Synthesis, Crystal Structure, and Chemical-Bonding Analysis of BaZn(NCN)2



1
Institute of Inorganic Chemistry, RWTH Aachen University, 52056 Aachen, Germany
2
Jülich-Aachen Research Alliance (JARA-HPC), RWTH Aachen University, 52056 Aachen, Germany
*
Author to whom correspondence should be addressed.
Inorganics 2018, 6(1), 1; https://doi.org/10.3390/inorganics6010001
Submission received: 1 December 2017
/
Revised: 20 December 2017
/
Accepted: 21 December 2017
/
Published: 24 December 2017
(This article belongs to the Section Inorganic Solid-State Chemistry)
Abstract
:The ternary carbodiimide BaZn(NCN)2 was prepared by a solid-state metathesis reaction between BaF2, ZnF2, and Li2NCN in a 1:1:2 molar ratio, and its crystal structure was determined from Rietveld refinement of X-ray data. BaZn(NCN)2 represents the aristotype of the LiBa2Al(NCN)4 structure which is unique to carbodiimide/cyanamide chemistry and is well regarded as being constructed from ZnN4 tetrahedra, sharing edges and vertices through NCN2− units to form corrugated layers with Ba2+ in the interlayer voids. Structural anomalies in the shape of the cyanamide units are addressed via IR spectrometry and DFT calculations, which suggest the presence of slightly bent N=C=N2− carbodiimide units with C2v symmetry. Moreover, chemical-bonding analysis within the framework of crystal orbital Hamilton population (COHP) reveals striking similarities between the bonding interactions in BaZn(NCN)2 and SrZn(NCN)2 despite their contrasting crystal structures. BaZn(NCN)2 is only the second example of a ternary post-transition metal carbodiimide, and its realization paves the way for the preparation of analogues featuring divalent transition metals at the tetrahedral Zn2+ site.
1. Introduction
Molecular cyanamide NCNH2, along with substituted derivatives, are commonplace in organic chemistry where they express an unusual duality in that they are capable of reacting as both electrophiles and nucleophiles. That said, the cyanamide moiety is not only the preserve of the organic chemist because deprotonation of NCNH2 can lead to the formation of salt-like inorganic compounds of NCNH− and NCN2−. For example, the doubly deprotonated NCN2− anion, found both in its symmetric N=C=N2− carbodiimide and asymmetric N–C≡N2− cyanamide forms, is particularly attractive as it may be regarded as an extended nitrogen-based pseudo-chalcogenide or pseudo-oxide.
CaNCN, a commonly used fertilizer, was the first solid-state carbodiimide to be discovered as early as 1895 [1]. Subsequent developments in the last couple of decades have realized the formation of binary, ternary, and even quaternary salt-like alkali, alkaline-earth, main group, and rare-earth carbodiimides [2,3,4,5,6]. The structural chemistry of transition-metal carbodiimides is, however, more modest with MNCN (M = Mn–Cu) and Cr2NCN3 binaries only accessible through synthetic routes featuring highly stable side products [7,8,9,10]. Notwithstanding their lack of structural and compositional diversity, transition metal carbodiimides show a variety of interesting physical properties, often with enhanced functionality over their oxide analogues [11]. Specifically, it has recently been shown that MNCN (M = Mn–Zn) and Cr2NCN3 binaries are electrochemically active in Li- and Na-ion batteries, and it is suggested that their improved performance over equivalent oxides is a direct consequence of their “softer” chemical character [12,13]. An intriguing future direction in this field is then the introduction of cation order into such phases, which should not only enrich the structural chemistry but may also yield new or enhanced properties.
Very recently, we have come close in this endeavor with the preparation of SrZn(NCN)2 [14], the first example of a ternary post-transition metal carbodiimide, which was targeted with inspiration from LiSr2M(NCN)4 (M = Al3+ and Ga3+) phases [15,16]. SrZn(NCN)2 adopts the versatile BaZnOS structure which, in oxychalcogenide analogues, can incorporate a range of different tetrahedrally coordinated divalent transition metals [17,18]. Likewise, this work describes the preparation of BaZn(NCN)2, a ternary analogue and high-symmetry modification of the quaternary LiBa2Al(NCN)4 phase in which tetrahedrally coordinated Li+ and Al3+ cations are replaced by Zn2+ [19]. This is the realization of only the second ternary post-transition or transition-metal carbodiimide and it is believed that through suitable compositional tuning transition-metal species may be introduced on to the tetrahedral sites, potentially leading to new functionality in this evolving family of pseudo-oxides.
2. Results
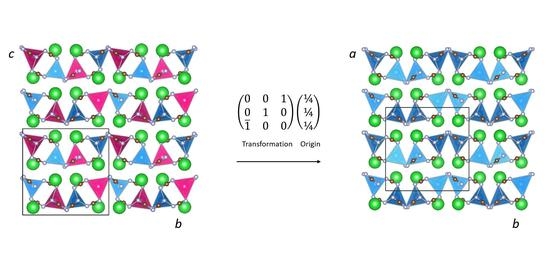
The powder X-ray diffraction pattern of a washed sample of BaZn(NCN)2 was indexed to a tetragonal unit cell (a = 11.9296 Å and c = 6.8451 Å) with lattice parameters very similar to those observed in orthorhombic LiBa2Al(NCN)4 (P212121; a = 6.843 Å, b = 11.828 Å and c = 11.857 Å) [19]. Indeed, replacing Al3+ and Li+ with Zn2+ at the 4a sites in a P212121 LiBa2Al(NCN)4-type model of BaZn(NCN)2 yields a simulated PXRD pattern which reproduces the observed data well. The presence of additional systematically absent reflections, however, suggests that BaZn(NCN)2 crystallizes in a high-symmetry variant, consistent with the presence of a single Zn2+ cation at the tetrahedral sites in BaZn(NCN)2, rather than the cation-ordered Li+/Al3+ distribution found in LiBa2Al(NCN)4 (Figure 1).
In the first instance, P41212 and P43212 were considered possible space groups as they are the only examples of tetragonal minimal supergroups of P212121. However, structure solution in these space groups, implemented in Expo2013 [20], did not result in any credible structural models. Moreover, the observed extinctions are inconsistent with any tetragonal space group and, instead, intimate the presence of additional glide planes in a metrically-tetragonal unit cell with orthorhombic symmetry. An unfortunate consequence of this metric tetragonality is, however, that the unambiguous determination of the extinction group is compromised since a number of different extinction groups can describe the observed reflections equally well. Considering then orthorhombic space groups that are minimal supergroups of P212121 but which also conform to the observed extinctions we arrive at two possibilities, namely Pbca and Pnma. Structure solution was then attempted in these two space groups, resulting, in the case of Pbca, in a structural model of BaZn(NCN)2 which reproduces the cation ordering scheme observed in LiBa2Al(NCN)4. The resultant structural model did not, however, describe regularly shaped NCN2− moieties. This is, presumably, a consequence of the weak X-ray scattering amplitudes of C and N relative to Ba, coupled with the positional freedom of N and C atoms at 8c general positions, which means that the NCN2− units are unconstrained by symmetry to adopt typical carbodiimide or cyanamide geometries.
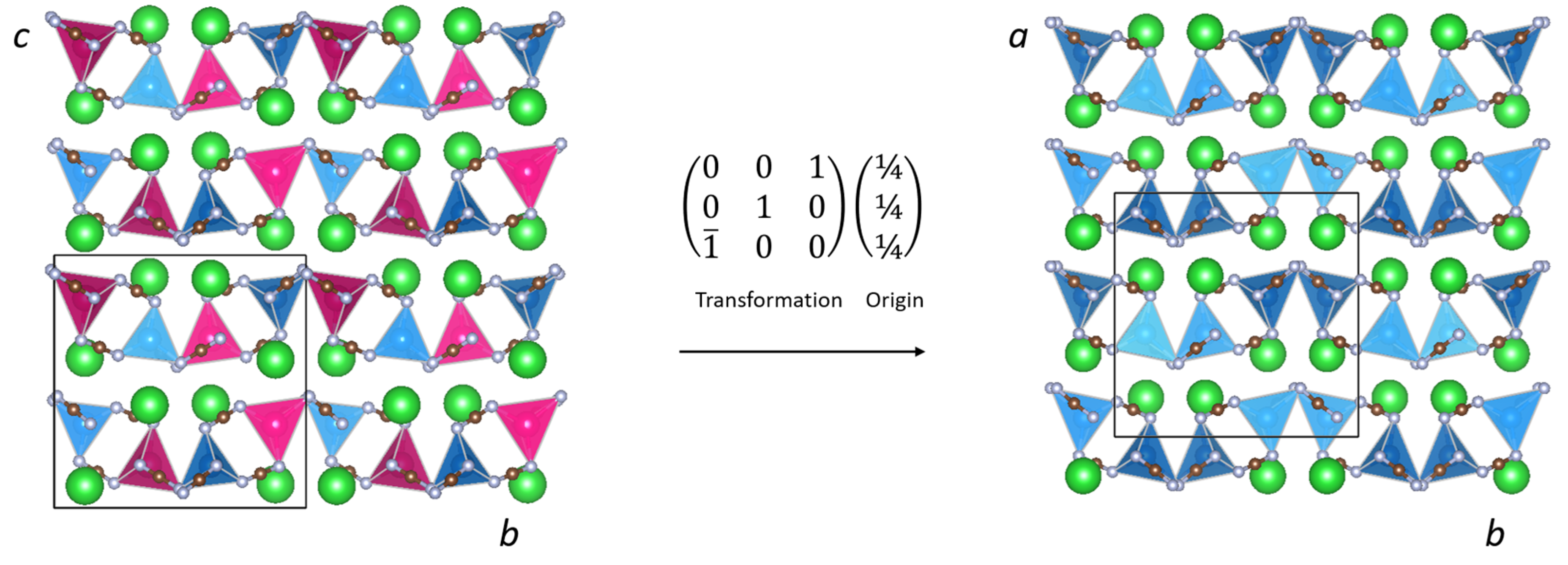
A starting Pbca model of BaZnNCN2 was therefore generated from the LiBa2Al(NCN)4 P212121 hettotype. This was achieved by performing an origin shift of (¼ ¼ ¼) and a cell transformation of [(0 0 1), (0 1 0), ( 0 0)] to the reported structure of LiBa2Al(NCN)4 as illustrated in Figure 1. Half of the 4a sites in LiBa2Al(NCN)4 were then selected to generate the atomic positions in BaZn(NCN)2, as they represent split positions derived from the 8c positions in Pbca. Preliminary Rietveld refinements were then performed, using the GSAS software suite [21], in which the fractional coordinates of all crystallographic sites were permitted to refine freely. Despite the resultant refinement reproducing the observed data well (Rwp = 6.25%), scrutiny of the fine details of the structural model reveals certain irregularities in the shape of the NCN2− units (Table 1). Specifically, the C(2)–N(1) bond length of 1.355(18) Å, described in Figure 2, is longer than is typically encountered in cyanamides and, more strikingly, both N–C–N bond angles deviate significantly from 180°. This model is, however, based on refinement against PXRD data and consequently, as alluded to earlier, the refined structural parameters of the relatively light C and N atoms are subject to significant errors. For this reason, powder neutron diffraction studies are planned to address these structural anomalies.
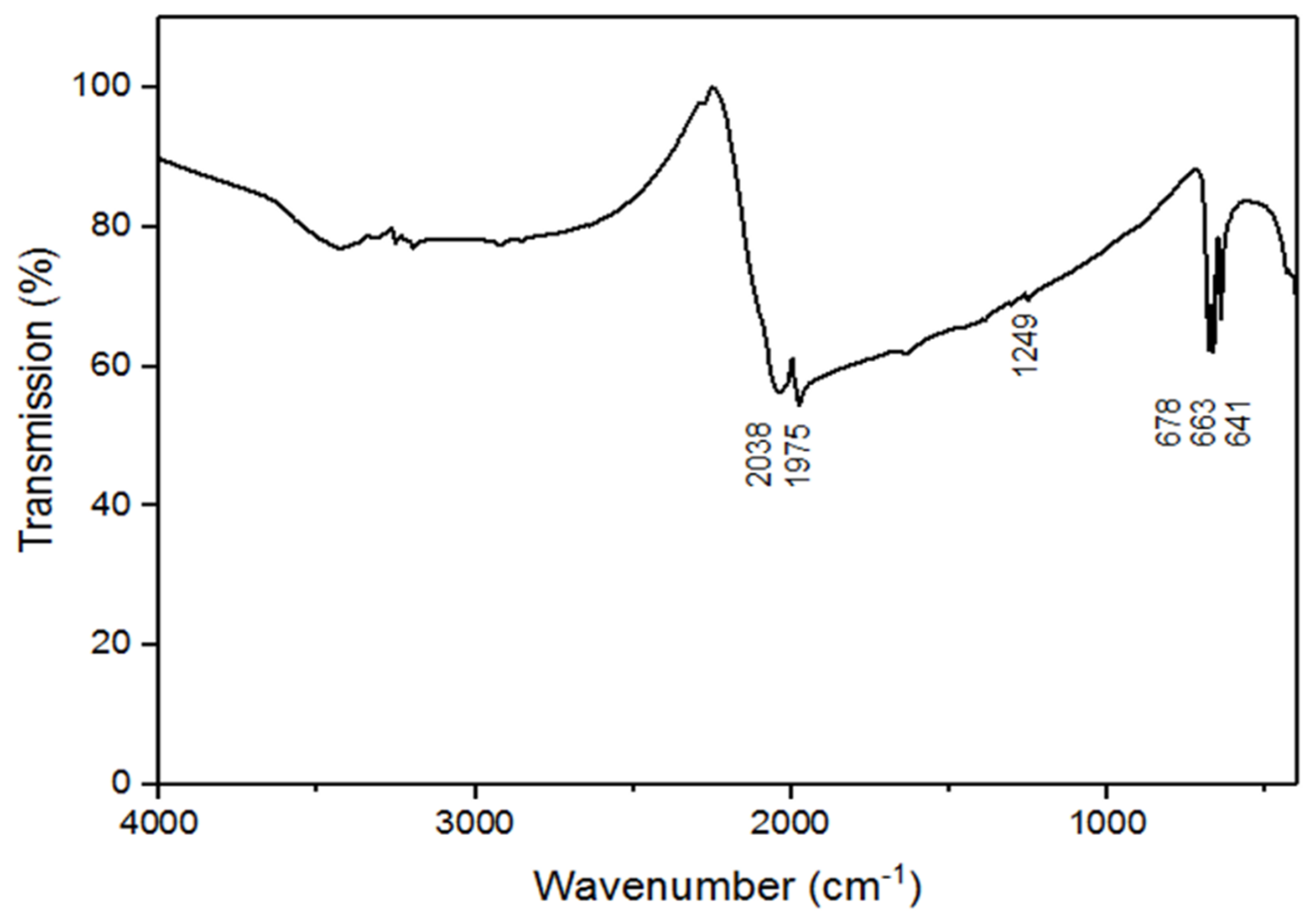
In the absence of powder neutron diffraction data, DFT(PBEsol) calculations were undertaken to help corroborate the structure of BaZn(NCN)2. The DFT-optimized structure of BaZn(NCN)2 is in good agreement with that found experimentally, though notably both NCN2− units have a much more regular carbodiimide character. That is, all C–N bond lengths come very close to 1.23 Å, a result strongly suggestive of C=N double-bond character, and N–C–N bond angles proximal to 180° (Table 1). It is, however, noted that DFT calculations are somewhat unreliable in accurately determining the energetics of cyanamide/carbodiimide shapes [22]. To gain further experimental insight into the character of the NCN2− units in BaZn(NCN)2, complementary IR data were collected, as shown in Figure 3. BaZn(NCN)2 expresses asymmetric vibrations (νas(NCN) = 2038 and 1975 cm−1) and deformation vibrations (δ(NCN) = 678, 663, and 641 cm−1) typical of metal carbodiimides/cyanamides [8]. A very weak absorption is also observed at 1249 cm−1, consistent with the presence of a νs(NCN) symmetric stretching mode [23], which indicates a degree of cyanamide character, since such a breathing mode is IR-forbidden for symmetric N=C=N2−.
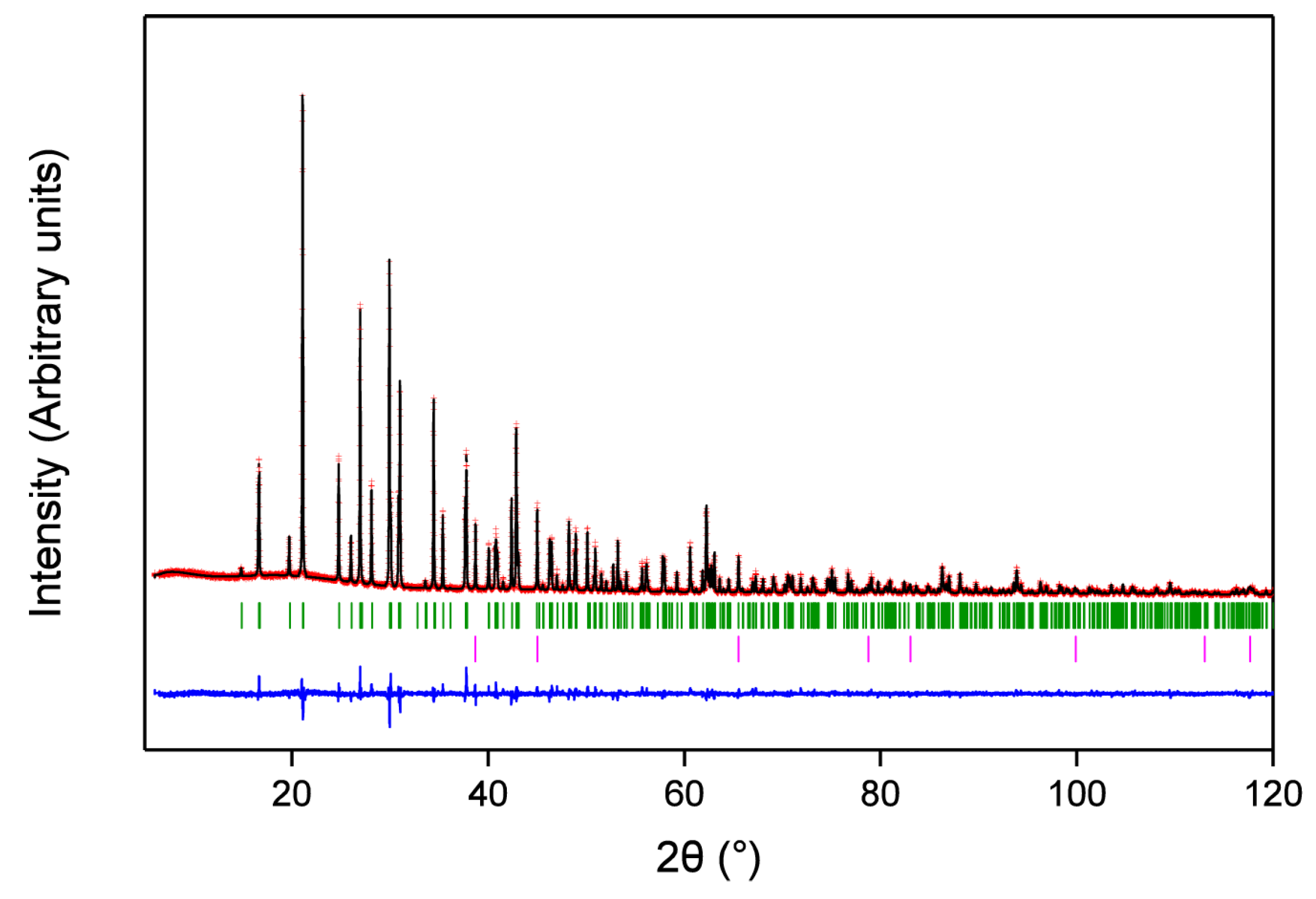
In light of these results, and given the weak X-ray scattering amplitudes of C and N relative to Ba, bond-angle and length restraints were introduced to the final cycles of least-squares refinement to dictate a strong degree of carbodiimide shape in the NCN2− units. Specifically, a soft restraint was applied to the C–N bond lengths, fixing the distances at 1.22 ± 0.02 Å, consistent with DFT calculations and literature data on D∞h carbodiimides [24]. Similarly, N–C–N bond-angle restraints were applied restraining the angles to 180° ± 5°. With these restraints in place the structural model was refined once more against PXRD data. The resultant refinement yielded a satisfactory structural model with very good agreement between the observed and calculated data as seen in the Rietveld fit in Figure 4. Crystallographic data are reported in Table 2 with selected bond lengths and angles detailed in Table 1 and cif data as supplementary materials.
3. Discussion
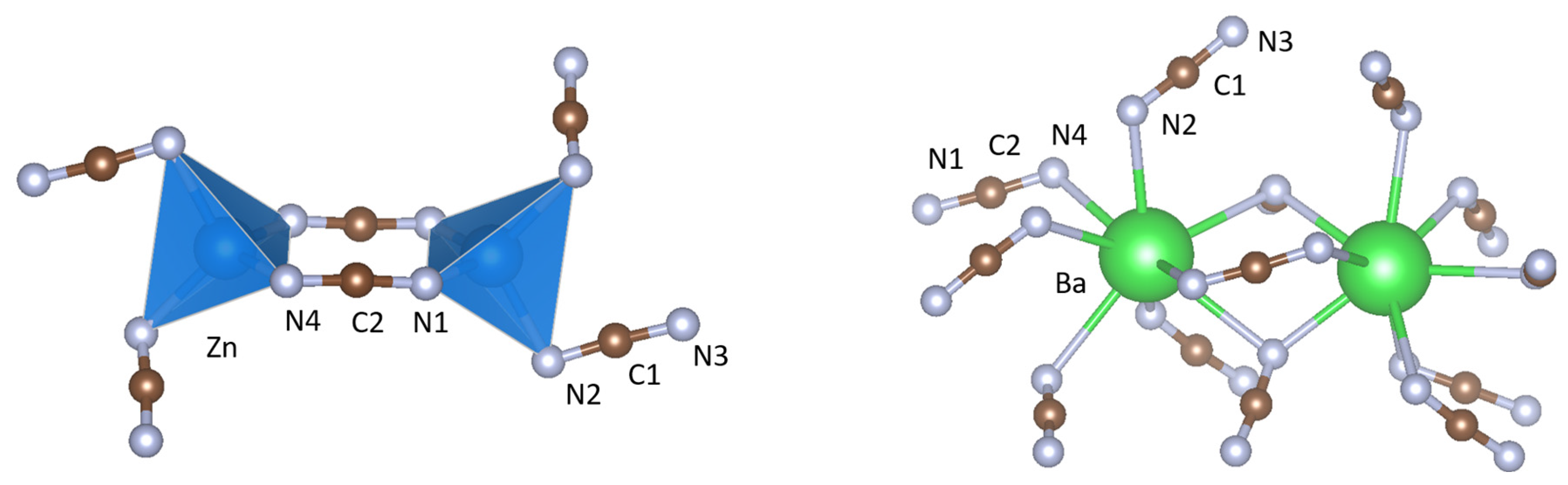
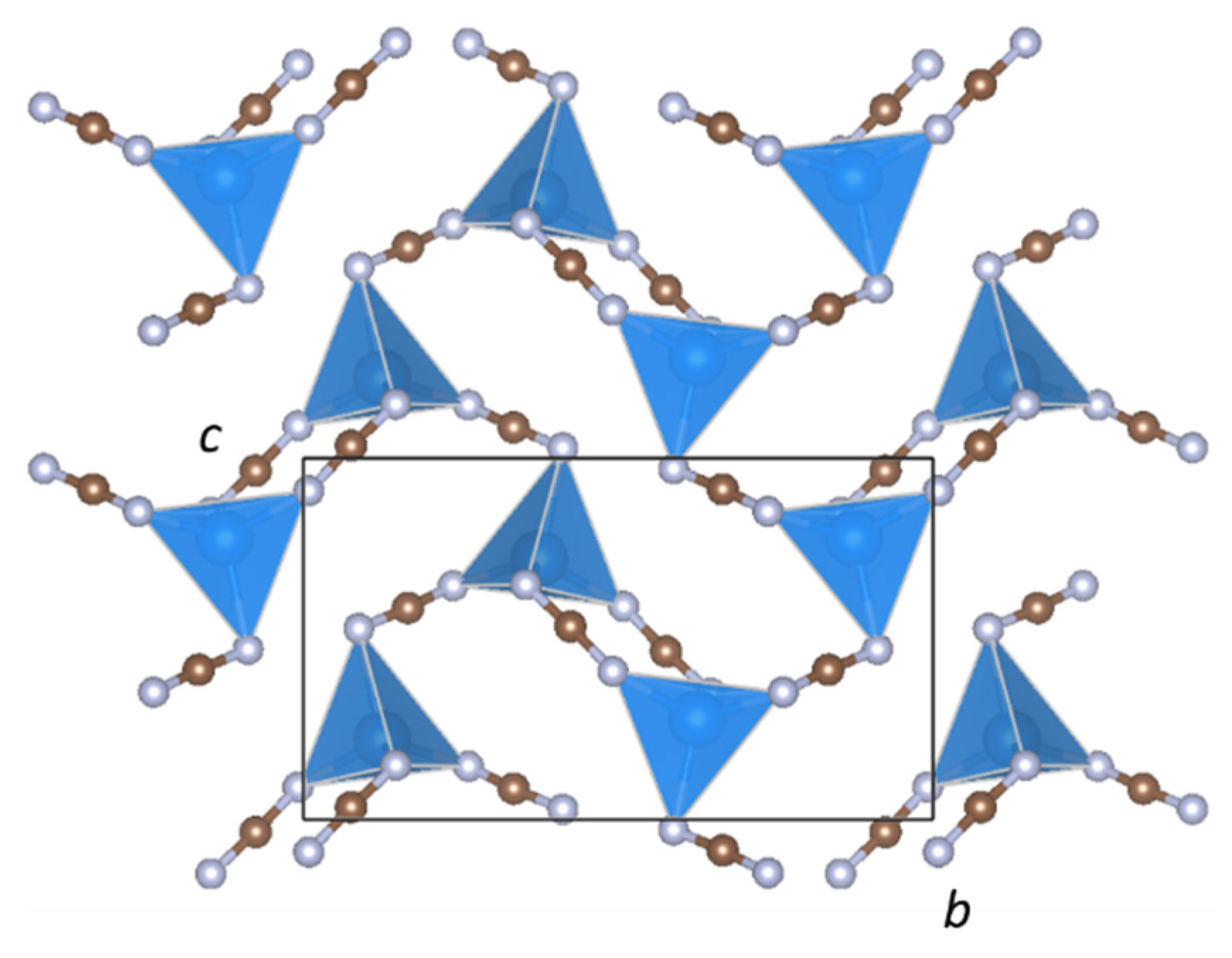
The crystal structure of BaZn(NCN)2 is well described as a two-dimensional network of ZnN4 tetrahedra (Figure 5) sharing edges through N(1)–C(2)–N(4) units and vertices through N(2)–C(1)–N(3) units, with Ba2+ cations in the interlayer voids (Figure 1). The ZnN4 tetrahedra are comprised of four distinct Zn–N bond lengths (Figure 2), with an average Zn–N bond distance of 2.080(8) Å comparable to those in SrZn(NCN)2 (dZn–N = 2.059(4) Å) and ZnNCN (dZn–N = 2.009(2) Å) [14,25], which also feature ZnN4 tetrahedra. Meanwhile, Ba2+ adopts an eight-fold coordination of nitrogen atoms, quite distinct from the irregular six-coordinate twisted trigonal prismatic environment observed in BaNCN [26], with Ba–N distances between 2.650(7) and 3.015(7) Å (Figure 2), and with an average Ba–N bond length dBa–N = 2.886(7) Å, similar to those observed in LiBa2Al(NCN)4 (dBa(1)–N = 2.889(8) and dBa(2)–N = 2.918(8) Å) [19]. The presence of soft restraints between N and C atoms in the two crystallographically distinct NCN2− units dictates a strong N=C=N2− carbodiimide character (Table 1), with all C–N bond lengths close to 1.22 Å and equal within experimental error. That said, the refined N–C–N bond angles and those from DFT calculations both deviate from 180° resulting in slightly bent N=C=N2− anions with C2v symmetry, as seen in HgNCN(I) [23]. However, a more comprehensive assessment of the character of the cyanamide anions in BaZn(NCN)2 awaits the results of powder neutron diffraction studies.
BaZn(NCN)2 represents only the second example of a ternary transition-metal or post-transition metal carbodiimide/cyanamide, following our recent discovery of SrZn(NCN)2 [14]. Although both phases adopt crystal structures that feature ZnN4 tetrahedra and large alkaline-earth metals, their topologies are quite different. This is in spite of the fact that SrZn(NCN)2 takes the BaZnOS structure [17], which is self-evidently capable of incorporating cations as large as Ba2+, and the layered nature of which should be favored by linear NCN2− units for packing reasons. It seems probable then that the trigonal bipyramidal site in the BaZnOS structure is under-bonded for Ba2+ unless the face capping anions are chalcogenides. This also helps to rationalize why the oxide analogue, BaZnO2, does not share this structure and preferentially forms a 3D network of corner-sharing ZnO4 tetrahedra in a distorted β-quartz-like array [27]. Such a network, however, is presumably disfavored for BaZn(NCN)2 as the extended nature of the NCN2− unit would generate voids much too large for Ba2+. Instead, BaZn(NCN)2 is the aristotype and only the second member of a structure type unique to metal cyanamide/carbodiimide chemistry.
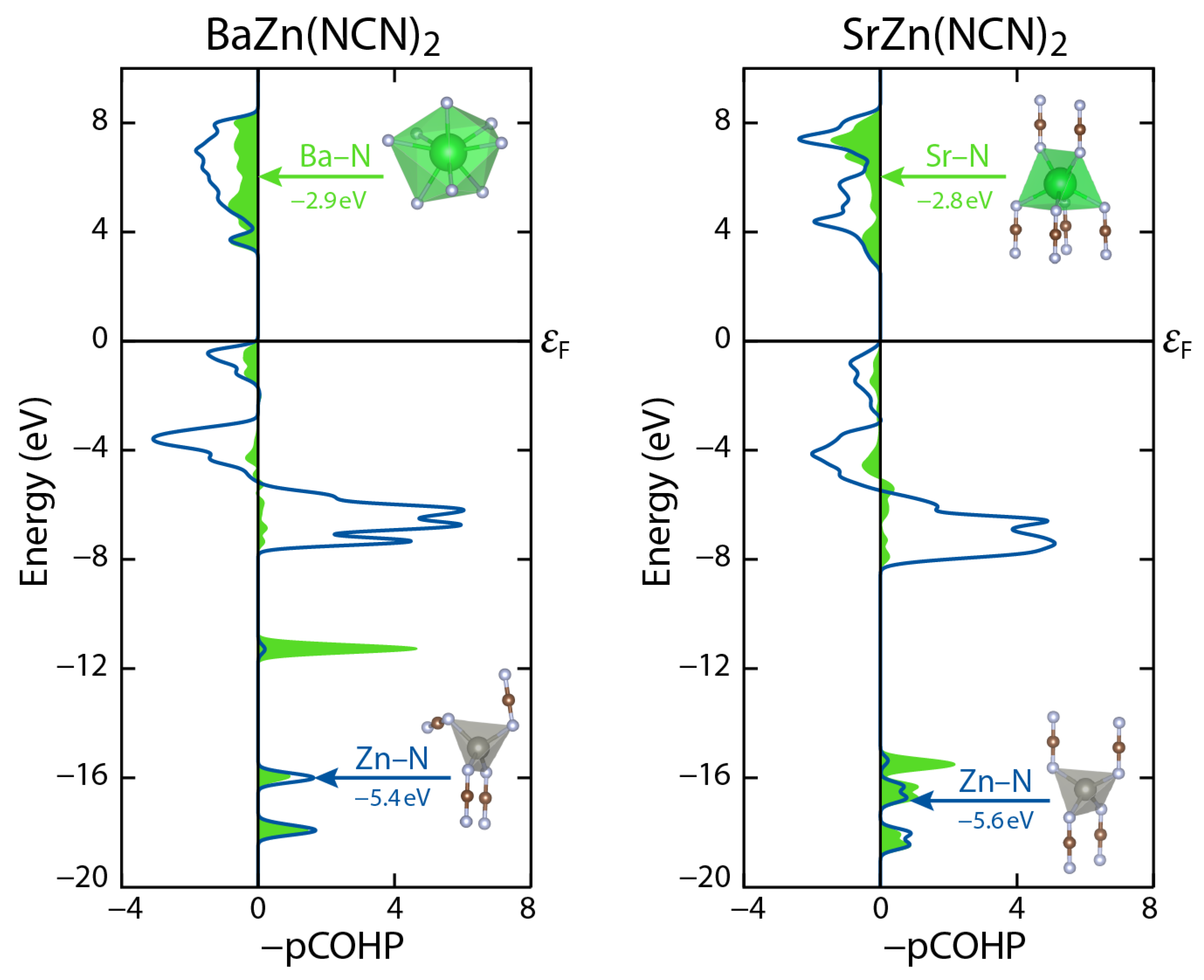
Despite these topological differences, quantum-chemical calculations reveal both BaZn(NCN)2 and SrZn(NCN)2 to be wide-band-gap semiconductors with similar band gaps of 3.6 and 3.0 eV, respectively. To gain a deeper insight in to the chemical bonding in the two phases, crystal orbital Hamilton population (COHP) analysis was performed using the LOBSTER program suite. Specifically, we compare the Zn–N and A–N interactions in the pCOHP plots shown in Figure 6. This comparison reveals striking similarities between the two phases, with Zn–N and A–N interactions of antibonding character in the vicinity of the Fermi level even though the network connectivities and A-site coordination environments are quite different. Similar, energetically disadvantageous M–N bonding interactions in the proximity of εF have recently also been witnessed for the case of MnNCN and may be looked upon as a typical fingerprint of the (late) 3d-metal carbodiimides [28]. Future work will look to extend this emerging AM(NCN)2 family by accessing analogues featuring divalent transition-metal cations at the tetrahedral site.
4. Experimental and Computational Details
4.1. Synthesis
BaZn(NCN)2 was synthesized on the 0.5 g scale in an argon-filled glove-box by a solid-state metathesis reaction between BaF2 (Alfa), ZnF2 (Alfa), and Li2NCN, which was prepared from a mixture of Li3N and C3N3(NH2)3, as described in reference [29]. A stoichiometric 1:1:2 molar ratio of the reactants, according to Equation (1), was homogenized using an agate pestle and mortar, and the reaction mixture was loaded into a dry glass capillary. The sample was then loaded into a glass ampoule and positioned in a tube furnace under flowing argon and heated to 550 °C for 24 h, with heating and cooling rates of 2 °C·min−1. The resulting white powder was then washed with water in an attempt to remove the poorly soluble LiF metathesis salt.
BaF2 + ZnF2 + 2 Li2NCN → BaZn(NCN)2 + 4 LiF
4.2. PXRD Analysis
As-made and washed samples of BaZn(NCN)2 were inspected and characterized by X-ray powder diffraction (STOE, Darmstadt, Germany) using a calibrated STOE STADI-P powder diffractometer with a flat sample holder (Cu Kα1, linear PSD, 2θ range = 5°–120° with an individual step size of 0.01°). The as-made sample contains the metathesis salt LiF along with reflections assigned to the title compound which match those in the washed sample, indicating that BaZn(NCN)2 is inert to water.
Structural refinements were performed using the Rietveld refinement suite GSAS [21]. In the final cycles of least squares refinement, lattice parameters, fractional coordinates, and isotropic thermal displacement parameters (with a single Uiso refined for all light atoms, C and N) were refined for BaZn(NCN)2 with a pseudo-Voigt profile function (CW profile 4). Details of bond angle and length restraints between C and N atoms are described in the main body of the text. A secondary phase was introduced to model reflections from leftover LiF metathesis salt. Further crystallographic details can be found in the supporting information or may be obtained from Fachinformationszentrum Karlsruhe, 76344 Eggenstein-Leopoldshafen, Germany, on quoting the deposition number CSD-433767.
4.3. IR Measurements
The infrared spectrum of BaZn(NCN)2 was measured on a Nicolet Avatar 369 FT-IR spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) in the range 400‒4000 cm−1 using KBr discs.
4.4. Computational Details
Density-functional theory (DFT) calculations employed the PBEsol functional [30,31], using plane-wave basis sets and the projector augmented wave (PAW) method [32] as implemented in the Vienna Ab Initio Simulation Package (VASP) [33,34,35]. The energy cutoff for the plane-wave expansion was 500 eV with an electronic convergence criterion of 10−7 eV. Structural optimization was performed until residual forces fell below 5 × 10−3 eV·Å−1. Reciprocal space was sampled on Γ-centered k-point grids with densities between 0.02 and 0.03 Å−1. Chemical bonding analysis within the framework of the crystal orbital Hamilton population (COHP) [36] was performed using LOBSTER [37,38]. Structural drawings were created using VESTA [39].
Supplementary Materials
The following are available online at www.mdpi.com/2304-6740/6/1/1/s1. Cif and cif-checked files.
Supplementary File 1Acknowledgments
We would like to thank the Deutsche Forschungsgemeinschaft (DFG) for funding, Björn Faßbänder for PXRD measurements, Irmgard Kalf for collecting IR spectra, and the Jülich-Aachen Research Alliance for computational resources (JARA-HPC project jara0033).
Author Contributions
Alex J. Corkett conceived and performed the experiments; Philipp M. Konze performed DFT calculations; Richard Dronskowski initialized and supervised the project. All authors were involved in writing the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Frank, A.; Garo, N. Verfahren zur Darstellung von Cyanverbindungen aus Carbiden. Ger. Pat. 8363, 1895. [Google Scholar]
- Down, M.G.; Haley, M.J.; Hubberstey, P.; Pulham, R.J.; Thunder, A.E. Synthesis of the dilithium salt of cyanamide in liquid lithium; X-ray crystal structure of Li2NCN. J. Chem. Soc. Chem. Commun. 1978, 52–53. [Google Scholar] [CrossRef]
- Krings, M.; Wessel, M.; Wilsmann, W.; Müller, P.; Dronskowski, R. Temperature-Dependent Synthetic Routes to and Thermochemical Ranking of α- and β-SrNCN. Inorg. Chem. 2010, 49, 2267–2272. [Google Scholar] [CrossRef] [PubMed]
- Neukirch, M.; Tragl, S.; Meyer, H.J. Syntheses and Structural Properties of Rare Earth Carbodiimides. Inorg. Chem. 2006, 45, 8188–8193. [Google Scholar] [CrossRef] [PubMed]
- Kubus, M.; Heinicke, R.; Ströbele, M.; Enseling, D.; Jüstel, T.; Meyer, H.J. Synthesis of new structurally related cyanamide compounds LiM(CN2)2 where M is Al3+, In3+ or Yb3+. Mater. Res. Bull. 2015, 62, 37–41. [Google Scholar] [CrossRef]
- Dolabdjian, K.; Schedel, C.; Enseling, D.; Jüstel, T.; Meyer, H.-J. Synthesis, Luminescence and Nonlinear Optical Properties of Homoleptic Tetracyanamidogermanates ARE[Ge(CN2)4] (A = K, Cs, and RE = La, Ce, Pr, Nd, Sm, Eu, Gd). Z. Anorg. Allg. Chem. 2017, 643, 488–494. [Google Scholar] [CrossRef]
- Tang, X.; Xiang, H.; Liu, X.; Speldrich, M.; Dronskowski, R. A Ferromagnetic Carbodiimide: Cr2(NCN)3. Angew. Chem. Int. Ed. 2010, 49, 4738–4742. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Krott, M.; Müller, P.; Hu, C.; Lueken, H.; Dronskowski, R. Synthesis, Crystal Structure, and Properties of MnNCN, the First Carbodiimide of a Magnetic Transition Metal. Inorg. Chem. 2005, 44, 3001–3003. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Stork, L.; Speldrich, M.; Lueken, H.; Dronskowski, R. FeNCN and Fe(NCNH)2: Synthesis, Structure, and Magnetic Properties of a Nitrogen-Based Pseudo-Oxide and -Hydroxide of Divalent Iron. Chem. Eur. J. 2009, 15, 1558–1561. [Google Scholar] [CrossRef] [PubMed]
- Krott, M.; Liu, X.; Fokwa, B.P.T.; Speldrich, M.; Lueken, H.; Dronskowski, R. Synthesis, Crystal-Structure Determination and Magnetic Properties of Two New Transition-Metal Carbodiimides: CoNCN and NiNCN. Inorg. Chem. 2007, 46, 2204–2207. [Google Scholar] [CrossRef] [PubMed]
- Ressnig, D.; Shalom, M.; Patscheider, J.; More, R.; Evangelisti, F.; Antonietti, M.; Patzke, G.R. Photochemical and electrocatalytic water oxidation activity of cobalt carbodiimide. J. Mater. Chem. A 2015, 3, 5072–5082. [Google Scholar] [CrossRef] [Green Version]
- Eguia-Barrio, A.; Castillo-Martinez, E.; Liu, X.; Dronskowski, R.; Armand, M.; Rojo, T. Carbodiimides: New materials applied as anode electrodes for sodium and lithium ion batteries. J. Mater. Chem. A 2016, 4, 1608–1611. [Google Scholar] [CrossRef]
- Sougrati, M.T.; Darwiche, A.; Liu, X.; Mahmoud, A.; Hermann, R.P.; Jouen, S.; Monconduit, L.; Dronskowski, R.; Stievano, L. Transition-Metal Carbodiimides as Molecular Negative Electrode Materials for Lithium- and Sodium-Ion Batteries with Excellent Cycling Properties. Angew. Chem. Int. Ed. 2016, 55, 5090–5095. [Google Scholar] [CrossRef] [PubMed]
- Corkett, A.J.; Konze, P.M.; Dronskowski, R. The Ternary Post-transition Metal Carbodiimide SrZn(NCN)2. Z. Anorg. Allg. Chem. 2017, 643, 1456–1461. [Google Scholar] [CrossRef]
- Unverfehrt, L.; Kalmutzki, M.; Strobele, M.; Meyer, H.J. Solid state synthesis of homoleptic tetracyanamidoaluminates. Dalton Trans. 2011, 40, 9921–9924. [Google Scholar] [CrossRef] [PubMed]
- Kalmutzki, M.; Ströbele, M.; Kroeker, S.; Wren, J.E.C.; Meyer, H.J. Synthesis and Characterization of the First Tetracyanamidogallate. Eur. J. Inorg. Chem. 2013, 2013, 6091–6096. [Google Scholar] [CrossRef]
- Broadley, S.; Gál, Z.A.; Corà, F.; Smura, C.F.; Clarke, S.J. Vertex-Linked ZnO2S2 Tetrahedra in the Oxysulfide BaZnOS: A New Coordination Environment for Zinc in a Condensed Solid. Inorg. Chem. 2005, 44, 9092–9096. [Google Scholar] [CrossRef] [PubMed]
- Salter, E.J.T.; Blandy, J.N.; Clarke, S.J. Crystal and Magnetic Structures of the Oxide Sulfides CaCoSO and BaCoSO. Inorg. Chem. 2016, 55, 1697–1701. [Google Scholar] [CrossRef] [PubMed]
- Unverfehrt, L.; Ströbele, M.; Meyer, H.J. The New Homoleptic Tetracyanamidoaluminate LiBa2[Al(CN2)4]. Z. Anorg. Allg. Chem. 2013, 639, 1722–1725. [Google Scholar] [CrossRef]
- Altomare, A.; Cuocci, C.; Giacovazzo, C.; Moliterni, A.; Rizzi, R.; Corriero, N.; Falcicchio, A. EXPO2013: A kit of tools for phasing crystal structures from powder data. J. Appl. Crystallogr. 2013, 46, 1231–1235. [Google Scholar] [CrossRef]
- Toby, B. EXPGUI, a graphical user interface for GSAS. J. Appl. Crystallogr. 2001, 34, 210–213. [Google Scholar] [CrossRef]
- Liu, X.; Müller, P.; Kroll, P.; Dronskowski, R.; Wilsmann, W.; Conradt, R. Experimental and Quantum-Chemical Studies on the Thermochemical Stabilities of Mercury Carbodiimide and Mercury Cyanamide. ChemPhysChem 2003, 4, 789. [Google Scholar] [CrossRef]
- Liu, X.; Müller, P.; Kroll, P.; Dronskowski, R. Synthesis, Structure Determination, and Quantum-Chemical Characterization of an Alternate HgNCN Polymorph. Inorg. Chem. 2002, 41, 4259–4265. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Decker, A.; Schmitz, D.; Dronskowski, R. Crystal Structure Refinement of Lead Cyanamide and the Stiffness of the Cyanamide Anion. Z. Anorg. Allg. Chem. 2000, 626, 103–105. [Google Scholar] [CrossRef]
- Becker, M.; Jansen, M. Zinc cyanamide, Zn(CN2). Acta Crystallogr. Sect. C 2001, 57, 347–348. [Google Scholar] [CrossRef]
- Berger, U.; Schnick, W. Syntheses, crystal structures, and vibrational spectroscopic properties of MgCN2, SrCN2, and BaCN2. J. Alloys Compd. 1994, 206, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Spitsbergen, U. The crystal structures of BaZnO2, BaCoO2 and BaMnO2. Acta Crystallogr. 1960, 13, 197–198. [Google Scholar] [CrossRef]
- Nelson, R.; Konze, P.M.; Dronskowski, R. First-Principles Chemical Bonding Study of Manganese Carbodiimide, MnNCN, As Compared to Manganese Oxide, MnO. J. Phys. Chem. A 2017, 121, 7778–7786. [Google Scholar] [CrossRef] [PubMed]
- Glaser, J.; Bettentrup, H.; Jüstel, T.; Meyer, H.J. Synthesis and Properties of Tetracyanamidosilicates ARE[Si(CN2)4]. Inorg. Chem. 2010, 49, 2954–2959. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 136406. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Dronskowski, R.; Blöchl, P.E. Crystal Orbital Hamilton Populations (COHP). Energy-Resolved Visualization of Chemical Bonding in Solids Based on Density-Functional Calculations. J. Phys. Chem. 1993, 97, 8617–8624. [Google Scholar] [CrossRef]
- Maintz, S.; Deringer, V.L.; Tchougréeff, A.L.; Dronskowski, R. Analytic Projection from Plane-Wave and PAW Wavefunctions and Application to Chemical-Bonding Analysis in Solids. J. Comput. Chem. 2013, 34, 2557–2567. [Google Scholar] [CrossRef] [PubMed]
- Maintz, S.; Deringer, V.L.; Tchougréeff, A.L.; Dronskowski, R. LOBSTER: A Tool to Extract Chemical Bonding from Plane-Wave Based DFT. J. Comput. Chem. 2016, 37, 1030–1035. [Google Scholar] [CrossRef] [PubMed]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
Figure 1.
Comparison of the crystal structures of (left) LiBa2Al(NCN)4 (P212121), and (right) BaZn(NCN)2 (Pbca).
Figure 1.
Comparison of the crystal structures of (left) LiBa2Al(NCN)4 (P212121), and (right) BaZn(NCN)2 (Pbca).
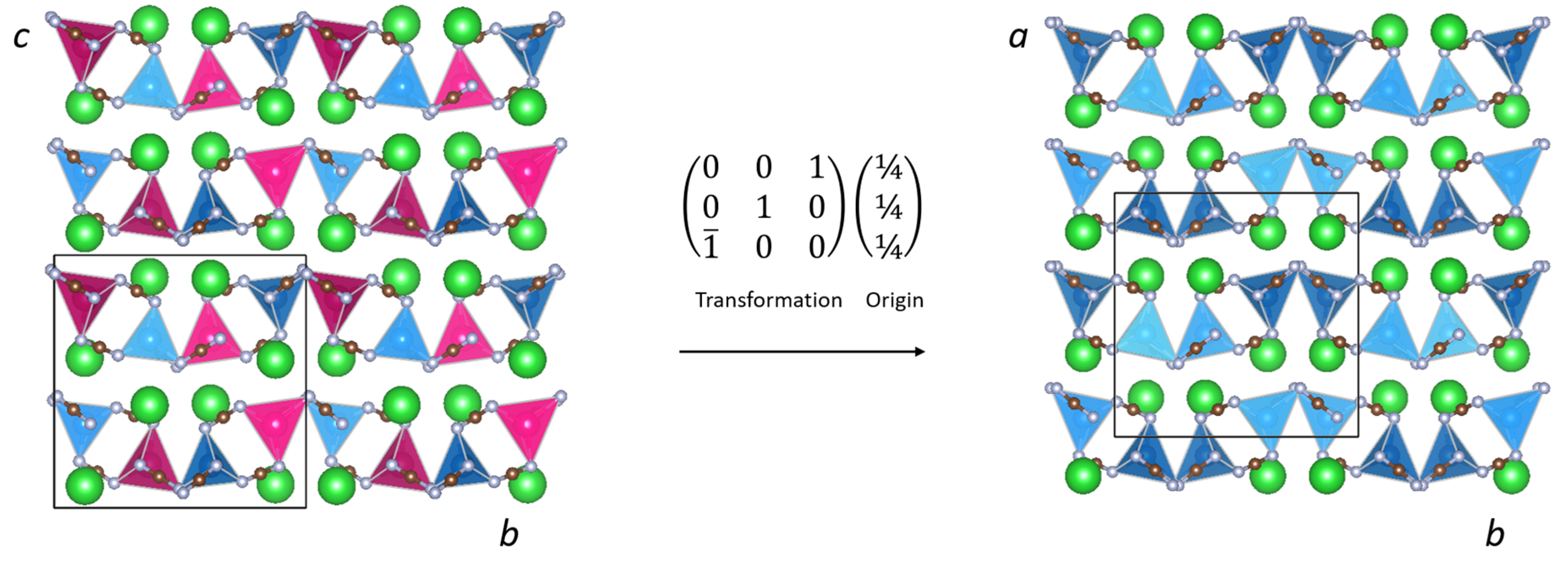
Figure 2.
Coordination environments of (left) zinc and (right) barium in BaZn(NCN)2.
Figure 3.
Infrared spectrum of BaZn(NCN)2.

Figure 4.
Rietveld fit of BaZn(NCN)2 to PXRD data, showing calculated (black), observed (red), and difference (blue) intensities. Bragg reflections for BaZn(NCN)2 and LiF are denoted by green and pink vertical markers, respectively.
Figure 4.
Rietveld fit of BaZn(NCN)2 to PXRD data, showing calculated (black), observed (red), and difference (blue) intensities. Bragg reflections for BaZn(NCN)2 and LiF are denoted by green and pink vertical markers, respectively.
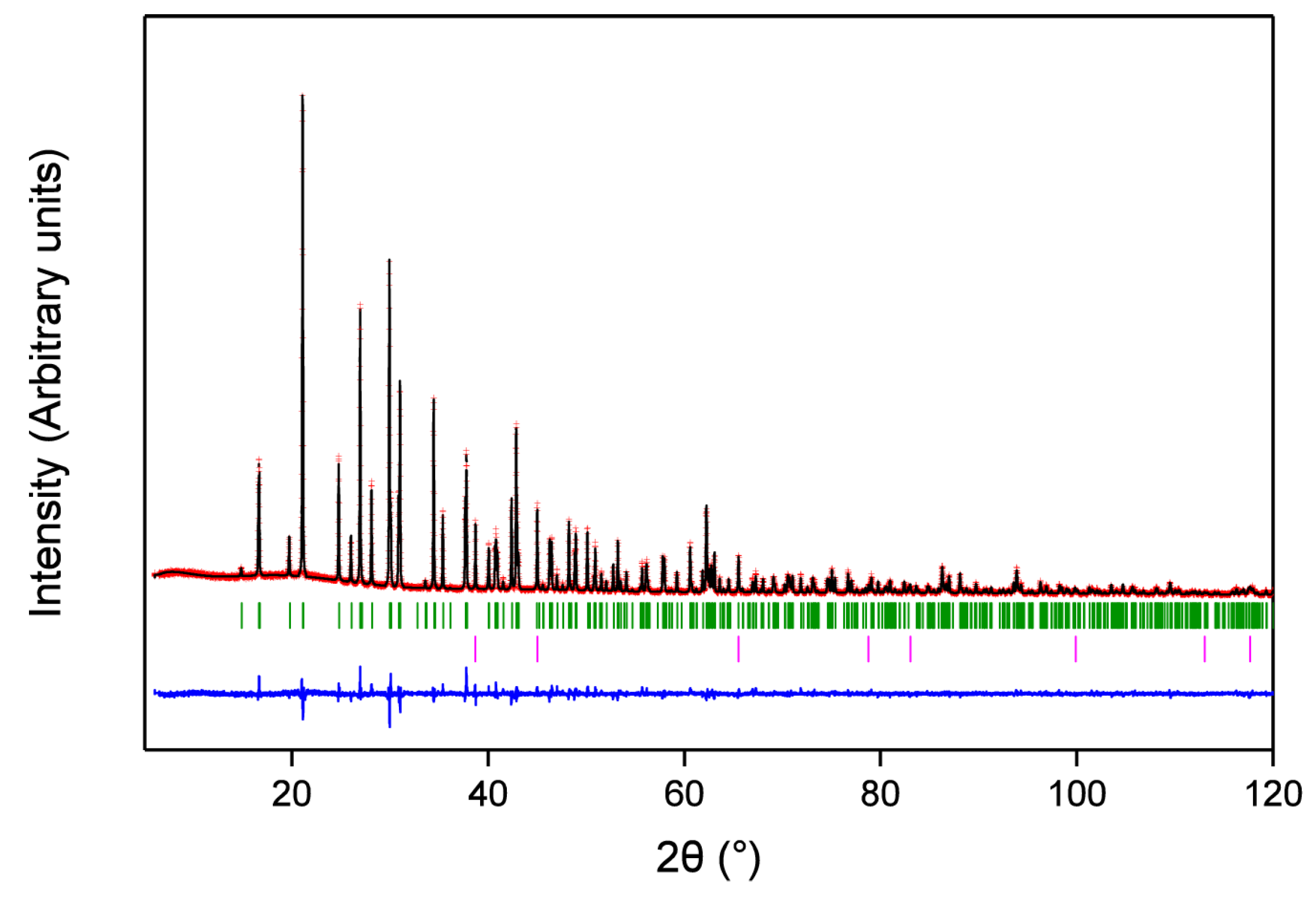
Figure 5.
Two-dimensional [Zn(NCN)2] network connectivity in BaZn(NCN)2.
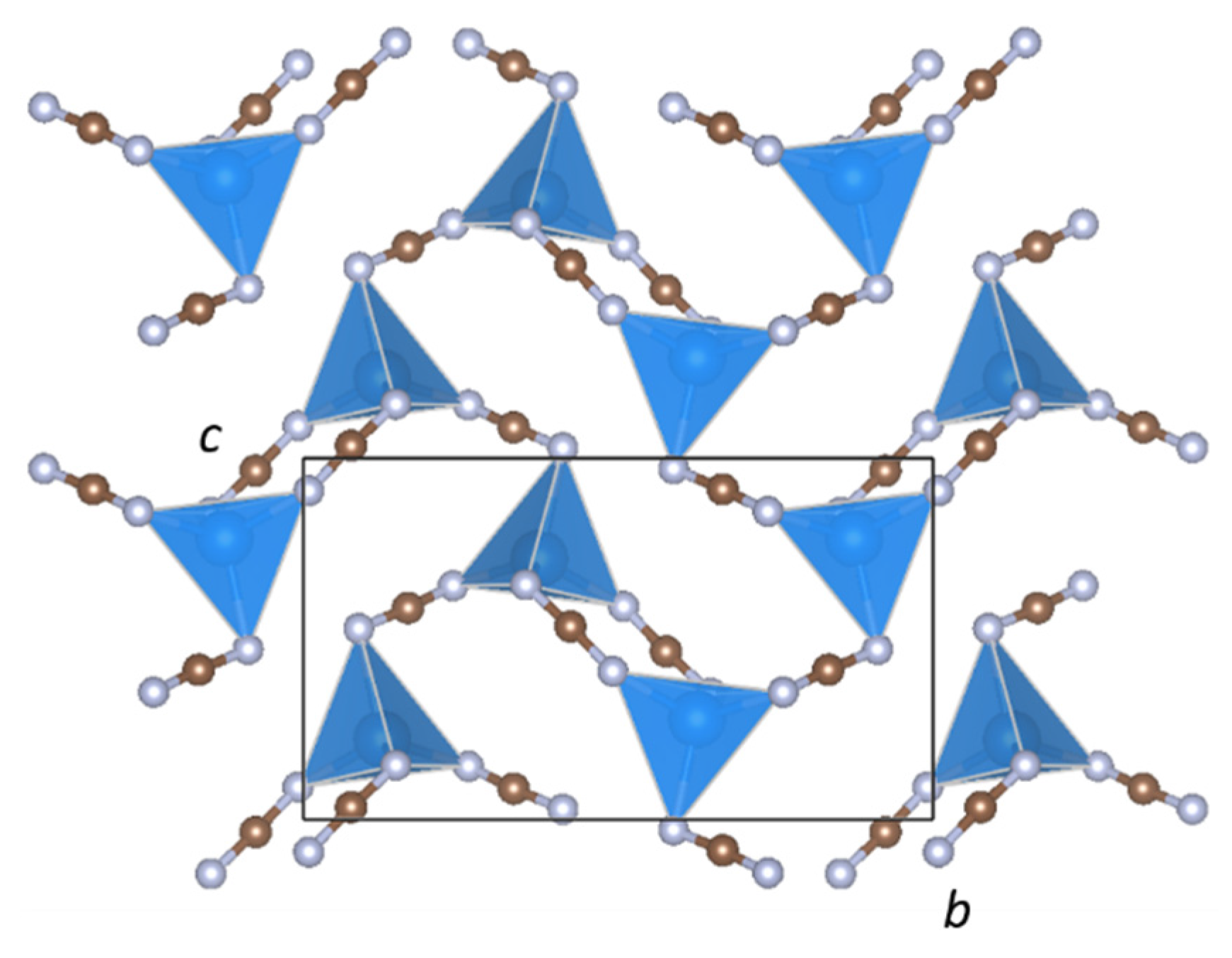
Figure 6.
Crystal orbital Hamilton population (COHP) analysis of BaZn(NCN)2 and SrZn(NCN)2. Both compounds show surprising similarities in the alkaline-earth–nitrogen bond, despite structural differences. Curves and integrated COHPs shown are for one polyhedra each (as shown) resulting in eight, six, and four bonds for each Ba–N, Sr–N, and Zn–N COHP, respectively.
Figure 6.
Crystal orbital Hamilton population (COHP) analysis of BaZn(NCN)2 and SrZn(NCN)2. Both compounds show surprising similarities in the alkaline-earth–nitrogen bond, despite structural differences. Curves and integrated COHPs shown are for one polyhedra each (as shown) resulting in eight, six, and four bonds for each Ba–N, Sr–N, and Zn–N COHP, respectively.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Comparison of bond lengths d and angles θ in BaZn(NCN)2 from both unrestrained (obs(unres)) and restrained (obs(res)) Rietveld refinements as well as DFT calculations (calc).
Table 1.
Comparison of bond lengths d and angles θ in BaZn(NCN)2 from both unrestrained (obs(unres)) and restrained (obs(res)) Rietveld refinements as well as DFT calculations (calc).
| dobs(unres) (Å) | dobs(res) (Å) | dcalc (Å) | |
|---|---|---|---|
| C1–N2 | 1.209(18) | 1.213(7) | 1.233 |
| C1–N3 | 1.146(18) | 1.212(8) | 1.236 |
| C2–N1 | 1.355(18) | 1.226(7) | 1.233 |
| C2–N4 | 1.213(18) | 1.228(8) | 1.239 |
| θobs(unres) (°) | θobs(res) (°) | θcalc (°) | |
| N2–C1–N3 | 165.1(13) | 174.7(5) | 176.7 |
| N1–C2–N4 | 153.4(14) | 176.1(5) | 179.2 |
Table 2.
Crystallographic data and fractional coordinates (all atoms on 8c) for BaZn(NCN)2. Standard deviations are given in parentheses.
Table 2.
Crystallographic data and fractional coordinates (all atoms on 8c) for BaZn(NCN)2. Standard deviations are given in parentheses.
| Atom | x | y | z | Uiso (102 × Å2) |
|---|---|---|---|---|
| Ba | 0.83999(9) | 0.87151(9) | 0.0404(1) | 1.54(1) |
| Zn | 0.9226(2) | 0.1252(2) | 0.2781(3) | 1.94(5) |
| C1 | 0.3632(4) | 0.1660(4) | 0.585(1) | 1.8(2) |
| C2 | 0.6117(7) | 0.4240(4) | 0.5343(7) | “ |
| N1 | 0.6432(7) | 0.4912(5) | 0.4103(8) | “ |
| N2 | 0.4196(5) | 0.0878(4) | 0.529(1) | “ |
| N3 | 0.3009(5) | 0.2379(5) | 0.648(1) | “ |
| N4 | 0.5771(7) | 0.3531(6) | 0.6495(9) | “ |
“: All values of Uiso for C and N atoms are equal.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Corkett, A.J.; Konze, P.M.; Dronskowski, R. Synthesis, Crystal Structure, and Chemical-Bonding Analysis of BaZn(NCN)2. Inorganics 2018, 6, 1. https://doi.org/10.3390/inorganics6010001
AMA Style
Corkett AJ, Konze PM, Dronskowski R. Synthesis, Crystal Structure, and Chemical-Bonding Analysis of BaZn(NCN)2. Inorganics. 2018; 6(1):1. https://doi.org/10.3390/inorganics6010001
Chicago/Turabian StyleCorkett, Alex J., Philipp M. Konze, and Richard Dronskowski. 2018. "Synthesis, Crystal Structure, and Chemical-Bonding Analysis of BaZn(NCN)2" Inorganics 6, no. 1: 1. https://doi.org/10.3390/inorganics6010001
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.