Decomposition of d- and f-Shell Contributions to Uranium Bonding from the Quantum Theory of Atoms in Molecules: Application to Uranium and Uranyl Halides



Abstract
:1. Introduction
2. Results
2.1. Identification of Model Chemistry
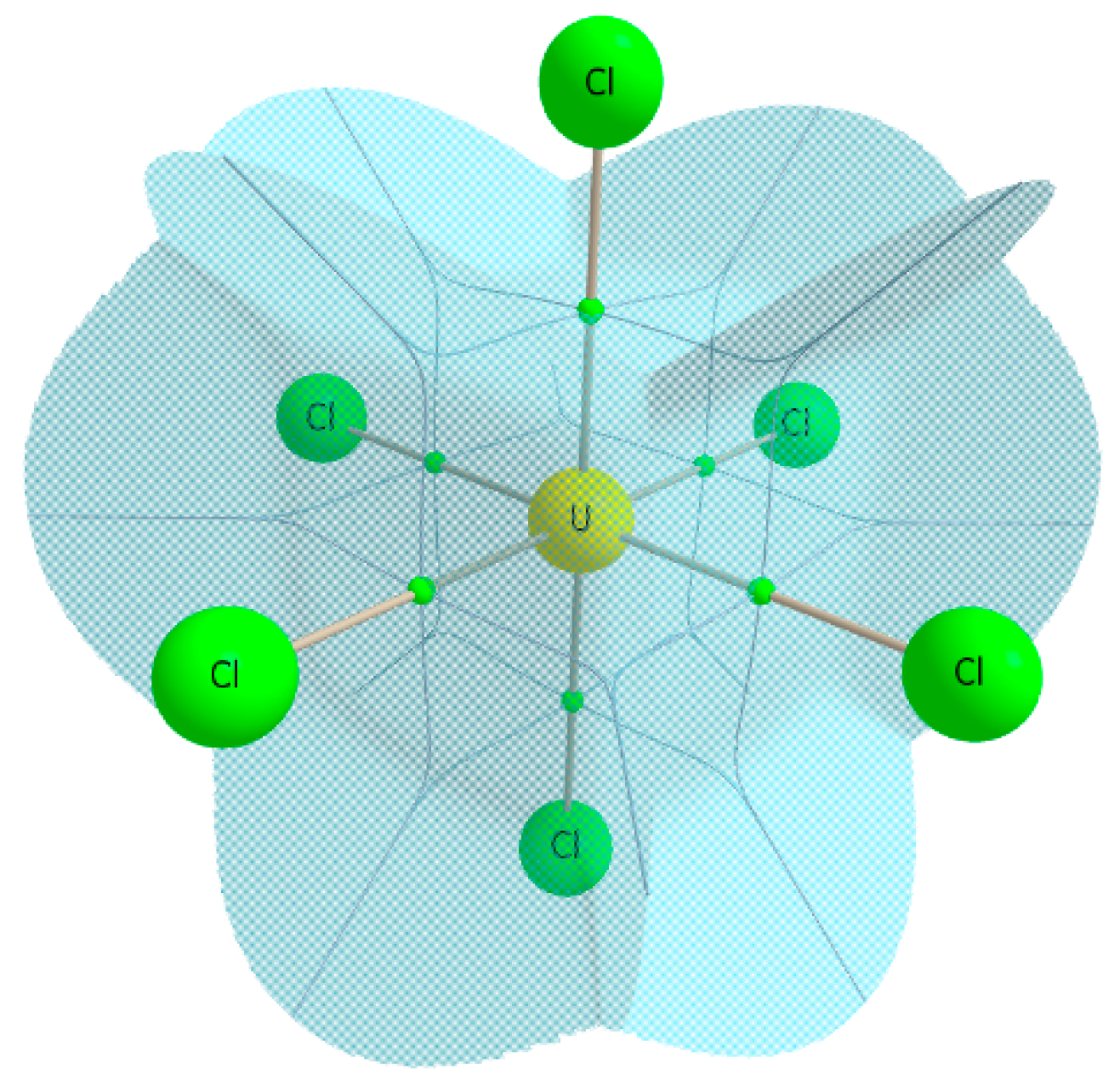
2.2. Homohalide Complexes of Uranium and Uranyl
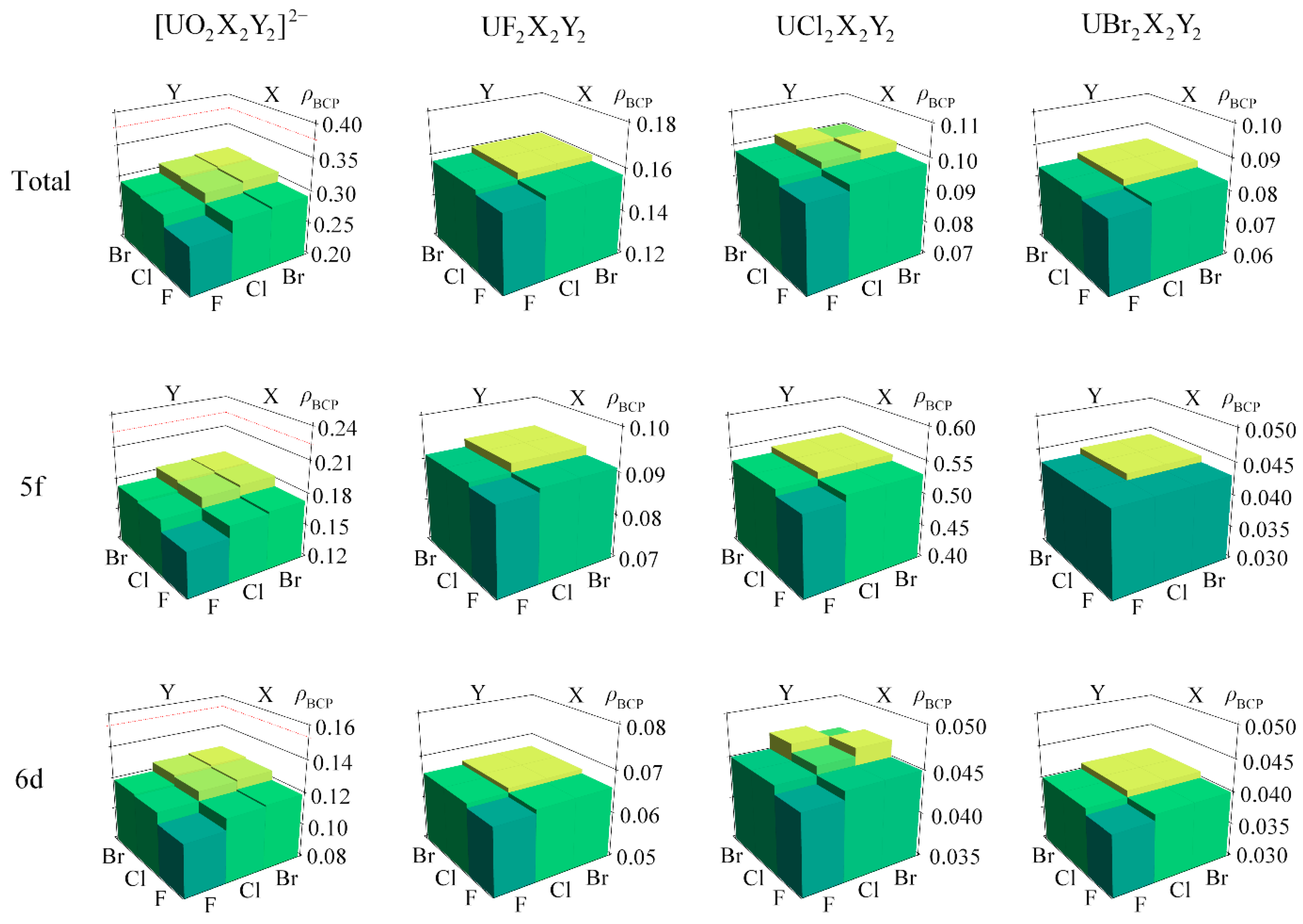
2.3. Heterohalide Complexes
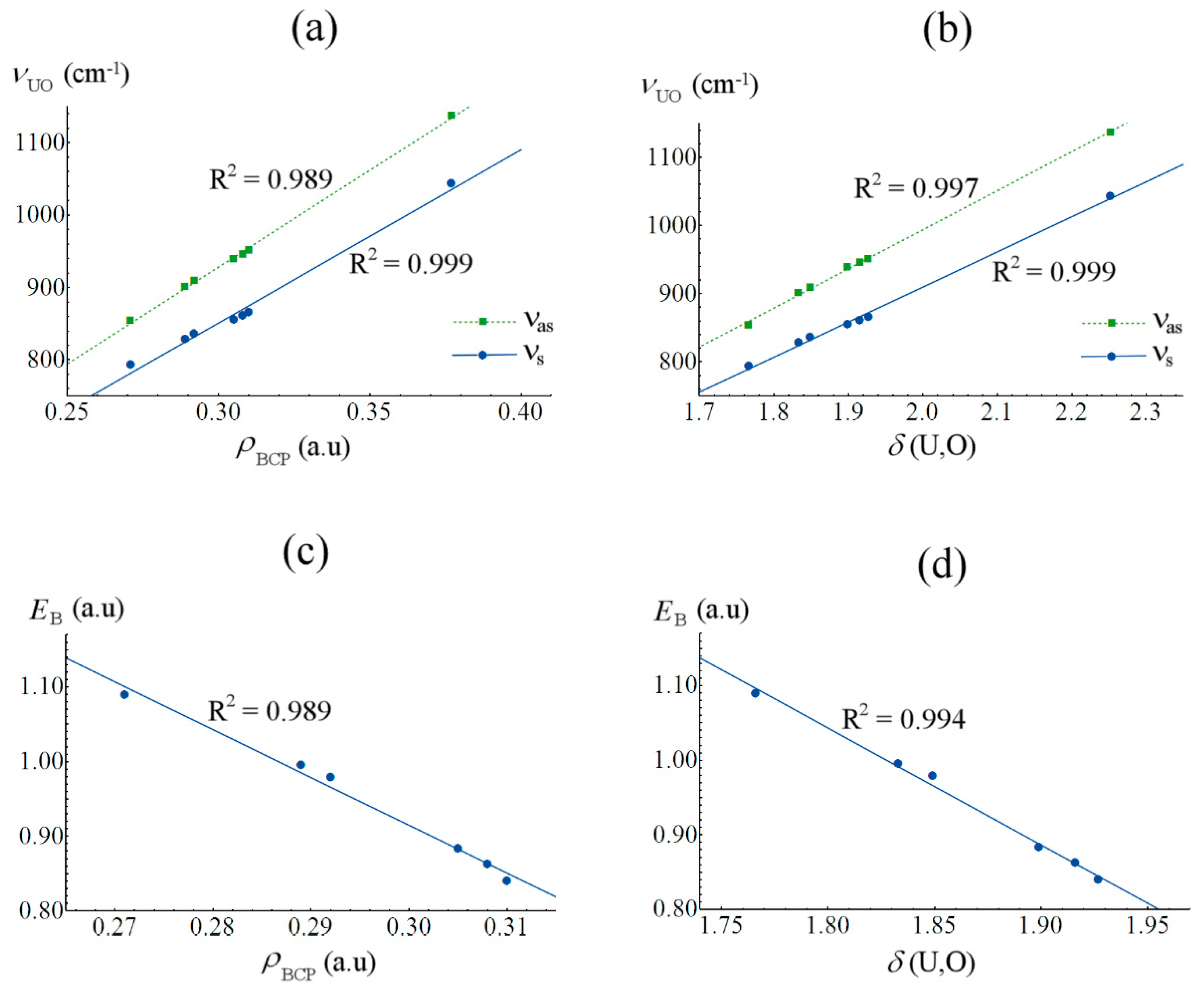
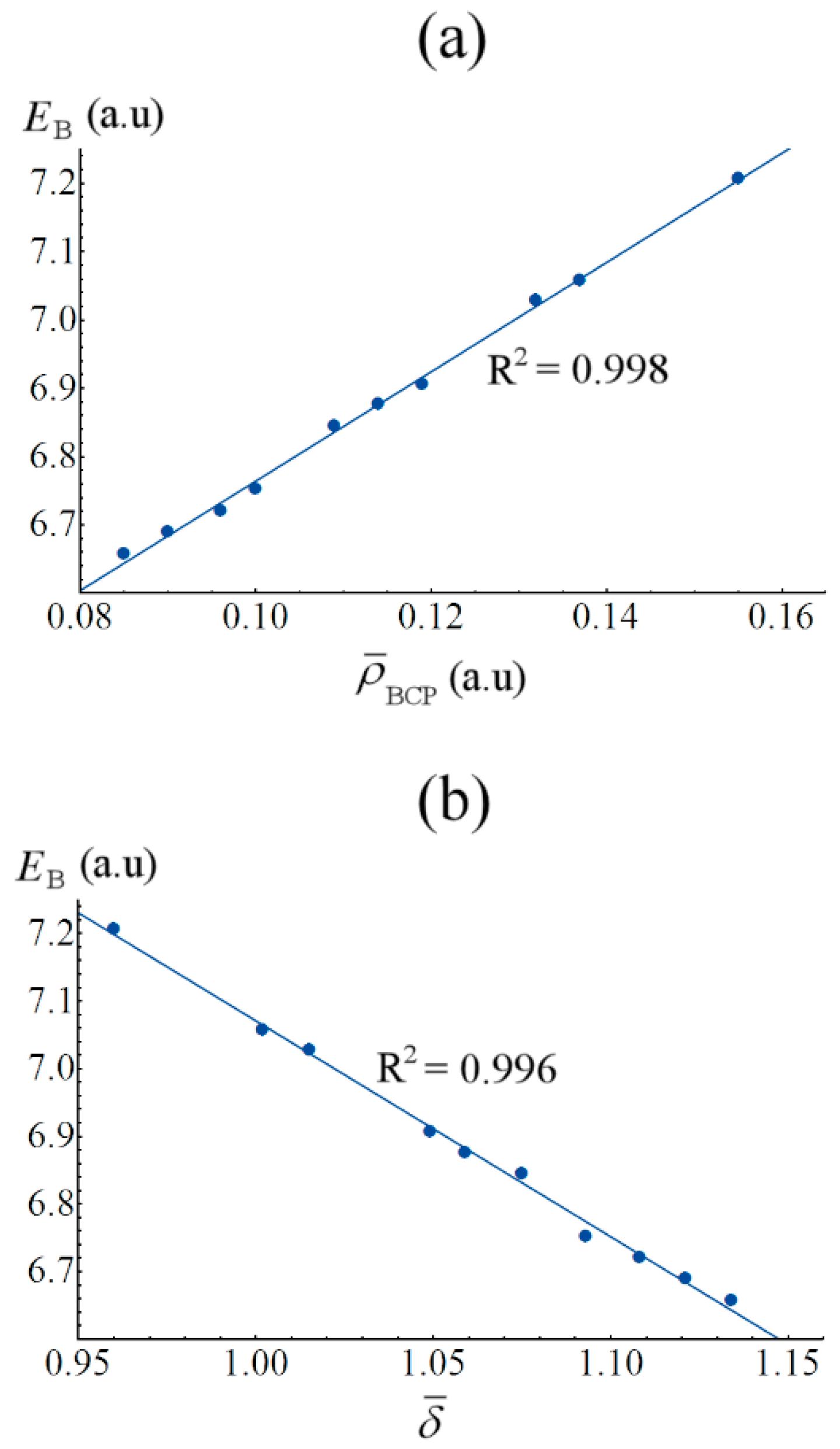
2.4. Covalency and Bond Stabilization
3. Computational Details
4. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jensen, M.P.; Bond, A.H. Comparison of covalency in the complexes of trivalent actinide and lanthanide cations. J. Am. Chem. Soc. 2002, 124, 9870–9877. [Google Scholar] [CrossRef] [PubMed]
- Gregson, M.; Lu, E.; Tuna, F.; McInnes, E.J.L.; Hennig, C.; Scheinost, A.C.; McMaster, J.; Lewis, W.; Blake, A.J.; Kerridge, A.; et al. Emergence of Comparable Covalency in Isostructural Cerium(IV)- and Uranium(IV)-Carbon Multiple Bonds. Chem. Sci. 2016, 7, 3286–3297. [Google Scholar] [CrossRef] [PubMed]
- Gregson, M.; Lu, E.; Mills, D.; Tuna, F.; McInnes, E.; Hennig, C.; Scheinost, A.; McMaster, J.; Lewis, W.; Blake, A.; et al. The inverse-trans-influence in tetravalent lanthanide and actinide bis(carbene) complexes. Nat. Commun. 2017, 8, 14137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozimor, S.A.; Yang, P.; Batista, E.R.; Boland, K.S.; Burns, C.J.; Clark, D.L.; Conradson, S.D.; Martin, R.L.; Wilkerson, M.P.; Wolfsberg, L.E. Trends in covalency for d- and f-element metallocene dichlorides identified using chlorine K-edge X-ray absorption spectroscopy and time-dependent density functional theory. J. Am. Chem. Soc. 2009, 131, 12125–12136. [Google Scholar] [CrossRef] [PubMed]
- Minasian, S.G.; Keith, J.M.; Batista, E.R.; Boland, K.S.; Clark, D.L.; Conradson, S.D.; Kozimor, S.A.; Martin, R.L.; Schwarz, D.E.; Shuh, D.K.; et al. Determining relative f and d orbital contributions to M–Cl covalency in MCl62− (M = Ti, Zr, Hf, U) and UOCl5− using Cl K-edge X-ray absorption spectroscopy and time-dependent density functional theory. J. Am. Chem. Soc. 2012, 134, 5586–5597. [Google Scholar] [CrossRef] [PubMed]
- Spencer, L.P.; Yang, P.; Minasian, S.G.; Jilek, R.E.; Batista, E.R.; Boland, K.S.; Boncella, J.M.; Conradson, S.D.; Clark, D.L.; Hayton, T.W.; et al. Tetrahalide complexes of the [U(NR)2]2+ ion: Synthesis, theory, and chlorine K-edge X-ray absorption spectroscopy. J. Am. Chem. Soc. 2013, 135, 2279–2290. [Google Scholar] [CrossRef] [PubMed]
- Minasian, S.G.; Keith, J.M.; Batista, E.R.; Boland, K.S.; Clark, D.L.; Kozimor, S.A.; Martin, R.L.; Shuh, D.K.; Tyliszczak, T. New evidence for 5f covalency in actinocenes determined from carbon K-edge XAS and electronic structure theory. Chem. Sci. 2014, 5, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Altman, A.B.; Pacold, J.I.; Wang, J.; Lukens, W.W.; Minasian, S.G. Evidence for 5d-σ and 5d-π covalency in lanthanide sesquioxides from oxygen K-edge X-ray absorption spectroscopy. Dalt. Trans. 2016, 45, 9948–9961. [Google Scholar] [CrossRef] [PubMed]
- Löble, M.W.; Keith, J.M.; Altman, A.B.; Stieber, S.C.E.; Batista, E.R.; Boland, K.S.; Conradson, S.D.; Clark, D.L.; Lezama Pacheco, J.; Kozimor, S.A.; et al. Covalency in Lanthanides. An X-ray Absorption Spectroscopy and Density Functional Theory Study of LnCl6x− (x= 3, 2). J. Am. Chem. Soc. 2015, 137, 2506–2523. [Google Scholar] [CrossRef] [PubMed]
- Minasian, S.G.; Batista, E.R.; Booth, C.H.; Clark, D.L.; Keith, J.M.; Kozimor, S.A.; Lukens, W.W.; Martin, R.L.; Shuh, D.K.; Stieber, S.C.E.; et al. Quantitative Evidence for Lanthanide-Oxygen Orbital Mixing in CeO2, PrO2, and TbO2. J. Am. Chem. Soc. 2017, 139, 18052–18064. [Google Scholar] [CrossRef] [PubMed]
- Dumas, T.; Guillaumont, D.; Fillaux, C.; Scheinost, A.; Moisy, P.; Petit, S.; Shuh, D.K.; Tyliszczak, T.; Auwer, C. Den. The nature of chemical bonding in actinide and lanthanide ferrocyanides determined by X-ray absorption spectroscopy and density functional theory. Phys. Chem. Chem. Phys. 2016, 18, 2887–2895. [Google Scholar] [CrossRef] [PubMed]
- Formanuik, A.; Ariciu, A.M.; Ortu, F.; Beekmeyer, R.; Kerridge, A.; Tuna, F.; McInnes, E.J.L.; Mills, D.P. Actinide covalency measured by pulsed electron paramagnetic resonance spectroscopy. Nat. Chem. 2017, 9, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Seaman, L.A.; Wu, G.; Edelstein, N.; Lukens, W.W.; Magnani, N.; Hayton, T.W. Probing the 5f orbital contribution to the bonding in a U(V) ketimide complex. J. Am. Chem. Soc. 2012, 134, 4931–4940. [Google Scholar] [CrossRef] [PubMed]
- Lukens, W.W.; Edelstein, N.M.; Magnani, N.; Hayton, T.W.; Fortier, S.; Seaman, L.A. Quantifying the σ and π interactions between U(V) f orbitals and halide, alkyl, alkoxide, amide and ketimide ligands. J. Am. Chem. Soc. 2013, 135, 10742–10754. [Google Scholar] [CrossRef] [PubMed]
- Lukens, W.W.; Speldrich, M.; Yang, P.; Duignan, T.J.; Autschbach, J.; Kögerler, P. The roles of 4f- and 5f-orbitals in bonding: A magnetochemical, crystal field, density functional theory, and multi-reference wavefunctional study. Dalt. Trans. 2016, 11508–11521. [Google Scholar] [CrossRef] [PubMed]
- Smiles, D.E.; Wu, G.; Hrobárik, P.; Hayton, T.W. Use of 77Se and 125Te NMR Spectroscopy to Probe Covalency of the Actinide-Chalcogen Bonding in [Th(En){N(SiMe3)2}3]−(E = Se, Te; n = 1, 2) and Their Oxo-Uranium(VI) Congeners. J. Am. Chem. Soc. 2016, 138, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Adam, C.; Kaden, P.; Beele, B.B.; Müllich, U.; Trumm, S.; Geist, A.; Panak, P.J.; Denecke, M.A. Evidence for covalence in a N-donor complex of americium(III). Dalton Trans. 2013, 42, 14068–14074. [Google Scholar] [CrossRef] [PubMed]
- Polinski, M.J.; Garner, E.B.; Maurice, R.; Planas, N.; Stritzinger, J.T.; Parker, T.G.; Cross, J.N.; Green, T.D.; Alekseev, E.V.; Van Cleve, S.M.; et al. Unusual structure, bonding and properties in a californium borate. Nat. Chem. 2014, 6, 387–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cary, S.K.; Vasiliu, M.; Baumbach, R.E.; Stritzinger, J.T.; Green, T.D.; Diefenbach, K.; Cross, J.N.; Knappenberger, K.L.; Liu, G.; Silver, M.A.; et al. Emergence of californium as the second transitional element in the actinide series. Nat. Commun. 2015, 6, 6827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, J.; Dau, P.D.; Liu, H.T.; Huang, D.L.; Wei, F.; Schwarz, W.H.E.; Li, J.; Wang, L.S. Photoelectron spectroscopy and theoretical studies of gaseous uranium hexachlorides in different oxidation states: UCl6q− (q = 0–2). J. Chem. Phys. 2015, 142, 134308. [Google Scholar] [CrossRef] [PubMed]
- Gianopoulos, C.G.; Zhurov, V.V.; Minasian, S.G.; Batista, E.R.; Jelsch, C.; Pinkerton, A.A. Bonding in Uranium(V) Hexafluoride Based on the Experimental Electron Density Distribution Measured at 20 K. Inorg. Chem. 2017, 56, 1775–1778. [Google Scholar] [CrossRef] [PubMed]
- Arnold, P.L.; Turner, Z.R.; Kaltsoyannis, N.; Pelekanaki, P.; Bellabarba, R.M.; Tooze, R.P. Covalency in CeIV and UIV halide and N-heterocyclic carbene bonds. Chem. Eur. J. 2010, 16, 9623–9629. [Google Scholar] [CrossRef] [PubMed]
- King, D.M.; Tuna, F.; McInnes, E.J.L.; McMaster, J.; Lewis, W.; Blake, A.J.; Liddle, S.T. Isolation and characterization of a uranium(VI)–nitride triple bond. Nat. Chem. 2013, 5, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.L.; Fortier, S.; Wu, G.; Kaltsoyannis, N.; Hayton, T.W. Synthesis and Spectroscopic and Computational Characterization of the Chalcogenido-Substituted Analogues of the Uranyl Ion, [OUE]2+ (E = S, Se). J. Am. Chem. Soc. 2013, 135, 5352–5355. [Google Scholar] [CrossRef] [PubMed]
- Arliguie, T.; Belkhiri, L.; Bouaoud, S.; Thuery, P.; Villiers, C.; Boucekkine, A.; Ephritikhine, M. Lanthanide(III) and Actinide(III) Complexes [M(BH4)2(THF)5][BPh4] and [M(BH4)2(18-crown-6)][BPh4] (M = Nd, Ce, U): Synthesis, Crystal Structure, and Density Functional Theory Investigation of the Covalent Contribution to Metal-Borohydride Bonding. Inorg. Chem. 2009, 48, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.B.; Gaunt, A.J.; Gordon, J.C.; Kaltsoyannis, N.; Neu, M.P.; Scott, B.L. Uncovering f-element bonding differences and electronic structure in a series of 1:3 and 1:4 complexes with a diselenophosphinate ligand. Chem. Sci. 2013, 4, 1189–1203. [Google Scholar] [CrossRef]
- Gregson, M.; Wooles, A.J.; Cooper, O.J.; Liddle, S.T. Covalent Uranium Carbene Chemistry. Comments Inorg. Chem. 2015, 35, 262–294. [Google Scholar] [CrossRef]
- Walensky, J.R.; Martin, R.L.; Ziller, J.W.; Evans, W.J. Importance of energy level matching for bonding in Th3+-Am3+ actinide metallocene amidinates, (C5Me5)2[iPrNC(Me)NiPr]An. Inorg. Chem. 2010, 49, 10007–10012. [Google Scholar] [CrossRef] [PubMed]
- Behrle, A.C.; Barnes, C.L.; Kaltsoyannis, N.; Walensky, J.R. Systematic investigation of thorium(IV)- and uranium(IV)-ligand bonding in dithiophosphonate, thioselenophosphinate, and diselenophosphonate complexes. Inorg. Chem. 2013, 52, 10623–10631. [Google Scholar] [CrossRef] [PubMed]
- Behrle, A.C.; Kerridge, A.; Walensky, J.R. Dithio- and Diselenophosphinate Thorium(IV) and Uranium(IV) Complexes: Molecular and Electronic Structures, Spectroscopy, and Transmetalation Reactivity. Inorg. Chem. 2015, 54, 11625–11636. [Google Scholar] [CrossRef] [PubMed]
- Kaltsoyannis, N. Does covalency increase or decrease across the actinide series? Implications for minor actinide partitioning. Inorg. Chem. 2013, 52, 3407–3413. [Google Scholar] [CrossRef] [PubMed]
- Tassell, M.J.; Kaltsoyannis, N. Covalency in AnCp4 (An = Th–Cm): A comparison of molecular orbital, natural population and atoms-in-molecules analyses. Dalt. Trans. 2010, 39, 6719–6725. [Google Scholar] [CrossRef] [PubMed]
- Kirker, I.; Kaltsoyannis, N. Does covalency really increase across the 5f series? A comparison of molecular orbital, natural population, spin and electron density analyses of AnCp3 (An = Th−Cm; Cp = η5-C5H5). Dalton Trans. 2011, 40, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.R.; Kingham, J.R.; Kaltsoyannis, N. The strength of actinide–element bonds from the quantum theory of atoms-in-molecules. Dalt. Trans. 2015, 44, 2554–2566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trumm, M.; Schimmelpfennig, B.; Geist, A. Structure and separation quality of various N- and O-donor ligands from quantum-chemical calculations. Nukleonika 2015, 60, 847–851. [Google Scholar] [CrossRef] [Green Version]
- Trumm, M.; Schimmelpfennig, B. Towards the origin of effective An(III)/Ln(III) separation by tridentate N-donor ligands: A theoretical study on atomic charges and polarisabilities for Cm(III)/Gd(III) separation. Mol. Phys. 2016, 114, 876–883. [Google Scholar] [CrossRef]
- Vallet, V.; Wahlgren, U.; Grenthe, I. Probing the nature of chemical bonding in uranyl(VI) complexes with quantum chemical methods. J. Phys. Chem. A 2012, 116, 12373–12380. [Google Scholar] [CrossRef] [PubMed]
- Szabo, Z.; Toraishi, T.; Vallet, V.; Grenthe, I. Solution coordination chemistry of actinides: Thermodynamics, structure and reaction mechanisms. Coord. Chem. Rev. 2006, 250, 784–815. [Google Scholar] [CrossRef]
- Di Pietro, P.; Kerridge, A. Assessing covalency in equatorial U–N bonds: Density based measures of bonding in BTP and isoamethyrin complexes of uranyl. Phys. Chem. Chem. Phys. 2016, 18, 16830–16839. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, P.; Kerridge, A. Ligand size dependence of U–N and U–O bond character in a series of uranyl hexaphyrin complexes: Quantum chemical simulation and density based analysis. Phys. Chem. Chem. Phys. 2017, 19, 7546–7559. [Google Scholar] [CrossRef] [PubMed]
- Fryer-Kanssen, I.; Austin, J.; Kerridge, A. Topological Study of Bonding in Aquo and Bis(triazinyl)pyridine Complexes of Trivalent Lanthanides and Actinides: Does Covalency Imply Stability? Inorg. Chem. 2016, 55, 10034–10042. [Google Scholar] [CrossRef] [PubMed]
- Woodall, S.D.; Swinburne, A.N.; Kerridge, A.; Di Pietro, P.; Adam, C.; Kaden, P.; Natrajan, L.S. Neptunyl(VI) centred visible LMCT emission directly observable in the presence of uranyl(VI). Chem. Commun. 2015, 51, 5402–5405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillaumont, D. Quantum Chemistry Study of Actinide(III) and Lanthanide(III) Complexes with Tridentate Nitrogen Ligands. J. Phys. Chem. A 2004, 108, 6893–6900. [Google Scholar] [CrossRef]
- Petit, L.; Joubert, L.; Maldivi, P.; Adamo, C. A comprehensive theoretical view of the bonding in actinide molecular complexes. J. Am. Chem. Soc. 2006, 128, 2190–2191. [Google Scholar] [CrossRef] [PubMed]
- Petit, L.; Adamo, C.; Maldivi, P. Toward a Clear-Cut Vision on the Origin of Insights from Theory. Society 2006, 45, 8517–8522. [Google Scholar]
- Lan, J.; Shi, W.; Yuan, L.; Zhao, Y.; Li, J.; Chai, Z. Trivalent Actinide and Lanthanide Separations by Tetradentate Nitrogen Ligands: A Quantum Chemistry Study. Inorg. Chem. 2011, 50, 9230–9237. [Google Scholar] [CrossRef] [PubMed]
- Roy, L.E.; Bridges, N.J.; Martin, L.R. Theoretical insights into covalency driven f element separations. Dalton Trans. 2013, 42, 2636–2642. [Google Scholar] [CrossRef] [PubMed]
- Dolg, M.; Cao, X.; Ciupka, J. Misleading evidence for covalent bonding from EuIIIX and AmIIIX density functional theory bond lengths. J. Electron Spectros. Relat. Phenomena 2014, 194, 8–13. [Google Scholar] [CrossRef]
- Wang, C.; Cheng, C.; Su, J.; Huai, P. Bonding nature of the actinide tetrafluorides AnF4 (An = Th−Cm). Mol. Phys. 2015, 113, 3450–3458. [Google Scholar] [CrossRef]
- Kaneko, M.; Miyashita, S.; Nakashima, S. Bonding Study on the Chemical Separation of Am(III) from Eu(III) by S-, N-, and O-Donor Ligands by Means of All-Electron ZORA-DFT Calculation. Inorg. Chem. 2015, 54, 7103–7109. [Google Scholar] [CrossRef] [PubMed]
- Bryantsev, V.S.; Hay, B.P. Theoretical prediction of Am(III)/Eu(III) selectivity to aid the design of actinide-lanthanide separation agents. Dalt. Trans. 2015, 7935–7942. [Google Scholar] [CrossRef] [PubMed]
- Duignan, T.; Autschbach, J. Impact of the Kohn-Sham Delocalization Error on the 4f Shell Localization and Population in Lanthanide Complexes. J. Chem. Theory Comput. 2016, 12, 3109–3121. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.X.; Li, W.L.; Dong, L.; Gibson, J.K.; Li, J. Crown ether complexes of actinyls: A computational assessment of AnO2 (15-crown-5)2+ (An = U, Np, Pu, Am, Cm). Dalt. Trans. 2017, 46, 12354–12363. [Google Scholar] [CrossRef] [PubMed]
- Madic, C.; Hudson, M.J.; Liljenzin, J.O.; Glatz, J.P.; Nannicini, R.; Facchini, A.; Kolarik, Z.; Odoj, R. Recent achievements in the development of partitioning processes of minor actinides from nuclear wastes obtained in the frame of the NEWPART European Programme (1996–1999). Prog. Nucl. Energy 2002, 40, 523–526. [Google Scholar] [CrossRef]
- Nilsson, M.; Nash, K.L. Review article: A review of the development and operational characteristics of the TALSPEAK process. Solvent Extr. Ion Exch. 2007, 25, 665–701. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Kerridge, A. Quantification of f-element covalency through analysis of the electron density: Insights from simulation. Chem. Commun. 2017, 53, 6685–6695. [Google Scholar] [CrossRef] [PubMed]
- Zhurov, V.V.; Zhurova, E.A.; Stash, A.I.; Pinkerton, A.A. Characterization of bonding in cesium uranyl chloride: Topological analysis of the experimental charge density. J. Phys. Chem. A 2011, 115, 13016–13023. [Google Scholar] [CrossRef] [PubMed]
- Kerridge, A. Oxidation state and covalency in f-element metallocenes (M = Ce, Th, Pu): A combined CASSCF and topological study. Dalton Trans. 2013, 42, 16428–16436. [Google Scholar] [CrossRef] [PubMed]
- Kerridge, A. f-orbital covalency in the actinocenes (An = Th–Cm): Multiconfigurational studies and topological analysis. RSC Adv. 2014, 4, 12078–12086. [Google Scholar] [CrossRef]
- Beekmeyer, R.; Kerridge, A. Assessing Covalency in Cerium and Uranium Hexachlorides: A Correlated Wavefunction and Density Functional Theory Study. Inorganics 2015, 3, 482–499. [Google Scholar] [CrossRef] [Green Version]
- Dau, P.D.; Su, J.; Liu, H.T.; Liu, J.B.; Huang, D.L.; Li, J.; Wang, L.S. Observation and investigation of the uranyl tetrafluoride dianion (UO2F42−) and its solvation complexes with water and acetonitrile. Chem. Sci. 2012, 3, 1137–1146. [Google Scholar] [CrossRef]
- Van den Bossche, G.; Spirlet, M.R.; Rebizant, J. Structure of Diammonium Tetrabromodioxouranate(VI) Dihydrate. Acta Crystallogr. C 1987, 43, 383–384. [Google Scholar] [CrossRef]
- Kimura, M.; Schomaker, V.; Smith, D.W.; Weinstock, B. Electron-Diffraction Investigation of the Hexafluorides of Tungsten, Osmium, Iridium, Uranium, Neptunium, and Plutonium. J. Chem. Phys. 1968, 48, 4001–4012. [Google Scholar] [CrossRef]
- Mcdowell, R.S.; Asprey, L.B.; Paine, R.T. Vibrational spectrum and force field of uranium hexafluoride. J. Chem. Phys. 1974, 61, 3571–3580. [Google Scholar] [CrossRef]
- Zachariasen, W.H. Crystal chemical studies of the 5f-series of elements. V. The crystal structure of uranium hexachloride. Acta Crystallogr. 1948, 1, 285–287. [Google Scholar] [CrossRef]
- Perdew, J.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef]
- Stephens, P.; Devlin, F.; Chabalowski, C.; Frisch, M. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Bauzá, A.; Quiñonero, D.; Deyaì, P.M.; Frontera, A. Is the use of diffuse functions essential for the properly description of noncovalent interactions involving anions? J. Phys. Chem. A 2013, 117, 2651–2655. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, P.; Kerridge, A. U-Oyl Stretching Vibrations as a Quantitative Measure of the Equatorial Bond Covalency in Uranyl Complexes: A Quantum-Chemical Investigation. Inorg. Chem. 2016, 55, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Watkin, D.J.; Denning, R.G.; Prout, K. Structure of dicaesium tetrachlorodioxouranium(VI). Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1991, 47, 2517–2519. [Google Scholar] [CrossRef] [Green Version]
- Bullock, J.I. Raman and infrared spectroscopic studies of the uranyl ion: The symmetric stretching frequency, force constants, and bond lengths. J. Chem. Soc. A 1969, 781–784. [Google Scholar] [CrossRef]
- Schreckenbach, G.; Hay, P.J.; Martin, R.L. Density functional calculations on actinide compounds: Survey of recent progress and application to [UO2X4]2− (X = F, Cl, OH) and AnF6 (An = U, Np, Pu). J. Comput. Chem. 1999, 20, 70–90. [Google Scholar] [CrossRef]
- Batista, E.R.; Martin, R.L.; Hay, P.J. Density functional investigations of the properties and thermochemistry of UFn and UCln (n = 1,...,6). J. Chem. Phys. 2004, 121, 11104–11111. [Google Scholar] [CrossRef] [PubMed]
- Grenthe, I.; Drożdżyński, J.; Fujino, T.; Buck, E.C.; Albrecht-Schmitt, T.E.; Wolf, S.F. Uranium. In The Chemistry of the Actinide and Transactinide Elements; Morss, L.R., Edelstein, N.M., Fuger, J., Katz, J.J., Eds.; Springer: Dordrecht, The Netherlands, 2006; pp. 253–698. [Google Scholar]
- Yun-guang, Z.; Yu-de, L. Relativistic density functional investigation of UX6 (X = F, Cl, Br and I). Chin. Phys. B 2010, 19, 033302. [Google Scholar] [CrossRef]
- TURBOMOLE, version 6.6, A development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH. 2014. Available online: http://www.turbomole.com (accessed on 29 August 2018).
- Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Küchle, W.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted pseudopotentials for the actinides. Parameter sets and test calculations for thorium and thorium monoxide. J. Chem. Phys. 1994, 100, 7535–7542. [Google Scholar] [CrossRef]
- Cao, X.; Dolg, M. Segmented contraction scheme for small-core actinide pseudopotential basis sets. J. Mol. Struct. THEOCHEM 2004, 673, 203–209. [Google Scholar] [CrossRef]
- De Jong, W.A.; Visscher, L.; Nieuwpoort, W.C. On the bonding and the electric field gradient of the uranyl ion. J. Mol. Struc. 1999, 458, 41–52. [Google Scholar] [CrossRef]
- Fryer-Kanssen, I.; Kerridge, A. Elucidation of the inverse trans influence in uranyl, its imido and carbene analogues via quantum chemical simulation. Chem. Commun. 2018, 54, 9761–9764. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Exp. a,b | BLYP | PBE | PBE0 | B3LYP | BHLYP | |
|---|---|---|---|---|---|---|---|
| rUO (Å) | 1.774 | 1.817 (+0.043) | 1.800 (+0.026) | 1.756 (−0.018) | 1.776 (+0.002) | 1.735 (−0.039) | |
| rUCl (Å) | 2.671 | 2.765 (+0.094) | 2.721 (+0.050) | 2.715 (+0.044) | 2.749 (+0.078) | 2.749 (+0.078) | |
| νUO (cm−1) | 834 | 776 (−58) | 803 (−31) | 896 (+62) | 855 (+21) | 953 (+119) | |
| 922 | 862 (−60) | 887 (−35) | 975 (+53) | 939 (+17) | 1023 (+101) | ||
| νUCl (cm−1) | 206 | 179 (−27) | 192 (−14) | 199 (−7) | 188 (−18) | 196 (−10) | |
| 236 | 197 (39) | 238 (+2) | 216 (−20) | 206 (−30) | 213 (−23) | ||
| 267 | 213 (−54) | 226 (−41) | 234 (−33) | 224 (−43) | 232 (−25) | ||
| Parameter | Exp. a,b | BLYP | PBE | PBE0 | B3LYP | BHLYP |
|---|---|---|---|---|---|---|
| rUF (Å) | 1.999 | 2.036 (+0.037) | 2.018 (+0.019) | 1.990 (−0.009) | 2.009 (+0.010) | 1.984 (−0.055) |
| νUF (cm−1) | 534 | 511 (−23) | 521 (−13) | 543 (+9) | 532 (−2) | 548 (+14) |
| 626 | 581 (−45) | 596 (−30) | 633 (+7) | 615 (−11) | 648 (+22) | |
| 667 | 613 (−54) | 630 (−37) | 682 (+15) | 660 (+7) | 710 (+43) |
| Complex | rUO (Å) | rUX (Å) | R = RU + RX (Å) | rUX – R (Å) |
|---|---|---|---|---|
| [UO2F4]2− | 1.824 | 2.229 | 0.73 + 1.33 = 2.06 | 0.169 |
| [UO2Cl4]2− | 1.776 | 2.749 | 0.73 + 1.81 = 2.54 | 0.209 |
| [UO2Br4]2− | 1.769 | 2.922 | 0.73 + 1.96 = 2.69 | 0.232 |
| UO22+ | 1.695 | - | - | - |
| Complex | rUX (Å) | R = RU + RX (Å) | rUX – R (Å) |
|---|---|---|---|
| UF6 | 2.009 | 0.73 + 1.33 = 2.06 | −0.051 |
| UCl6 | 2.470 | 0.73 + 1.81 = 2.54 | −0.070 |
| UBr6 | 2.639 | 0.73 + 1.96 = 2.69 | −0.051 |
| Irrep ( Γ) | Ag | B1g | B2g | B3g | Au | B1u | B2u | B3u |
|---|---|---|---|---|---|---|---|---|
| Component | 6dσ, 6dδ | 6dδ | 6dπ | 6dπ | 5fδ | 5fσ, 5fδ | 5fπ, 5fφ | 5fπ, 5fφ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanti, J.; Lincoln, M.; Kerridge, A. Decomposition of d- and f-Shell Contributions to Uranium Bonding from the Quantum Theory of Atoms in Molecules: Application to Uranium and Uranyl Halides. Inorganics 2018, 6, 88. https://doi.org/10.3390/inorganics6030088
Tanti J, Lincoln M, Kerridge A. Decomposition of d- and f-Shell Contributions to Uranium Bonding from the Quantum Theory of Atoms in Molecules: Application to Uranium and Uranyl Halides. Inorganics. 2018; 6(3):88. https://doi.org/10.3390/inorganics6030088
Chicago/Turabian StyleTanti, Jonathan, Meghan Lincoln, and Andy Kerridge. 2018. "Decomposition of d- and f-Shell Contributions to Uranium Bonding from the Quantum Theory of Atoms in Molecules: Application to Uranium and Uranyl Halides" Inorganics 6, no. 3: 88. https://doi.org/10.3390/inorganics6030088