Metallated [3]Ferrocenophanes Containing P3M Bridges (M = Li, Na, K) §


Institut für Chemie und CINSaT, Universität Kassel, Heinrich-Plett-Straße 40, 34132 Kassel, Germany
*
Author to whom correspondence should be addressed.
§
Dedicated to Prof. Dr. Dietmar Stalke on the occasion of his 60th birthday.
Inorganics 2018, 6(3), 67; https://doi.org/10.3390/inorganics6030067
Submission received: 19 June 2018
/
Revised: 4 July 2018
/
Accepted: 6 July 2018
/
Published: 11 July 2018
(This article belongs to the Special Issue Organometallic Macrocycles and Their Applications)
Abstract
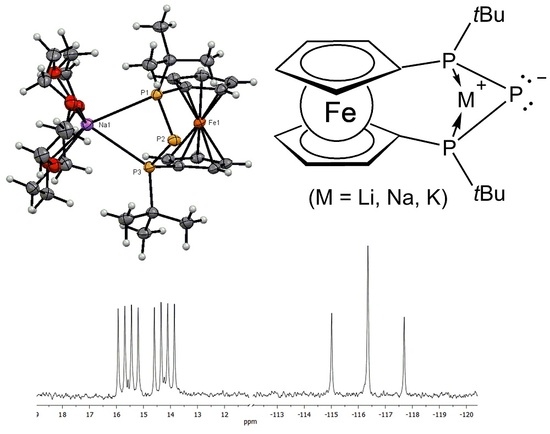
:Alkali-metal phosphanides can be embedded into a [3]ferrocenophane scaffold giving rise to bicyclic ferrocenophanes [MFe(C5H4PtBu)2P] (M = Li, Na, K). Coordination of the alkali-metal ions takes place via the terminal phosphorus atoms adopting a puckered P3M four-membered ring. All compounds were characterized via single-crystal X-ray diffraction and multinuclear NMR spectroscopy (1H, 31P, 7Li), whereas 13C NMR data could only be recorded for the Li derivative, owing to the limited solubility of its heavier congeners in unreactive solvents.
1. Introduction
Alkali-metal phosphanides are versatile reagents for connecting phosphorus centers to various functionalities [1,2]. Apart from this role as important synthetic tools, metal phosphanides show a rich structural diversity [1,3]. While metal-rich phosphanides are well known [4], research on saturated phosphorus-rich phosphanides just started emerging [5,6]. In a similar vein, unsaturated phosphorus-rich phosphanides were employed as building blocks for remarkable supramolecular architectures at the limits of molecular chemistry [7,8,9]. Based on our own efforts in the chemistry of phosphorus-rich ferrocenophanes, we became interested in exploring the metallation of a P–H functionalized bridging unit in a fully phosphorus-bridged [3]ferrocenophane [10,11,12,13]. Herein, we report the results of our investigation targeting the metallation of a stereochemically predefined secondary triphosphane constrained by the [3]ferrocenophane scaffold.
2. Results and Discussion
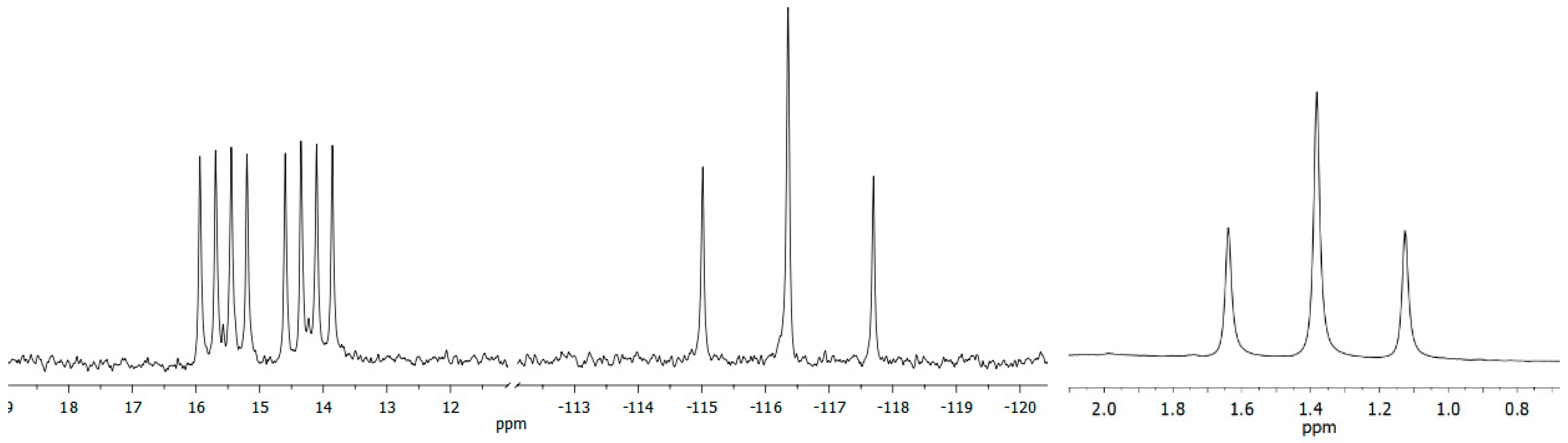
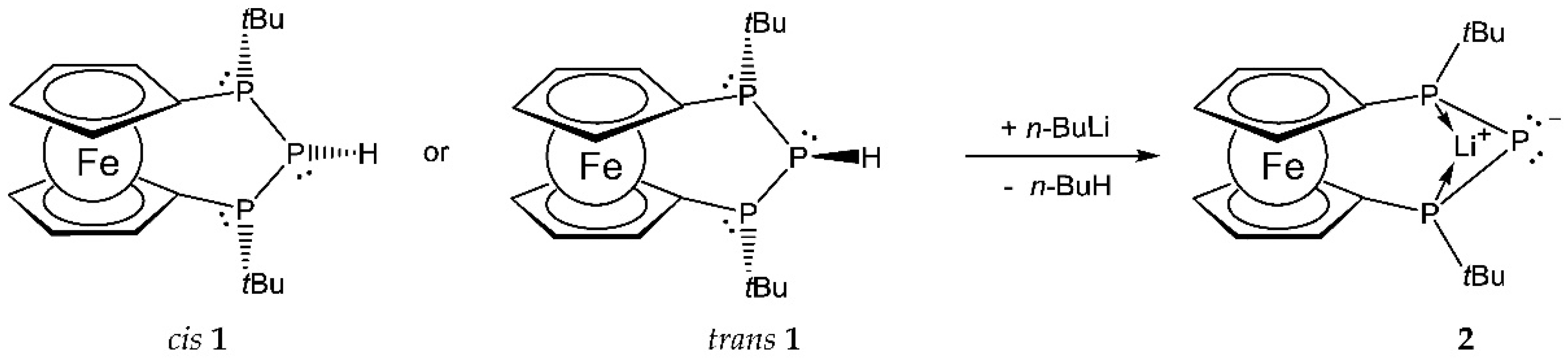
Secondary triphosphane (1, Scheme 1) was obtained over the course of our investigations dealing with phosphorus-rich ferrocenophanes. It is stereochemically predefined in a way that only the optically inactive meso forms, cis and trans 1, are obtained, with the terminal bridgehead phosphorus atoms showing opposite absolute configuration, and both t-butyl groups pointing to the same side of the molecule [10]. Reaction of a diastereomeric mixture of cis and trans 1 with n-butyl lithium in hexane in the presence of tetramethyl ethylene diamine (TMEDA) afforded the envisaged lithium phosphanide (2, Scheme 1) in the form of a single diastereomer (Scheme 1). In the 31P and 7Li NMR spectra, 2 was characterized by a characteristic signal splitting (Figure 1). The product showed a 31P NMR signal at −116.4 ppm (C6D6) for the central phosphorus atom, split into a triplet owing to coupling with the two chemically equivalent terminal phosphorus atoms (1JPP = 271 Hz) resonating at 14.9 ppm. In addition to the already mentioned P–P coupling, the doublet of the latter resonance is split into a quartet of equal intensity due to coupling with 7Li. In line with this, the 7Li resonance showed a triplet with 1JLiP = 50 Hz at 1.4 ppm, indicating symmetric bridging of the two terminal phosphorus atoms via the lithium atom.
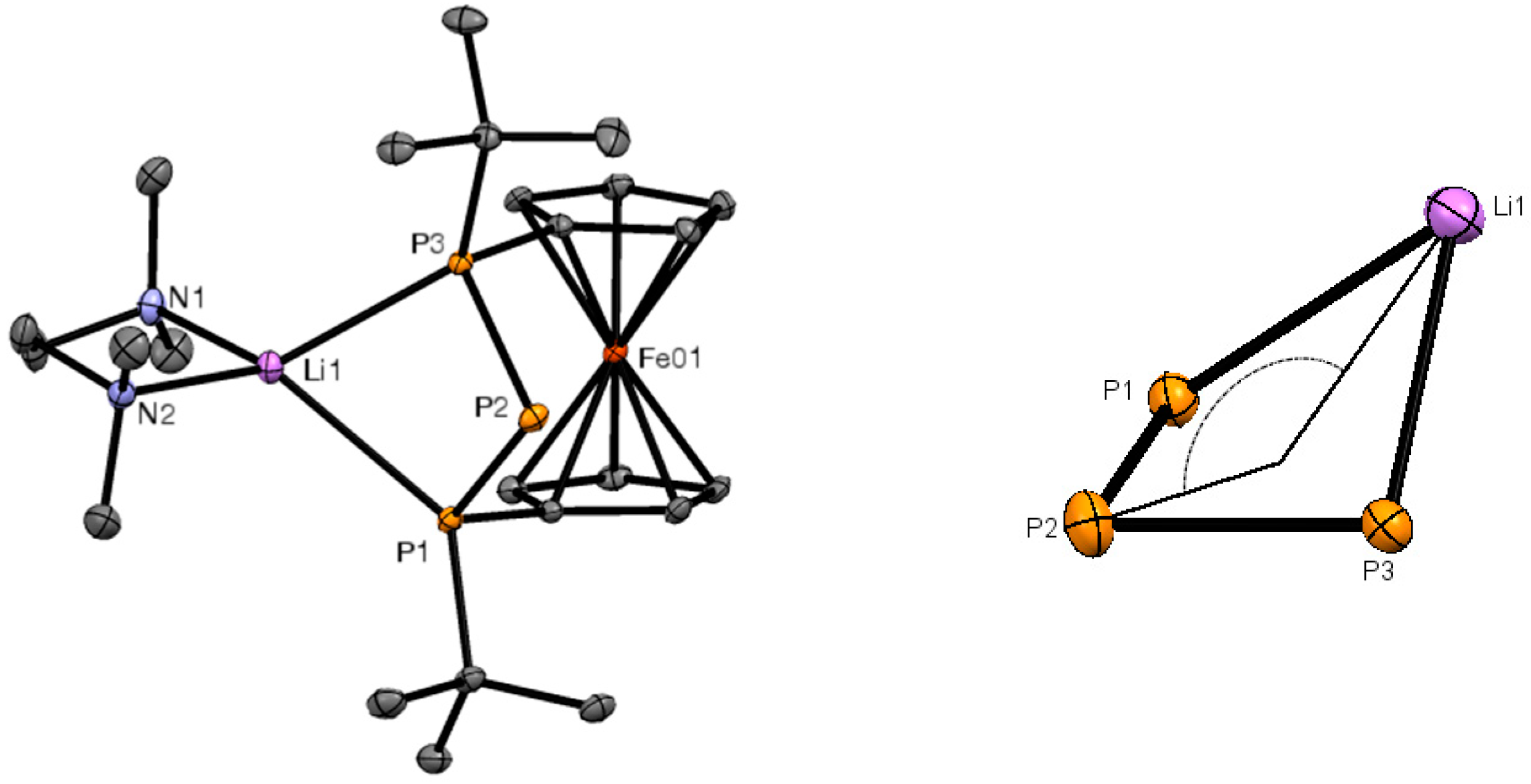
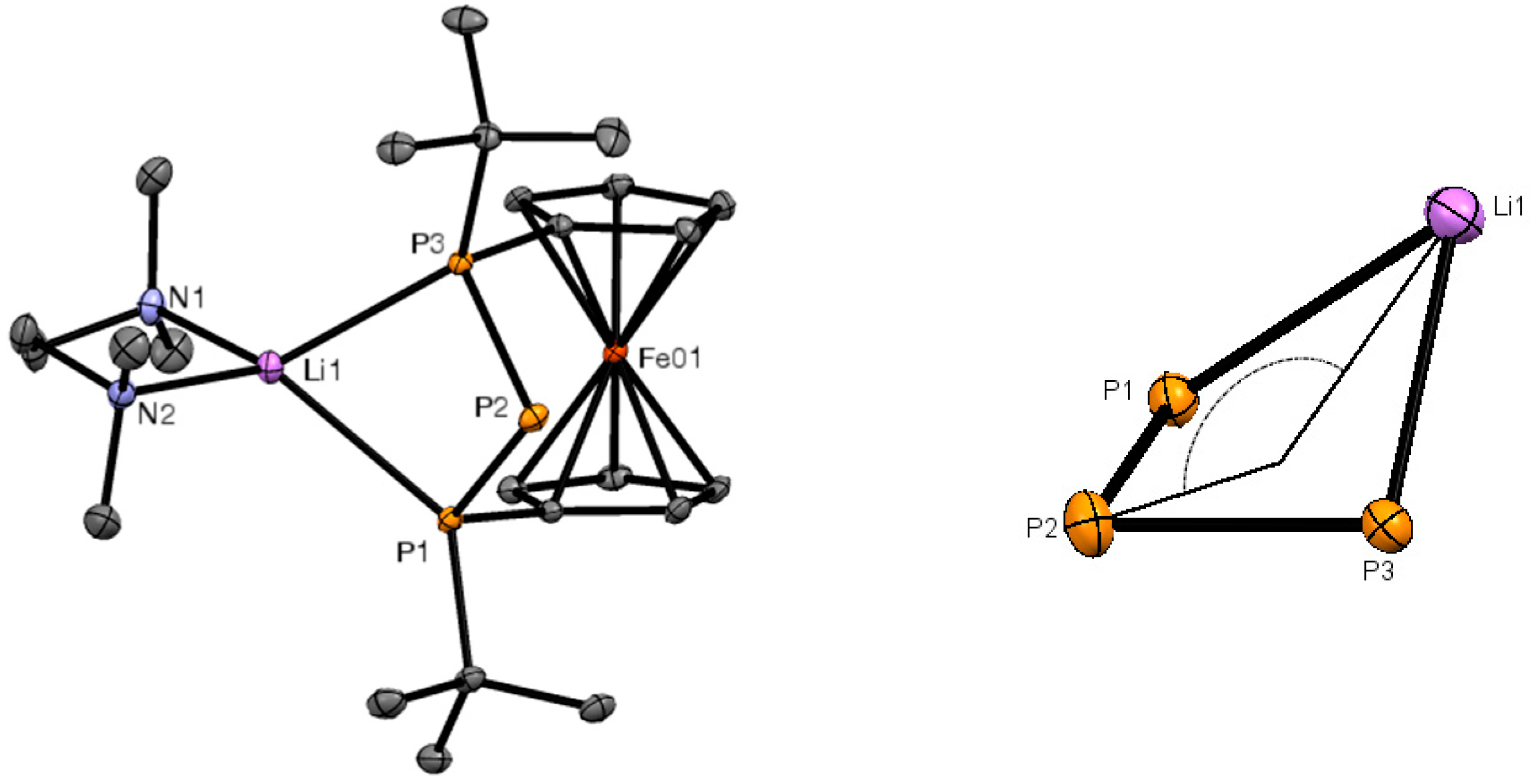
The structural motif of a P3Li four-membered ring was further corroborated using X-ray diffraction on single-crystalline 2. It crystallized in a monoclinic space group (C2/c) with half a molecule of hexane in the asymmetric unit. The P3Li unit formed a diamond-like puckered ring, bridging the Cp-rings in this bicyclic [3]ferrocenophane (Figure 2). The lithium atom showed symmetric contacts to the adjacent phosphorus atoms (2.662(3) Å and 2.548(3) Å), and a distance of 3.455(3) Å to the formal phosphanide center. The P–Li distances in 2 were close to those observed in anionic 1,3-diphospha-2-silaallylic systems with lithium as a counter ion [14]. The P–P bond lengths in 2, between the central phosphorus and the adjacent terminal phosphorus atoms, were 2.1710(8) Å and 2.1670(8) Å. This triphospha metallacycle folded with an angle of 135.89(6)° (Figure 2). The coordination sphere of lithium was completed by one molecule of TMEDA. The respective Li–N distances were placed with 2.113(4) Å and 2.080(4) Å in the expected range known from comparable compounds [15,16]. Owing to steric effects, the coordination geometry around lithium differed from an ideal tetrahedron with angles of 119.8(1)° to 135.6(2)°, showing larger values toward the ferrocene backbone. The Cp rings of the ferrocene unit showed a nearly ecliptic conformation (torsion angle of about 2°), and opened an interplanar angle of 3.3(1)° toward the lithium TMEDA adduct.
The structural situation is closely related to the dilithiated bisphosphanide [Fe(Cp-P(Li)tBu)2], where two phosphorus atoms are symmetrically bridged by two lithium atoms forming a P2Li2 four-membered ring in the solid state and in solution [11,15]. Similarly, the spectroscopic data are comparable to the latter bisphosphanide, as well as to other dimeric lithium phosphanides [5,17]. Phosphanide 2 was very sensitive to air and moisture, and was stable only in hydrocarbon solvents.
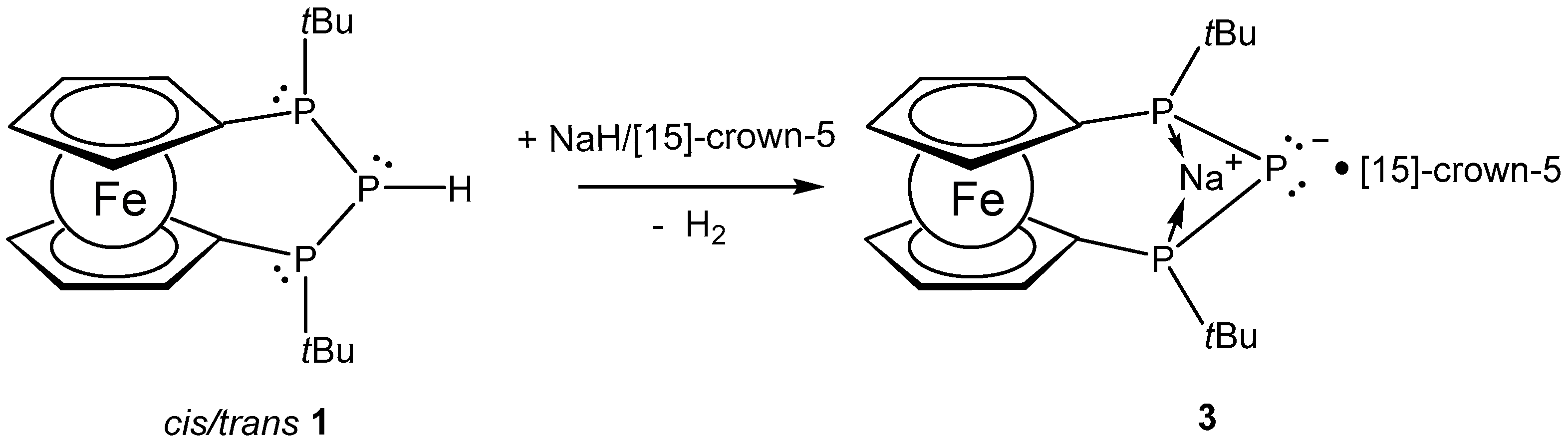
In order to explore the accessibility of the sodium analog of 2, we reacted 1 with sodium hydride in the presence of [15]-crown-5 in tetrahydrofuran (THF) solution. The solution turned light-red over a period of 12 h, indicating the formation of sodium phosphanide (3, Scheme 2). The mixture was stirred for an additional 24 h for completion. In the 31P NMR spectra, 3 was characterized by a resonance at −100.3 ppm (THF-d8) for the central phosphorus atom, which splits into a pseudo triplet with a pseudo coupling constant of 1JPP = 277 Hz. The two chemically equivalent terminal phosphorus atoms showed a single doublet at 21.3 ppm, with a matching coupling constant to the central phosphorus atom.
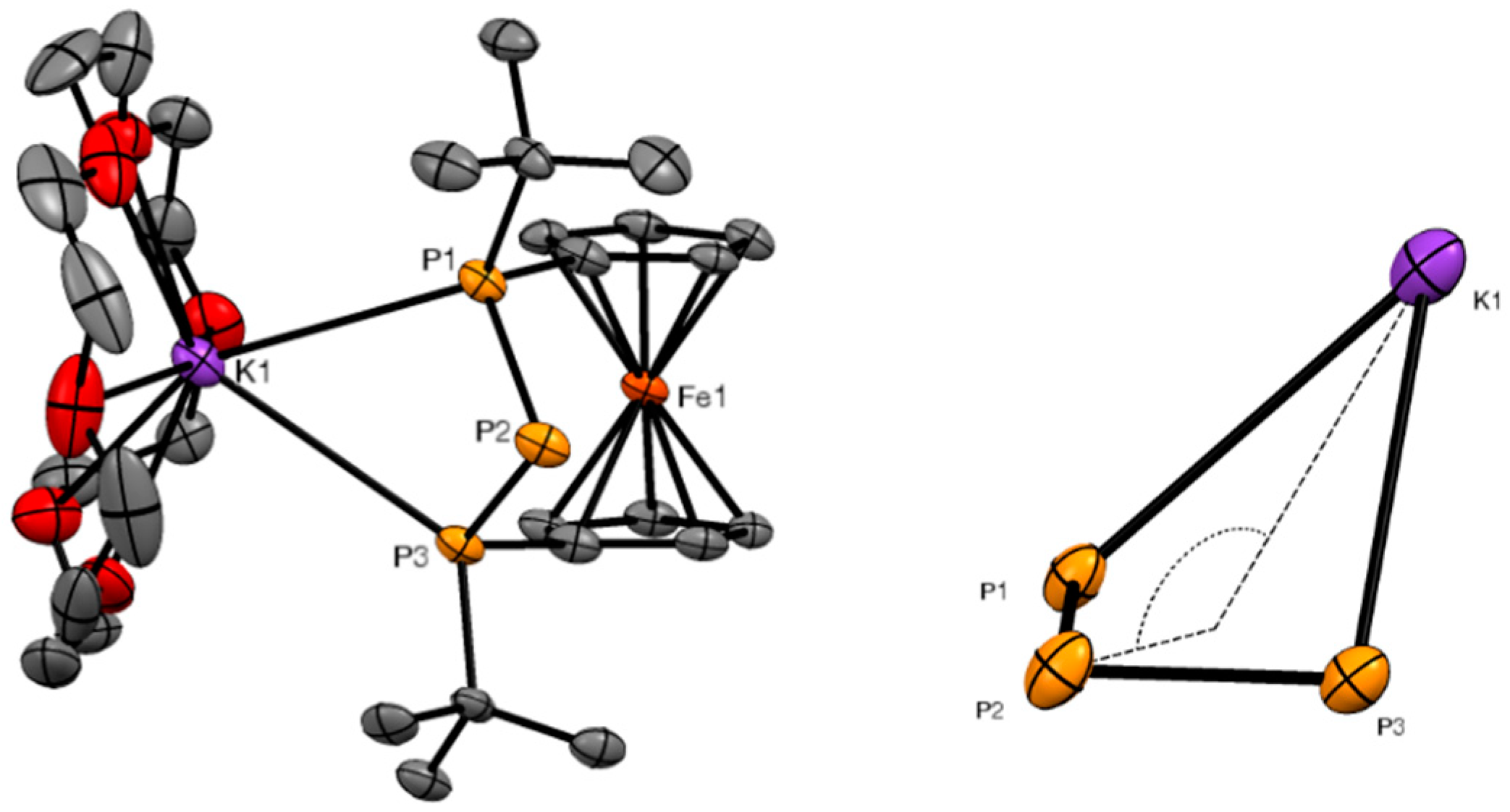
Again, single crystals suitable for X-ray diffraction could be obtained, confirming the presence of the P3Na motif also in this heavier alkali phosphanide (Figure 3). Compound 3 crystallized in a monoclinic space group (P21/c), with a somewhat smaller interplanar angle (3.1(1)°) compared to 2, and a bigger torsion angle of the ferrocene backbone (about 3°). Similar to 2, the P3Na unit formed a diamond-like puckered ring which was more folded (129.63(3)°) than in its lithium congener. The sodium atom showed symmetric contacts to the adjacent phosphorus atoms (3.037(2) Å and 3.107(2) Å), and a distance of 3.868(1) Å to the formal phosphanide center. The P–P bond lengths between the latter and the adjacent phosphorus atoms were 2.184(2) Å and 2.174(2) Å. The coordination sphere of sodium was completed by one molecule of [15]-crown-5. The respective Na–O distances lay in the range of 2.453(2) Å to 2.607(2) Å, with an average distance of 2.498(2) Å, which matched values known in literature [18,19,20,21,22,23]. In contrast to the number of published potassium triphosphanides, characterized sodium triphosphanides are scarce. Nevertheless, the available 31P chemical shifts in the literature fit our results quite well [24]. Furthermore, Grützmacher et al. described a P3 phosphanediide ion with similar bond distances between the sodium cation and the phosphanide centers, as well as similar described phosphorus–phosphorus distances [25]. Even the Na–P distances in triphosphaallylic sodium were close to those found in 3, despite the quite different bonding situation of the π-conjugated species [26].
To amend this series of closely related alkali-metal triphosphanides, we set out to prepare the corresponding potassium compound, 4 (Scheme 3). While the deprotonation of 1 with potassium hydride was precluded by the intrinsically low solubility of this reagent, we were able to achieve deprotonation using potassium t-butoxide (KOtBu). Owing to the limited solubility of KOtBu in neat hydrocarbons, the reaction had to be carried out in coordinating solvents such as THF (Scheme 3). The formation of 4 was indicated by an instantaneous color change showing a deep-red solution. In the 31P NMR spectra, 4 was characterized by a resonance of the central phosphanide atom as a triplet at −107.3 ppm (THF-d8) with a 1JPP coupling of 268 Hz. The corresponding doublet was located at 16.8 ppm.
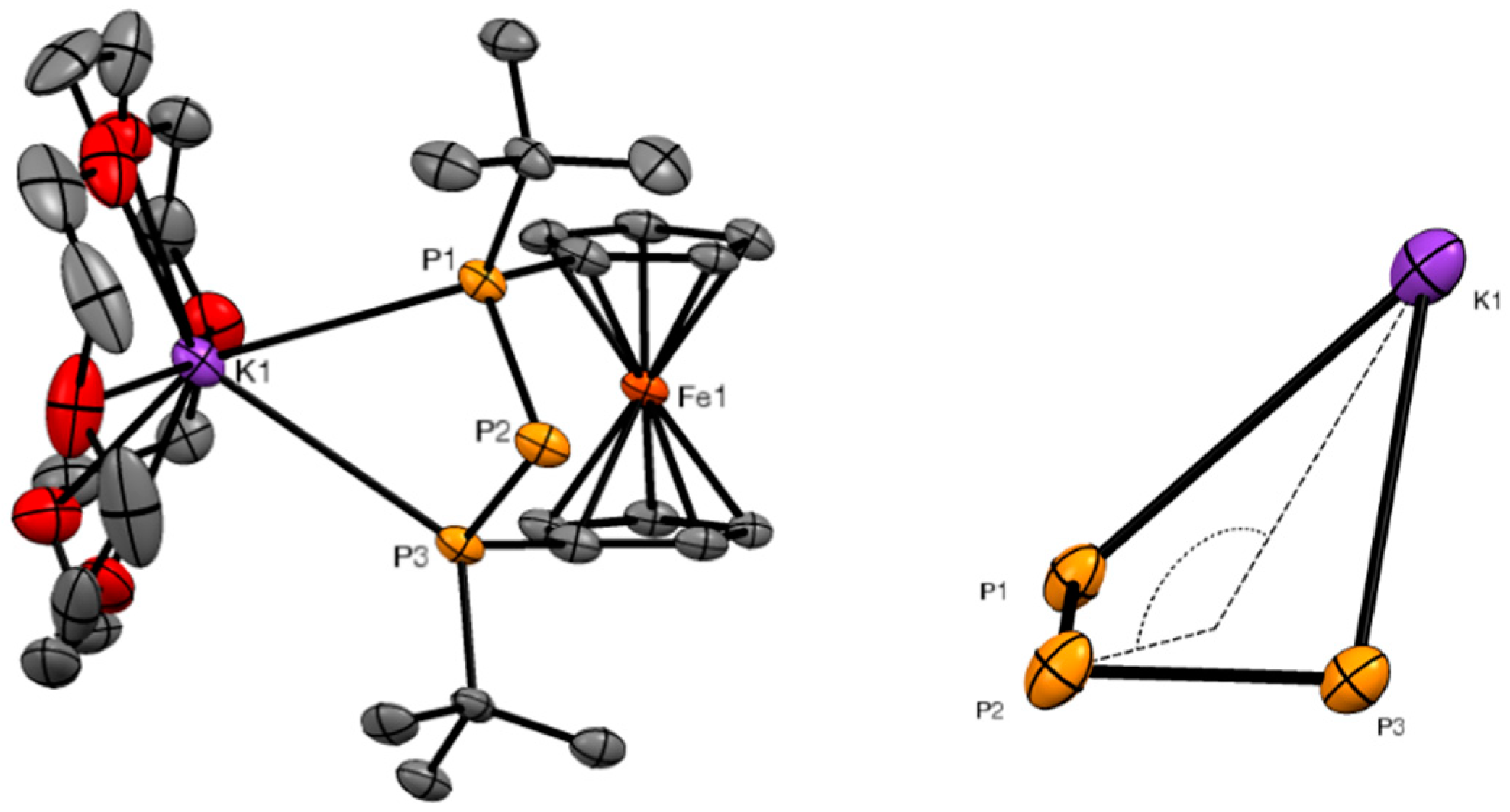
Microcrystalline and poorly soluble 4 was recrystallized in the presence of [18]-crown-6 affording single-crystalline samples suitable for X-ray diffraction. The heaviest alkali metal of our series showed a similar P3K motif to that in its lighter analogs (Figure 4). Compound 4·[18]-crown-6 crystallized in a triclinic space group (P−1), and showed a less strained (interplanar angle 2.1(5)°) but more staggered (torsion angle about 5°) structure. Similar to 2 and 3, the P3K unit formed a diamond-like puckered ring which was more folded (125.5(1)°) than in the Li/Na congeners, possibly caused by the introduction of a sterically more demanding crown ether. The potassium atom showed slight asymmetric contacts to the adjacent phosphorus atoms (3.581(3) Å and 3.333(2) Å), and a distance of 4.227(3) Å to the formal phosphanide center. The P–P bond lengths between the latter and the adjacent phosphorus atoms were 2.187(2) Å and 2.192(4) Å, and the folding angle of the puckered ring was smallest in this series. The coordination sphere of potassium was completed by one molecule of [18]-crown-6, adding up to a total coordination number of eight around the potassium atom. The respective K–O distances lay in the range of 2.748(8) Å to 3.039(8) Å, showing an average distance of 2.88(1) Å. This was in line with similar distances in the literature [18,19,27,28]. The crown ether showed disorder at the carbon and oxygen atoms, in which ellipsoids stretched along the ring, indicating different positions of the crown in the crystal by rotation about the potassium center. This disorder limited the R value to 0.1104.
As already mentioned, phosphanides 2–4 were extremely sensitive to air and moisture, and traces of residual water or reactive solvents available for deprotonation, such as THF, led to protonation of 2–4, resulting in the formation of 1, which precluded the isolation of 2–4 in an analytically pure form. Interestingly, the protonation of isostructural phosphanides 2–4 resulted exclusively in the formation of trans 1. We assume that this selectivity most likely originated from the preferential attack of water from the metal side for kinetic reasons, since the energy difference between cis and trans 1 was only small in favor of the trans isomer (0.1 kcal/mol at the B3LYP/6-311+G** level of theory) [10].
The heteronuclear NMR data did not follow a clear trend along the series of alkali-metal cations from Li+ to K+ in line with observations reported in the literature [24]. Compared with the parent hydrophosphane, 1, the formal phosphanide centers in deprotonated 2–4 were more shielded with respect to cis 1, but deshielded with respect to trans 1. The adjacent phosphorus atoms were shifted downfield upon interaction with the alkali-metal cations (δ(31P): cis 1: −74.3/−9.8 ppm; trans 1: −125.1/−6.5 ppm [10]; 2: −116.4/14.9 ppm; 3: −100.3/21.3 ppm; 4: −107.3/16.8 ppm). The 1JPP coupling constants of 2–4 were quite similar (2: 271 Hz; 3: 277 Hz; 4: 268 Hz), with values between those observed for cis and trans 1 (cis: 295/289 Hz; trans: 107 Hz), indicating reduced P–P interaction compared to the cis isomer 1, in which the lone pairs of all phosphorus atoms were located on the same side of the molecule [10]. In turn, the additional electron pair at the formal phosphanide center in 2–4 increased the interaction compared with trans 1 [29,30].
With increasing size of the alkali metal, the diamond-shaped ring was more folded, possibly owing to repulsive interactions between the crown ether and the ferrocene backbone. As a consequence of this steric effect, the torsion angle of the ferrocene unit slightly increased within the series, whereas the interplanar angle of the Cp rings decreased. Similar structural effects could be observed in triphospha [3]ferrocenophanes substituted with sterically demanding alkyl groups [10]. The M–P2 distances in 2–4 were all slightly below the sum of the Van-der-Waals radii [31]. However, the interaction between the alkali metal and P2 was most likely electrostatic in nature. The distances of the formal phosphanide atom to the iron center of the ferrocene backbone were almost identical for 2–4 (ca. 3.9 Å), and matched the value for the parent phosphane, 1, to which they were isostructural [10].
3. Experimental Details
All manipulations were carried out under inert argon atmosphere using a standard Schlenk technique. All solvents were dried and freshly distilled over an Na/K-alloy where applicable. Compound 1 was prepared according to a previously published procedure [10]. All other reagents were purchased and used without further purification. 1H, 13C, and 31P NMR spectra were recorded on a Varian VNMRS-500 MHz spectrometer (Varian Inc., Palo Alto, CA, USA) at room temperature using tetramethylsilane (TMS) as the external reference for 1H and 13C nuclei. 13C–1H correlation was used for the detailed chemical shift assignments in 2. For multiplets, chemical shifts were reported as the center of the respective multiplet.
3.1. Synthesis of Compound 2
To a stirred mixture of 0.16 g (0.4 mmol) of 1 and 0.1 mL (0.7 mmol) of TMEDA in 5 mL hexane, 0.16 mL (0.4 mmol; 2.5 M) of nBuLi in hexane was added dropwise at room temperature. After 2 h, the resulting orange precipitate was filtered off and washed with 1 mL of pentane. The product was obtained as an orange solid. Single crystals suitable for X-ray diffraction analysis could be obtained via recrystallization from hexane. Yield: 0.18 g (89%). 1H NMR (500 MHz, C6D6): δ = 1.52 (m, 18H, tBu), 1.68 (br s, 4H, CH2), 2.01 (br s, 12H, N(CH3)2), 4.04 (m, 2H, Cp), 4.28 (m, 2H, Cp), 4.35 (m, 2H, Cp), 5.15 (m, 2H, Cp) ppm; 13C{1H} NMR (126 MHz, C6D6): δ = 31.1 (m, tBu Cq), 31.3 (m, tBu CH3), 46.5 (s, N(CH3)2), 56.6 (s, CH2), 67.6 (m, Cp), 71.0 (s, Cp), 72.3 (m, Cp), 77.0 (m, Cp), 91.5 (m, Cp Cipso) ppm; 31P NMR (202 MHz, C6D6): δ = −116.4 (t, 1P, 1JPP = 271 Hz, P−), 14.9 (dq, 2P, 1JPP = 271 Hz, 1JPLi = 50 Hz, P tBu). 7Li NMR (194 MHz, C6D6): δ = 1.4 (t, 1JLiP = 50 Hz).
3.2. Synthesis of Compound 3
Firstly, 0.078 g (0.2 mmol) of 1, 0.005 g (0.2 mmol) of sodium hydride, and 0.022 g (0.2 mmol) of 15-crown-5 were dissolved in THF (2 mL), and stirred for two days at room temperature. A red solid precipitated from the former orange solution, which was separated by filtration. The remaining solid was washed with pentane (2 × 5 mL), and gave 3 as a light-red solid. Recrystallization from THF afforded single crystals suitable for X-ray diffraction analysis. Yield: 0.10 g (78%). 1H NMR (500 MHz, THF-d8): δ = 1.17 (m, 18H, tBu), 3.59 (m, 20H, 15-crown-5), 3.76 (m, 2H, Cp), 4.07 (m, 2H, Cp), 4.29 (m, 2H, Cp), 4.66 (m, 2H, Cp) ppm; 31P NMR (202 MHz, THF-d8): δ = −100.3 (pseudo t, 1P, 1JPP = 277 Hz, P−), 21.3 (d, 1P, 1JPP = 277 Hz, P tBu).
3.3. Synthesis of Compound 4
To a stirred dry mixture of 0.118 g (0.3 mmol) of 1 and 0.034 g (0.3 mmol) of KOtBu, 3 mL of THF was added at room temperature. The solution immediately turned deep red, and stirring was continued for an additional 30 min. All volatiles were removed under reduced pressure, and the residue was kept under vacuum for at least 1 h. Then, 2 mL of THF and 0.080 g (0.3 mmol) of 18-crown-6 were added to the residue, and stirred for 5 min. After removing the solvent in vacuo, the product was obtained as a red solid. Recrystallization from THF afforded crystals suitable for X-ray diffraction analysis. Yield: 0.14 g (65%). 1H NMR (500 MHz, THF-d8): δ = 1.13 (m, 18H, tBu), 3.14–3.50 (br s, 24H, 18-crown-6), 3.80 (m, 2H, Cp), 4.09 (m, 2H, Cp), 4.13 (m, 2H, Cp), 4.64 (m, 2H, Cp) ppm; 31P NMR (202 MHz, THF-d8): δ = −107.3 (t, 1P, 1JPP = 268 Hz, P−), 16.8 (d, 2P, 1JPP = 268 Hz, P tBu).
3.4. X-ray Crystallography
X-ray diffraction measurements were performed on a Stoe IPDS 2 diffractometer (3) (STOE & Cie. GmbH, Darmstadt, Germany) with an image plate detector and monochromated (graded multilayer mirror) Mo Kα radiation (Mo Genix), or on a Stoe StadiVari diffractometer (2 and 4) with a Dectris Pilatus 200 K detector using monochromated (graded multilayer) Cu Kα radiation (Cu Genix) (Xenocs, Sassenage, France). The datasets were recorded with ω-scans, and corrected for Lorentz, polarization, and absorption effects. The structures were solved using direct methods and refined without restraints by full-matrix least-squares techniques against F2 (SHELXT and SHELXL-2014/7) [32]. Details of the structure determinations and refinement for 2–4 are summarized in Table 1. Further programs used for analysis and visualization of structural information included WinGX and Mercury [33,34]. CCDC 1847287–1847289 contains the supplementary crystallographic data for this paper (See Supplementary Materials). These data are provided free of charge by the Cambridge Crystallographic Data Centre.
4. Conclusions
In summary, we demonstrated that secondary triphosphanides of the alkali metals, M = Li, Na, K, could be embedded into a [3]ferrocenophane scaffold, giving rise to bicyclic ferrocenophanes with a two-fold intramolecular P→M donor stabilization as part of puckered P3M rings. The folding of these rings became more pronounced as the alkali-metal cation increased in size. Compounds 3 and 4 were limited in solubility to organic polar solvents, such as THF, while 2 was soluble in nonpolar solvents as well. All alkali-metal cations required coordination of additional donors to complete their coordination spheres. Nevertheless, all compounds were highly sensitive to air and moisture in solution and in the solid state. It is worth noting that the formally hydridic P–H bond (according to a difference in electronegativity) clearly behaved in a protic nature with all reagents under investigation, even though secondary triphosphane lacked any electronegative substituents at phosphorus. Remarkably, the protonation of phosphanides 2–4 led to the stereoselective formation of trans 1, which was otherwise not accessible.
Supplementary Materials
The following are available online at https://www.mdpi.com/2304-6740/6/3/67/s1. Cif and checkcif files of complexes 2–4.
Author Contributions
I.S. and L.-M.F. performed the experiments, and analyzed the spectroscopic data; C.B. performed the X-ray interpretation; R.P. provided the materials and wrote the paper.
Funding
German Science fund (DFG, PI 353/8-1 and CRC 1319).
Acknowledgments
The authors would like to thank the German Science fund (DFG, PI 353/8-1 and CRC 1319) for financial support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Izod, K. Group I complexes of P- and as-donor ligands. Adv. Inorg. Chem. 2000, 50, 33–107. [Google Scholar]
- Fritz, G.; Scheer, P. Silylphosphanes: Developments in Phosphorus Chemistry. Chem. Rev. 2000, 100, 3341–3402. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, L. Metal complexes of planar PR2 ligands: Examining the carbene analogy. Coord. Chem. Rev. 2012, 256, 606–626. [Google Scholar] [CrossRef]
- Driess, M.; Nöth, H. Molecular Clusters of the Main Group Elements; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Wolf, R.; Schisler, A.; Lönnecke, P.; Jones, C.; Hey-Hawkins, E. Syntheses and Molecular Structures of Novel Alkali Metal Tetraorganylcyclopentaphosphanides and Tetraorganyltetraphosphane-1, 4-diides. Eur. J. Inorg. Chem. 2004, 16, 3277–3286. [Google Scholar] [CrossRef]
- Wolf, R.; Gomez-Ruiz, S.; Reinhold, J.; Boehlmann, W.; Hey-Hawkins, E. The (P4HMes4)− anion: Lability, fluxionality, and structural ambiguity (Mes = 2,4,6-Me3C6H2). Inorg. Chem. 2006, 45, 9107–9113. [Google Scholar] [CrossRef] [PubMed]
- Dielmann, F.; Heindl, C.; Hastreiter, F.; Peresypkina, E.V.; Virovets, A.V.; Gschwind, R.M.; Scheer, M. A nano-sized Supramolecule beyond the Fullerene Topology. Angew. Chem. Int. Ed. 2014, 53, 13605–13608. [Google Scholar] [CrossRef] [PubMed]
- Scheer, M.; Schindler, A.; Merkle, R.; Johnson, B.P.; Linseis, M.; Winter, R.; Anson, C.E.; Virovets, A.V. Fullerene C60 as an Endohedral Molecule within an Inorganic Supramolecule. J. Am. Chem. Soc. 2007, 129, 13386–13387. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Virovets, A.V.; Scheer, M. Synthesis of Inorganic Fullerene-Like Molecules. Science 2003, 300, 781–783. [Google Scholar] [CrossRef] [PubMed]
- Borucki, S.; Kelemen, Z.; Maurer, M.; Bruhn, C.; Nyulászi, L.; Pietschnig, R. Stereochemical alignment in triphospha-[3]ferrocenophanes. Chem. Eur. J. 2017, 23, 10438–10450. [Google Scholar] [CrossRef] [PubMed]
- Kargin, D.; Kelemen, Z.; Krekić, K.; Maurer, M.; Bruhn, C.; Nyulászi, L.; Pietschnig, R. [3]Ferrocenophanes with bisphosphanotetryl bridge: Inorganic rings on the way to tetrylenes. Dalton Trans. 2016, 45, 2180–2189. [Google Scholar] [CrossRef] [PubMed]
- Moser, C.; Belaj, F.; Pietschnig, R. Phosphorus-Rich Ferrocenophanes. Phosphorus Sulfur Silicon Relat. Elem. 2015, 190, 837–844. [Google Scholar] [CrossRef]
- Moser, C.; Belaj, F.; Pietschnig, R. Diphospha[2]ferrocenophane alias (1,4-dihydrotetraphosphaneoxide): Stereoselective formation via hydrolytic P-P bond formation. Chem. Eur. J. 2009, 15, 12589–12591. [Google Scholar] [CrossRef] [PubMed]
- Lange, D.; Klein, E.; Bender, H.; Niecke, E.; Nieger, M.; Pietschnig, R.; Schoeller, W.W.; Ranaivonjatovo, H. 1,3-Diphospha-2-silaallylic Lithium Complexes and Anions: Synthesis, Crystal Structures, Reactivity, and Bonding Properties. Organometallics 1998, 17, 2425–2432. [Google Scholar] [CrossRef]
- Hitzel, S.; Färber, C.; Bruhn, C.; Siemeling, U. Phosphido complexes derived from 1,1’-ferrocenediyl-bridged secondary diphosphines. Dalton Trans. 2017, 46, 6333–6348. [Google Scholar] [CrossRef] [PubMed]
- Less, R.J.; Naseri, V.; Wright, D.S. Formation of an organometallic phosphanediide via main-group dehydrocoupling. Organometallics 2009, 28, 1995–1997. [Google Scholar] [CrossRef]
- Reich, H.J.; Dykstra, R.R. Solution Structure of Lithium Benzeneselenolate and Lithium Diphenylphosphide: NMR Identification of Cyclic Dimers and Mixed Dimers. Organometallics 1994, 13, 4578–4585. [Google Scholar] [CrossRef]
- Poonia, N.S.; Bajaj, A.V. Coordination chemistry of alkali and alkaline earth cations. Chem. Rev. 1979, 79, 389–445. [Google Scholar] [CrossRef]
- Bajaj, A.V.; Poonia, N.S. Comprehensive coordination chemistry of alkali and alkaline earth cations with macrocyclic multidentates: Latest position. Coord. Chem. Rev. 1988, 87, 55–213. [Google Scholar] [CrossRef]
- Berry, A.; Green, M.L.H.; Bandy, J.A.; Prout, K. Transition Metal-Hydrogen-Alkali Metal Bonds: Synthesis and Crystal Structures of [K(18-crown-6)][W(PMe3)3H5], [Na(15-crown-5)][W(PMe3)3H5] and [{W(PMe3)3H5Li}4] and Related Studies. J. Chem. Soc. Dalton Trans. 1991, 2185–2206. [Google Scholar] [CrossRef]
- Gjikaj, M.; Adam, A. Complexation of Alkali Triflates by Crown Ethers: Synthesis and Crystal Structure of [Na(12-crown-4)2][SO3CF3], [Na(15-crown-5)][SO3CF3], [Rb(18-crown-6)][SO3CF3], and [Cs(18-crown-6)][SO3CF3]. Z. Anorg. Allg. Chem. 2006, 632, 2475–2480. [Google Scholar] [CrossRef]
- Nöth, H.; Warchhold, M. Sodium Hydro(isothiocyanato)borates: Synthesis and Structures. Eur. J. Inorg. Chem. 2004, 2004, 1115–1124. [Google Scholar] [CrossRef]
- Cole, M.L.; Jones, C.; Junk, P.C. Ether and crown ether adduct complexes of sodium and potassium cyclopentadienide and methylcyclopentadienide—Molecular structures of [Na(dme)Cp]∞, [K(dme)0.5Cp]∞, [Na(15-crown-5)Cp], [Na(18-crown-6)CpMe] and the “naked Cp–” complex [K(15-crown-5)2][Cp]. J. Chem. Soc. Dalton Trans. 2002, 896–905. [Google Scholar] [CrossRef]
- Schmidpeter, A.; Burget, G. Die Reaktion Von Alkaliphosphiden Mit Weissem Phosphor, 11 Bildung Von 1,1,3,3-Tetraphenyl-Triphosphid, 2,3,4,5-Tetraphenyl-Cyclopentaphosphid und Phenyl-Tricycloheptaphosphid. Phosphorus Sulfur Silicon Relat. Elem. 1985, 22, 323–335. [Google Scholar] [CrossRef]
- Geier, J.; Ruegger, H.; Worle, M.; Grützmacher, H. Sodium oligophosphanediide ions in the PhPCl2/Na system: Syntheses and structural characterization. Angew. Chem. Int. Ed. 2003, 42, 3951–3954. [Google Scholar] [CrossRef] [PubMed]
- Wiberg, N.; Wörner, A.; Lerner, H.W.; Karaghiosoff, K.; Fenske, D.; Baum, G.; Dransfeld, A.; von Ragué Schleyer, P. The Triphosphide (tBu3Si)2P3Na: Formation, X-ray and Ab initio Structure Analyses, Protonation and Oxidation to Triphosphane (tBu3Si)2P3H and Hexaphosphanes (tBu3Si)4P6. Eur. J. Inorg. Chem. 1998, 1998, 833–841. [Google Scholar] [CrossRef]
- Thiele, G.; Vondung, L.; Donsbach, C.; Pulz, S.; Dehnen, S. Organic Cation and Complex Cation-Stabilized (Poly-)Selenides, [Cation]x(Sey)z: Diversity in Structures and Properties. Z. Anorg. Allg. Chem. 2014, 640, 2684–2700. [Google Scholar] [CrossRef]
- Kobrsi, I.; Zheng, W.; Knox, J.E.; Heeg, M.J.; Schlegel, H.B.; Winter, C.H. Experimental and theoretical study of the coordination of 1, 2, 4-triazolato, tetrazolato, and pentazolato ligands to the [K(18-crown-6)]+ fragment. Inorg. Chem. 2006, 45, 8700–8710. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J. Higher order 31P NMR Spectra of Polyphosphorus Compounds. In Phosphorus-31 NMR Spectroscopy in Stereochemical Analysis; Verkade, J.G., Quin, L.D., Eds.; VCH: Weinheim, Germany, 1987; pp. 331–364. [Google Scholar]
- Albrand, J.P.; Faucher, H.; Gagnaire, D.; Robert, J.B. Calculation of the 1 J(PP) angular dependence in P2H4. Chem. Phys. Lett. 1976, 38, 521–523. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van der Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
Scheme 1.
Formation of 2 via lithiation of diastereomeric 1.
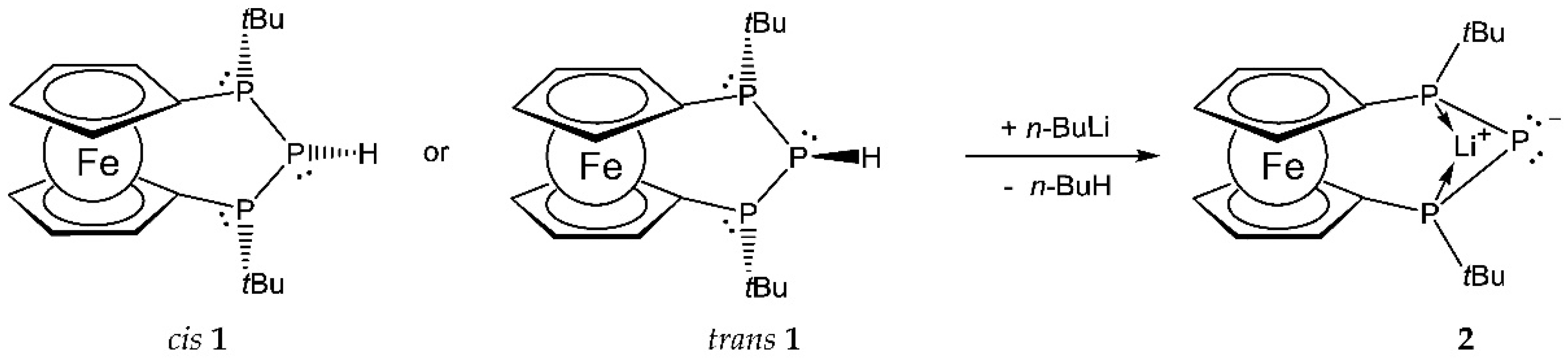
Figure 1.
Expansion from the 31P ((left) and (mid), 202 MHz) and 7Li ((right), 194 MHz) NMR spectra of 2 (Scheme 1) in benzene-d6 solution at room temperature.
Figure 1.
Expansion from the 31P ((left) and (mid), 202 MHz) and 7Li ((right), 194 MHz) NMR spectra of 2 (Scheme 1) in benzene-d6 solution at room temperature.
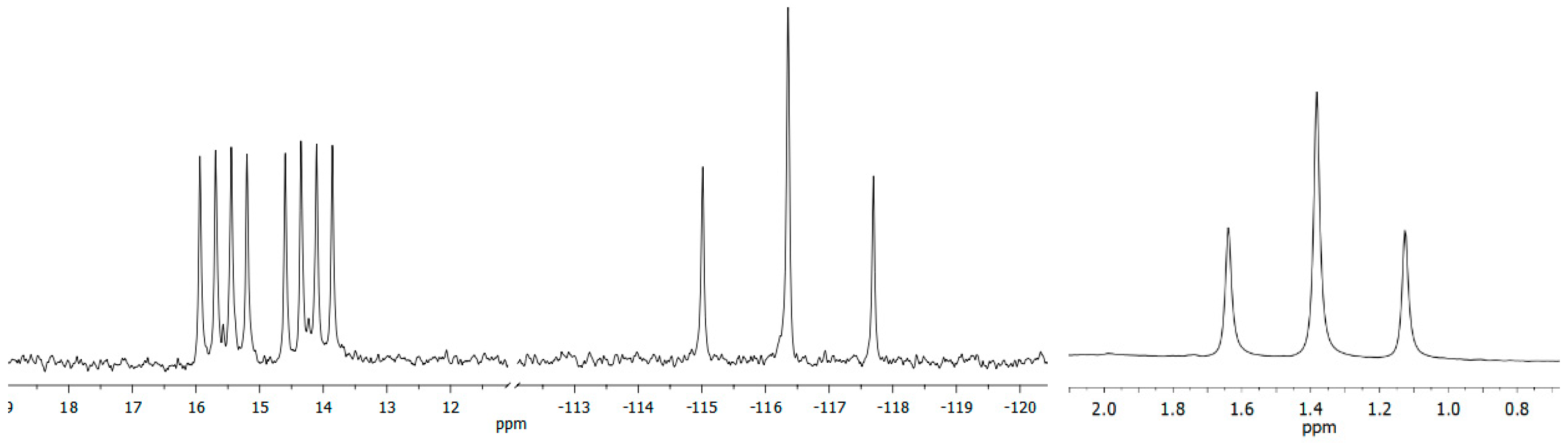
Figure 2.
ORTEP plot of the molecular structure of 2 (left), and expansion of the folded P3Li ring (right). Ellipsoids were drawn at the 30% probability level. Hydrogen atoms were omitted for clarity.
Figure 2.
ORTEP plot of the molecular structure of 2 (left), and expansion of the folded P3Li ring (right). Ellipsoids were drawn at the 30% probability level. Hydrogen atoms were omitted for clarity.
Scheme 2.
Formation of 3 via deprotonation of 1.

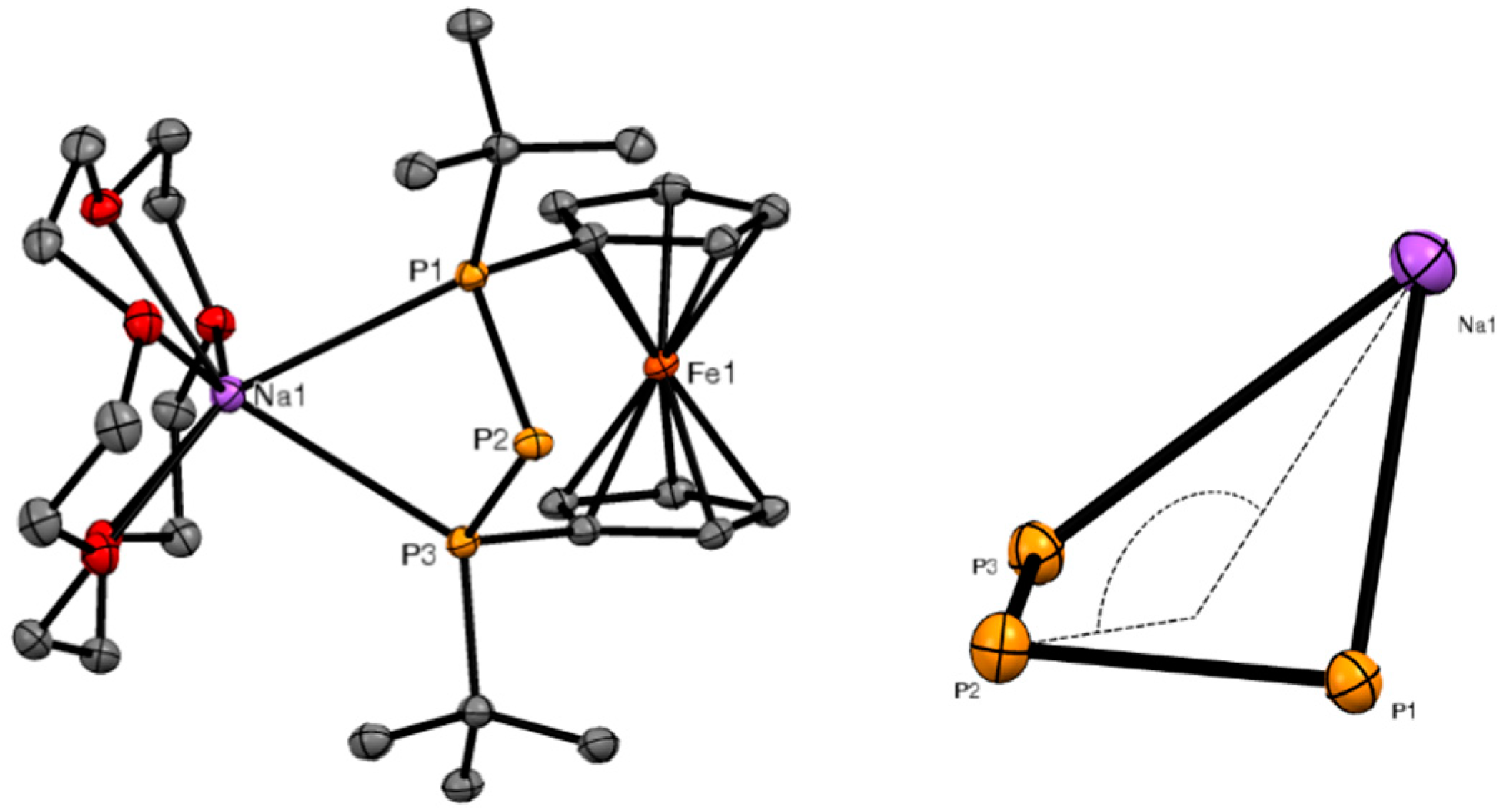
Figure 3.
ORTEP plot of the molecular structure of 3 (Scheme 2; (left)) and expansion of the folded P3Na ring (right). Ellipsoids were drawn at the 30% probability level. Hydrogen atoms were omitted for clarity.
Figure 3.
ORTEP plot of the molecular structure of 3 (Scheme 2; (left)) and expansion of the folded P3Na ring (right). Ellipsoids were drawn at the 30% probability level. Hydrogen atoms were omitted for clarity.
Scheme 3.
Formation of 4 via direct metallation of 1.
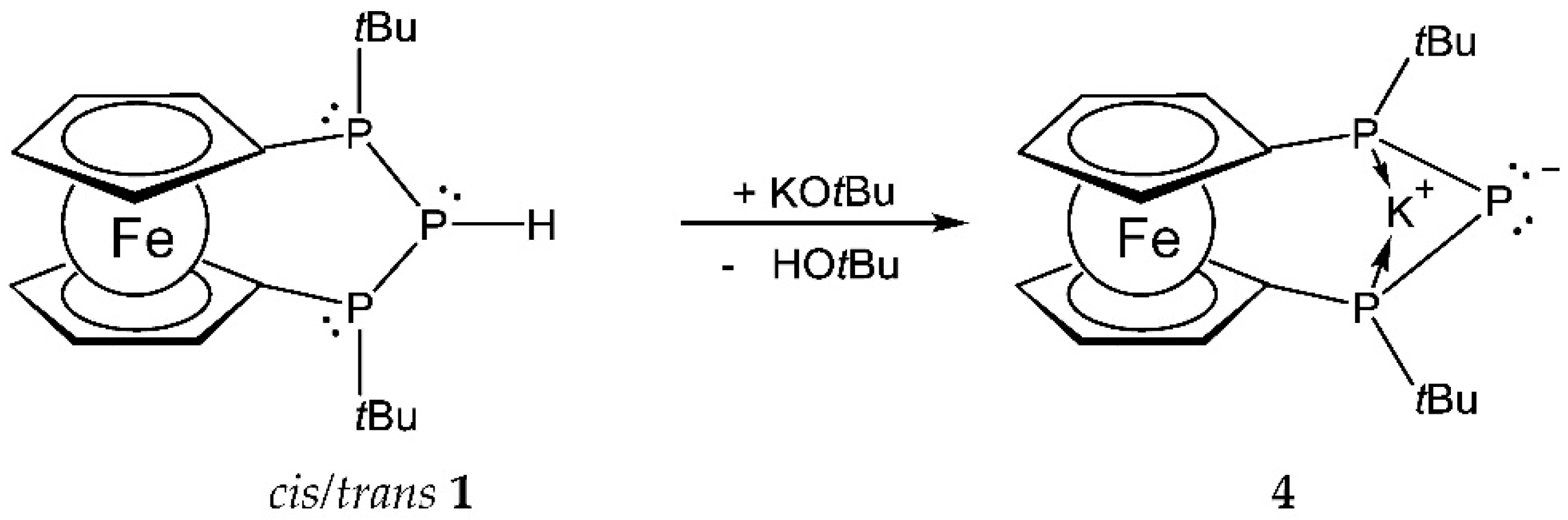
Figure 4.
ORTEP plot of 4·[18]-crown-6 (Scheme 3; (left)) and expansion of the folded P3K ring (right). Ellipsoids were drawn at the 30% probability level. Hydrogen atoms were omitted for clarity.
Figure 4.
ORTEP plot of 4·[18]-crown-6 (Scheme 3; (left)) and expansion of the folded P3K ring (right). Ellipsoids were drawn at the 30% probability level. Hydrogen atoms were omitted for clarity.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2 | 3 | 4 | |
|---|---|---|---|
| Formula | C24H42FeLiN2P3,0.5(C6H14) | C28H46FeNaO5P3 | C30H50FeKO6P3 |
| Formular weight | 557.38 | 634.40 | 694.56 |
| Temperature (K) | 100 | 100 | 100 |
| Wavelength (Å) | 1.54186 | 0.71073 | 1.54186 |
| Crystal system | monoclinic | monoclinic | triclinic |
| Space group | C2/c | P21/c | P−1 |
| Unit-cell dimensions: | |||
| a (Å) | 10.2172(3) | 14.2758(6) | 10.791(1) |
| b (Å) | 17.3386(6) | 10.3266(3) | 10.7753(10) |
| c (Å) | 34.6955(9) | 21.4828(9) | 16.8369(16) |
| α (°) | 90 | 90 | 73.904(7) |
| β (°) | 90.735(2) | 99.536(3) | 71.663(7) |
| γ (°) | 90 | 90 | 69.620(7) |
| Volume (Å3) | 6145.9(3) | 3123.2(2) | 1710.8(3) |
| Z | 8 | 4 | 2 |
| Calculated density (mg/m3) | 1.205 | 1.349 | 1.348 |
| μ (mm−1) | 5.526 | 0.685 | 6.265 |
| Θ-range for data collected (°) | 5.100–68.990 | 1.446–25.498 | 2.815–68.998 |
| Data/parameters | 5340/318 | 5814/349 | 6198/376 |
| Goodness-of-fit on F2 | 1.021 | 1.047 | 1.024 |
| R1 (observed data) | 0.0309 | 0.0531 | 0.1104 |
| wR2 (all data) | 0.0733 | 0.1559 | 0.3201 |
| Rint | 0.0240 | 0.0654 | 0.0772 |
| r.e.d. min/max (e·Å−3) | 0.539/0.834 | −0.944/0.776 | 0.451/0.887 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Isenberg, S.; Frenzel, L.-M.; Bruhn, C.; Pietschnig, R.
Metallated [3]Ferrocenophanes Containing P3M Bridges (M = Li, Na, K)
AMA Style
Isenberg S, Frenzel L-M, Bruhn C, Pietschnig R.
Metallated [3]Ferrocenophanes Containing P3M Bridges (M = Li, Na, K)
Isenberg, Stefan, Lisa-Marie Frenzel, Clemens Bruhn, and Rudolf Pietschnig.
2018. "Metallated [3]Ferrocenophanes Containing P3M Bridges (M = Li, Na, K)
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.