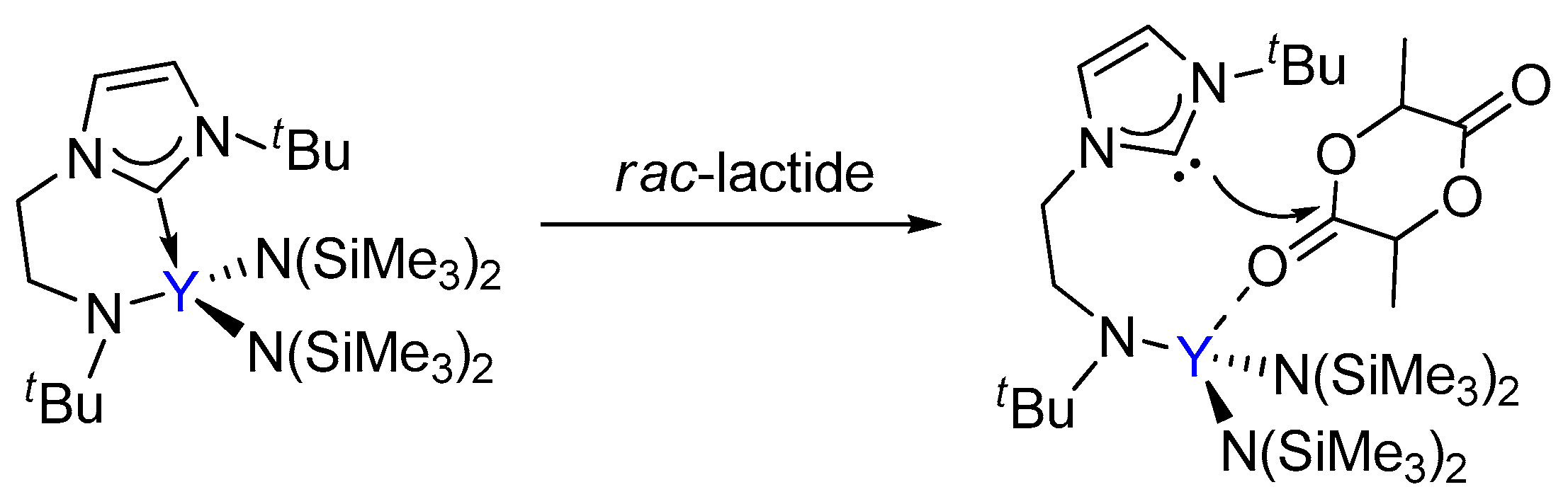
Arnold had reported that the ROP of lactide catalyzed a yttrium complex featuring an NHC tethered moiety. Such a yttrium complex showed high activity and stereoselectivity toward the ROP of rac-lactide, resulting in a heterotactic based PLA (Scheme 1) [35]. In the same work, it was proposed that the polymerization occurs through a bifunctional mechanism involving electrophilic activation of the monomer by Lewis acidic metal centre Y and nucleophilic attack of the labile NHC fragment on the activated monomer (Scheme 2). However, the influence of the bifunctional catalyst on the mechanism of this reaction and the origin of stereocontrol, which is interesting and important for catalyst design, has remained unclear.
Theoretical calculations have shown their ability to efficiently describe and explain the ROP mechanism of cyclic esters, such as lactones and lactide at the molecular level [36,37,38,39,40,41]. For instance, Hormnirun et al. conducted a DFT study on the rac-lactide ring-opening mechanism initiated by a series of bis(pyrrolidene) Schiff-base aluminium complexes to reveal the correlation between the structure of backbone linker and the polymerization activity and stereoselectivity [42]. Rzepa et al. computationally studied both the mechanism of ring-opening and the origin of heterotactic stereocontrol in the β-diketiminate magnesium system [43]. They found that the stereoselectivity is determined by the ring-opening transition state and apparently arises from the minimization of several steric interactions and possibly from weak attractive C–H···π interactions. Maron et al. reported a DFT study of the ROP of lactide induced by a dinuclear indium catalyst [44]; the mechanism is proposed to involve the dinuclear species rather than a mononuclear complex because the dissociation energy of the dimer is too high. When rac-lactide is used, no clear preference for (R,R)-lactide vs. (S,S)-lactide insertion was found, therefore atactic polymer formation is predicted. A thorough DFT study of latide ROP initiated by an NHC zinc complex was also reported and showed the assistance of the second metal center [45]. These calculations provided valuable information on the design and development of homogeneous transition and main-group metal polymerization catalysts. In comparison, there are relatively few theoretical studies on lactide polymerization catalyzed by rare-earth metal complexes. Xu et al. conducted experimental and computational studies on the ROP of rac-lactide catalyzed by a novel yttrium bis(phenolate) ether complex, which showed high activity and excellent isotactic selectivity [46]. They found that the formation of an isotactic polymer originated chiefly from interactions between the methyl groups of the monomer units in the chain and the auxiliary ligand.
To the best of our knowledge, there is no report on DFT studies on the polymerization of rac-lactide initiated by bifunctional NHC-ligated rare-earth metal catalysts, such as that shown in Scheme 1. Simulated by our previous computational studies on olefin polymerization catalyzed by rare-earth metal complexes [47,48,49,50], we became interested in the ROP of cyclic ester catalyzed by rare-earth metal complexes. To obtain better insight into the mechanism of the reaction shown in Scheme 1 as well as to establish the role of the bifunctional catalyst in the ROP of lactide, DFT calculations were performed for the initiation and propagation of lactide ROP catalyzed by an NHC-ligated yttrium complex (Scheme 1). It was found that the carbonyl of lactide is attacked by the N(SiMe3)2 group rather than the NHC moiety. The steric repulsion between the incoming monomer and the ligand framework plays an important role in the heterotactic selectivity.
2. Computational Details
All calculations were performed with Gaussian 09 program [51]. The DFT method of B3PW91 [52,53] was utilized for geometry optimization and subsequent frequency calculations. The 6-31G(d) basis set was used for C, H, O, and N atoms; the LANL2DZ basis set together with the associated effective core potential (ECP) was utilized for Si and Y atoms [54]. One d-polarization function (exponent of 0.284) was augmented for the basis set of Si atoms [55]. Such a computational strategy has been widely used for the study of transition metal-containing systems [56,57,58,59,60,61]. The transition states were ascertained by a single imaginary frequency for the correct mode. The solvation effects were considered through single-point calculations with the SMD [62] solvation model. These single-point calculations are based on the optimized structures and carried out at the level of B3PW91-D3 (B3PW91with Grimme’s DFT-D3 correction [63]. In the single-point calculations, the 6-311g(d,p) basis set for C, H, O, N, and Si atoms in addition to the Stuttgart/Dresden ECP together with the MWB28 basis set for Y were utilized. Toluene (ε = 2.37) was employed as a solvent in the SMD calculations. The free energy (ΔG, 298.15 K, 1 atm) in solution was obtained from the solvation single-point calculation, and the gas-phase Gibbs free energy correction was included. The sum of free energies of the isolated complex and the corresponding free lactide molecule was set to be an energy reference point in the overall reaction. The 3D molecular structure displayed in this paper was drawn by using CYLview [64].
3. Results and Discussion
3.1. The Chain Initiation Step of Lactide ROP Mediated by the Bifunctional Yttrium Complex
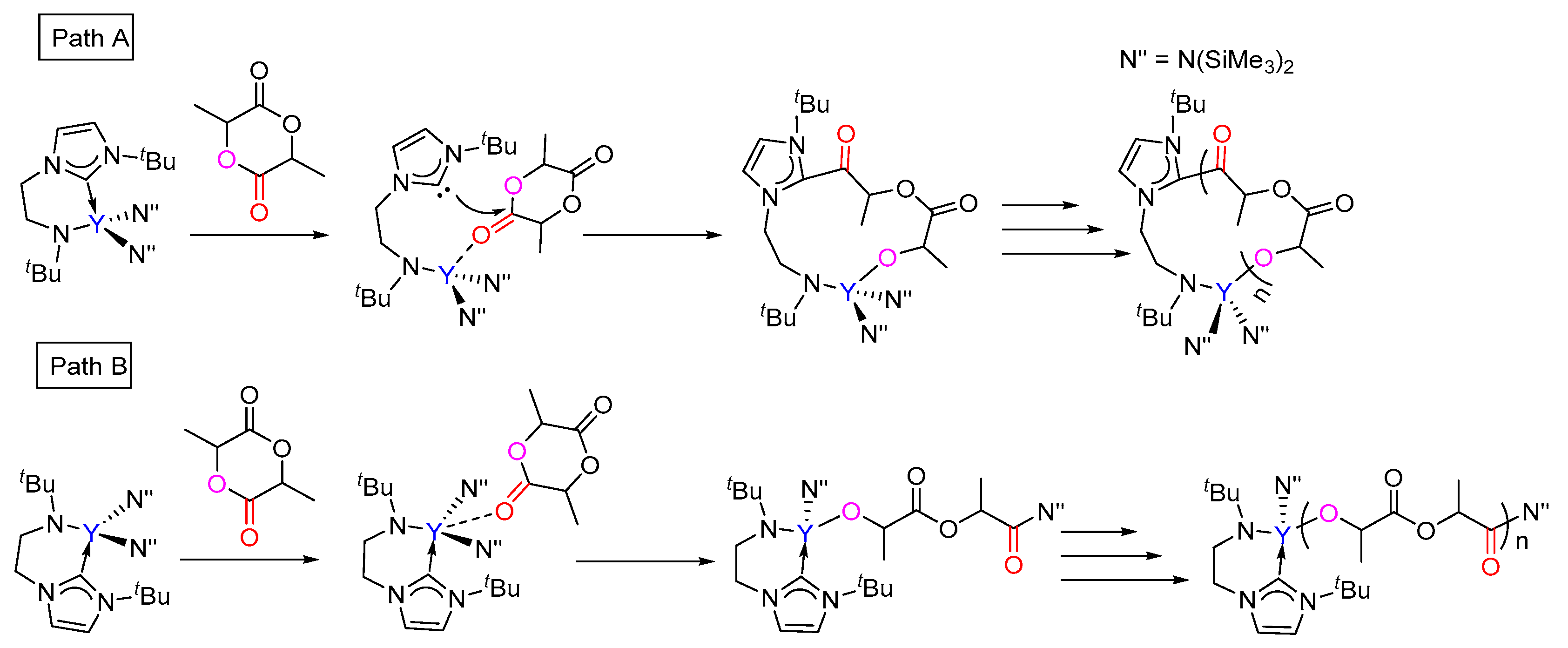
The coordination-insertion mechanism, which is generally accepted for the ROP of a cyclic ester, was considered for the current reaction. To address the mechanism of the ROP of rac-lactide initiated by bifunctional yttrium NHC catalyst in detail, the insertion and ring-opening of the (S,S)-lactide and (R,R)-lactide monomer were first examined, respectively. There are two possible different pathways for the chain initiation of the polymerization of rac-lactide catalyzed by the bifunctional yttrium catalyst. As shown in Scheme 3, (i) in path A, the lactide coordinates to the metal center, then the nucleophilic attack of the NHC group to the carbonyl C atom of the coordinating lactide takes place, leading to a cyclic insertion product; (ii) in path B, the nucleophilic attack of the N(SiMe3)2 group to the carbonyl C atom of the coordinating lactide occurs, yielding a linear insertion product.
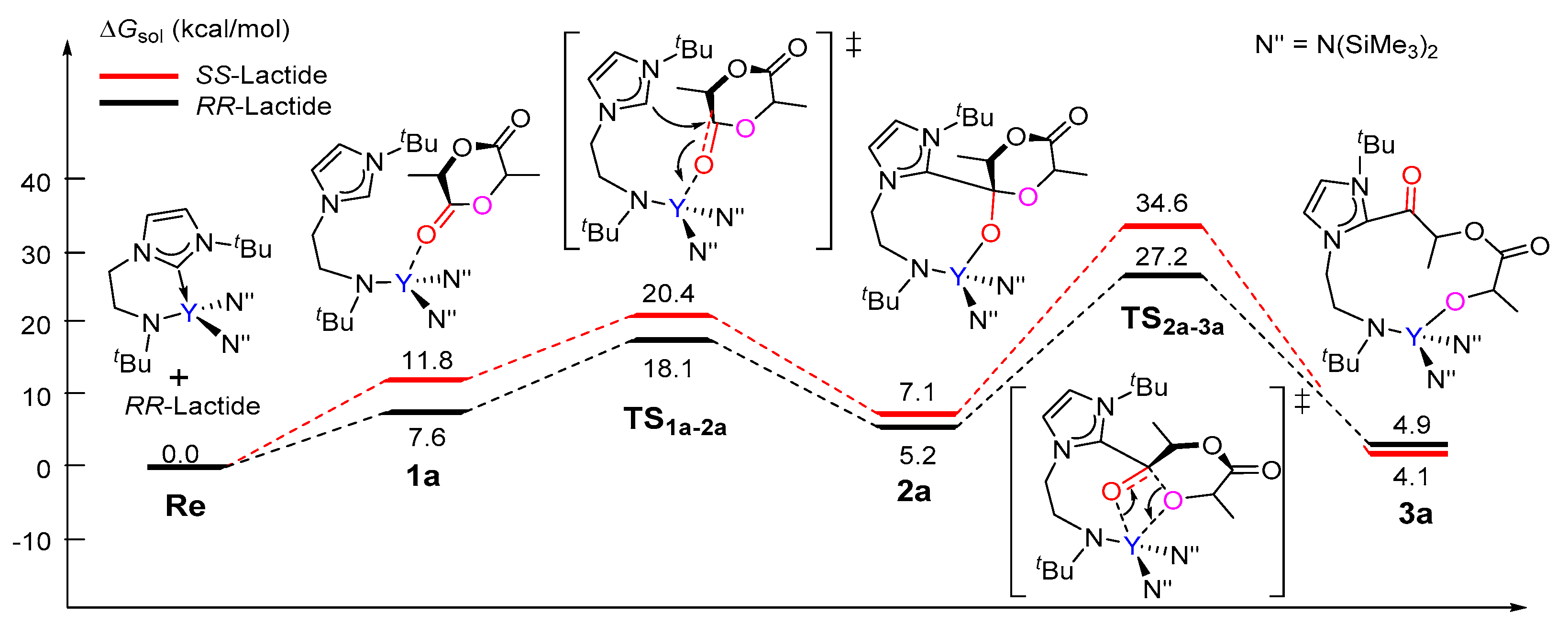
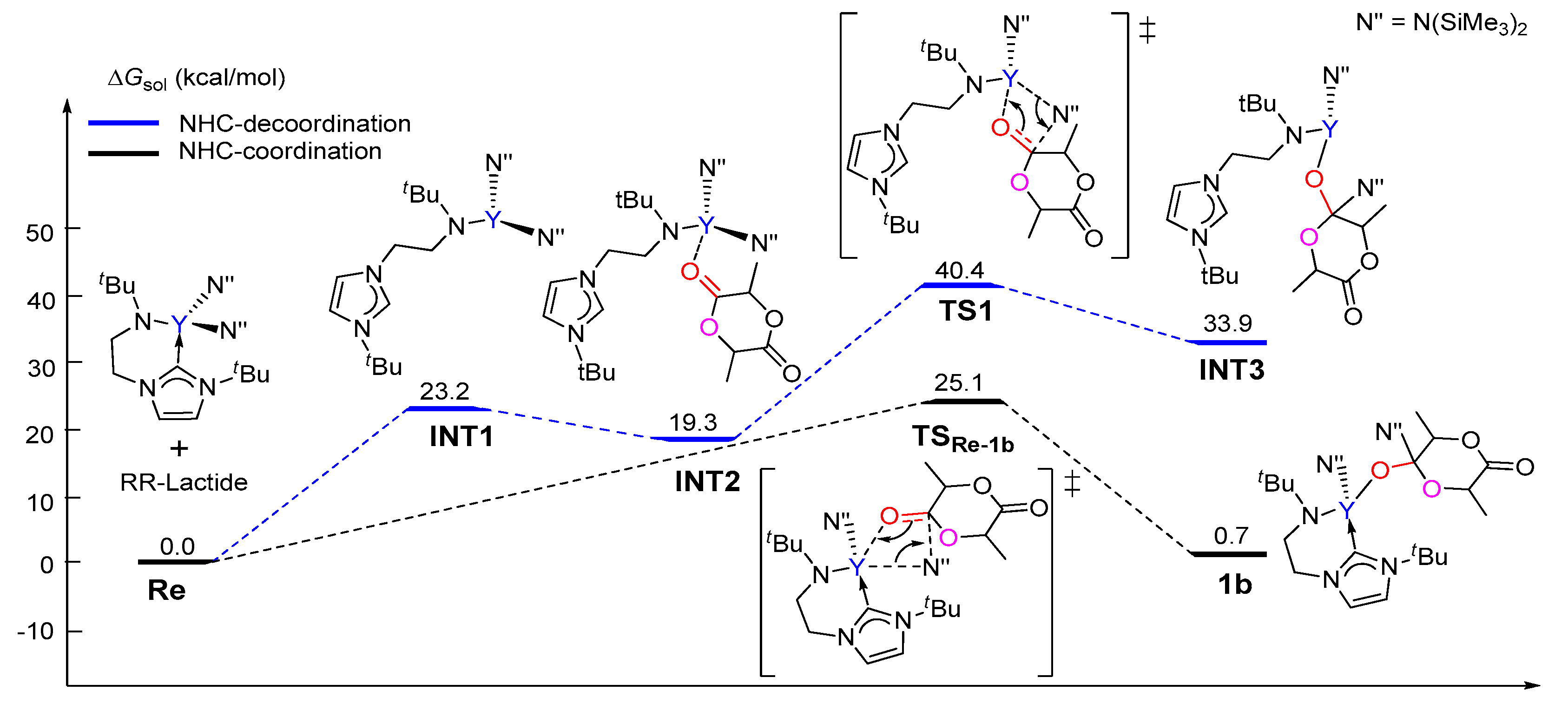
The calculated free energy profile for the chain initiation shown in path A is presented in Figure 1. As shown in this figure, the lactide coordinates to the metal center, which results in the (S,S)-lactide or (R,R)-lactide coordinating intermediate 1a (Y···O = 2.39 and 2.41 Å, respectively), destabilized by 11.8 kcal/mol and 7.6 kcal/mol with respect to the catalyst and lactide monomer. In the insertion transition state TS1a-2a, the carbonyl C atom of the coordinating lactide undergoes a change in hybridization from sp2 to sp3 due to the nucleophilic attack by the NHC species. The activation barriers for this step are 20.4 and 18.1 kcal/mol for the (S,S)- and (R,R)-lactide cases, respectively. The intermediate 2a with a newly formed C–C bond is therefore obtained. The second step proceeds through the transition state TS2a-3a, in which the lactide acyl–O bond is elongated (2.02 for (S,S) and 1.94 Å for (R,R) cases) with respect to the free lactide (1.35 Å) in concomitance with the formation of a new yttrium–alkoxide bond. The carbonyl carbon atom undergoes a change in hybridization from sp3 to sp2. This step results in the ring-opening of the lactide moiety and has an activation barrier of 34.6 kcal/mol for (S,S)-lactide and 27.2 kcal/mol for (R,R)-lactide. Moreover, the whole initiation step is endergonic by more than 4 kcal/mol. From an energy point of view, such high energy barriers and endergonic character suggest that path A is unlikely to be feasible pathway under the experimental conditions. This drove us to further investigate path B, as shown in Scheme 3.
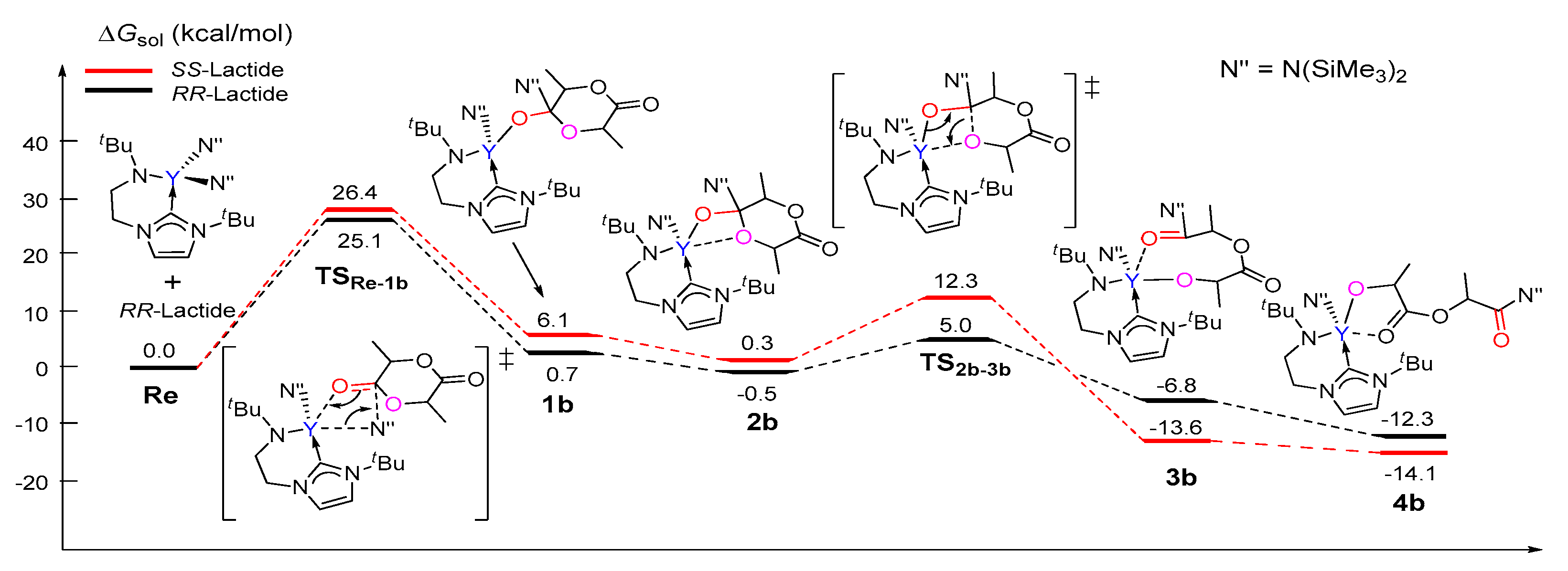
Unlike that in path A, the coordination complex of the monomer with the catalyst was not found on the energy profile of path B, possibly due to the coordinative saturation around the metal center (Figure 2). A nucleophilic attack of one N(SiMe3)2 group on the carbonyl group of (S,S)-lactide and (R,R)-lactide proceeds via the transition state TSRe-1b with an activation barrier of 26.4 kcal/mol and 25.1 kcal/mol, respectively. This step represents the rate-determining step for the first monomer insertion along with path B. The resulting tetrahedral intermediate 1b rearranges to give intermediate 2b. Then, 2b overcomes the transition state TS2b-3b to accomplish the ring-opening of the lactide moiety, accompanied with the concerted cleavage of the acyl–oxygen bond and the formation of the new metal-alkoxyl bond. After the ring-opening, the chain reorganization occurs, affording a more stable five-membered metallacyclic product 4b. The calculated energy barriers for the ring-opening step are less than that of the insertion step. As a whole, the conversion of the reactant into a product is exergonic by 14.1 and 12.3 kcal/mol for (S,S)-lactide and (R,R)-lactide, respectively (Figure 2). A comparison of the energy profiles for paths A and B (Figure 1 and Figure 2) indicates that path B is more kinetically favorable than path A (an energy barrier of 34.6 kcal/mol and 27.2 kcal/mol vs. 26.4 kcal/mol and 25.1 kcal/mol), in addition to being more thermodynamically favorable (4.9 kcal/mol and 4.1 kcal/mol vs. −12.3 kcal/mol and −14.1 kcal/mol). It is also obvious that the reaction of (R,R)-lactide is more kinetically favorable than that of (S,S)-lactide (Figure 1 and Figure 2). These results suggest that the NHC moiety could not directly participate in the insertion and ring-opening processes and the (R,R)-lactide could be favorably involved at the chain initiation step.
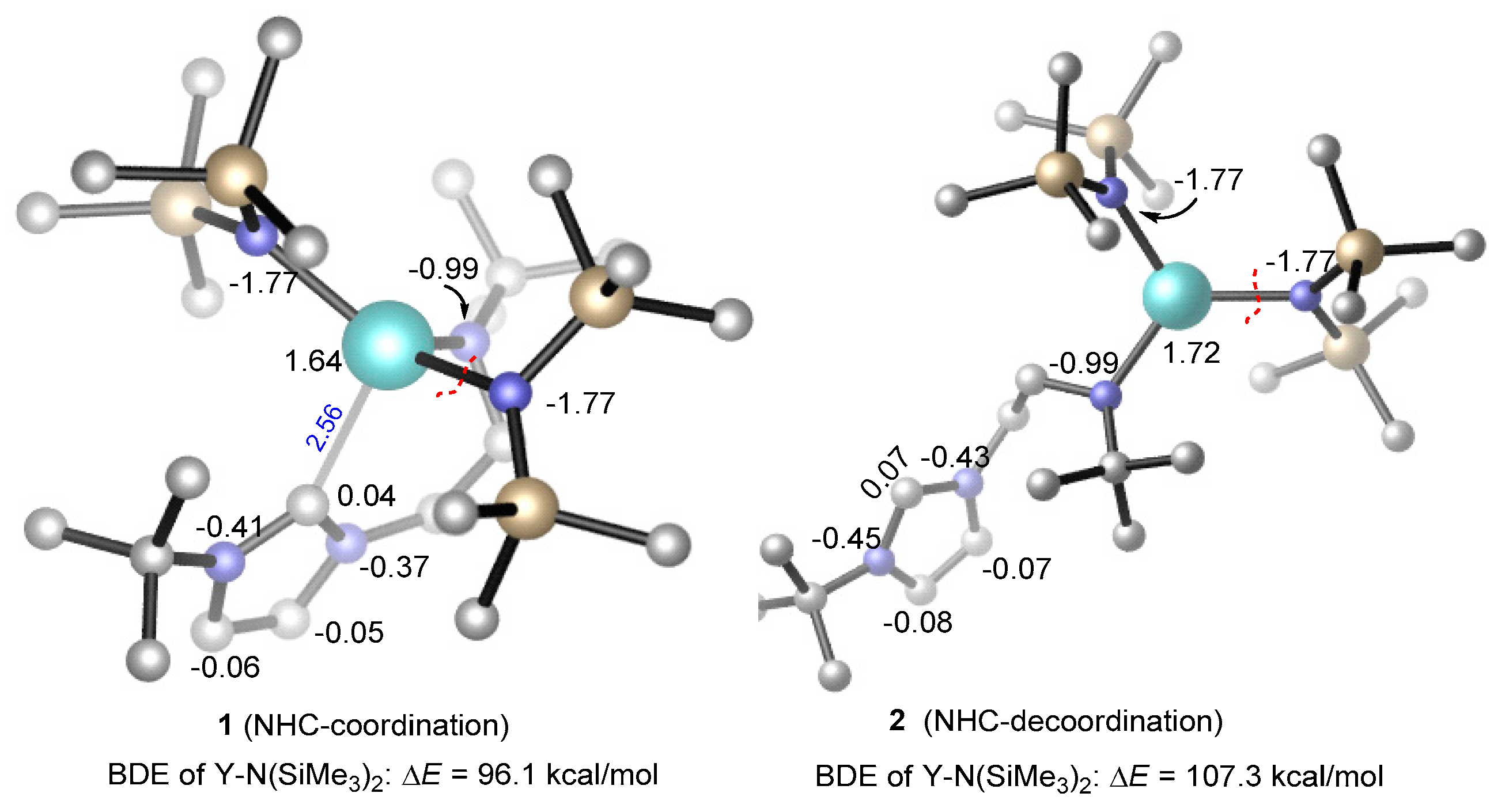
The reasonable chain initiation pathway was studied, as shown above. However, the role of NHC is unclear in the process of the ROP of rac-lactide. In order to further investigate the role of NHC in the chain initiation step, we modelled the de-coordination of the NHC moiety from the metal center via a single-bond rotation in the complex. The dissociation energies of a Y–N(SiMe3)2 bond were cleaved for the monomer insertion as well as the insertion of the first (R,R)-lactide monomer before (1) and after (2) the rotation was calculated (Figure 3 and Figure 4). Obviously, in the case of NHC de-coordination, the dissociation energy of the Y–N(SiMe3)2 bond (ΔE = 107.3 kcal/mol) is larger than that for the NHC-coordination case (ΔE = 96.1 kcal/mol, Figure 3), because the NHC moiety serves as a good electron donor and could decrease the Lewis acidity of the metal center (NBO (Natural bond orbital) charge of 1.64 vs. 1.72, Figure 3). One may suppose that the increase in the Y–N(SiMe3)2 bond dissociation energy could increase the insertion energy barrier of the monomer. Actually, the calculated energy profiles support this hypothesis. As shown in Figure 4, in the case of NHC de-coordination, the insertion energy barrier is much higher than that for the coordination case (40.4 vs. 25.1 kcal/mol). Moreover, the former case is significantly endergonic, while the latter is an almost an isoenergetic process. These results suggest that the NHC ligation could accelerate the monomer insertion and therefore improve the polymerization activity.
3.2. Selectivity of rac-Lactide Polymerization
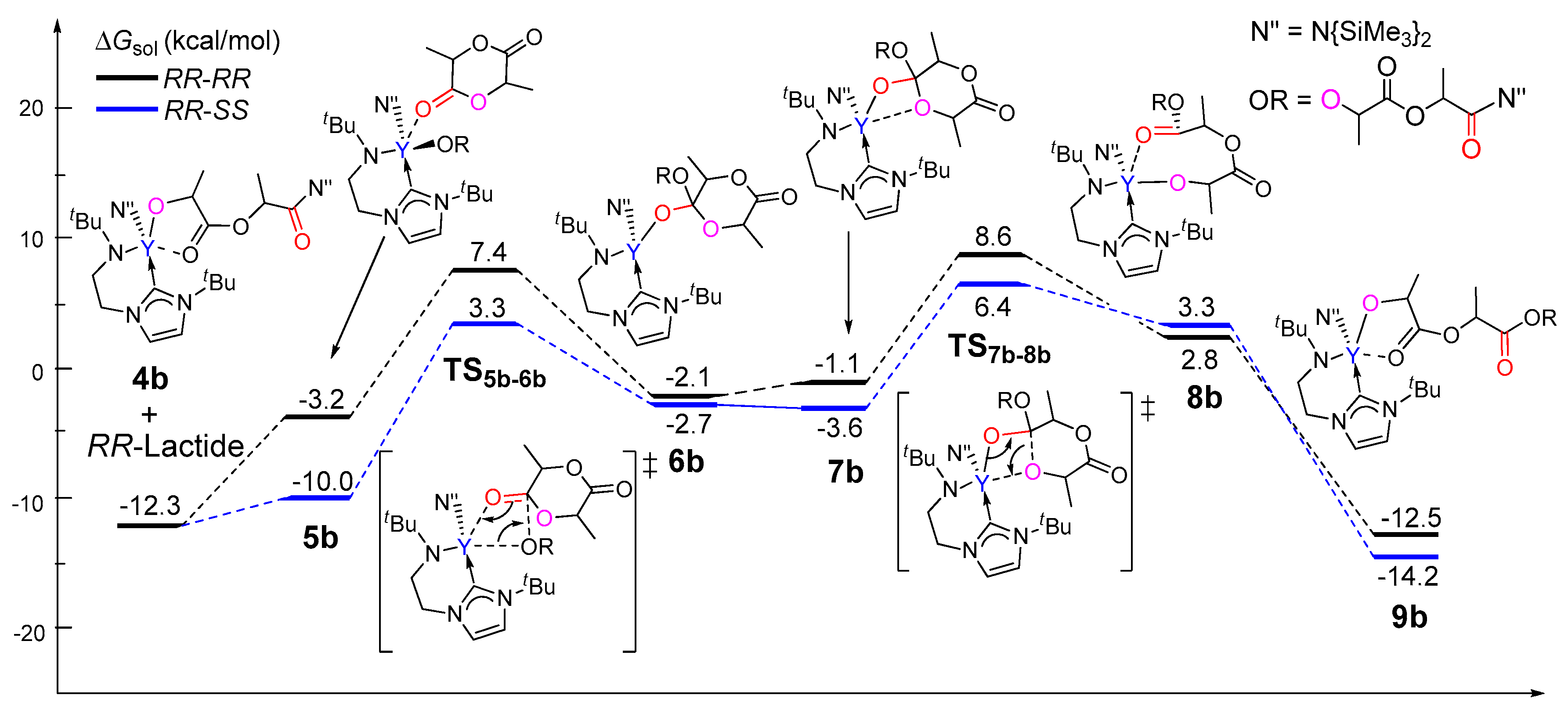
As aforementioned, in the chain initiation step, the reaction of (R,R)-lactide is more kinetically favorable than that of (S,S)-lactide. To explore the origin of stereocontrol in the rac-lactide polymerization, the insertions of both (R,R)- and (S,S)-lactide monomers were calculated in order to model the chain propagation on the basis of the resulting insertion product of the (R,R)-lactide 4b. As shown in Figure 5, the incoming monomer coordinates to 4b to form 5b, which could undergo a migratory insertion and subsequent ring-opening to finally give 9b, accompanied by structure isomerizations. A comparison of the coordination complex 4b indicates that the (S,S)-lactide is more thermodynamically favorable to coordinate to the metal center than its stereoisomers (relative energies of −10.0 vs. −3.2 kcal/mol). Kinetically, the ring-opening via TS7b-8b is the rate-determining step for both cases. Also, the free energy barrier for the case of (S,S)-lactide is 18.7 kcal/mol relative to 4b and the free monomer, which is lower than that for the case of its enantiomeric form (20.9 kcal/mol). These results suggest that the reaction of 4b with (S,S)-lactide is more favorable compared with (R,R)-lactide. This results in the (R,R)-(S,S) sequence, which is in line with the experimentally observed heterotactic selectivity. Considering that an achiral initiator cannot not lead any enantioselectivity in the chain initiation step, the case of (S,S)-enantiomer is also used for modeling the chain propagation. Actually, on the basis of the (S,S)-lactide-initiated product 4b(S,S), the subsequent reaction of the (R,R)-monomer is also more favorable than (S,S)-lactide, yielding the (S,S)-(R,R) heterotactic sequence (Figure S1).
To further elucidate the origin of stereoselectivity, an energy decomposition analysis [65,66,67,68] was performed for the coordination complex 5b, which shows a significant difference in energy between the two stereoisomers. In the energy decomposition analysis, 5b could be divided into two fragments, viz., lactide moiety (A) and the remaining part of the metal complex (B). The energies of the fragments A and B in the geometry of 5b were evaluated by single-point calculations. Such single-point energies of the fragments and the energy (corrected by the basis set superposition error) of 5b were used to estimate the interaction energy ΔEint. These energies, together with the energy of each fragment of their optimal geometries, allow for the estimation of the deformation energies of the two fragments, ΔEdef(A) and ΔEdef(B). As the total energy of 5b, ΔE5b is evaluated with respect to the energy of two separated fragments, and the relation ΔE5b = ΔEint + ΔEdef(A) + ΔEdef(B) holds. As shown in Table 1, the deformation energy of 5b(RR,SS) is similar to that of 5b(RR,RR), but the interaction energy is more favorable for 5b(RR,SS) (−32.1 kcal/mol) as compared to that of (RR,RR) (−26.9 kcal/mol), resulting in the lower ΔE5b for 5b(RR,SS) (−12.8 kcal/mol) in comparison with that for 5b(RR,RR) (−7.0 kcal/mol). It is obvious that the smaller ΔEint (more negative) for 5b(RR,SS) accounts for the lower energy barrier of 5b(RR,SS) in comparison with 5b(RR,RR). Therefore, the stability of the coordination complex 5b is controlled by the interaction energy between the monomer and the metal complex moiety.
Considering that the energies of TS5b-6b and TS7b-8b are close (Figure 5), and that the latter is unsuitable for such a fragment division, an energy decomposition analysis was also carried out for TS5b-6b to add better understanding to the stereoselectivity. It was found that the stronger interaction (more negative ΔEint) between the monomer and the metal could account for the lower energy barrier of TS5b-6b(RR,SS) in comparison with TS5b-6b(RR,RR). This suggests that the electronic factor plays an important role during the formation of the heterotactic sequence (Table S1).
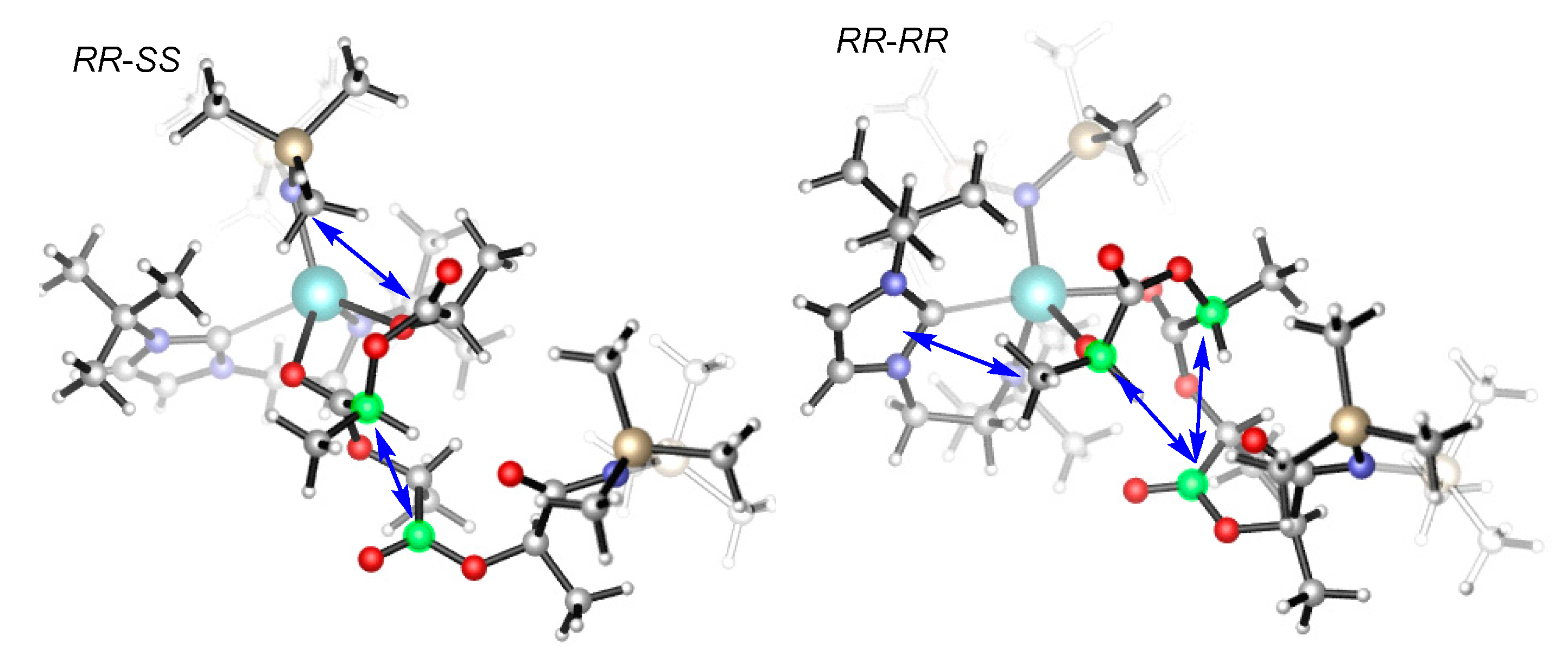
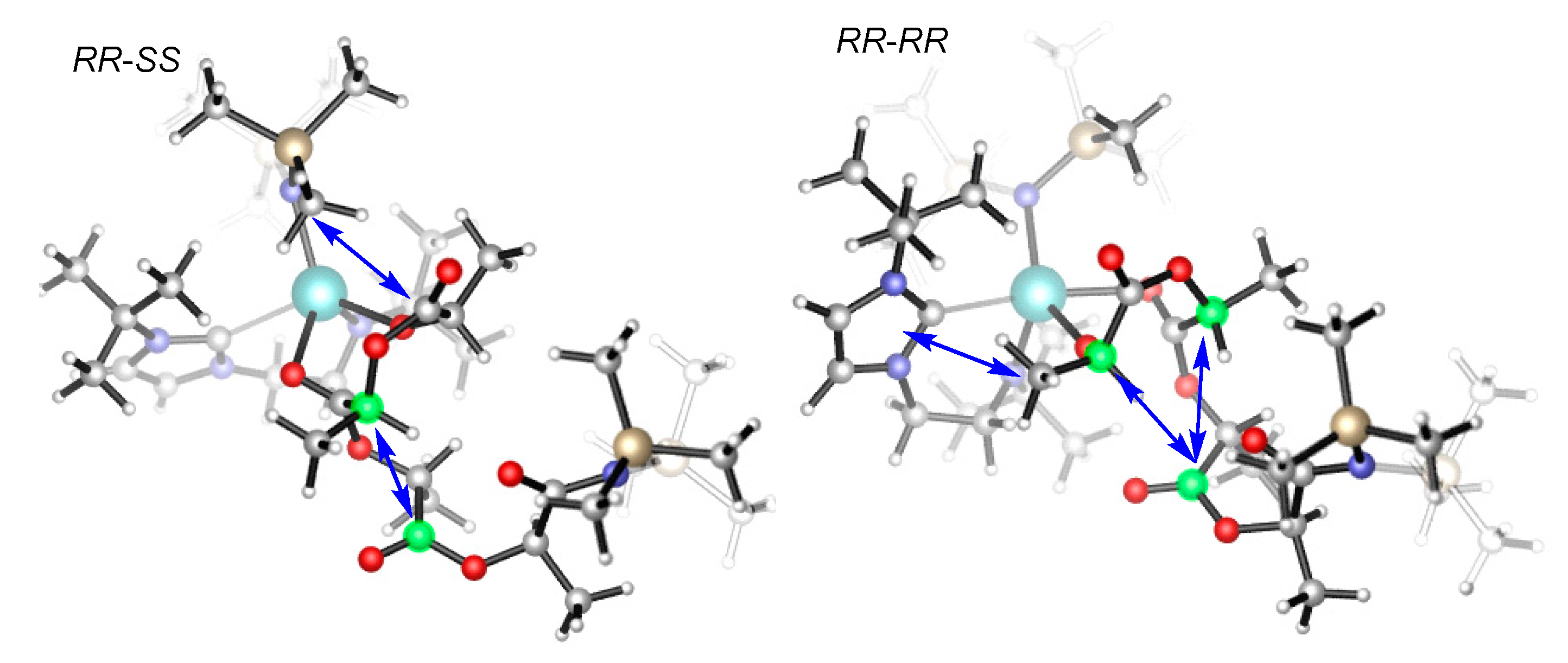
A closer examination of the structure of the ring-opening transition state (TS7b-8b) indicates that the (RR,RR)-sequenced structure shows a strong repulsion between the methyl of the incoming monomer and the ligand framework (Figure 6). The two chiral carbons of the incoming monomer sterically interact with the carbonyl group of the pre-enchained unit. However, the structure with the (RR,SS) sequence shows a repulsive interaction between the carbonyl group of the incoming monomer and the ligand framework. Furthermore, one chiral carbon of the incoming monomer sterically interacts with the carbonyl group of the pre-enchained unit. These structural features suggest that the steric clash between the incoming monomer and the ancillary ligand as well as the pre-enchained monomer unit could account for the stereoselectivity, in accordance with the ligand-assisted chain-end controlled mechanism [43]. Also, we know that the charge dispersion is conducive to the stability of a structure. In general, the smaller the values of |Q| and S, the more stable the structure [69]. The charge analyses indicate that the |Q| = 1.035 and S = 0.526 for the TS7b-8b(RR,SS) are smaller than those (|Q| = 1.038 and S = 0.558) for the TS7b-8b(RR,RR). Therefore, the charge dispersion also suggests that the former is more stable than the later (Figure S2).
4. Conclusions
The stereoselective polymerization of rac-lactide by bifunctional yttrium NHC catalyst LY[N(SiMe3)2]2 (L = tBuNCH2CH2(1-C{NCHCHNtBu})) was studied by using density functional theory. At the chain initiation stage, it was found that the NHC group could not be directly involved in the polymerization process and the N(SiMe3)2 group nucleophilically attacks the carbonyl C atom of the coordinating lactide instead. However, computational modelling suggests that the NHC moiety to be a good electron-donor that could accelerate the carbonyl insertion and thus improve the polymerization activity. The subsequent reaction of the second monomer was also calculated in order to model the chain propagation. Having achieved an agreement between the experimental and theoretical results, the origin of the observed stereoselectivity was further investigated. It was found that the steric repulsion between the incoming monomer and ancillary ligand as well as the pre-enchained monomer unit could account for the stereoselectivity, in accordance with the ligand-assisted chain-end controlled mechanism. In addition, the electronic factor may play an important role in the monomer insertion reaction to form a heterotactic sequence. The results reported in this work are expected to shed light on the development of stereoselective catalysts for the ring-opening polymerization of lactides.
Supplementary Materials
The following are available online at www.mdpi.com/2304-6740/5/3/46/s1, Figure S1: Calculated free energy profile for the ring-opening polymerization of the second rac-lcatide monomer on the basis of the resulting insertion product of the (S,S)-lactide 4b; Figure S2: the charge dispersion analyses of TS7b-8b; Table S1: The energy decomposition of TS5b-6b. Optimized Cartesian coordinates (XYZ) with the self-consistent field (SCF) energies and the imaginary frequencies of transition states.
Acknowledgments
This work was partially supported by the NSFC (Nos. 21429201, 21674014). The authors also thank the Fundamental Research Funds for the Central Universities (DUT2016TB08) and the Network and Information Center of the Dalian University of Technology for part of computational resources.
Author Contributions
Yincheng Wang and Yi Luo conceived and designed the calculations; Yincheng Wang performed the calculations; Yincheng Wang and Yi Luo analyzed the data and wrote the paper, with contributions from Yanan Zhao; Andleeb Mehmood polished the English sentence and grammar; Jingping Qu participated in the discussion; Yi Luo directed the project.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Uhrich, K.E.; Cannizzaro, S.M.; Langer, R.S. Polymeric systems for controlled drug release. Chem. Rev. 1999, 99, 3181–3198. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.P.; Kumar, V. New emerging trends in synthetic biodegradable polymers—Polylactide: A critique. Eur. Polym. J. 2007, 43, 4053–4074. [Google Scholar] [CrossRef]
- Albertsson, A.C.; Varma, I.K. Recent developments in ring opening polymerization of lactones for biomedical applications. Biomacromolecules 2003, 4, 1466–1486. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Liu, X.; Shang, X. Achiral lanthanide alkyl complexes bearing N,O multidentate ligands. Synthesis and catalysis of highly heteroselective ring-opening polymerization of rac-lactide. Organometallics 2007, 26, 2747–2757. [Google Scholar]
- Ragauskas, A.J.; Williams, C.K.; Davison, B.H.; Britovsek, G.; Cairney, J.; Eckert, C.A.; Frederick, W.J., Jr.; Hallett, J.P.; Leak, D.J.; Liotta, C.L. The path forward for biofuels and biomaterials. Science 2006, 311, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Platel, R.H.; Hodgson, L.M.; Williams, C.K. Biocompatible initiators for lactide polymerization. Polym. Rev. 2008, 48, 11–63. [Google Scholar] [CrossRef]
- Stanford, M.J.; Dove, A.P. Stereocontrolled ring-opening polymerisation of lactide. Chem. Soc. Rev. 2010, 39, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Rosen, T.; Goldberg, I.; Venditto, K.M. Tailor-made stereoblock copolymers of poly(lactic acid) by a truly living polymerization catalyst. J. Am. Chem. Soc. 2016, 138, 12041–12044. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Schmid, T.E.; Richard, V.; Haquette, P.; Raman, S.K.; Rager, M.N.; Gauvin, R.M.; Morin, Y.; Trivelli, X.; Guerineau, V. Mechanistic aspects of the polymerization of lactide using a highly efficient aluminum(III) catalytic system. J. Am. Chem. Soc. 2017, 139, 6217–6225. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.M. Stereocontrolled ring-opening polymerization of cyclic esters: Synthesis of new polyester microstructures. Chem. Soc. Rev. 2010, 39, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.A.; Crisci, A.G.D.; Hedrick, J.L.; Waymouth, R.M. Amidine-mediated zwitterionic polymerization of lactide. ACS Macro Lett. 2012, 1, 1113–1115. [Google Scholar] [CrossRef]
- Dove, A.P. Organic Catalysis for Ring-Opening Polymerization. ACS Macro Lett. 2012, 1, 1409–1412. [Google Scholar] [CrossRef]
- Dechy-Cabaret, O.; Martin-Vaca, B.; Bourissou, D. Controlled ring-opening polymerization of lactide and glycolide. Chem. Rev. 2004, 104, 6147–6176. [Google Scholar] [CrossRef] [PubMed]
- Dove, A.P. Controlled ring-opening polymerisation of cyclic esters: Polymer blocks in self. Chem. Commun. 2008, 48, 6446–6470. [Google Scholar] [CrossRef] [PubMed]
- Ajellal, N.; Carpentier, J.F.; Guillaume, C.; Guillaume, S.M.; Heloua, M.; Poiriera, V.; Sarazina, Y.; Trifonovb, A. Metal-catalyzed immortal ring-opening polymerization of lactones, lactides and cyclic carbonates. Dalton Trans. 2010, 39, 8363–8376. [Google Scholar] [CrossRef] [PubMed]
- Platel, R.H.; White, A.J.P.; Williams, C.K. Bis(phosphinic)diamido yttrium amide, alkoxide, and aryloxide complexes: An evaluation of lactide ring-opening polymerization initiator efficiency. Inorg. Chem. 2011, 50, 7718–7728. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Yu, T.L.; Chen, C.T.; Lin, C.C. Recent developments in main group metal complexes catalyzed/initiated polymerization of lactides and related cyclic esters. Coord. Chem. Rev. 2006, 250, 602–626. [Google Scholar] [CrossRef]
- Wheaton, C.A.; Hayes, P.G.; Ireland, B.J. Complexes of Mg, Ca and Zn as homogeneous catalysts for lactide polymerization. Dalton Trans. 2009, 4832–4846. [Google Scholar] [CrossRef] [PubMed]
- Amgoune, A.; Thomas, A.M.; Carpentier, J.F. Controlled rong-opening polymerization of lactide by group 3 metal complexes. Pure Appl. Chem. 2007, 79, 2013–2030. [Google Scholar] [CrossRef]
- Sauer, A.; Kapelski, A.; Fliedel, C.; Dagorne, S.; Kol, M.; Okuda, J. Structurally well-defined group 4 metal complexesas initiators for ring-opening polymerization of lactide monomers. Dalton Trans. 2013, 42, 9007–9023. [Google Scholar] [CrossRef] [PubMed]
- Le Roux, E. Recent advances on tailor-made titanium catalysts for biopolymer synthesis. Coord. Chem. Rev. 2016, 306, 65–85. [Google Scholar] [CrossRef]
- Dagorne, S.; Normand, M.; Kirillov, E.; Carpentier, J.F. Gallium and Indium complexes for ring-opening polymerization of cyclic ethers, esters and carbonates. Coord. Chem. Rev. 2013, 257, 1869–1886. [Google Scholar] [CrossRef] [Green Version]
- Connor, E.F.; Nyce, G.W.; Myers, M.; Möck, K.; Hedrick, J.L. First example of N-heterocyclic carbenes as catalysts for living polymerization: Organocatalytic ring-opening polymerization of cyclic esters. J. Am. Chem. Soc. 2002, 124, 914–915. [Google Scholar] [CrossRef] [PubMed]
- Kamber, N.E.; Jeong, W.; Waymouth, R.M. Organocatalytic ring-opening polymerization. Chem. Rev. 2007, 107, 5813–5840. [Google Scholar] [CrossRef] [PubMed]
- Kiesewetter, M.K.; Shin, E.J.; Hedrick, J.L.; Waymouth, R.M. Organocatalysis: Opportunities and challenges for polymer synthesis. Macromolecules 2010, 43, 2093–2107. [Google Scholar] [CrossRef]
- Acharya, A.K.; Chang, Y.A.; Jones, G.O.; Rice, J.E.; Hedrick, J.L.; Horn, H.W.; Waymouth, R.M. Experimental and computational studies on the mechanism of zwitterionic ring-opening polymerization of δ-valerolactone with N-heterocyclic carbenes. J. Phys. Chem. B 2014, 118, 6553–6560. [Google Scholar] [CrossRef] [PubMed]
- Suriano, F.; Coulembier, O.; Hedrick, J.L.; Dubois, P. Functionalized cyclic carbonates: From synthesis and metal-free catalyzed ring-opening polymerization to applications. Polym. Chem. 2011, 2, 528–533. [Google Scholar] [CrossRef]
- Piedra-Arroni, E.; Amgoune, A.; Bourissou, D. Dual catalysis: New approaches for the polymerization of lactones and polar olefins. Dalton Trans. 2013, 42, 9024–9029. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Zhang, H. Combining transition metal catalysis and organocatalysis: A broad new concept for catalysis. Chem. Soc. Rev. 2009, 40, 2745–2755. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Shi, X. When organocatalysis meets transition-metal catalysis. Eur. J. Org. Chem. 2010, 16, 2999–3025. [Google Scholar] [CrossRef]
- Piedra-Arroni, E.; Brignou, P.; Amgoune, A.; Guillaume, S.M.; Carpentier, J.F.; Bourissou, D. A dual organic/organometallic approach for catalytic ring-opening polymerization. Chem. Commun. 2011, 47, 9828–9830. [Google Scholar] [CrossRef] [PubMed]
- Brignou, P.; Guillaume, S.M.; Roisnel, T.; Bourissou, D.; Carpentier, J.-F. Discrete cationic zinc and magnesium complexes for dual organic/organometallic-catalyzed ring-opening polymerization of trimethylene carbonate. Chem. Eur. J. 2012, 18, 9360–9370. [Google Scholar] [CrossRef] [PubMed]
- Schnee, G.; Fliedel, C.; Aviles, T.; Dagrone, S. Neutral and cationic N-heterocyclic carbene zinc adducts and the BnOH/Zn(C6F5)2 binary mixture characterization and use in the ring-opening polymerization of β-Butyrolactone, lactides and trimethylene carbonate. Eur. J. Inorg. Chem. 2013, 3699–3709. [Google Scholar] [CrossRef]
- Piedra-Arroni, E.; Ladaviere, C.; Amgoune, A.; Bourissou, D. Ring-opening polymerization with Zn(C6F5)2-based lewis pairs: Original and efficient approach to cyclic polyesters. J. Am. Chem. Soc. 2013, 135, 13306–13309. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Liddle, S.T.; Mungur, S.A.; Rodden, M.; Blake, A.J.; Arnold, P.L. Bifunctional yttrium(III) and titanium(IV) NHC catalysts for lactide polymerization. Chem. Commun. 2006, 1124–1126. [Google Scholar] [CrossRef] [PubMed]
- Vieira, I.S.; Whitelaw, E.L.; Jones, M.D.; Pawlis, S.H. Synergistic empirical and theoretical study on the stereoselective mechanism for the aluminum salalen complex mediated polymerization of rac-lactide. Chem. Eur. J. 2013, 19, 4712–4716. [Google Scholar] [CrossRef] [PubMed]
- Dyer, H.E.; Huijser, S.; Susperregui, N.; Bonnet, F.; Schwarz, A.D.; Duchateau, R.; Maron, L.; Mountford, P. Ring-opening polymerization of rac-Lactide by bis(phenolate)amine-supported samarium borohydride complexes: An experimental and DFT Study. Organometallics 2010, 29, 3602–3621. [Google Scholar] [CrossRef]
- Broderick, E.M.; Guo, N.; Wu, T.; Vogel, C.S.; Xu, C.; Sutter, J.; Miller, J.T.; Meyer, K.; Cantat, T.; Diaconescu, P.L. Redox control of a polymerization catalyst by changing the oxidation state of the metal center. Chem. Commun. 2011, 47, 9897–9899. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Walshe, A.; Maron, L.; Baker, R.J. Ring-opening polymerization of epoxides catalyzed by uranyl complexes: An experimental and theoretical study of the reaction mechanism. Inorg. Chem. 2012, 51, 9132–9140. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Tschan, M.J.-L.; Roisnel, T.; Trivelli, X.; Gauvin, R.M.; Thomas, C.M.; Maron, L. Yttrium catalysts for syndioselective b-butyrolactone polymerization: On the origin of ligand-induced stereoselectivity. Polym. Chem. 2013, 4, 360–367. [Google Scholar] [CrossRef]
- Rosal, I.D.; Brignou, P.; Guillaume, S.M.; Carpentier, J.F.; Maron, L. DFT investigations on the ring-opening polymerization of substituted cyclic carbonates catalyzed by zinc-{β-diketiminate} complexes. Polym. Chem. 2015, 6, 3336–3352. [Google Scholar] [CrossRef] [Green Version]
- Tabthong, S.; Nanok, T.; Sumrit, P.; Kongsaeree, P.; Prabpai, S.; Chuawong, P.; Hormnirun, P. Bis(pyrrolidene) schiff base aluminum complexes as isoselective-biased initiators for the controlled ring-opening polymerization of rac-lactide: Experimental and theoretical Studies. Macromolecules 2015, 48, 6846–6861. [Google Scholar] [CrossRef]
- Marshall, E.L.; Gibson, V.C.; Rzepa, H.S. A computational analysis of the ring-opening polymerization of rac-lactide initiated by single-site β-diketiminate metal complexes: Defining the mechanistic pathway and the origin of stereocontrol. J. Am. Chem. Soc. 2005, 127, 6048–6051. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Yu, I.; Mehrkhodavandi, P.; Maron, L. Theoretical investigation of lactide ring-opening polymerization induced by a dinuclear indium catalyst. Organometallics 2013, 32, 6950–6956. [Google Scholar] [CrossRef]
- Fliedel, C.; Vila-Vicosa, D.; Calhorda, M.J.; Dagorne, S.; Aviles, T. Dinuclear Zinc–N-Heterocyclic carbine complexes either the controlled ring-opening polymerization of lactide or the controlled degradation of polylactide under mild conditions. ChemCatChem 2014, 6, 1357–1367. [Google Scholar] [CrossRef]
- Xu, T.Q.; Yang, G.W.; Liu, C.; Lu, X.B. Highly robust yttrium bis(phenolate) ether catalysts for excellent isoselective ring-opening polymerization of racemic lactide. Macromolecules 2017, 50, 515–522. [Google Scholar] [CrossRef]
- Kang, X.H.; Song, Y.M.; Luo, Y.; Li, G.; Hou, Z.M.; Qu, J.P. Computational studies on isospecific polymerization of 1-hexene catalyzed by cationic rare-earth metal alkyl complex bearing a C3 iPr-trisox ligand. Macromolecules 2012, 45, 640–651. [Google Scholar] [CrossRef]
- Kang, X.H.; Yamamoto, A.; Nishiura, M.; Luo, Y.; Hou, Z.M. Computational analyses of the effect of lewis bases on styrene polymerization catalyzed by cationic scandium half-sandwich complexes. Organometallics 2015, 34, 5540–5548. [Google Scholar] [CrossRef]
- Kang, X.H.; Zhou, G.L.; Wang, X.B.; Luo, Y.; Hou, Z.M.; Qu, J.P. Alkyl effects on the chain initiation efficiency of olefin polymerization by cationic half-sandwich scandium catalysts: A DFT study. Organometallics 2016, 35, 913–920. [Google Scholar] [CrossRef]
- Kang, X.H.; Luo, Y.; Zhou, G.L.; Wang, X.B.; Yu, X.R.; Hou, Z.M.; Qu, J.P. Theoretical mechanistic studies on the trans-1,4-specific polymerization of isoprene catalyzed by a cationic La–Al binuclear complex. Macromolecules 2014, 47, 4596–4606. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, Y.; Mennucci, B.; Petersson, G.A. Gaussian 09, Revision A.02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5653. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Yang, Y.; Weaver, M.N.; Merz, K.M., Jr. Assessment of the “6-31 + G** + LANL2DZ” mixed basis set coupled with density functional theory methods and the effective core potential: Prediction of heats of formation and ionization potentials for first-row-transition-metal complexes. J. Phys. Chem. A 2009, 113, 9843–9851. [Google Scholar] [CrossRef] [PubMed]
- Hoellwarth, A.; Boehme, M.; Dapprich, S.; Ehlers, A.W.; Gobbi, A.; Jonas, V.; Köhler, K.F.; Stegmann, R.; Veldkamp, A.; Frenking, G. A set of d-polarization functions for pseudo-potential basis sets of the main group elements Al–Bi and f-type polarization functions for Zn, Cd, Hg. Chem. Phys. Lett. 1993, 208, 237–240. [Google Scholar]
- Liu, Y.; Liu, Y.; Drew, M.G.B. Correlation between regioselectivity and site charge in propene polymerisation catalysed by metallocene. Struct. Chem. 2010, 21, 21–28. [Google Scholar] [CrossRef]
- Zhang, C.; Yu, S.; Zhang, L.; Li, H.Y.; Wang, Z.X. DFT mechanistic study of the H2-assisted chain transfer copolymerization of propylene and p-methylstyrene catalyzed by zirconocene complex. J. Polym. Sci. Part A Polym. Chem. 2015, 53, 576–585. [Google Scholar] [CrossRef]
- Valente, A.; Zinck, P.; Mortreux, A.; Visseauxa, M.; Mendes, P.J.G.; Silva, T.J.L.; Garcia, M.H. Polymerization of ε-caprolactone using ruthenium(II) mixed metallocene catalysts and isopropyl alcohol: Living character and mechanistic study. J. Mol. Catal. A Chem. 2011, 346, 102–110. [Google Scholar] [CrossRef]
- Jitonnom, J.; Molloy, R.; Punyodom, W.; Meelua, W. Theoretical studies on aluminum trialkoxide-initiated lactone ring-opening polymerizations: Roles of alkoxide substituent and monomer ring structure. Comput. Theor. Chem. 2016, 1097, 25–32. [Google Scholar] [CrossRef]
- Jitonnom, J.; Meelua, W. Effects of silicon-bridge and π-ligands on the electronic structures and related properties of dimethyl zirconocene polymerization catalysts: A comparative theoretical study. Chiang Mai J. Sci. 2014, 41, 1220–1229. [Google Scholar]
- Jitonnom, J.; Sontag, C. Catalytic oxidation of glucose with hydrogen peroxide and colloidal gold as pseudo-homogenous catalyst: A combined experimental and theoretical investigation. Chiang Mai J. Sci. 2016, 43, 825–833. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H–Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Legault, C.Y. CLYview, Version 1.0b; University of California: Los Angeles, CA, USA, 2007. [Google Scholar]
- Kitaura, K.; Morokuma, K. A new energy decomposition scheme for molecular interactions within the Hartree-Fock approximation. Int. J. Quantum Chem. 1976, 10, 325–340. [Google Scholar] [CrossRef]
- Pan, Y.; Xu, X.; Wei, N.N.; Hao, C.; Zhu, X.D.; He, G.H. DFT study on 1,7-octadiene polymerization catalyzed by non-bridged half-titanocene system. RSC Adv. 2016, 6, 69939–69946. [Google Scholar] [CrossRef]
- Li, Y.; Qi, X.; Lei, Y.; Lan, Y. Mechanism and selectivity for zinc-mediated cycloaddition of azides with alkynes: A computational study. RSC Adv. 2015, 5, 49802–49808. [Google Scholar] [CrossRef]
- Bhattacharjee, R.; Nijamudheen, A.; Datta, A. Mechanistic insights into the synergistic catalysis by Au(I), Ga(III), and counterions in the nakamura reaction. Org. Biomol. Chem. 2015, 13, 7412–7420. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Luo, G.; Hou, Z.M.; Luo, Y. Mechanistic insights into scandium-catalyzed hydroaminoalkylation of olefins with amines: Origin of regioselectivity and charge-based prediction model. Organometallics 2017, 36, 1557–1565. [Google Scholar] [CrossRef]
Scheme 1.
The ring-opening polymerization of rac-lactide by the bifunctional yttrium catalyst.
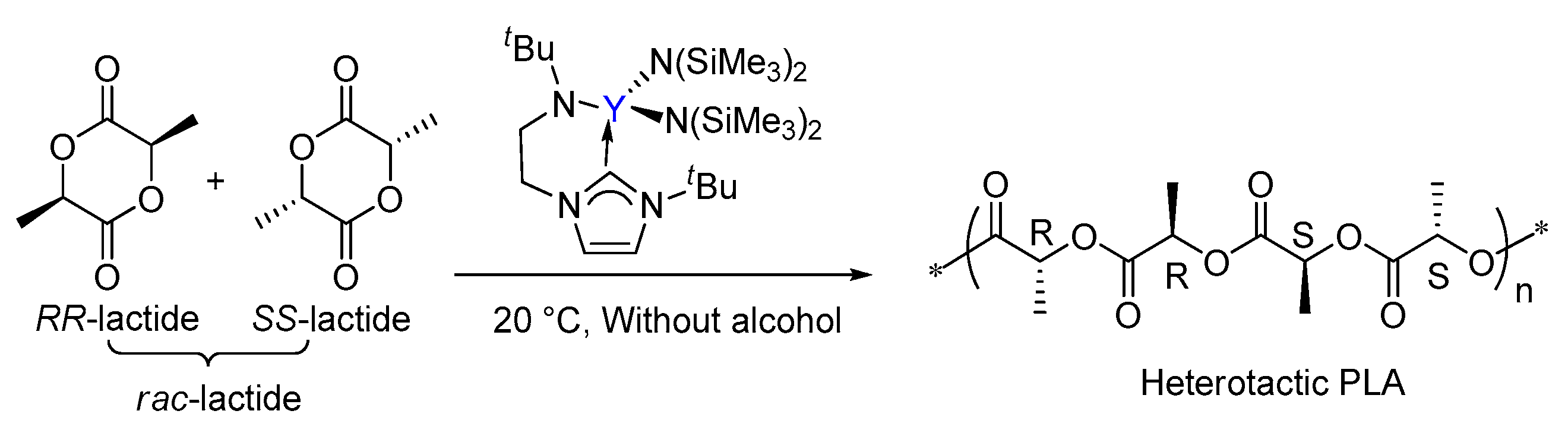
Scheme 2.
The previously proposed bifunctional activation mode of rac-lactide by the bifunctional yttrium complex.
Scheme 2.
The previously proposed bifunctional activation mode of rac-lactide by the bifunctional yttrium complex.
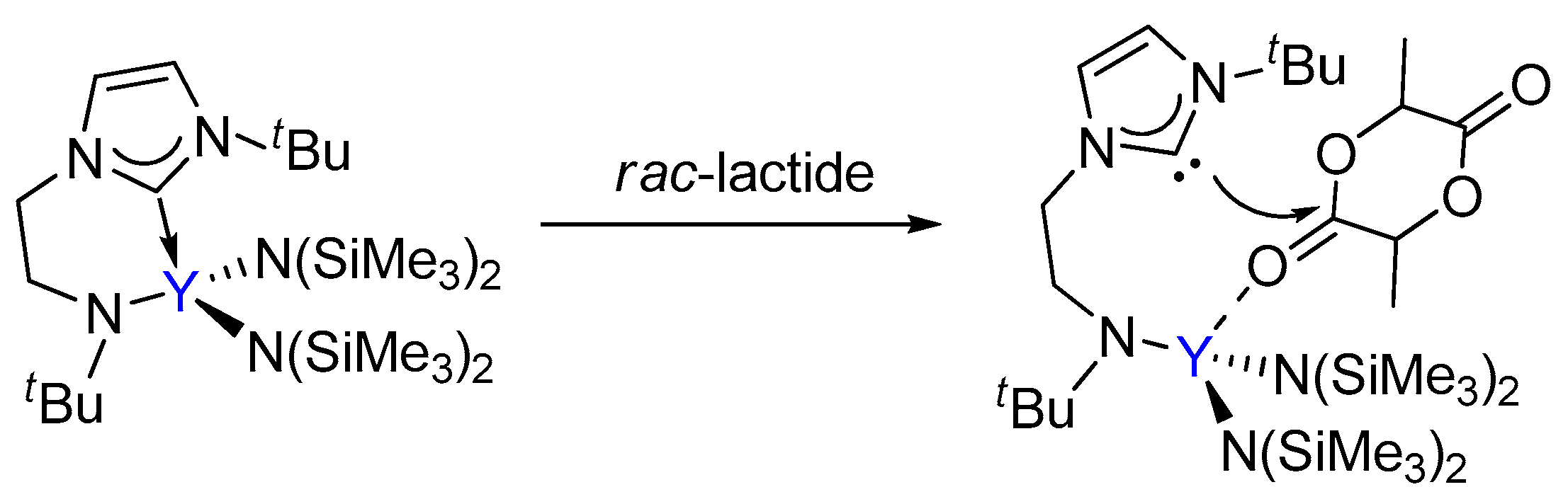
Scheme 3.
Possible pathways for the ROP of lactide catalyzed by the bifunctional yttrium complex.
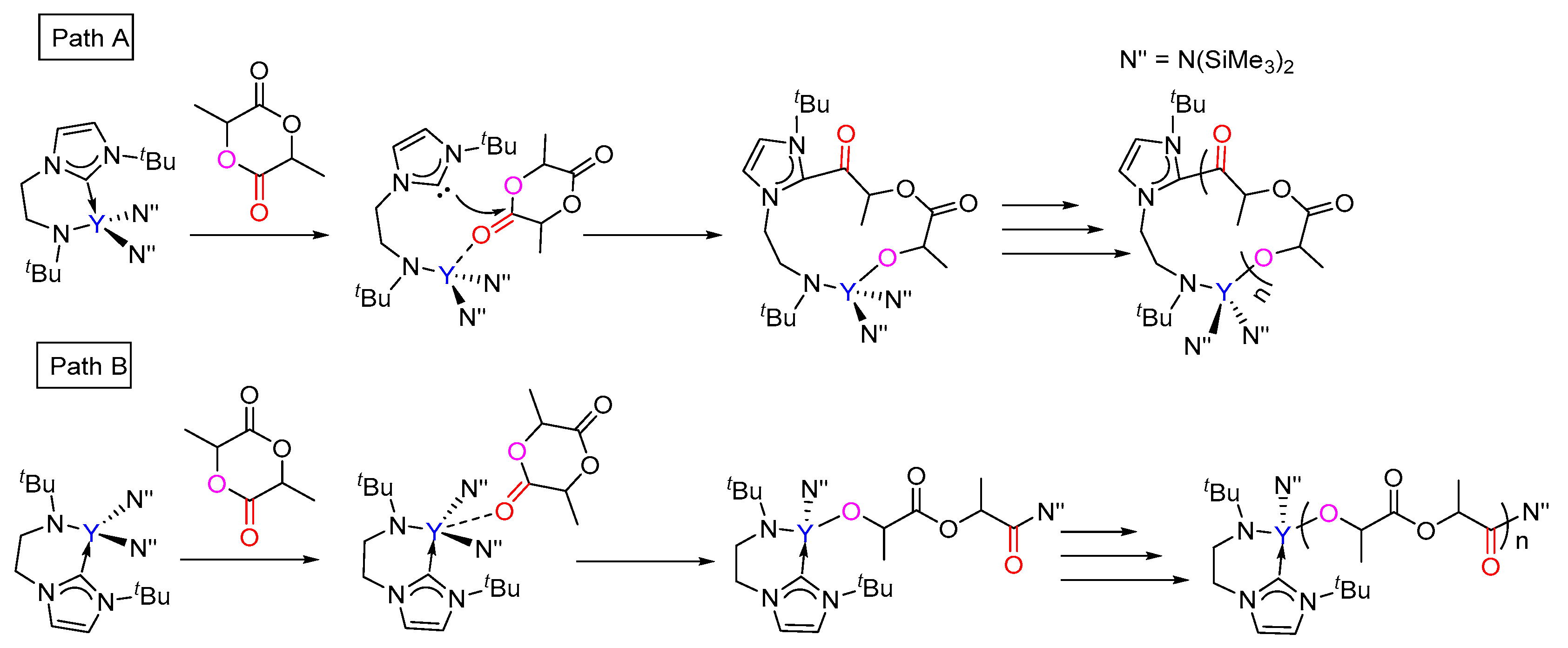
Figure 1.
Calculated free energy profile for the chain initiation of the ROP of rac-lactide along path A. TS1a-2a, nucleophilic addition transition state; TS2a-3a, ring-opening transition state.
Figure 1.
Calculated free energy profile for the chain initiation of the ROP of rac-lactide along path A. TS1a-2a, nucleophilic addition transition state; TS2a-3a, ring-opening transition state.

Figure 2.
Calculated free energy profile for the chain initiation of the ROP of rac-lactide along path B. TSRe-1b, nucleophilic addition transition state; TS2b-3b, ring-opening transition state.
Figure 2.
Calculated free energy profile for the chain initiation of the ROP of rac-lactide along path B. TSRe-1b, nucleophilic addition transition state; TS2b-3b, ring-opening transition state.
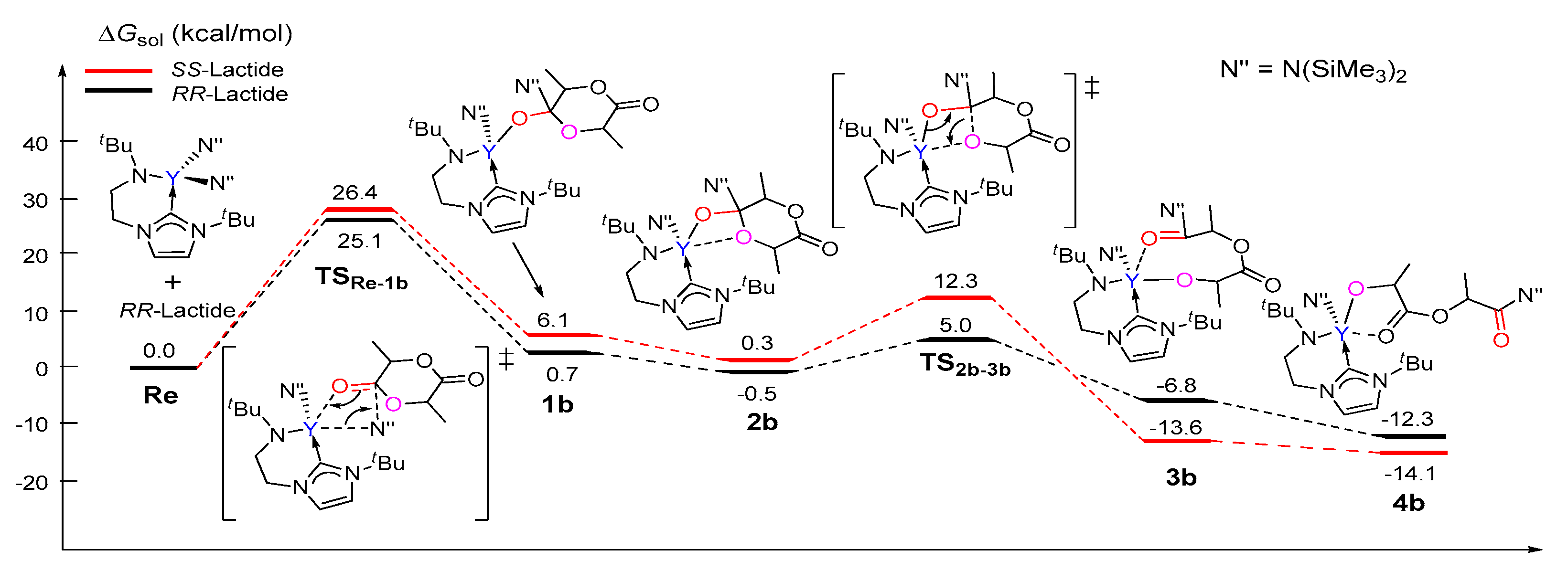
Figure 3.
The bond dissociation energy of Y–N(SiMe3)2 in the coordination and de-coordination of the N-heterocyclic carbine (NHC) moiety. NBO (Natural bond orbital) atomic charges are given in black. All H atoms are omitted for clarity.
Figure 3.
The bond dissociation energy of Y–N(SiMe3)2 in the coordination and de-coordination of the N-heterocyclic carbine (NHC) moiety. NBO (Natural bond orbital) atomic charges are given in black. All H atoms are omitted for clarity.
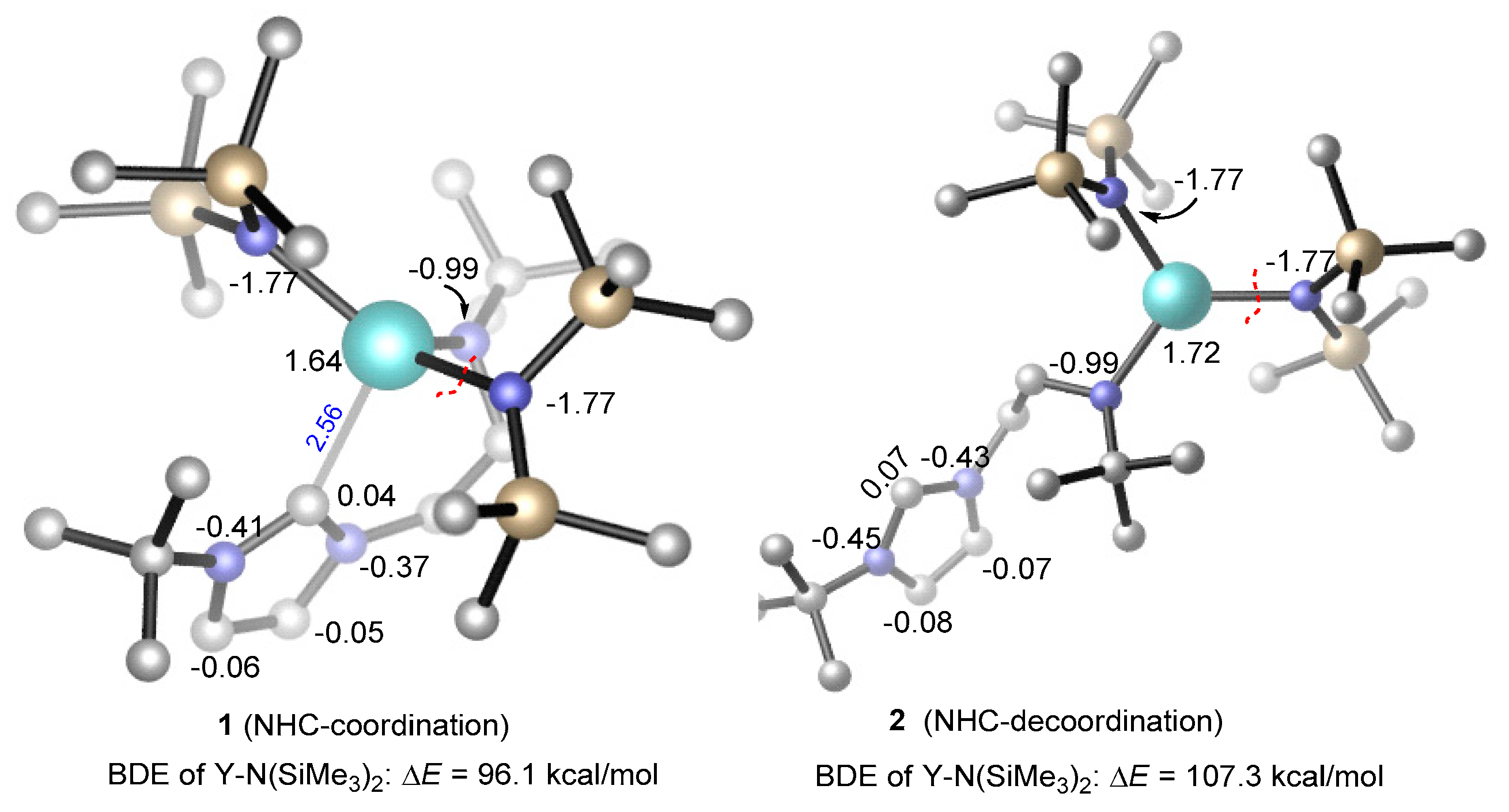
Figure 4.
Calculated free energy profiles for the insertion of the (R,R)-lactide monomer in the cases of NHC de-coordination and coordination.
Figure 4.
Calculated free energy profiles for the insertion of the (R,R)-lactide monomer in the cases of NHC de-coordination and coordination.
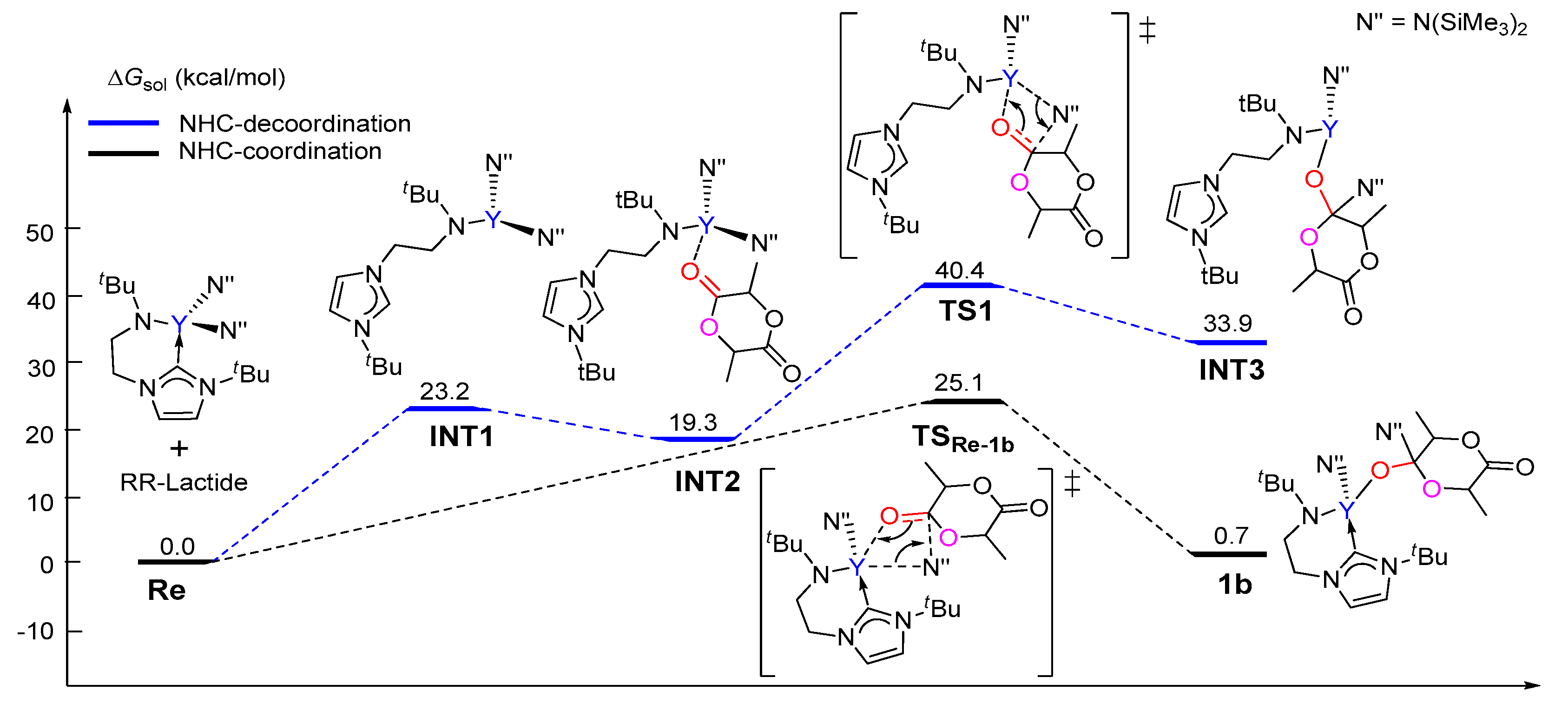
Figure 5.
Calculated free energy profile for the ring-opening polymerization of the second rac-lactide monomer.
Figure 5.
Calculated free energy profile for the ring-opening polymerization of the second rac-lactide monomer.
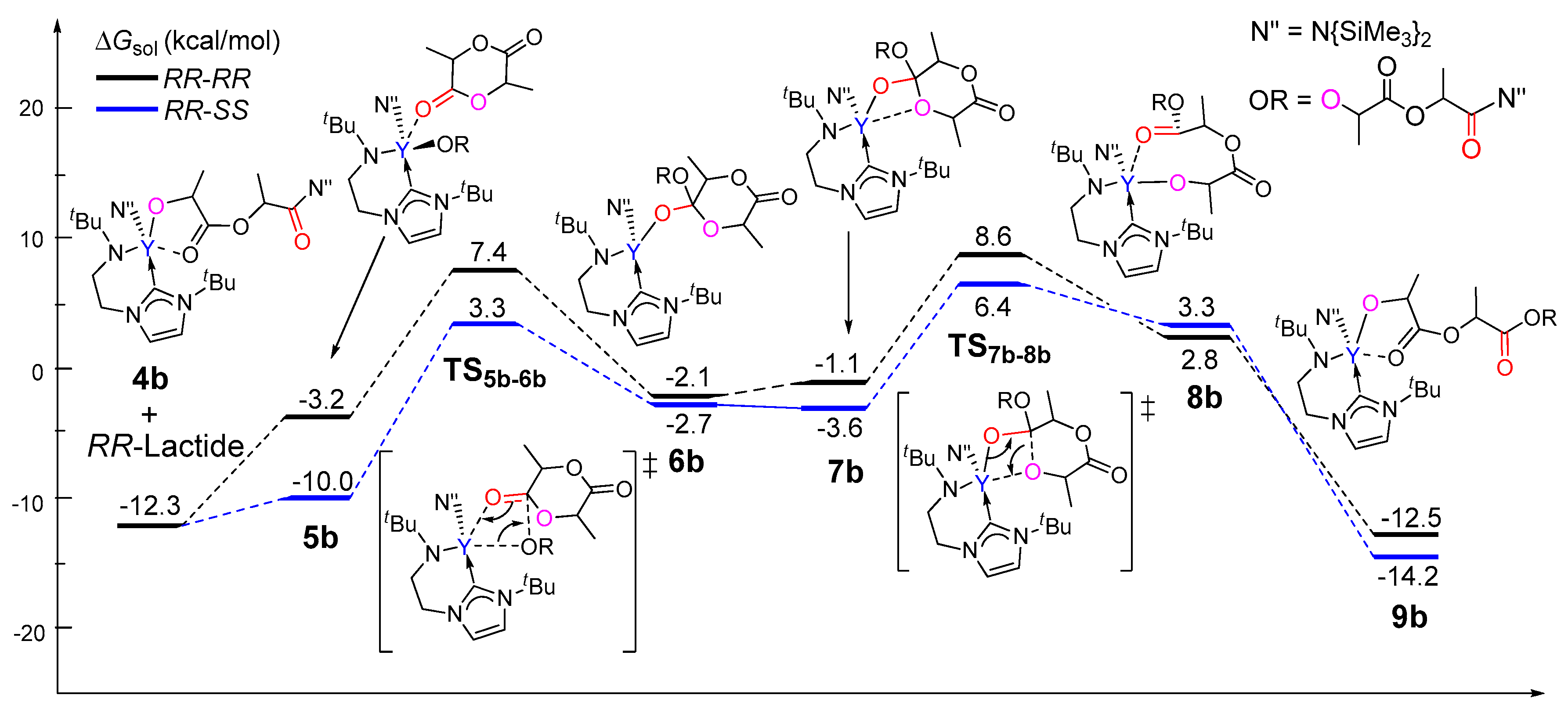
Figure 6.
Optimized geometries of TS7b-8b; the arrows show repulsive interaction.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
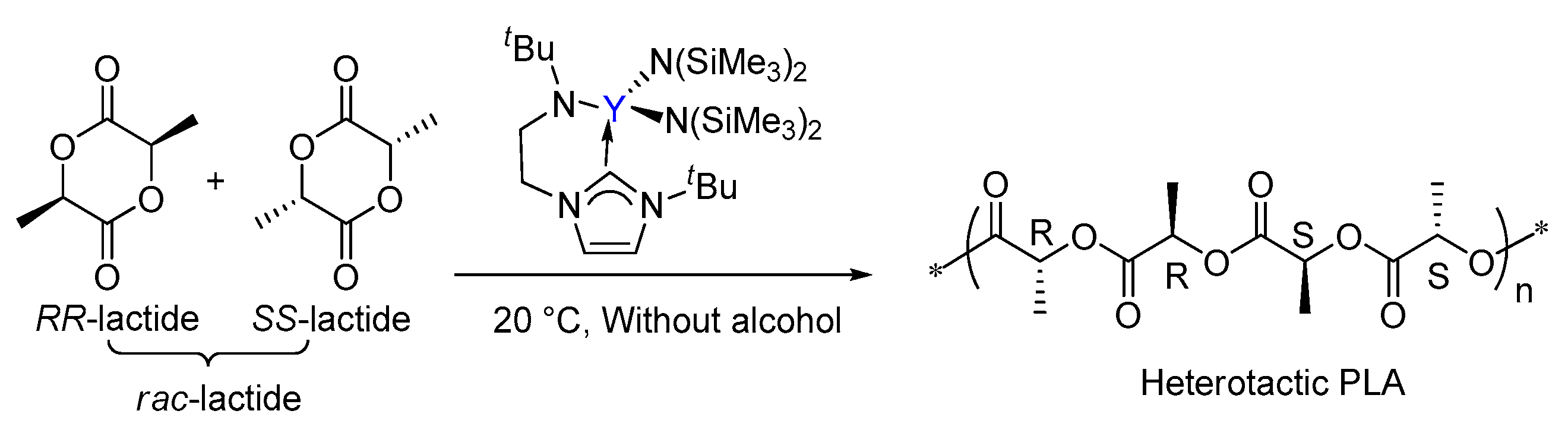{kind=link}
{kind=link}
{kind=link}
Table 1.
The energy decomposition of 5b (kcal/mol).
| Structure | ΔEint | ΔEdef(A) 1 | ΔEdef(B) 2 | ΔEdef | ΔE5b |
|---|---|---|---|---|---|
| 5b (RR,RR) | −26.9 | 0.9 | 19 | 19.9 | −7 |
| 5b (RR,SS) | −32.1 | 1.2 | 18.1 | 19.3 | −12.8 |
1 Deformation energy of the lactide monomer fragment in 5b; 2 Deformation energy of the remaining part of 5b.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wang, Y.; Mehmood, A.; Zhao, Y.; Qu, J.; Luo, Y. Computational Studies on the Selective Polymerization of Lactide Catalyzed by Bifunctional Yttrium NHC Catalyst. Inorganics 2017, 5, 46. https://doi.org/10.3390/inorganics5030046
AMA Style
Wang Y, Mehmood A, Zhao Y, Qu J, Luo Y. Computational Studies on the Selective Polymerization of Lactide Catalyzed by Bifunctional Yttrium NHC Catalyst. Inorganics. 2017; 5(3):46. https://doi.org/10.3390/inorganics5030046
Chicago/Turabian StyleWang, Yincheng, Andleeb Mehmood, Yanan Zhao, Jingping Qu, and Yi Luo. 2017. "Computational Studies on the Selective Polymerization of Lactide Catalyzed by Bifunctional Yttrium NHC Catalyst" Inorganics 5, no. 3: 46. https://doi.org/10.3390/inorganics5030046
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.