Abstract
A series of unsymmetrically substituted oligosilanes were synthesized via stepwise introduction of different substituents to α-chloro-ω-hydrooligosilanes. The reactions of α-chloro-ω-hydrooligosilanes with organolithium or Grignard reagents gave hydrooligosilanes having various alkyl, alkenyl, alkynyl and aryl groups. Thus-obtained hydrooligosilanes were converted into alkoxyoligosilanes by ruthenium-catalyzed dehydrogenative alkoxylation with alcohols.
1. Introduction
Recently, various functionalized oligosilanes have attracted growing interests due to their unique photophysical and electronic properties [1,2,3,4,5,6,7,8,9,10,11,12,13]. In the synthesis of such compounds, introduction of functionality to oligosilanes is an important process. Oligosilanes having both chlorosilane and hydrosilane moieties are especially interesting because two different functionalities could be introduced to the oligosilanes. For example, α-chloro-ω-hydrooligosilanes are potential precursors for such modification.
A problem to be overcome is limitation of facile functionalization of the hydrosilane moiety in the oligosilanes. Thus far, halogenation [14,15,16], Lewis acid-catalyzed reactions [17,18,19] and radical-promoted reactions [20,21,22,23,24] have been used for the Si–H modification of hydrooligosilanes. Transition metal-catalyzed reactions of hydrosilanes are well known as Si–H conversion methods, including hydrosilylation [25,26,27], dehydrogenative silylation of OH and NH groups [28,29,30,31], silylation of aromatic rings [32,33,34,35,36] and so on. However, most of them are applicable only to hydromonosilanes. When hydrooligosilanes are subjected to these transition metal-catalyzed reactions, major products come from the cleavage of Si–Si bonds. Although the transition metal-catalyzed reactions are utilized as valuable organic synthesis methodologies [37,38,39,40], it is not the case for the transformation of hydrooligosilanes. As a rare example, Yamanoi and Nishihara have reported the palladium-catalyzed arylation of hydrooligosilanes with aryl iodides to afford the corresponding arylated oligosilanes with preservation of the Si–Si bonds [41,42].
Previously, we reported titanium-catalyzed synthesis of the hydrogen-terminated oligosilanes from α,ω-dichlorooligosilanes [43]. This method enabled us to synthesize α-chloro-ω-hydrooligo-silanes with high selectivity. The α-chloro-ω-hydrooligosilanes are good precursors of variously substituted hydrooligosilanes via nucleophilic substitution of the chlorosilane moiety. Furthermore, we have recently found ruthenium-catalyzed dehydrogenative alkoxylation of a hydrodisilane with alcohols [44]. Interestingly, the reactions proceed with preserving the Si–Si bond despite its susceptible nature to transition metals [37,38,39,40]. Combination of these two methods leads to convenient synthesis of unsymmetrically substituted oligosilanes. In this paper, we report the synthesis of various substituted hydrooligosilanes from α-chloro-ω-hydrooligosilanes and transformation of thus-obtained hydrooligosilanes to the corresponding alkoxy oligosilanes.
2. Results and Discussion
2.1. Synthesis of 1-Hydrooligosilanes
As reported previously, α-chloro-ω-hydrooligosilanes 1–3 are synthesized by the selective monoreduction of α,ω-dichlorooligosilanes with alkylmagnesium chlorides in the presence of a catalytic amount of TiCl4 (Scheme 1) [43]. The remaining chlorosilane moiety is possible functionality for introduction of various substituents.
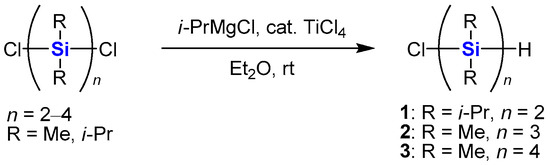
Scheme 1.
Monoreduction of α,ω-dichlorooligosilanes with i-PrMgCl in the presence of a catalytic amount of TiCl4.
As shown in Scheme 2, the chlorosilane moiety of 1 was smoothly substituted by organolithium reagents. When 1 was treated with 2-thienyllithium, 2-thienyldisilane 4 was obtained in high yield. 4-Methoxyphenyl group was also introduced to afford 4-methoxyphenyldisilane 5.
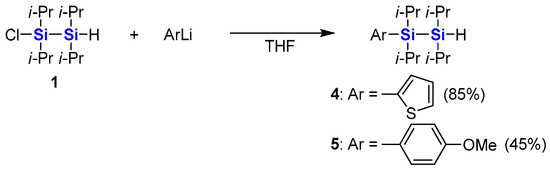
Scheme 2.
Synthesis of substituted hydrodisilanes from 1-chloro-2-hydrodisilane.
In the cases of polymethylated oligosilanes 2 and 3, Grignard reagents are more suitable for the substitution of the chlorosilane moiety. The Grignard reagents were prepared in the presence of lithium chloride and DIBAL-H to activate magnesium metal [45]. As shown in Scheme 3, various alkyl, alkenyl, alkynyl and aryl groups were introduced, and substituted hydrooligosilanes were successfully synthesized. As alkyl group installation, 5-hexenyl- and 2-phenylethylmagnesium reagents were used to afford hydrotrisilanes 6 and 7. Mono- and disubstituted alkenyl groups such as styryl and 2-buten-2-yl ones were also introduced to the trisilane skeleton successfully. The stereochemistry of the alkene moieties of trisilanes 8 and 9 was determined by 1H NMR spectroscopy. The E geometry of the styryl group in 8 was confirmed by the J value of the vinylic coupling (19 Hz). The major Z isomer of 9 was confirmed by NOE experiment. Three types of alkynyl Grignard reagents with aliphatic, aryl and silyl substituents were subjected to the reaction to afford alkynyltrisilanes 10–12. Various aryl groups were also introduced to 2 to afford phenyl-, 4-methoxyphenyl-, 4-dimethylaminophenyl-, and 2-thienyltrisilanes 13–16. The similar arylation was also applicable to 3, and 2-thienyltetrasilane 17 was obtained. All of these reactions proceed without loss of the hydrosilane moieties, which can be used for further modification of the hydrooligosilanes. The reason for low yields in some cases is attributed to loss during the isolation procedure by column chromatography over silica gel.
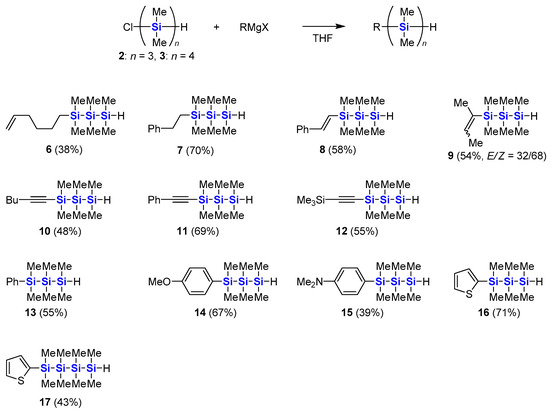
Scheme 3.
Synthesis of substituted hydrooligosilanes from α-chloro-ω-hydrooligosilanes.
2.2. Dehydrogenative Alkoxylation of Hydrooligosilanes with Alcohols
As mentioned above, we have reported ruthenium-catalyzed dehydrogenative alkoxylation of a hydrodisilane with alcohols without Si–Si bond cleavage [44]. The reactions are also applicable to various hydrotrisilanes. As shown in Scheme 4, some of the hydrotrisilanes synthesized above were subjected to the ruthenium-catalyzed dehydrogenative alkoxylation with methanol. All reactions proceeded smoothly in toluene at room temperature in the presence of 2.5 mol % of [RuCl2(p-cymene)]2 to afford methoxytrisilanes 18–21 in good yields. It is worth noting that the alkenyl and alkynyl moieties tolerate the reactions. Neither hydrosilylation nor bis-silylation occurred under these reaction conditions.
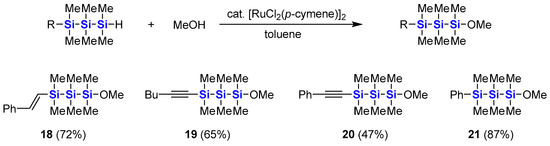
Scheme 4.
Synthesis of 1-methoxytrisilanes from 1-hydrotrisilanes.
To gain further insight into the substituent effects in the ruthenium-catalyzed alkoxylation, ethyl-substituted hydrodisilane 22 and 2-hydrotrisilane 26 were used for the reactions with methanol. The results are summarized in Table 1 and Table 2. Even though an excess amount of methanol (10 equiv) was used in the reaction of 22, the reaction rate is much slower than that of PhMe2SiSiMe2H, which finished within 2 h under the same reaction conditions [44]. When bulkier alcohols are used, the reaction rate becomes slower. The reaction of 22 with ethanol needed heating at 50 °C to be completed within one day. The reaction with a large excess of 2-propanol is much more sluggish. Even though the reaction was carried out on heating, more than 40 h were needed for complete consumption of 22.

Table 1.
Reactions of 22 with various alcohols in the presence of [RuCl2(p-cymene)]2.
Table 2.
Reactions of 26 with methanol in the presence of various ruthenium catalysts.
For the alkoxylation of 26, optimization of the ruthenium catalyst was necessary, as shown in Table 2. The reaction of 26 with methanol in the presence of the (p-cymene)ruthenium catalyst gave the desired 2-methoxytrisilane 27 in low yield along with monosilane 28, which was produced by Si–Si bond cleavage (Entry 1). Changing the aromatic ligand to mesitylene slightly improved the formation of 27, but the Si–Si bond cleavage still occurred significantly (Entry 2). In contrast, the (benzene)ruthenium catalyst gave 27 more selectively and suppressed the formation of 28 (Entry 3). Compared with the (arene)ruthenium complexes, RuHCl(CO)(PPh3)3 showed little catalytic activity, and no alkoxylation product was detected in the reaction mixture (Entry 4).
The superior performance of the (benzene)ruthenium catalyst over the (p-cymene)ruthenium catalyst might be attributed to the less steric hindrance around the coordinated arenes. The intermediate of the reaction might be the hydrosilane-bound ruthenium complex via σ-coordination or oxidative addition of the Si–H bond to the ruthenium atom. Nucleophilic attack of methanol to the silicon atom having the Si–H bond produces 27. At this step, more crowded p-cymene prevents methanol from attacking the central silicon atom. As a result, methanol attacks the terminal silicon atom of 26 to afford 28 via Si–Si bond cleavage.
3. Materials and Methods
All reactions were carried out under an argon atmosphere using standard Schlenk techniques unless otherwise noted. THF and diethyl ether were distilled from sodium benzophenone ketyl under a nitrogen atmosphere. Toluene was distilled from sodium under a nitrogen atmosphere. Cl(i-Pr)2SiSi(i-Pr)2H (1) [43], Cl(SiMe2)3H (2) [43], Cl(SiMe2)4H (3) [43], PhMe2SiSi(H)MeSiMe2Ph (26) [46] and Et3SiSiEt3 [47] were prepared according to the reported procedures. Silica gel for column chromatography (Kanto Chemical, silica gel 60N, spherical, neutral, particle size 100–210 μm) was purchased and used as received. The other chemicals were purchased (Kanto Chemical, Tokyo, Japan; Kishida Chemical, Osaka, Japan; Sigma-Aldrich Japan, Tokyo, Japan; Tokyo Chemical Industry, Tokyo, Japan; Wako Pure Chemical Industries, Osaka, Japan) and used without further purification.
GC analysis was performed on a Shimadzu GC-8A gas chromatograph equipped with packed columns containing 10% silicone SE-30 on Uniport B (GL Sciences, Tokyo, Japan) and a Shimadzu (Kyoto, Japan) C-R8A Chromatopack integrator. 1H, 13C and 29Si NMR spectra were measured with JEOL (Akishima, Japan) JNM-ECA600, JNM-ECS400 and JNM-ECS300 spectrometers. Dodecane, tricosane or mesitylene were used as internal standards for NMR yield estimation. IR spectra were recorded on Shimadzu FTIR-8700, JASCO (Hachioji, Japan) FT/IR-4600 and Hitachi (Tokyo, Japan) FTIR 270-50 spectrophotometers. Mass spectra and high-resolution mass spectra were recorded on Shimadzu GCMS-QP2010 SE and JEOL JMS-T100GCV mass spectrometers. Spectral data of all new compounds are showed in Figure S1–S113 in supplementary materials.
3.1. Synthesis of 1,1,2,2-Tetraisopropyl-1-(2′-thienyl)disilane (4)
A 1.64 M solution of butyllithium in hexane (1.5 mL, 2.5 mmol) was added dropwise to a solution of thiophene (212 mg, 2.52 mmol) in THF (8 mL) at 0 °C, and the mixture was stirred at 0 °C for 1 h. Compound 1 (536 mg, 2.02 mmol) was added, and the mixture was stirred for 2 days at room temperature. The reaction was quenched by adding 1.0 M hydrochloric acid. The reaction mixture was extracted with hexane. The organic layer was washed with water and brine, and dried over anhydrous sodium sulfate. After evaporation of the solvents, the residue was distilled with a Kugelrohr distillation apparatus (100–130 °C/1 mmHg) to give 4 (537 mg, 85%) as a yellow oil.
4. 1H NMR (301 MHz, CDCl3): δ 1.07 (d, 6H, J = 6.9 Hz), 1.10 (d, 6H, J = 6.9 Hz), 1.12 (d, 6H, J = 7.2 Hz), 1.14 (d, 6H, J = 8.1 Hz), 1.16–1.31 (m, 2H), 1.40 (sept, 2H, J = 7.3 Hz), 3.73 (t, 1H, J = 3.2 Hz), 7.20 (dd, 1H, J = 4.7, 3.5 Hz), 7.31 (dd, 1H, J = 3.5, 1.1 Hz), 7.61 (dd, 1H, J = 4.7, 1.1 Hz). 13C NMR (76 MHz, CDCl3): δ 11.7, 13.7, 19.3, 19.5, 20.8, 21.6, 128.0, 130.6, 134.8, 136.2. 29Si NMR (60 MHz, CDCl3): δ −14.7, −9.0. IR (NaCl): 2940, 2860, 2070, 1460, 1210, 1000, 880, 750, 700 cm−1. MS (EI, 70 eV): m/z 312 (M+, 16), 269 (100), 227 (37), 197 (16), 185 (25), 155 (17), 141 (15), 127 (20). HRMS (EI): found 312.1774, calcd for C16H32SSi2 312.1763.
3.2. Synthesis of 1,1,2,2-Tetraisopropyl-1-(4′-methoxyphenyl)disilane (5)
A 1.53 M solution of tert-butyllithium in pentane (3.7 mL, 5.7 mmol) was added dropwise to a solution of 4-iodoanisole (260 mg, 1.11 mmol) in THF (5 mL) at −78 °C, and the mixture was stirred at −78 °C for 1 h. After warming to room temperature, 1 (244 mg, 0.921 mmol) was added, and the mixture was stirred at room temperature for 24 h. The reaction was quenched by adding 1.0 M hydrochloric acid. The reaction mixture was extracted with hexane. The organic layer was washed with water and brine, and dried over anhydrous sodium sulfate. After evaporation of the solvents, the residue was distilled with a Kugelrohr distillation apparatus (190–220 °C/1 mmHg) to give 5 (141 mg, 45%) as a yellow oil.
5. 1H NMR (301 MHz, CDCl3): δ 1.03 (d, 6H, J = 7.2 Hz), 1.05 (d, 6H, J = 7.2 Hz), 1.08 (d, 6H, J = 7.2 Hz), 1.09 (d, 6H, J = 7.2 Hz), 1.14–1.27 (m, 2H), 1.40 (sept, 2H, J = 7.2 Hz), 3.73 (t, 1H, J = 3.3 Hz), 3.81 (s, 3H), 6.89 (d, 2H, J = 8.7 Hz), 7.45 (d, 2H, J = 8.7 Hz). 13C NMR (76 MHz, CDCl3): δ 11.7, 12.7, 19.2, 19.4, 21.0, 21.7, 55.0, 113.4, 126.5, 137.2, 160.1. 29Si NMR (99 MHz, CDCl3): δ −15.0, −6.1. IR (NaCl): 2940, 2860, 2070, 1590, 1500, 1460, 1280, 1250, 1180, 1100 cm−1. MS (EI, 70 eV): m/z 336 (M+, 43), 293 (100), 251 (48), 221 (77), 209 (38), 179 (43), 167 (19), 165 (16), 151 (27), 137 (14). HRMS (EI): found 336.2305, calcd for C19H36OSi2 336.2305.
3.3. Representative Procedure: Synthesis of 1,1,2,2,3,3-Hexamethyl-1-(2′-thienyl)trisilane (16)
A 1.64 M solution of butyllithium in hexane (4.8 mL, 7.9 mmol) was added dropwise to a solution of chlorotrimethylsilane (847 mg, 7.80 mmol) in THF (15 mL) at −78 °C, and the mixture was stirred at −78 °C for 10 min. After warming to room temperature, magnesium turnings (392 mg, 16.1 mmol) and a 1.0 M solution of DIBAL-H in toluene (0.1 mL, 0.1 mmol) were added. After stirring for 5 min, the mixture was cooled to −10 °C, 2-bromothiophene (974 mg, 5.97 mmol) was added. After stirring for 1 h, 2 (876 mg, 4.15 mmol) was added, and the mixture was stirred at room temperature overnight. The reaction was quenched with 1.0 M hydrochloric acid. The reaction mixture was extracted with hexane. The organic layer was washed with water and brine, and dried over anhydrous sodium sulfate. After evaporation of the solvents, the residue was separated by column chromatography over silica gel with hexane to give 16 (758 mg, 71%) as a colorless oil.
16. 1H NMR (600 MHz, CDCl3): δ 0.09 (d, 6H, J = 4.5 Hz), 0.15 (s, 6H), 0.41 (s, 6H), 3.73 (sept, 1H, J = 4.5 Hz), 7.19 (dd, 1H, J = 4.5, 3.2 Hz), 7.21 (dd, 1H, J = 3.2, 0.8 Hz), 7.59 (dd, 1H, J = 4.5, 0.8 Hz). 13C NMR (151 MHz, CDCl3): δ −6.6, −5.9, −1.8, 128.3, 130.6, 134.2, 139.1. 29Si NMR (119 MHz, CDCl3): δ −47.2, −36.3, −20.5. IR (NaCl): 2950, 2090, 1400, 1250, 1030, 880, 830, 770 cm−1. MS (EI, 70 eV): m/z 258 (M+, 5), 243 (59), 199 (21), 173 (26), 170 (23), 141 (82), 116 (52), 73 (100). HRMS (EI): found 257.0669, calcd for C10H21SSi3 (M+ − H) 257.0672.
3.4. Synthesis of 1-(5′-Hexenyl)-1,1,2,2,3,3-hexamethyltrisilane (6)
Synthesis of 6 was carried out by the same procedure as 16 using THF (4 mL), chlorotrimethylsilane (178 mg, 1.64 mmol), a 1.64 M solution of butyllithium in hexane (1.14 mL, 1.87 mmol), magnesium turnings (95 mg, 3.9 mmol), a 1.0 M solution of DIBAL-H in toluene (0.024 mL, 0.024 mmol), 6-bromo-1-hexene (234 mg, 1.44 mmol) and 2 (178 mg, 0.844 mmol). Separation by column chromatography over silica gel with hexane gave 6 (84 mg, 38%) as a colorless oil.
6. 1H NMR (600 MHz, CDCl3): δ 0.06 (s, 6H), 0.12 (s, 6H), 0.15 (d, 6H, J = 4.5 Hz), 0.59–0.64 (m, 2H), 1.30–1.37 (m, 2H), 1.40–1.44 (m, 2H), 2.03–2.07 (m, 2H), 3.72 (sept, 1H, J = 4.5 Hz), 4.92–4.94 (m, 1H), 4.98–5.01 (m, 1H), 5.77–5.84 (m, 1H). 13C NMR (151 MHz, CDCl3): δ −6.4, −5.8, −3.4, 15.5, 24.3, 33.1, 33.7, 114.3, 139.2. 29Si NMR (119 MHz, CDCl3): δ −48.0, −36.1, −13.5. IR (NaCl): 3080, 2950, 2920, 2090, 1640, 1411, 1250, 910, 880, 830, 790 cm−1. MS (EI, 70 eV): m/z 258 (M+, 9), 215 (20), 141 (64), 127 (18), 117 (20), 116 (19), 73 (100), 59 (51). HRMS (EI): found 258.1649, calcd for C12H30Si3 258.1655.
3.5. Synthesis of 1,1,2,2,3,3-Hexamethyl-1-(2′-phenylethyl)trisilane (7)
Synthesis of 7 was carried out by the same procedure as 16 using THF (20 mL), chlorotrimethylsilane (1.01 g, 9.32 mmol), a 1.60 M solution of butyllithium in hexane (6.0 mL, 9.6 mmol), magnesium turnings (472 mg, 19.4 mmol), a 1.0 M solution of DIBAL-H in toluene (0.12 mL, 0.12 mmol), (2-bromoethyl)benzene (1.30 g, 7.04 mmol) and 2 (1.01 g, 4.79 mmol). Separation by column chromatography over silica gel with hexane containing 1% triethylamine gave 7 (939 mg, 70%) as a colorless oil.
7. 1H NMR (600 MHz, CDCl3): δ 0.16 (s, 6H), 0.19 (s, 6H), 0.21 (d, 6H, J = 4.5 Hz), 1.01–1.05 (m, 2H), 2.65–2.70 (m, 2H), 3.81 (sept, 1H, J = 4.5 Hz), 7.17–7.26 (m, 3H), 7.29–7.33 (m, 2H). 13C NMR (151 MHz, CDCl3): δ −6.4, −5.8, −3.5, 17.9, 30.9, 125.7, 127.9, 128.5, 145.5. 29Si NMR (119 MHz, CDCl3): δ −47.9, −36.2, −13.3. IR (NaCl): 3030, 2950, 2890, 2090, 1600, 1490, 1450, 1410, 1250, 880, 830, 790 cm−1. MS (EI, 70 eV): m/z 265 (M+−CH3, 11), 221 (76), 163 (71), 135 (100), 117 (84), 116 (38), 73 (78), 59 (84). HRMS (EI): found 265.1263, calcd for C13H25Si3 (M+ − CH3) 265.1264.
3.6. Synthesis of 1,1,2,2,3,3-Hexamethyl-1-(E)-styryltrisilane (8)
Synthesis of 8 was carried out by the same procedure as 16 using THF (8 mL), chlorotrimethylsilane (407 mg, 3.75 mmol), a 1.64 M solution of butyllithium in hexane (2.3 mL, 3.8 mmol), magnesium turnings (188 mg, 7.73 mmol), a 1.0 M solution of DIBAL-H in toluene (0.064 mL, 0.064 mmol), β-bromostyrene (527 mg, 2.88 mmol) and 2 (430 mg, 2.04 mmol). Separation by column chromatography over silica gel with hexane gave 8 (332 mg, 58%) as a colorless oil.
8. 1H NMR (600 MHz, CDCl3): δ 0.09 (s, 6H), 0.10 (d, 6H, J = 4.5 Hz), 0.20 (s, 6H), 3.70 (sept, 1H, J = 4.5 Hz), 6.45 (d, 1H, J = 19 Hz), 6.78 (d, 1H, J = 19 Hz), 7.16–7.19 (m, 1H), 7.24–7.28 (m, 2H), 7.33–7.38 (m, 2H). 13C NMR (151 MHz, CDCl3): δ −6.5, −5.8, −3.3, 126.4, 128.0, 128.7 (2 peaks are overlapped.), 138.6, 143.8. 29Si NMR (119 MHz, CDCl3): δ −47.2, −36.1, −20.2. IR (NaCl): 3020, 2950, 2890, 2090, 1710, 1600, 1570, 1490, 1450, 1400, 1250, 990, 880, 840, 790, 730 cm−1. MS (EI, 70 eV): m/z 278 (M+, 4), 219 (18), 204 (22), 161 (26), 145 (79), 135 (32), 117 (25), 116 (91), 102 (17), 73 (100), 59 (68). HRMS (EI): found 278.1334, calcd for C14H26Si3 278.1342.
3.7. Synthesis of 1-(2-Buten-2-yl)-1,1,2,2,3,3-hexamethyltrisilane (9)
Synthesis of 9 was carried out by the same procedure as 16 using THF (8 mL), chlorotrimethylsilane (407 mg, 3.75 mmol), a 1.64 M solution of butyllithium in hexane (2.3 mL, 3.8 mmol), magnesium turnings (188 mg, 7.73 mmol), a 1.0 M solution of DIBAL-H in toluene (0.048 mL, 0.048 mmol), 2-bromo-2-butene (388 mg, 2.87 mmol) and 2 (420 mg, 1.99 mmol). Separation by column chromatography over silica gel with hexane gave 9 (248 mg, 54%, E/Z = 32/68) as a colorless oil.
(Z)-9. 1H NMR (600 MHz, CDCl3): δ 0.15 (s, 6H), 0.15 (d, 6H, J = 4.8 Hz), 0.25 (s, 6H), 1.67–1.71 (m, 3H), 1.74–1.76 (m, 3H), 3.74 (sept, 1H, J = 4.2 Hz), 6.07–6.11 (m, 1H). 13C NMR (151 MHz, CDCl3): δ −5.9, −5.8, −1.9, 18.4, 25.3, 134.8, 136.4. 29Si NMR (119 MHz, CDCl3): δ −46.6, −35.7, −22.3.
(E)-9. 1H NMR (600 MHz, CDCl3): δ 0.13 (d, 6H, J = 4.2 Hz), 0.15 (s, 6H), 0.25 (s, 6H), 1.68–1.71 (m, 3H), 1.74–1.75 (m, 3H), 3.71 (sept, 1H, J = 4.8 Hz), 5.77–5.80 (m, 1H). 13C NMR (151 MHz, CDCl3): δ −6.1, −5.9, −3.7, 14.4, 15.4, 133.9, 136.2. 29Si NMR (119 MHz, CDCl3): δ −47.8, −35.7, −17.3.
Mixture of (Z)-9 and (E)-9. IR (NaCl): 2950, 2900, 2090, 1250, 880, 830, 790 cm−1. MS (EI, 70 eV): m/z 230 (M+, 7), 156 (14), 141 (16), 131 (19), 117 (35), 116 (82), 97 (19), 73 (100), 59 (41). HRMS (EI): found 230.1335, calcd for C10H26Si3 230.1342.
3.8. Synthesis of 1-(1′-Hexynyl)-1,1,2,2,3,3-hexamethyltrisilane (10)
A 0.90 M solution of isopropylmagnesium chloride in THF (1.11 mL, 1.0 mmol) was added to a solution of 1-hexyne (83 mg, 1.0 mmol) in THF (5 mL), and the mixture was stirred at 40 °C for 1 h. After cooling to room temperature, 2 (316 mg, 1.50 mmol) was added, and the mixture was stirred at room temperature for 1 h. The reaction was quenched with 1.0 M hydrochloric acid. The reaction mixture was extracted with hexane. The organic layer was washed with water, saturated aqueous sodium hydrogencarbonate and brine, and dried over anhydrous sodium sulfate. After evaporation of the solvents, the residue was separated by column chromatography over silica gel with hexane to give 10 (123 mg, 48%) as a colorless oil.
10. 1H NMR (600 MHz, CDCl3): δ 0.16 (s, 6H), 0.18 (d, 6H, J = 4.5 Hz), 0.20 (s, 6H), 0.90 (t, 3H, J = 7.2 Hz), 1.37–1.44 (m, 2H), 1.46–1.51 (m, 2H), 2.24 (t, 2H, J = 6.9 Hz), 3.75 (sept, 1H, J = 4.5 Hz). 13C NMR (151 MHz, CDCl3): δ −6.7, −5.8, −1.8, 13.7, 19.9, 22.0, 31.0, 82.9, 110.3. 29Si NMR (119 MHz, CDCl3): δ −46.8, −36.2, −34.9. IR (NaCl): 2960, 2170, 2090, 1710, 1260, 800 cm−1. MS (EI, 70 eV): m/z 256 (M+, 3), 241 (14), 197 (16), 141 (38), 139 (22), 117 (37), 116 (100), 101 (14), 83 (22), 73 (99), 59 (36). HRMS (EI): found 256.1491, calcd for C12H28Si3 256.1499.
3.9. Synthesis of 1,1,2,2,3,3-Hexamethyl-1-(phenylethynyl)trisilane (11)
Synthesis of 11 was carried out by the same procedure as 10 using THF (25 mL), phenylacetylene (811 mg, 7.94 mmol), a 0.81 M solution of isopropylmagnesium chloride in THF (9.5 mL, 7.7 mmol) and 2 (1.55 g, 7.36 mmol). Separation by column chromatography over silica gel with hexane containing 1% triethylamine gave 11 (1.41 g, 69%) as a colorless oil.
11. 1H NMR (600 MHz, CDCl3): δ 0.22 (d, 6H, J = 4.5 Hz), 0.23 (s, 6H), 0.31 (s, 6H), 3.81 (sept, 1H, J = 4.5 Hz), 7.27–7.30 (m, 3H), 7.44–7.46 (m, 2H). 13C NMR (151 MHz, CDCl3): δ −6.6, −5.8, −2.1, 93.3, 107.8, 123.6, 128.3, 128.5, 132.0. 29Si NMR (119 MHz, CDCl3): δ −46.3, −36.2, −33.7. IR (NaCl): 2950, 2360, 2150, 1490, 1250 cm−1. MS (EI, 70 eV): m/z 276 (M+, 14), 261 (15), 217 (63), 203 (27), 159 (64), 135 (26), 116 (71), 73 (100). HRMS (EI): found 276.1178, calcd for C14H24Si3 276.1186.
3.10. Synthesis of 1,1,2,2,3,3-Hexamethyl-1-(trimethylsilylethynyl)trisilane (12)
Synthesis of 12 was carried out by the same procedure as 10 using THF (5 mL), trimethylsilylacetylene (98 mg, 1.0 mmol), a 0.90 M solution of isopropylmagnesium chloride in THF (1.11 mL, 1.0 mmol) and 2 (316 mg, 1.50 mmol). Separation by column chromatography over silica gel with hexane gave 12 (149 mg, 55%) as a colorless oil.
12. 1H NMR (600 MHz, CDCl3): δ 0.16 (s, 9H), 0.17 (s, 6H), 0.19 (d, 6H, J = 4.5 Hz), 0.22 (s, 6H), 3.77 (sept, 1H, J = 4.5 Hz). 13C NMR (151 MHz, CDCl3): δ −6.7, −5.8, −2.2, 0.1, 113.0, 116.9. 29Si NMR (119 MHz, CDCl3): δ −46.6, −36.1, −34.7, −18.9. IR (NaCl): 2960, 2090, 1260, 840, 800 cm−1. MS (EI, 70 eV): m/z 272 (M+, 4), 257 (12), 213 (41), 155 (18), 117 (16), 116 (100), 73 (80). HRMS (EI): found 272.1267, calcd for C11H28Si4 272.1268.
3.11. Synthesis of 1,1,2,2,3,3-Hexamethyl-1-phenyltrisilane (13)
Synthesis of 13 was carried out by the same procedure as 16 using THF (10 mL), chlorotrimethylsilane (0.7 mL, 6 mmol), a 1.60 M solution of butyllithium in hexane (3.4 mL, 5.4 mmol), magnesium turnings (290 mg, 11.9 mmol), a 1.0 M solution of DIBAL-H in toluene (0.073 mL, 0.073 mmol), bromobenzene (0.44 mL, 4.2 mmol) and 2 (609 mg, 2.89 mmol). Separation by column chromatography over silica gel with hexane containing 1% triethylamine gave 13 (404 mg, 55%) as a colorless oil. The 1H NMR spectrum is identical to the reported data [48].
13. 1H NMR (600 MHz, CDCl3): δ 0.08 (d, 6H, J = 4.5 Hz), 0.13 (s, 6H), 0.40 (s, 6H), 3.73 (sept, 1H, J = 4.5 Hz), 7.33–7.36 (m, 3H), 7.46–7.48 (m, 2H). 13C NMR (151 MHz, CDCl3): δ −6.5, −5.9, −3.2, 127.9, 128.5, 133.9, 139.8. 29Si NMR (119 MHz, CDCl3): δ −47.4, −36.1, −18.0.
3.12. Synthesis of 1-(4′-Methoxyphenyl)-1,1,2,2,3,3-hexamethyltrisilane (14)
Synthesis of 14 was carried out by the same procedure as 16 using THF (5 mL), chlorotrimethylsilane (301 mg, 2.77 mmol), a 1.64 M solution of butyllithium in hexane (1.7 mL, 2.8 mmol), magnesium turnings (127 mg, 5.22 mmol), a 1.0 M solution of DIBAL-H in toluene (0.06 mL, 0.06 mmol), 4-bromoanisole (380 mg, 2.03 mmol) and 2 (301 mg, 1.43 mmol). Separation by column chromatography over silica gel with hexane gave 14 (270 mg, 67%) as a colorless oil.
14. 1H NMR (600 MHz, CDCl3): δ 0.08 (d, 6H, J = 4.5 Hz), 0.11 (s, 6H), 0.35 (s, 6H), 3.71 (sept, 1H, J = 4.5 Hz), 3.82 (s, 3H), 6.90 (d, 2H, J = 8.7 Hz), 7.37 (d, 2H, J = 8.7 Hz). 13C NMR (151 MHz, CDCl3): δ −6.4, −5.9, −2.9, 55.1, 113.7, 130.3, 135.2, 160.2. 29Si NMR (119 MHz, CDCl3): δ −47.5, −36.1, −18.4. IR (NaCl): 2950, 2090, 1590, 1500, 1280, 1250, 1180, 1110, 880, 830, 780 cm−1. MS (EI, 70 eV): m/z 282 (M+, 8), 281 (20), 193 (36), 165 (100), 135 (16), 116 (54), 73 (29). HRMS (EI): found 281.1213, calcd for C13H25OSi3 (M+ − H) 281.1213.
3.13. Synthesis of 1-(4′-Dimethylaminophenyl)-1,1,2,2,3,3-hexamethyltrisilane (15)
Synthesis of 15 was carried out by the same procedure as 16 using THF (5 mL), chlorotrimethylsilane (290 mg, 2.67 mmol), a 1.64 M solution of butyllithium in hexane (1.6 mL, 2.6 mmol), magnesium turnings (122 mg, 5.02 mmol), a 1.0 M solution of DIBAL-H in toluene (0.06 mL, 0.06 mmol), 4-bromo-N,N-dimethylaniline (400 mg, 2.00 mmol) and 2 (277 mg, 1.31 mmol). The crude product was separated by column chromatography over silica gel with hexane–ethyl acetate (5:1). Compound 15 (150 mg, 39%) was obtained as a colorless oil.
15. 1H NMR (600 MHz, CDCl3): δ 0.09 (d, 6H, J = 4.5 Hz), 0.11 (s, 6H), 0.33 (s, 6H), 2.95 (s, 6H), 3.71 (sept, 1H, J = 4.5 Hz), 6.73 (d, 2H, J = 8.4 Hz), 7.32 (d, 2H, J = 8.4 Hz). 13C NMR (151 MHz, CDCl3): δ –6.4, –5.8, –2.8, 40.4, 112.3, 124.5, 135.0, 150.8. 29Si NMR (119 MHz, CDCl3): δ −47.5, −36.0, −19.0. IR (NaCl): 2950, 2890, 2080, 1600, 1510, 1350, 1240, 1110, 880, 830, 790 cm−1. MS (EI, 70 eV): m/z 295 (M+, 15), 178 (100), 162 (9), 134 (12), 116 (9), 102 (12), 73 (9). HRMS (EI): found 294.1528, calcd for C14H28NSi3 (M+ − H) 294.1529.
3.14. Synthesis of 1,1,2,2,3,3,4,4-Octamethyl-1-(2′-thienyl)tetrasilane (17)
Synthesis of 17 was carried out by the same procedure as 16 using THF (5 mL), chlorotrimethylsilane (282 mg, 2.60 mmol), a 1.64 M solution of butyllithium in hexane (1.6 mL, 2.6 mmol), magnesium turnings (127 mg, 5.22 mmol), a 1.0 M solution of DIBAL-H in toluene (0.08 mL, 0.08 mmol), 2-bromothiophene (324 mg, 1.99 mmol) and 3 (381 mg, 1.42 mmol). Separation by column chromatography over silica gel with hexane gave 17 (191 mg, 43%) as a colorless oil.
17. 1H NMR (600 MHz, CDCl3): δ 0.09 (s, 6H), 0.12 (d, 6H, J = 4.5 Hz), 0.15 (s, 6H), 0.42 (s, 6H), 3.72 (sept, 1H, J = 4.5 Hz), 7.19 (dd, 1H, J = 4.5, 3.3 Hz), 7.20 (dd, 1H, J = 3.0, 0.6 Hz), 7.59 (dd, 1H, J = 4.2, 1.2 Hz). 13C NMR (151 MHz, CDCl3): δ −5.9, −5.81, −5.79, −1.4, 128.3, 130.5, 134.1, 139.3. 29Si NMR (119 MHz, CDCl3): δ −44.1, −43.9, −35.9, −19.9. IR (NaCl): 2950, 2890, 2090, 1400, 1250, 1210, 990, 880, 830, 780, 700 cm−1. MS (EI, 70 eV): m/z 316 (M+, 1), 257 (100), 173 (17), 167 (34), 159 (19), 141 (39), 116 (18), 73 (93), 59 (15). HRMS (EI): found 301.0753, calcd for C11H25SSi4 (M+ − CH3) 301.0754.
3.15. Synthesis of 1-Methoxy-1,1,2,2,3,3-hexamethyl-3-(E)-styryltrisilane (18)
[RuCl2(p-cymene)]2 (15 mg, 0.024 mmol) was added to a solution of 8 (271 mg, 0.973 mmol) and methanol (65 μL, 1.6 mmol) in toluene (4 mL) at 0 °C. The mixture was stirred with gradual warming to room temperature overnight. After evaporation of the solvent, the residue was distilled with a Kugelrohr distillation apparatus (146–152 °C/0.90–1.4 mmHg) to give 18 (216 mg, 72%) as a colorless oil.
18. 1H NMR (600 MHz, CDCl3): δ 0.19 (s, 6H), 0.25 (s, 6H), 0.28 (s, 6H), 3.43 (s, 3H), 6.54 (d, 1H, J = 18.9 Hz), 6.86 (d, 1H, J = 18.9 Hz), 7.23–7.25 (m, 1H), 7.31–7.35 (m, 2H), 7.42–7.45 (m, 2H). 13C NMR (151 MHz, CDCl3): δ −6.4, −3.4, −0.1, 51.4, 126.4, 127.9, 128.58, 128.64, 138.6, 143.8. 29Si NMR (119 MHz, CDCl3): δ −50.2, −20.7, 21.3. IR (NaCl): 2950, 1590, 1570, 1490, 1450, 1400, 1250, 1080, 990, 830, 780, 730, 690 cm−1. MS (EI, 70 eV): m/z 308 (M+, 10), 293 (44), 219 (48), 173 (52), 145 (72), 135 (65), 133 (37), 117 (41), 116 (57), 89 (39), 73 (100), 59 (71). HRMS (FD): found 308.1449, calcd for C15H28OSi3 308.1448.
3.16. Synthesis of 1-(1′-Hexynyl)-3-methoxy-1,1,2,2,3,3-hexamethyltrisilane (19)
Synthesis of 19 was carried out by the same procedure as 18 using toluene (4 mL), 10 (264 mg, 1.03 mmol), methanol (65 μL, 1.6 mmol) and [RuCl2(p-cymene)]2 (15 mg, 0.024 mmol). The crude product was distilled with a Kugelrohr distillation apparatus (112–116 °C/1.2–1.3 mmHg) to give 19 (191 mg, 65%) as a colorless oil.
19. 1H NMR (600 MHz, CDCl3): δ 0.17 (s, 6H), 0.20 (s, 6H), 0.25 (s, 6H), 0.89 (t, 3H, J = 7.2 Hz), 1.35–1.42 (m, 2H), 1.45–1.50 (m, 2H), 2.22 (t, 2H, J = 7.2 Hz), 3.43 (s, 3H). 13C NMR (151 MHz, CDCl3): δ −6.6, −1.9, −0.1, 13.7, 19.9, 22.0, 30.9, 51.4, 82.9, 110.2. 29Si NMR (119 MHz, CDCl3): δ −49.9, −35.1, 21.2. IR (NaCl): 2960, 2170, 1250, 1090, 1040, 830, 780 cm−1. MS (EI, 70 eV): m/z 286 (M+, 1), 257 (19), 229 (16), 133 (20), 117 (35), 116 (54), 89 (41), 73 (100), 59 (61). HRMS (FD): found 286.1612, calcd for C13H30OSi3 286.1604.
3.17. Synthesis of 1-Methoxy-1,1,2,2,3,3-hexamethyl-3-(phenylethynyl)trisilane (20)
Synthesis of 20 was carried out by the same procedure as 18 using toluene (4 mL), 11 (280 mg, 1.01 mmol), methanol (65 μL, 1.6 mmol) and [RuCl2(p-cymene)]2 (15 mg, 0.024 mmol). The crude product was distilled with a Kugelrohr distillation apparatus (134–135 °C/0.90–0.93 mmHg) to give 20 (147 mg, 47%) as a colorless oil.
20. 1H NMR (600 MHz, CDCl3): δ 0.25 (s, 6H), 0.30 (s, 6H), 0.33 (s, 6H), 3.46 (s, 3H), 7.29–7.30 (m, 3H), 7.43–7.45 (m, 2H). 13C NMR (151 MHz, CDCl3): δ −6.5, −2.1, 0.0, 51.5, 93.3, 107.8, 123.5, 128.3, 128.5, 132.0. 29Si NMR (119 MHz, CDCl3): δ −49.5, −33.9, 21.0. IR (NaCl): 2970, 2940, 2900, 2150, 1490, 1250, 1080, 840, 780, 690 cm−1. MS (EI, 70 eV): m/z 306 (M+, 7), 305 (23), 193 (41), 159 (37), 116 (34), 89 (30), 73 (100), 59 (64). HRMS (FD): found 306.1293, calcd for C15H26OSi3 306.1291.
3.18. Synthesis of 1-Methoxy-1,1,2,2,3,3-hexamethyl-3-phenyltrisilane (21)
Synthesis of 21 was carried out by the same procedure as 18 using toluene (4 mL), 13 (258 mg, 1.02 mmol), methanol (60 μL, 1.5 mmol) and [RuCl2(p-cymene)]2 (15 mg, 0.024 mmol). The crude product was distilled with a Kugelrohr distillation apparatus (153–157 °C/11 mmHg) to give 21 (251 mg, 87%) as a colorless oil. The NMR data are identical to the reported data [48].
21. 1H NMR (600 MHz, CDCl3): δ 0.156 (s, 6H), 0.160 (s, 6H), 0.42 (s, 6H), 3.35 (s, 3H), 7.32–7.37 (m, 3H), 7.46–7.49 (m, 2H).13C NMR (151 MHz, CDCl3): δ −6.3, −3.2, −0.2, 51.3, 127.9, 128.5, 133.9, 139.6. 29Si NMR (119 MHz, CDCl3): δ −50.2, −18.4, 21.3. IR (NaCl): 2950, 2890, 1250, 1080, 830, 780, 730, 700 cm−1. MS (EI, 70 eV): m/z 282 (M+, 2), 267 (38), 193 (14), 135 (54), 116 (100), 89 (15), 73 (23), 59 (17). HRMS (FD): found 282.1300, calcd for C13H26OSi3 282.1291.
3.19. Synthesis of 1,2-Dichloro-1,1,2,2-tetraethyldisilane
Freshly distilled acetyl chloride (14.0 mL, 197 mmol) was added dropwise to the mixture of aluminum chloride (26.04 g, 195 mmol) and hexaethyldisilane (21.8 mL, 79.5 mmol). The flask was immersed in a water bath to prevent raising the reaction temperature too much. After stirring for 3 h, the reaction mixture was distilled under reduced pressure (bp 100–131 °C/9 mmHg) to give 1,2-dichloro-1,1,2,2-tetraethyldisilane (16.47 g, 85%) as a colorless oil. The 1H NMR spectrum is identical to the reported data [14].
3.20. Synthesis of 1,1,2,2-Tetraethyl-1-phenyldisilane (22)
Phenylmagnesium bromide was prepared from magnesium turnings (2.17 g, 89.4 mmol), bromobenzene (11.72 g, 74.6 mmol) and diethyl ether (42 mL). The Grignard reagent was added dropwise to a solution of 1,2-dichloro-1,1,2,2-tetraethyldisilane (16.47 g, 67.7 mmol) in diethyl ether (30 mL) at 0 °C. The mixture was stirred at room temperature for 12 h. After filtration of the reaction mixture, the filtrate was evaporated to give a crude product of 1-chloro-1,1,2,2-tetraethyl-2-phenyldisilane (19.29 g).
A solution of 1-chloro-1,1,2,2-tetraethyl-2-phenyldisilane in diethyl ether (60 mL) was added dropwise to a mixture of lithium aluminum hydride (1.21 g, 32.0 mmol) and diethyl ether (100 mL) at 0 °C. The mixture was stirred with gradual warming to room temperature overnight. The reaction was quenched with 1.0 M hydrochloric acid. The reaction mixture was extracted with hexane. The organic layer was washed with brine and dried over anhydrous sodium sulfate. After evaporation of the solvents, the residue was distilled under reduced pressure according to a normal distillation procedure (bp 129–131 °C/5 mmHg) to give 22 (12.30 g, 73% in two steps) as a colorless oil.
22. 1H NMR (600 MHz, CDCl3): δ 0.69–0.76 (m, 4H), 0.95–1.02 (m, 10H), 1.03–1.06 (m, 6H), 3.67 (quin, 1H, J = 3.8 Hz), 7.33–7.36 (m, 3H), 7.48–7.50 (m, 2H). 13C NMR (151 MHz, CDCl3): δ 2.2, 4.4, 8.4, 10.2, 127.9, 128.6, 134.6, 137.6. 29Si NMR (119 MHz, CDCl3): δ −26.4, −13.1. IR (NaCl): 3070, 2950, 2910, 2870, 2080, 1460, 1430, 1230, 1100, 1010, 970, 800, 770, 700 cm−1. MS (EI, 70 eV): m/z 250 (M+, 13), 163 (61), 135 (100), 107 (55). HRMS (FD): found 250.1574, calcd for C14H26Si2 250.1573.
3.21. Synthesis of 1,1,2,2-Tetraethyl-1-methoxy-2-phenyldisilane (23)
[RuCl2(p-cymene)]2 (16 mg, 0.026 mmol) was added to a solution of 22 (260 mg, 1.04 mmol) and methanol (338 mg, 10.5 mmol) in toluene (4 mL) at 0 °C. The mixture was stirred with gradual warming to room temperature for 1 day. After evaporation of the solvent, the residue was distilled with a Kugelrohr distillation apparatus (81–117 °C/0.5 mmHg) to give 23 (241 mg, 83%) as a colorless oil.
23. 1H NMR (600 MHz, CDCl3): δ 0.70–0.81 (m, 4H), 0.95–1.02 (m, 10H), 1.06–1.10 (m, 6H), 3.41 (s, 3H), 7.31–7.36 (m, 3H), 7.51–7.53 (m, 2H). 13C NMR (151 MHz, CDCl3): δ 4.4, 7.0, 7.3, 8.3, 51.7, 127.9, 128.5, 134.8, 137.7. 29Si NMR (119 MHz, CDCl3): δ −17.3, 19.2. IR (NaCl): 3070, 2960, 2880, 1460, 1430, 1090, 1010, 700 cm−1. MS (EI, 70 eV): m/z 280 (M+, 40), 279 (100), 265 (21), 251 (20), 237 (40), 223 (27), 209 (28), 195 (23), 135 (39), 117 (25), 107 (61), 89 (35), 59 (21). HRMS (FD): found 280.1670, calcd for C15H28OSi2 280.1678.
3.22. Synthesis of 1-Ethoxy-1,1,2,2-tetraethyl-2-phenyldisilane (24)
Synthesis of 24 was carried out by the almost same procedure as 23 using toluene (4 mL), 22 (253 mg, 1.01 mmol), ethanol (476 mg, 10.3 mmol) and [RuCl2(p-cymene)]2 (15 mg, 0.024 mmol). The mixture was stirred at 50 °C for 1 day. The solvent was evaporated, and the residue was distilled with a Kugelrohr distillation apparatus (95–110 °C/0.5 mmHg) to give 24 (135 mg, 45%) as a colorless oil.
24. 1H NMR (600 MHz, CDCl3): δ 0.69–0.80 (m, 4H), 0.94–1.01 (m, 10H), 1.05–1.08 (m, 6H), 1.14 (t, 3H, J = 7.0 Hz), 3.61 (q, 2H, J = 7.0 Hz), 7.32–7.35 (m, 3H), 7.49–7.53 (m, 2H). 13C NMR (151 MHz, CDCl3): δ 4.4, 7.1, 7.6, 8.3, 18.8, 59.5, 127.9, 128.5, 134.8, 137.9. 29Si NMR (119 MHz, CDCl3): δ −17.4, 16.5. IR (NaCl): 2960, 2880, 1460, 1430, 1100, 1080, 1000, 700 cm−1. MS (EI, 70 eV): m/z 294 (M+, 2), 293 (4), 265 (21), 237 (37), 209 (33), 135 (36), 131 (47), 107 (100), 105 (39), 103 (63), 87 (19), 75 (35), 59 (23). HRMS (FD): found 294.1849, calcd for C16H30OSi2 294.1835.
3.23. Synthesis of 1,1,2,2-Tetraethyl-1-isopropoxy-2-phenyldisilane (25)
[RuCl2(p-cymene)]2 (17 mg, 0.028 mmol) was added to a solution of 22 (265 mg, 1.06 mmol) and 2-propanol (675 mg, 11.2 mmol) in toluene (4 mL) at 0 °C. The mixture was stirred with gradual warming to room temperature for 12 h. Additional 2-propanol (5.84 g, 97.1 mmol) was added, and the mixture was heated at 50 °C for 1 day. After cooling to room temperature, the solvent and excess 2-propanol were evaporated, and the residue was distilled with a Kugelrohr distillation apparatus (91–111 °C/0.5 mmHg) to give 25 (188 mg, 58%) as a colorless oil.
25. 1H NMR (600 MHz, CDCl3): δ 0.71–0.75 (m, 4H), 0.93–1.00 (m, 10H), 1.04–1.07 (m, 6H), 1.09 (d, 6H, J = 6.0 Hz), 3.90 (sept, 1H, J = 6.0 Hz), 7.31–7.34 (m, 3H), 7.51–7.53 (m, 2H). 13C NMR (151 MHz, CDCl3): δ 4.4, 7.3, 8.2, 8.4, 26.1, 66.0, 127.8, 128.4, 134.8, 138.0. 29Si NMR (119 MHz, CDCl3): δ −17.5, 14.1. IR (NaCl): 2960, 2880, 1460, 1430, 1380, 1120, 1020, 700 cm−1. MS (EI, 70 eV): m/z 308 (M+, 0.2), 265 (73), 237 (84), 221 (17), 209 (54), 193 (25), 181 (14), 165 (15), 145 (41), 135 (41), 107 (100), 105 (35), 103 (79), 75 (75). HRMS (FD): found 308.1997, calcd for C17H32OSi2 308.1991.
3.24. Synthesis of 2-Methoxy-1,1,2,3,3-pentamethyl-1,3-diphenyltrisilane (27)
[RuCl2(benzene)]2 (13 mg, 0.026 mmol) was added to a solution of 26 (317 mg, 1.01 mmol) and methanol (328 mg, 10.2 mmol) in toluene (4 mL) at 0 °C. The mixture was stirred with gradual warming to room temperature for 1 day and at 50 °C for 5 days. After cooling to room temperature, the solvent was evaporated, and the residue was distilled with a Kugelrohr distillation apparatus (128–149 °C/0.5 mmHg) to give 27 (100 mg, 29%) as a colorless oil.
27. 1H NMR (600 MHz, CDCl3): δ 0.32 (s, 6H), 0.34 (s, 6H), 0.37 (s, 3H), 3.32 (s, 3H), 7.32–7.33 (m, 6H), 7.42–7.43 (m, 4H). 13C NMR (151 MHz, CDCl3): δ −3.8, −2.90, −2.87, 53.2, 127.9, 128.7, 134.1, 139.1. 29Si NMR (119 MHz, CDCl3): δ −21.8, 12.6. IR (NaCl): 3070, 2950, 1430, 1250, 1070, 770, 730, 700 cm−1. MS (EI, 70 eV): m/z 344 (M+, 5), 329 (25), 251 (14), 209 (17), 197 (10), 193 (13), 179 (33), 178 (54), 163 (24), 135 (100), 122 (11), 117 (11), 107 (11), 105 (12), 59 (17). HRMS (FD): found 344.1460, calcd for C18H28OSi3 344.1447.
4. Conclusions
We found that α-chloro-ω-hydrooligosilanes, synthesized by the titanium-catalyzed monoreduction of α,ω-dichlorooligosilanes, are good precursors for the synthesis of unsymmetrically substituted oligosilanes. Each functional group, the chlorosilane and hydrosilane moieties, is independently substituted by the reactions with organolithium or Grignard reagents and the ruthenium-catalyzed alkoxylations. In both steps, little or no cleavage of the Si–Si bond occurred. Further studies on these synthetic reactions are now in progress.
Supplementary Materials
The following are available online at http://www.mdpi.com/2304-6740/6/3/99/s1, Figures S1–S113: spectral data of all new compounds.
Author Contributions
Conceptualization, K.-i.K.; Experiments, Y.A., Y.N., M.I. and K.K.; Project Administration, S.K.; Preparation of the manuscript, K.-i.K. and S.K.
Funding
This work was supported in part by Grants-in-Aid for Scientific Research (Nos. 23550044 and 26410036) from the Japan Society for the Promotion of Science and the Element Innovation Project of Gunma University, Japan.
Acknowledgments
Some of the spectral data of the new compounds were measured in the Instrumental Analysis Division, Equipment Management Center, Creative Research Institution, Hokkaido University, Japan.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Miller, R.D.; Michl, J. Polysilane high polymers. Chem. Rev. 1989, 89, 1359–1410. [Google Scholar] [CrossRef]
- Beckmann, J. Oligosilanes. In Comprehensive Organometallic Chemistry III; Crabtree, R.H., Mingos, D.M.P., Eds.; Elsevier: Oxford, UK, 2007; Volume 3, pp. 409–512. [Google Scholar]
- Marschner, C. Oligosilanes. In Functional Molecular Silicon Compounds I: Regular Oxidation States; Scheschkewitz, D., Ed.; Springer: Cham, Switzerland, 2014; pp. 163–228. [Google Scholar]
- Mignani, G.; Krämer, A.; Puccetti, G.; Ledoux, I.; Soula, G.; Zyss, J.; Meyrueix, R. A new class of silicon compounds with interesting nonlinear optical effects. Organometallics 1990, 9, 2640–2643. [Google Scholar] [CrossRef]
- Mignani, G.; Krämer, A.; Puccetti, G.; Ledoux, I.; Zyss, J.; Soula, G. Effect of a weak donor on the intramolecular charge transfer of molecules containing two neighboring silicon atoms. Organometallics 1991, 10, 3656–3659. [Google Scholar] [CrossRef]
- Mignani, G.; Barzoukas, M.; Zyss, J.; Soula, G.; Balegroune, F.; Grandjean, D.; Josse, D. Improved transparency–efficiency trade-off in a new class of nonlinear organosilicon compounds. Organometallics 1991, 10, 3660–3668. [Google Scholar] [CrossRef]
- Tsuji, H.; Sasaki, M.; Shibano, Y.; Toganoh, M.; Kataoka, T.; Araki, Y.; Tamao, K.; Ito, O. Photoinduced electron transfer of dialkynyldisilane-linked zinc porphyrin–[60]fullerene dyad. Bull. Chem. Soc. Jpn. 2006, 79, 1338–1346. [Google Scholar] [CrossRef]
- Shibano, Y.; Sasaki, M.; Tsuji, H.; Araki, Y.; Ito, O.; Tamao, K. Conformation effect of oligosilane linker on photoinduced electron transfer of tetrasilane-linked zinc porphyrin–[60]fullerene dyads. J. Organomet. Chem. 2007, 692, 356–367. [Google Scholar] [CrossRef]
- Sasaki, M.; Shibano, Y.; Tsuji, H.; Araki, Y.; Tamao, K.; Ito, O. Oligosilane chain-length dependence of electron transfer of zinc porphyrin–oligosilane–fullerene molecules. J. Phys. Chem. A 2007, 111, 2973–2979. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, H.; Horiuchi, H.; Takanoha, Y.; Matsumoto, H.; Yoshihara, T.; Okutsu, T.; Negishi, K.; Kyushin, S.; Matsumoto, H. Excited-state property of 1-(4-cyanophenyl)-2-(4-methoxyphenyl)-1,1,2,2-tetramethyl- disilane. Chem. Lett. 2007, 36, 1168–1169. [Google Scholar] [CrossRef]
- Iwamoto, T.; Tsushima, D.; Kwon, E.; Ishida, S.; Isobe, H. Persilastaffanes: Design, synthesis, structure, and conjugation between silicon cages. Angew. Chem., Int. Ed. 2012, 51, 2340–2344. [Google Scholar] [CrossRef] [PubMed]
- Surampudi, S.; Yeh, M.-L.; Siegler, M.A.; Hardigree, J.F.M.; Kasl, T.A.; Katz, H.E.; Klausen, R.S. Increased carrier mobility in end-functionalized oligosilanes. Chem. Sci. 2015, 6, 1905–1909. [Google Scholar] [CrossRef] [PubMed]
- Shimada, M.; Yamanoi, Y.; Matsushita, T.; Kondo, T.; Nishibori, E.; Hatakeyama, A.; Sugimoto, K.; Nishihara, H. Optical properties of disilane-bridged donor–acceptor architectures: Strong effect of substituents on fluorescence and nonlinear optical properties. J. Am. Chem. Soc. 2015, 137, 1024–1027. [Google Scholar] [CrossRef] [PubMed]
- Kunai, A.; Kawakami, T.; Toyoda, E.; Ishikawa, M. Highly selective synthesis of chlorosilanes from hydrosilanes. Organometallics 1992, 11, 2708–2711. [Google Scholar] [CrossRef]
- Kunai, A.; Ochi, T.; Iwata, A.; Ohshita, J. Synthesis of bromohydrosilanes: Reactions of hydrosilanes with CuBr2 in the presence of CuI. Chem. Lett. 2001, 1228–1229. [Google Scholar] [CrossRef]
- Kunai, A.; Ohshita, J. Selective synthesis of halosilanes from hydrosilanes and utilization for organic synthesis. J. Organomet. Chem. 2003, 686, 3–15. [Google Scholar] [CrossRef]
- Harrison, D.J.; McDonald, R.; Rosenberg, L. Borane-catalyzed hydrosilylation of thiobenzophenone: A new route to silicon–sulfur bond formation. Organometallics 2005, 24, 1398–1400. [Google Scholar] [CrossRef]
- Kato, N.; Tamura, Y.; Kashiwabara, T.; Sanji, T.; Tanaka, M. AlCl3-catalyzed hydrosilylation of alkynes with hydropolysilanes. Organometallics 2010, 29, 5274–5282. [Google Scholar] [CrossRef]
- Oestreich, M.; Hermeke, J.; Mohr, J. A unified survey of Si–H and H–H bond activation catalysed by electron-deficient boranes. Chem. Soc. Rev. 2015, 44, 2202–2220. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C. Organosilanes in Radical Chemistry; Wiley: Chichester, UK, 2004. [Google Scholar]
- Chatgilialoglu, C. (Me3Si)3SiH: Twenty years after its discovery as a radical-based reducing agent. Chem. Eur. J. 2008, 14, 2310–2320. [Google Scholar] [CrossRef] [PubMed]
- Urenovitch, J.V.; West, R. Pentamethyldisilane and 1,1,2,2-tetramethyldisilane and their addition to olefins. J. Organomet. Chem. 1965, 3, 138–145. [Google Scholar] [CrossRef]
- Sakurai, H.; Kishida, T.; Hosomi, A.; Kumada, M. Decomposition of some free radical initiators in hexamethyldisilane. J. Organomet. Chem. 1967, 8, 65–68. [Google Scholar] [CrossRef]
- Hsiao, Y.-L.; Waymouth, R.M. Free-radical hydrosilylation of poly(phenylsilane): Synthesis of functional polysilanes. J. Am. Chem. Soc. 1994, 116, 9779–9780. [Google Scholar] [CrossRef]
- Ojima, I. The hydrosilylation reaction. In The Chemistry of Organic Silicon Compounds; Patai, S., Rappoport, Z., Eds.; Wiley: Chichester, UK, 1989; pp. 1479–1526. [Google Scholar]
- Ojima, I.; Li, Z.; Zhu, J. Recent advances in the hydrosilylation and related reactions. In The Chemistry of Organic Silicon Compounds; Rappoport, Z., Apeloig, Y., Eds.; Wiley: Chichester, UK, 1998; Volume 2, pp. 1687–1792. [Google Scholar]
- Marciniec, B.; Maciejewski, H.; Pietraszuk, C.; Pawluć, P. Hydrosilylation: A Comprehensive Review on Recent Advances; Marciniec, B., Ed.; Springer: Berlin, Germany, 2009. [Google Scholar]
- Lukevics, E.; Dzintara, M. The alcoholysis of hydrosilanes. J. Organomet. Chem. 1985, 295, 265–315. [Google Scholar] [CrossRef]
- Corey, J.Y. Dehydrogenative coupling reactions of hydrosilanes. In Advances in Silicon Chemistry; Larson, G.L., Ed.; JAI Press: Greenwich, CT, USA, 1991; Volume 1, pp. 327–387. [Google Scholar]
- Gauvin, F.; Harrod, J.F.; Woo, H.G. Catalytic dehydrocoupling: A general strategy for the formation of element–element bonds. Adv. Organomet. Chem. 1998, 42, 363–405. [Google Scholar]
- Reichl, J.A.; Berry, D.H. Recent progress in transition metal-catalyzed reactions of silicon, germanium, and tin. Adv. Organomet. Chem. 1999, 43, 197–265. [Google Scholar]
- Murata, M.; Suzuki, K.; Watanabe, S.; Masuda, Y. Synthesis of arylsilanes via palladium(0)-catalyzed silylation of aryl halides with hydrosilane. J. Org. Chem. 1997, 62, 8569–8571. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, N.; Hartwig, J.F. Intermolecular and intramolecular, platinum-catalyzed, acceptorless dehydrogenative coupling of hydrosilanes with aryl and aliphatic methyl C–H bonds. J. Am. Chem. Soc. 2005, 127, 5022–5023. [Google Scholar] [CrossRef] [PubMed]
- Yamanoi, Y. Palladium-catalyzed silylations of hydrosilanes with aryl halides using bulky alkyl phosphine. J. Org. Chem. 2005, 70, 9607–9609. [Google Scholar] [CrossRef] [PubMed]
- Murata, M.; Yamasaki, H.; Ueta, T.; Nagata, M.; Ishikura, M.; Watanabe, S.; Masuda, Y. Synthesis of aryltriethoxysilanes via rhodium(I)-catalyzed cross-coupling of aryl electrophiles with triethoxysilane. Tetrahedron 2007, 63, 4087–4094. [Google Scholar] [CrossRef]
- Yamanoi, Y.; Nishihara, H. Direct and selective arylation of tertiary silanes with rhodium catalyst. J. Org. Chem. 2008, 73, 6671–6678. [Google Scholar] [CrossRef] [PubMed]
- Horn, K.A. Regio- and stereochemical aspects of the palladium-catalyzed reactions of silanes. Chem. Rev. 1995, 95, 1317–1350. [Google Scholar] [CrossRef]
- Sharma, H.K.; Pannell, K.H. Activation of the Si–Si bond by transition metal complexes. Chem. Rev. 1995, 95, 1351–1374. [Google Scholar] [CrossRef]
- Suginome, M.; Ito, Y. Transition-metal-catalyzed additions of silicon–silicon and silicon–heteroatom bonds to unsaturated organic molecules. Chem. Rev. 2000, 100, 3221–3256. [Google Scholar] [CrossRef] [PubMed]
- Beletskaya, I.; Moberg, C. Element–element additions to unsaturated carbon–carbon bonds catalyzed by transition metal complexes. Chem. Rev. 2006, 106, 2320–2354. [Google Scholar] [CrossRef] [PubMed]
- Lesbani, A.; Kondo, H.; Sato, J.; Yamanoi, Y.; Nishihara, H. Facile synthesis of hypersilylated aromatic compounds by palladium-mediated arylation reaction. Chem. Commun. 2010, 46, 7784–7786. [Google Scholar] [CrossRef] [PubMed]
- Inubushi, H.; Hattori, Y.; Yamanoi, Y.; Nishihara, H. Structures and optical properties of tris(trimethylsilyl)silylated oligothiophene derivatives. J. Org. Chem. 2014, 79, 2974–2979. [Google Scholar] [CrossRef] [PubMed]
- Kanno, K.; Niwayama, Y.; Kyushin, S. Selective catalytic monoreduction of dichlorooligosilanes with Grignard reagents. Tetrahedron Lett. 2013, 54, 6940–6943. [Google Scholar] [CrossRef]
- Kanno, K.; Aikawa, Y.; Kyushin, S. Ruthenium-catalyzed alkoxylation of a hydrodisilane without Si–Si bond cleavage. Tetrahedron Lett. 2017, 58, 9–12. [Google Scholar] [CrossRef]
- Piller, F.M.; Metzger, A.; Schade, M.A.; Haag, B.A.; Gavryushin, A.; Knochel, P. Preparation of polyfunctional arylmagnesium, arylzinc, and benzylic zinc reagents by using magnesium in the presence of LiCl. Chem. Eur. J. 2009, 15, 7192–7202. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.G.; Guveli, T.; Kariuki, B.M.; Snaith, J.S. Synthesis and characterisation of two new binaphthyl trisilanes. J. Organomet. Chem. 2009, 694, 137–141. [Google Scholar] [CrossRef]
- Ahmed, M.A.K.; Wragg, D.S.; Nilsen, O.; Fjellvåg, H. Synthesis and properties of ethyl, propyl, and butyl hexa-alkyldisilanes and tetrakis(tri-alkylsilyl)silanes. Z. Anorg. Allg. Chem. 2014, 640, 2956–2961. [Google Scholar] [CrossRef]
- Hoffmann, F.; Wagler, J.; Roewer, G. Selective synthesis of functional alkynylmono- and -trisilanes. Eur. J. Inorg. Chem. 2010, 1133–1142. [Google Scholar] [CrossRef]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).