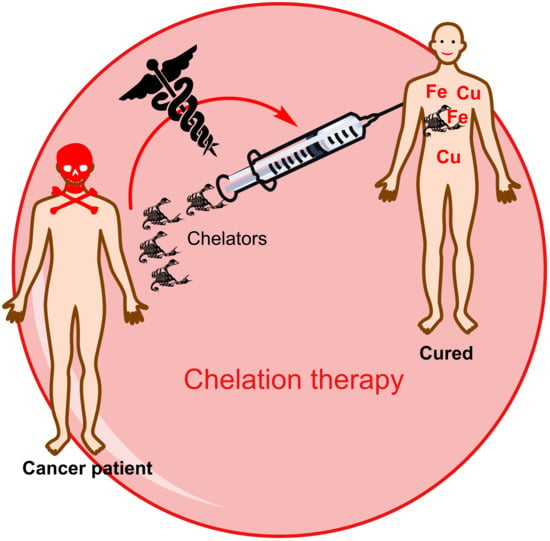
Iron and Copper Intracellular Chelation as an Anticancer Drug Strategy

 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
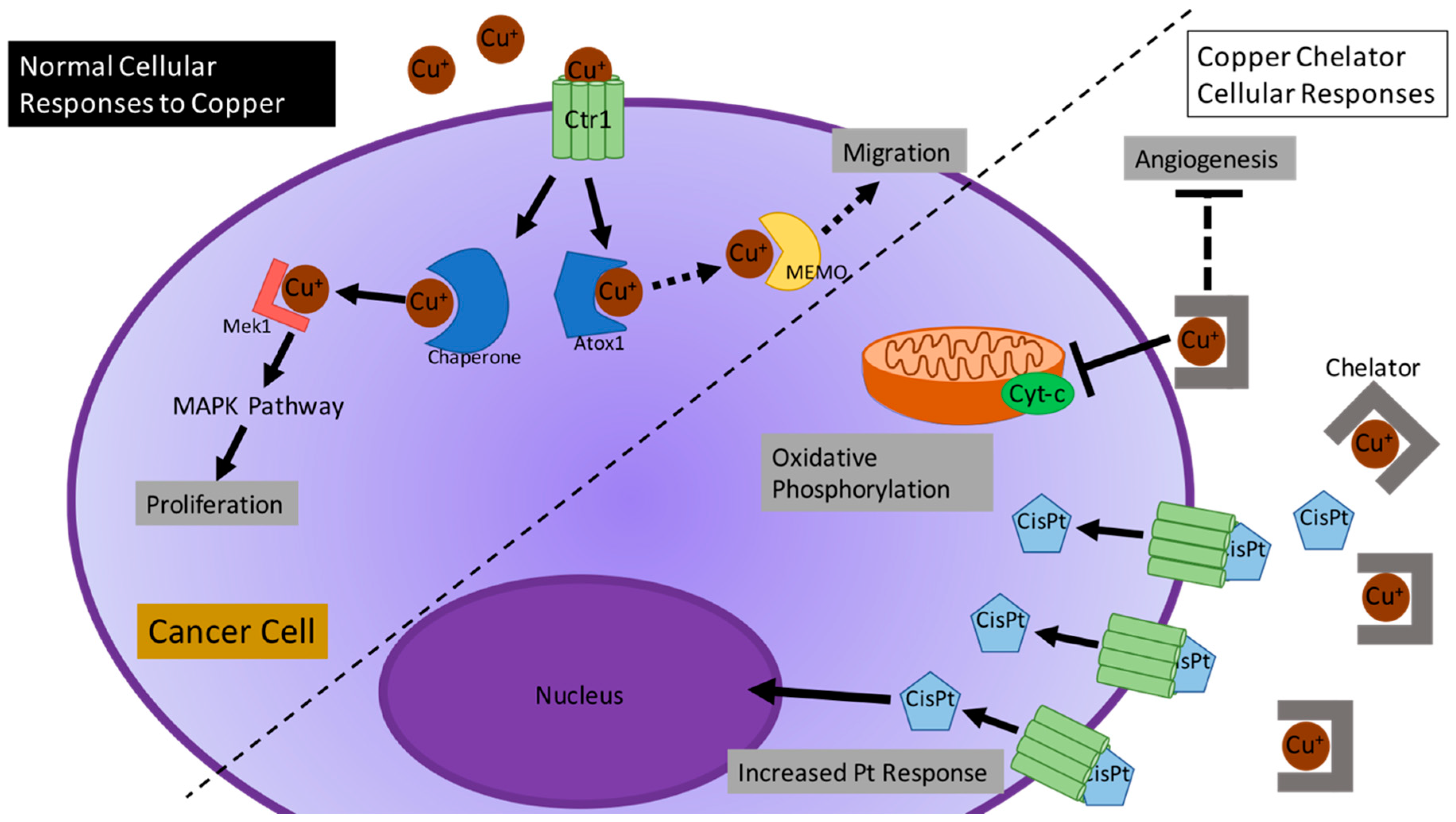
2. The Role of Cu in Cancer
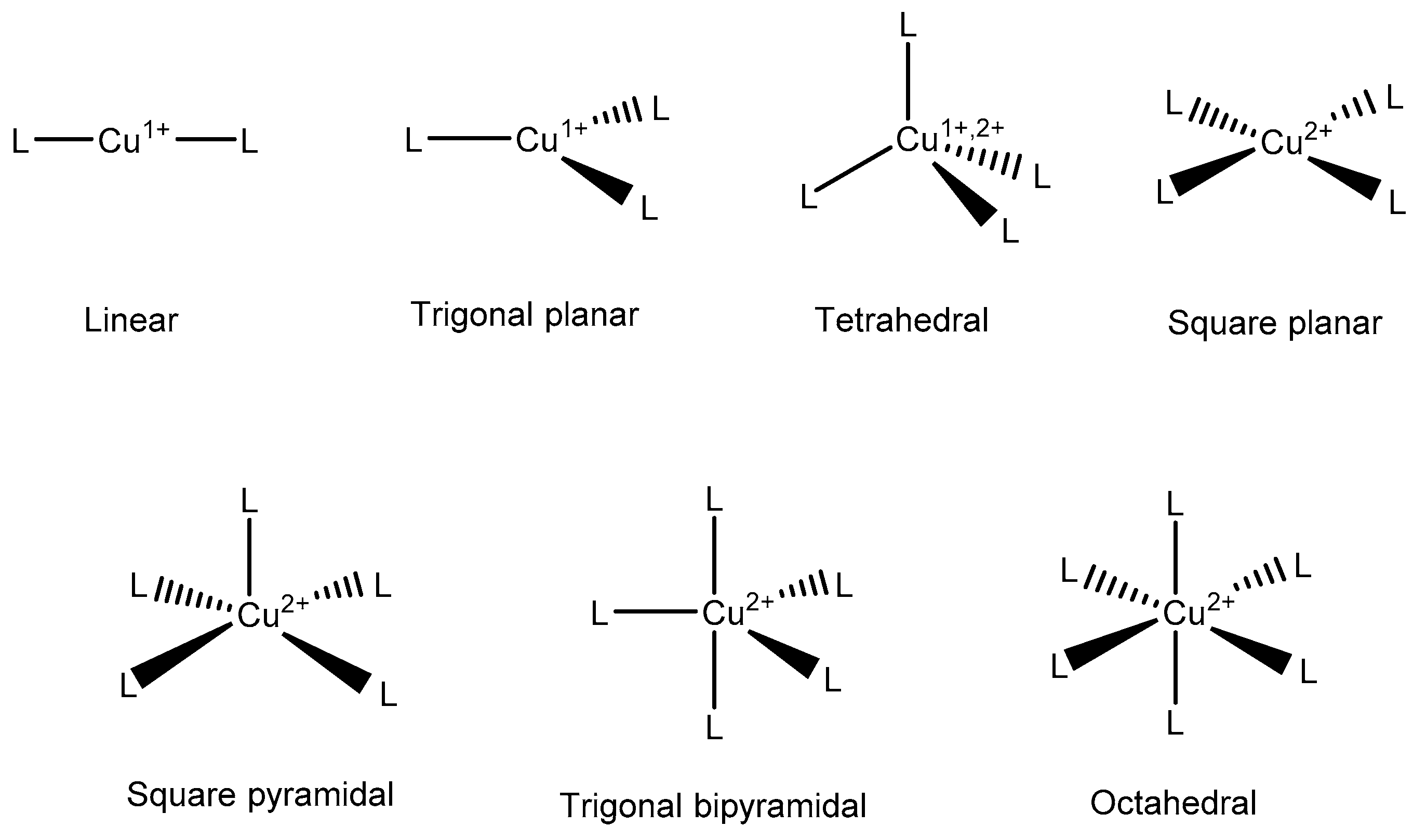
2.1. Cu as an Important Biological Co-Factor
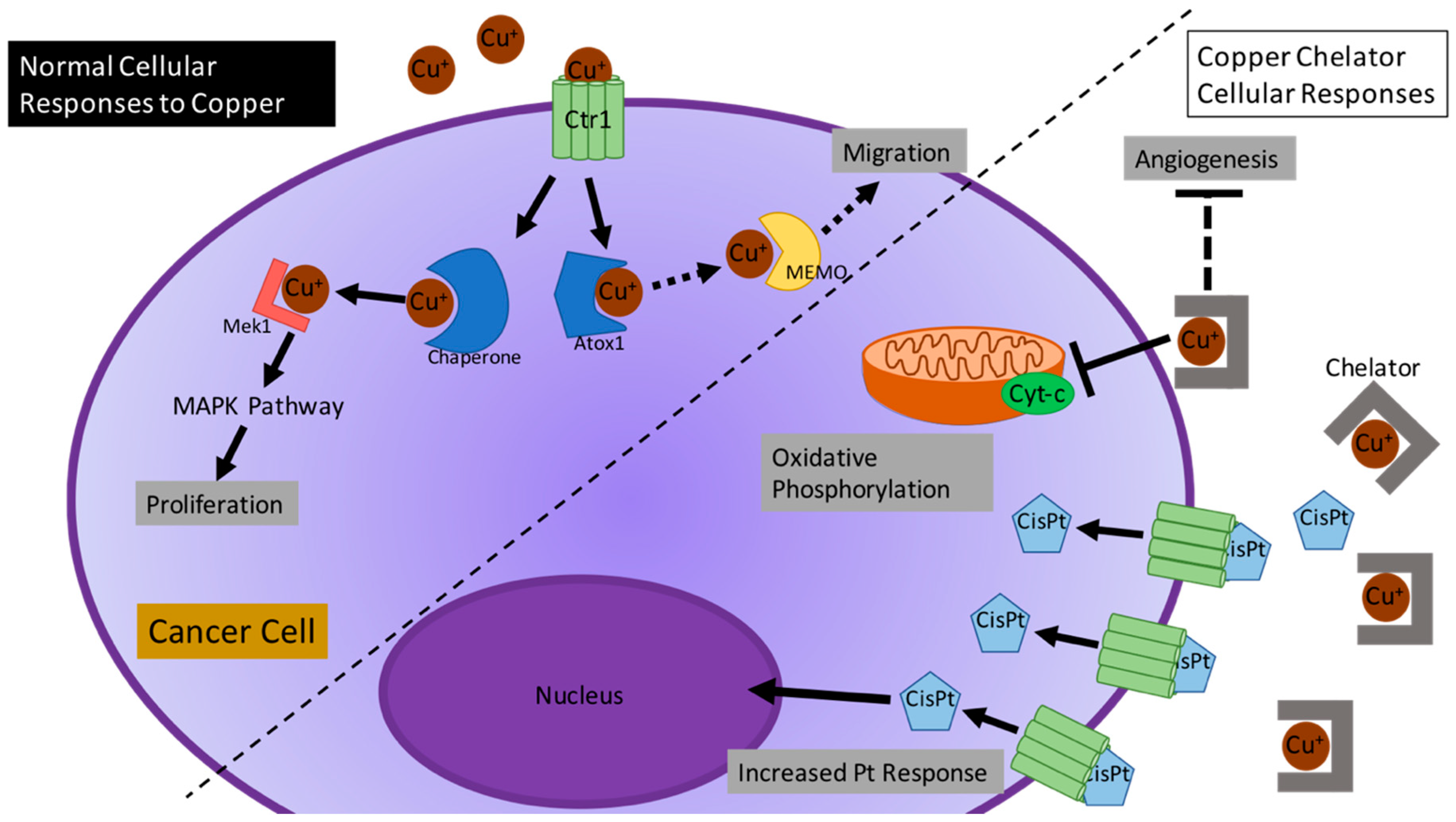
2.2. Current Studies of Cu in Different Cancer-Altered Biological Processes
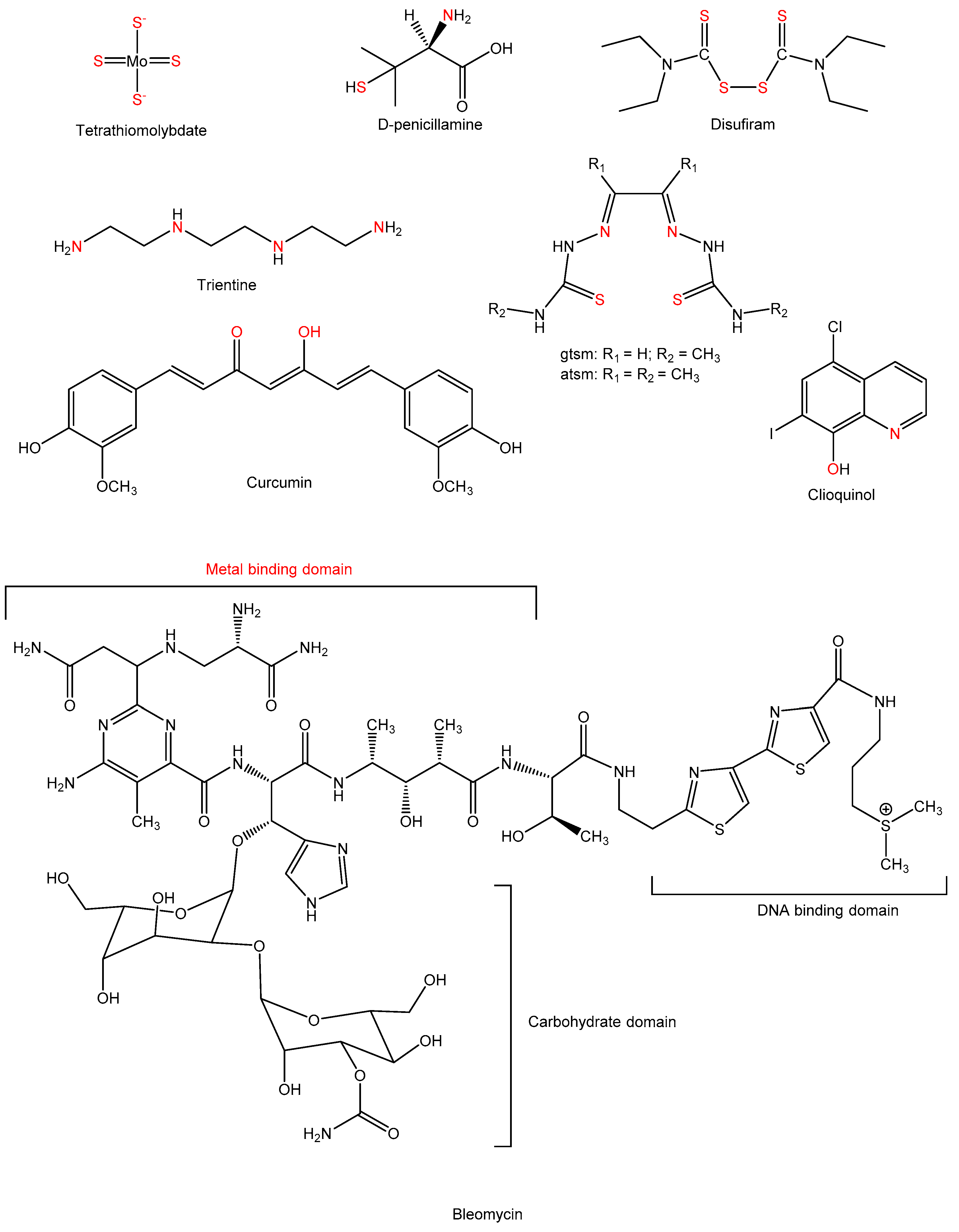
2.3. Chelation Strategies
2.3.1. D-Penicillamine
2.3.2. Tetrathiomolybdate
2.3.3. Trientine
2.3.4. Bleomycin
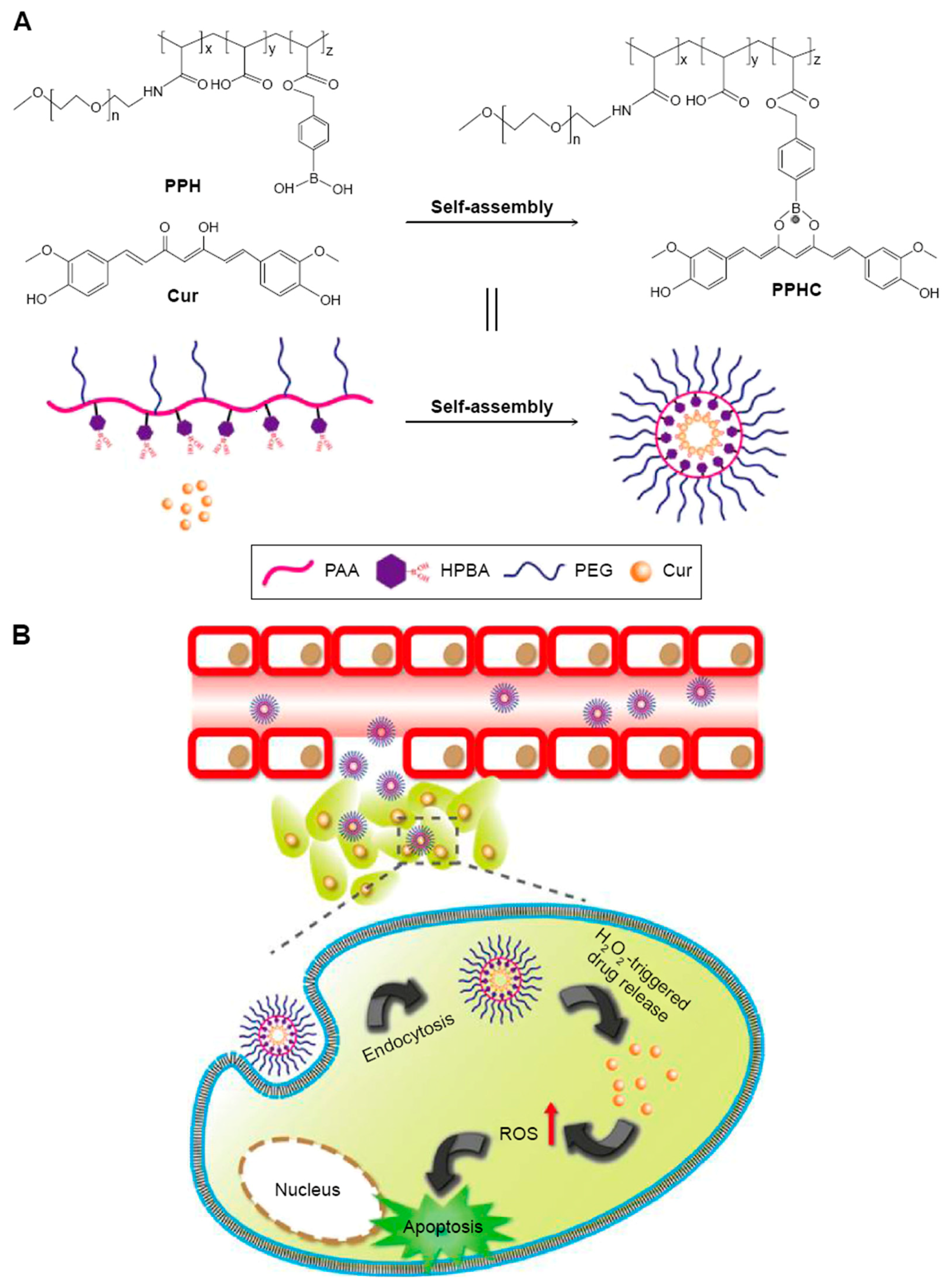
2.3.5. Curcumin
2.3.6. Ionophores
2.4. Efforts to Optimize Drug Delivery and Efficacy of Cu Chelator Agents
3. The Role of Fe in Cancer
3.1. Fe Transport and Regulation
3.2. The Role of Fe in Cancer and Its Progression
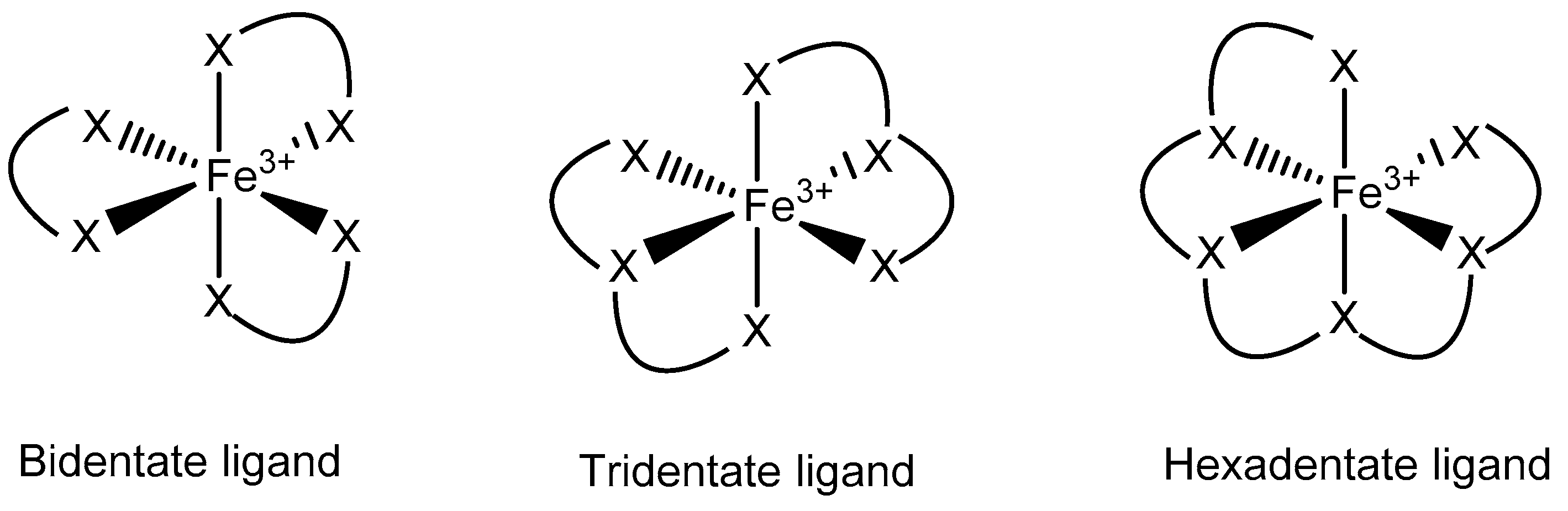
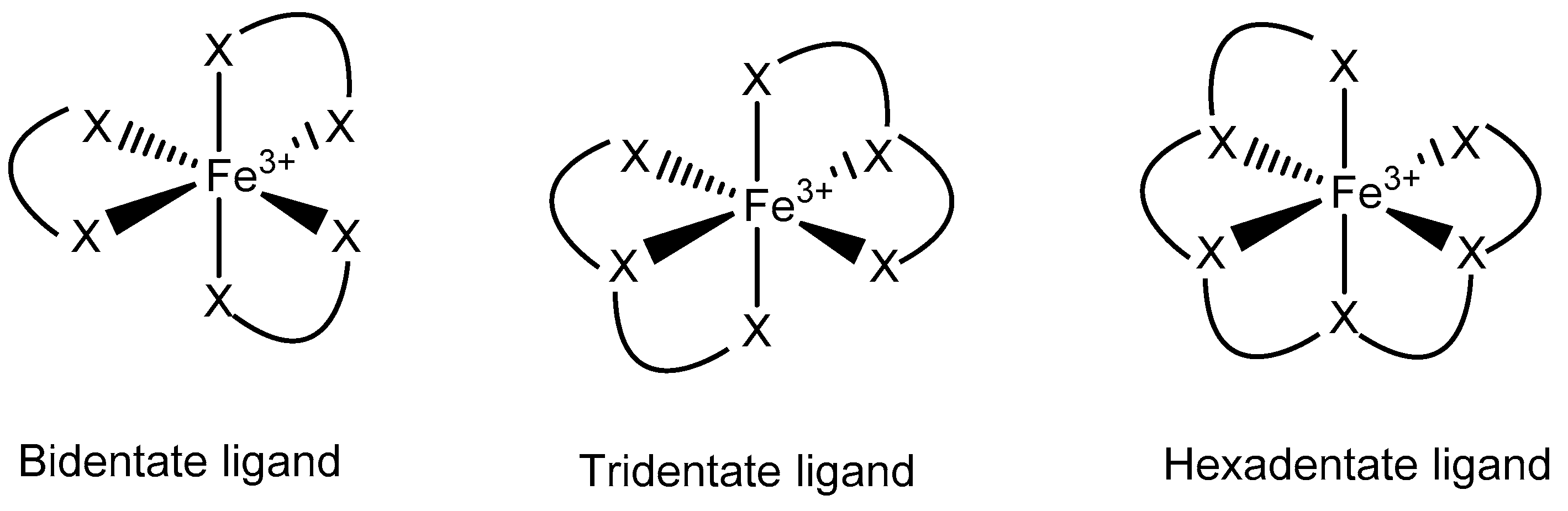
3.3. Fe Chelators in Cancer Treatment
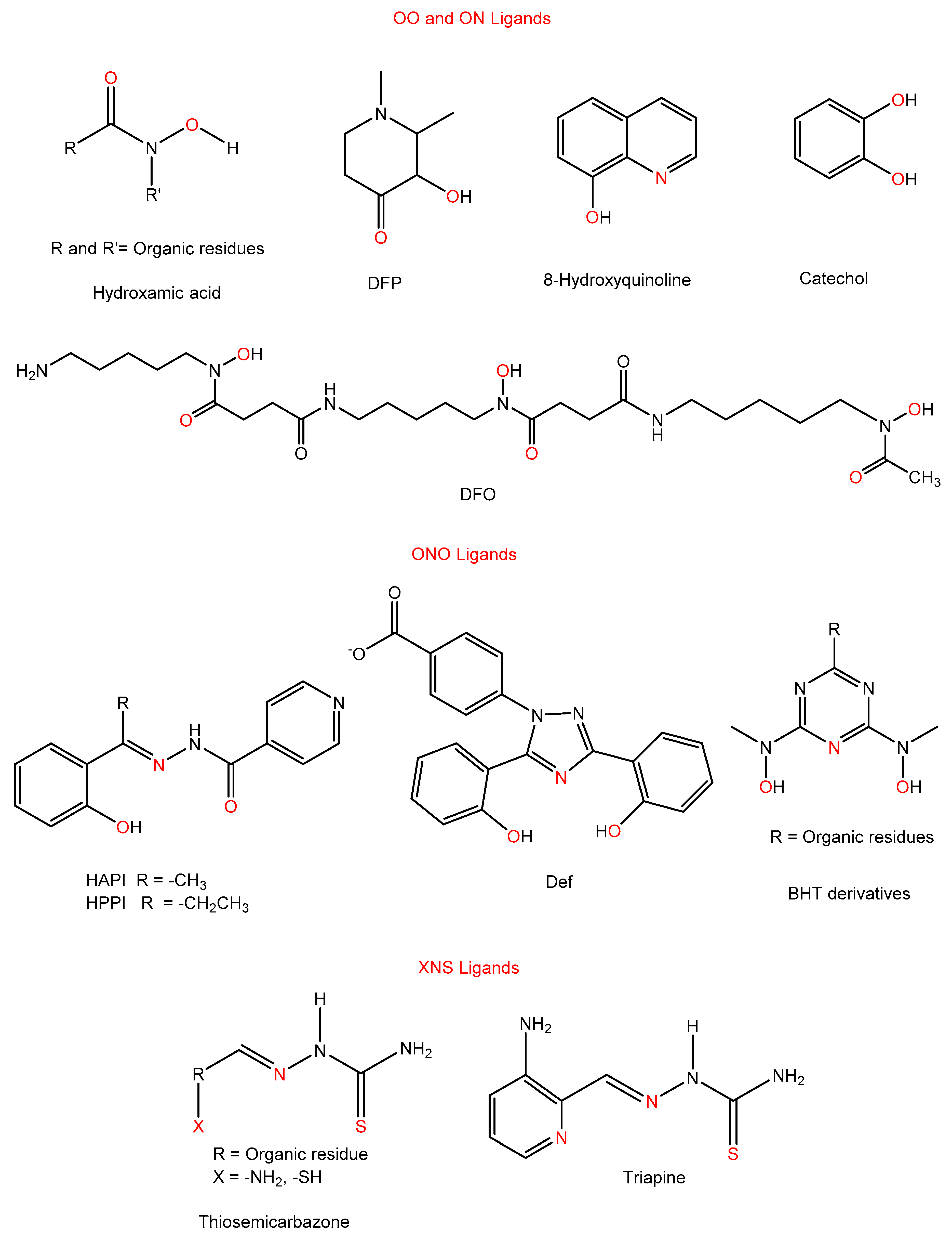
3.3.1. OO and ON Ligands
3.3.2. ONO Ligands
3.3.3. XNS Ligands
3.4. Efforts to Optimize Drug Delivery and Efficacy of Fe Chelator Agents
3.4.1. Nano-Approaches for Delivering Fe Chelators
3.4.2. Liposomes for Delivering Fe Chelators
3.4.3. Prochelation
3.4.4. Synergistic Treatment
4. Transmetalation as a New Anticancer Strategy to Target Cu and Fe Chelation
5. Analytical Tools to Quantify and Track Cu and Fe
5.1. Techniques to Quantify Cu and Fe Levels
5.2. Techniques to Track Cu and Fe in Vitro/ex Vivo and in Vivo
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Centers for Disease Control and Prevention: Cancer Prevention and Control. Available online: https://www.cdc.gov/ (accessed on 1 September 2018).
- Weinberg, R. The Biology of Cancer; Garland Science: New York, NY, USA, 2013. [Google Scholar]
- Roychowdhury, S.; Chinnaiyan, A.M. Translating cancer genomes and transcriptomes for precision oncology. CA Cancer J. Clin. 2016, 66, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.H.; Snyder, M. Omics Profiling in Precision Oncology. Mol. Cell. Proteom. 2016, 15, 2525–2536. [Google Scholar] [CrossRef] [PubMed]
- Mancini, R.J.; Stutts, L.; Ryu, K.A.; Tom, J.K.; Esser-Kahn, A.P. Directing the Immune System with Chemical Compounds. ACS Chem. Biol. 2014, 9, 1075–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, K.; Singha, S.; Clemente-Casares, X.; Tsai, S.; Yang, Y.; Santamaria, P. Nanoparticle-Based Immunotherapy for Cancer. ACS Nano. 2015, 9, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Ma, Y.; Yu, L.; Jiang, J.; Shen, S.; Hou, Y.; Wang, T. Cancer Immunotherapy: A Focus on the Regulation of Immune Checkpoints. Int. J. Mol. Sci. 2018, 19, 1389. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Gutierrez, E.; Kovacevic, Z.; Saletta, F.; Obeidy, P.; Suryo Rahmanto, Y.; Richardson, D.R. Iron Chelators for the Treatment of Cancer. Curr. Med. Chem. 2012, 19, 2689–2702. [Google Scholar] [CrossRef] [PubMed]
- Telleria, C.M. Drug Repurposing for Cancer Therapy. J. Cancer Sci. Ther. 2012, 4, ix–xi. [Google Scholar] [CrossRef] [PubMed]
- Mevada, S.T.; AlDhuli, A.S.; Al-Rawas, A.H.; Al-Khabori, M.K.; Nazir, H.; Zachariah, M.; Wali, Y. Liver Enzymes Changes and Safety Profile of Deferasirox Iron Chelator in Omani Children with Thalassemia Major. Blood 2014, 124, 4903. [Google Scholar]
- Grubman, A.; White, A.R. Copper as a key regulator of cell signalling pathways. Expert Rev. Mol. Med. 2014, 16, E11. [Google Scholar] [CrossRef] [PubMed]
- Blockhuys, S.; Wittung-Stafshede, P. Copper chaperone ATOX1 plays role in breast cancer cell migration. Biochem. Biophys. Res. Commun. 2017, 483, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; New, E. What has fluorescent sensing told us about copper and brain malfunction? Metallomics 2014, 7, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Denoyer, D.; Masaldan, S.; La Fontaine, S.; Cater, M.A. Targeting copper in cancer therapy: ‘Copper That Cancer’. Metallomics 2015, 7, 1459–1476. [Google Scholar] [CrossRef] [PubMed]
- Blockhuys, S.; Wittung-Stafshede, P. Roles of Copper-Binding Proteins in Breast Cancer. Int. J. Mol. Sci. 2017, 18, 871. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, J.; Yang, H.; Wu, C.; Dang, X.; Liu, Y. Copper depletion inhibits CoCl2-induced aggressive phenotype of MCF-7 cells via downregulation of HIF-1 and inhibition of Snail/Twist-mediated epithelial-mesenchymal transition. Sci. Rep. 2015, 5, 12410. [Google Scholar] [CrossRef] [PubMed]
- Shukla, V.; Skuntz, S.; Pant, H.C. Deregulated Cdk5 activity is involved in inducing Alzheimer’s disease. Arch. Med. Res. 2012, 43, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, S.; Endoh, D.; Okui, T.; Hayashi, M. Trientine, a copper-chelating agent, induced apoptosis in murine fibrosarcoma cells by activation of the p38 MAPK pathway. J. Vet. Med. Sci. 2009, 71, 1541–1544. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Luo, C.; Shan, C.; You, Q.; Lu, J.; Elf, S.; Zhou, Y.; Wen, Y.; Vinkenborg, J.L.; Fan, J. Inhibition of human copper trafficking by a small molecule significantly attenuates cancer cell proliferation. Nat. Chem. 2015, 7, 968–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamza, I.; Faisst, A.; Prohaska, J.; Chen, J.; Gruss, P.; Gitlin, J.D. The metallochaperone ATOX1 plays a critical role in perinatal copper homeostasis. Proc. Natl. Acad. Sci. USA 2001, 98, 6848–6852. [Google Scholar] [CrossRef] [PubMed]
- Barresi, V.; Spampinato, G.; Musso, N.; Salinaro, A.T.; Rizzarelli, E.; Condorelli, D.F. ATOX1 gene silencing increases susceptibility to anticancer therapy based on copper ionophores or chelating drugs. J. Inorg. Biochem. 2016, 156, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Ding, P.; Zhang, X.; Jin, S.; Duan, B.; Chu, P.; Zhang, Y.; Chen, Z.-N.; Xia, B.; Song, F. CD147 functions as the signaling receptor for extracellular divalent copper in hepatocellular carcinoma cells. Oncotarget 2017, 8, 51151. [Google Scholar] [CrossRef] [PubMed]
- Ishida, S.; Andreux, P.; Poitry-Yamate, C.; Auwerx, J.; Hanahan, D. Bioavailable copper modulates oxidative phosphorylation and growth of tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 19507–19512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turski, M.L.; Brady, D.C.; Kim, H.J.; Kim, B.-E.; Nose, Y.; Counter, C.M.; Winge, D.R.; Thiele, D.J. A novel role for copper in Ras/mitogen-activated protein kinase signaling. Mol. Cell. Biol. 2012, 32, 1284–1295. [Google Scholar] [CrossRef] [PubMed]
- Brady, D.C.; Crowe, M.S.; Greenberg, D.N.; Counter, C.M. Copper Chelation Inhibits BRAFV600E-driven Melanomagenesis and Counters Resistance to BRAFV600E and MEK1/2 Inhibitors. Cancer Res. 2017, 77, 6240–6252. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Urao, N.; Ashino, T.; Sudhahar, V.; McKinney, R.D.; Hamakubo, T.; Iwanari, H.; Ushio-Fukai, M.; Fukai, T. Novel Role of Copper Transport Protein Antioxidant-1 in Neointimal Formation after Vascular InjurySignificance. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 805–813. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, G.; Nalvarte, I.; Smirnova, T.; Vecchi, M.; Aceto, N.; Doelemeyer, A.; Frei, A.; Lienhard, S.; Wyckoff, J.; Hess, D.; et al. Memo is a copper-dependent redox protein with an essential role in migration and metastasis. Sci. Signal. 2014, 7, ra56. [Google Scholar] [CrossRef] [PubMed]
- Sammons, S.; Brady, D.; Vahdat, L.; Salama, A.K. Copper suppression as cancer therapy: The rationale for copper chelating agents in BRAF V600 mutated melanoma. Melanoma Manag. 2016, 3, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Barresi, V.; Trovato-Salinaro, A.; Spampinato, G.; Musso, N.; Castorina, S.; Rizzarelli, E.; Condorelli, D.F. Transcriptome analysis of copper homeostasis genes reveals coordinated upregulation of SLC31A1, SCO1, and COX11 in colorectal cancer. FEBS Open Bio 2016, 6, 794–806. [Google Scholar] [CrossRef] [PubMed]
- Gupte, A.; Mumper, R.J. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat. Rev. 2009, 35, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.Q.; Lind, S.E. Metal ionophores—An emerging class of anticancer drugs. IUBMB Life 2009, 61, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Denoyer, D.; Pearson, H.B.; Clatworthy, S.A.; Smith, Z.M.; Francis, P.S.; Llanos, R.M.; Volitakis, I.; Phillips, W.A.; Meggyesy, P.M.; Masaldan, S.; et al. Copper as a target for prostate cancer therapeutics: Copper-ionophore pharmacology and altering systemic copper distribution. Oncotarget. 2016, 7, 37064. [Google Scholar] [CrossRef] [PubMed]
- Weekley, C.M.; He, C. Developing drugs targeting transition metal homeostasis. Curr. Opin. Chem. Biol. 2017, 37, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Housecroft, C.E.; Sharpe, A.G. Inorganic Chemistry, 4th ed.; Pearson: Cambridge, UK, 2012. [Google Scholar]
- Walshe, J. Penicillamine, a new oral therapy for Wilson’s disease. Am. J. Med. 1956, 21, 487–495. [Google Scholar] [CrossRef]
- Goldberg, A.; Smith, J.A.; Lochhead, A.C. Treatment of lead-poisoning with oral penicillamine. Br. Med. J. 1963, 1, 1270. [Google Scholar] [CrossRef] [PubMed]
- Walshe, J.M. The story of penicillamine: A difficult birth. Mov. Disord. 2003, 18, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Walshe, J. Disturbances of aminoacid metabolism following liver injury: A study by means of paper chromatography. Q. J. Med. 1953, 22, 483–505. [Google Scholar] [PubMed]
- Weigert, W.M.; Offermanns, H.; Degussa, P.S. D-Penicillamine—Production and Properties. Angew. Chem. Int. Ed. Engl. 1975, 14, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Metodiewa, D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic. Biol. Med. 1999, 27, 322–328. [Google Scholar] [CrossRef]
- Held, K.D.; Biaglow, J.E. Mechanisms for the oxygen radical-mediated toxicity of various thiol-containing compounds in cultured mammalian cells. Radiat. Res. 1994, 139, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J.; Askari, F.K. Wilson’s disease: Clinical management and therapy. J. Hepatol. 2005, 42, S13–S21. [Google Scholar] [CrossRef] [PubMed]
- Khandpur, S.; Jain, N.; Singla, S.; Chatterjee, P.; Behari, M. D-penicillamine induced degenerative dermopathy. Indian J. Dermatol. 2015, 60, 406. [Google Scholar] [CrossRef] [PubMed]
- Levy, R.S.; Fisher, M.; Alter, J.N. Penicillamine: Review and cutaneous manifestations. J. Am. Acad. Dermatol. 1983, 8, 548–558. [Google Scholar] [CrossRef]
- Shiokawa, Y.; Horiuchi, Y.; Honma, M.; Kageyama, T.; Okada, T.; Azuma, T. Clinical evaluation of D-penicillamine by multicentric double-blind comparative study in chronic rheumatoid arthritis. Arthritis Rheumatol. 1977, 20, 1464–1472. [Google Scholar] [CrossRef] [Green Version]
- Brem, S.; Grossman, S.A.; Carson, K.A.; New, P.; Phuphanich, S.; Alavi, J.B.; Mikkelsen, T.; Fisher, J.D. Phase 2 trial of copper depletion and penicillamine as antiangiogenesis therapy of glioblastoma. Neuro Oncol. 2005, 7, 246–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, D.; Ikeda, Y.; Nakazawa, S. Suppression of 9L gliosarcoma growth by copper depletion with copper-deficient diet and D-penicillamine. J. Neurooncol. 1993, 17, 91–97. [Google Scholar] [CrossRef]
- Feli, A.; Jazayeri, S.; Bitaraf, M.A.; Dodaran, M.S.; Parastouei, K.; Hosseinzadeh-Attar, M.J. Combination Therapy with Low Copper Diet, Penicillamine and Gamma Knife Radiosurgery Reduces VEGF and IL-8 In Patients with Recurrent Glioblastoma. Asian Pac. J. Cancer Prev. 2017, 18, 1999. [Google Scholar] [PubMed]
- Matsubara, T.; Saura, R.; Hirohata, K.; Ziff, M. Inhibition of human endothelial cell proliferation in vitro and neovascularization in vivo by D-penicillamine. J. Clin. Investig. 1989, 83, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, S.; Mumper, R.J. D-penicillamine and other low molecular weight thiols: Review of anticancer effects and related mechanisms. Cancer Lett. 2013, 337, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Starkebaum, G.; Root, R. D-Penicillamine: Analysis of the mechanism of copper-catalyzed hydrogen peroxide generation. J. Immunol. 1985, 134, 3371–3378. [Google Scholar] [PubMed]
- Gupte, A.; Mumper, R.J. Copper chelation by D-penicillamine generates reactive oxygen species that are cytotoxic to human leukemia and breast cancer cells. Free Radic. Biol. Med. 2007, 43, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Gupte, A.; Wadhwa, S.; Mumper, R.J. Enhanced Intracellular Delivery of the Reactive Oxygen Species (ROS)-Generating Copper Chelator D-Penicillamine via a Novel Gelatin—D-Penicillamine Conjugate. Bioconjug. Chem. 2008, 19, 1382–1388. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J.; Askari, F.; Lorincz, M.T.; Carlson, M.; Schilsky, M.; Kluin, K.J.; Hedera, P.; Moretti, P.; Fink, J.K.; Tankanow, R.; et al. Treatment of Wilson disease with ammonium tetrathiomolybdate: IV. Comparison of tetrathiomolybdate and trientine in a double-blind study of treatment of the neurologic presentation of Wilson disease. Arch. Neurol. 2006, 63, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J.; Johnson, V.; Dick, R.D.; Kluin, K.J.; Fink, J.K.; Brunberg, J.A. Treatment of Wilson disease with ammonium tetrathiomolybdate: II. Initial therapy in 33 neurologically affected patients and follow-up with zinc therapy. Arch. Neurol. 1996, 53, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J.; Dick, R.D.; Johnson, V.; Wang, Y.; Yuzbasiyan-Gurkan, V.; Kluin, K.; Fink, J.K.; Aisen, A. Treatment of Wilson’s disease with ammonium tetrathiomolybdate: I. Initial therapy in 17 neurologically affected patients. Arch. Neurol. 1994, 51, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J.; Dick, R.; Ullenbruch, M.R.; Jin, H.; Phan, S.H. Inhibition of key cytokines by tetrathiomolybdate in the bleomycin model of pulmonary fibrosis. J. Inorg. Biochem. 2004, 98, 2160–2167. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J. Copper lowering therapy with tetrathiomolybdate as an antiangiogenic strategy in cancer. Curr. Cancer Drug Targets 2005, 5, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Kleer, C.G.; Van Golen, K.L.; Irani, J.; Bottema, K.M.; Bias, C.; De Carvalho, M.; Mesri, E.A.; Robins, D.M.; Dick, R.D. Copper deficiency induced by tetrathiomolybdate suppresses tumor growth and angiogenesis. Cancer Res. 2002, 62, 4854–4859. [Google Scholar] [PubMed]
- Brewer, G.J.; Dick, R.D.; Grover, D.K.; LeClaire, V.; Tseng, M.; Wicha, M.; Pienta, K.; Redman, B.G.; Jahan, T.; Sondak, V.K.; et al. Treatment of metastatic cancer with tetrathiomolybdate, an anticopper, antiangiogenic agent: Phase I study. Clin. Cancer Res. 2000, 6, 1–10. [Google Scholar] [PubMed]
- Redman, B.G.; Esper, P.; Pan, Q.; Dunn, R.L.; Hussain, H.K.; Chenevert, T.; Brewer, G.J.; Merajver, S.D. Phase II trial of tetrathiomolybdate in patients with advanced kidney cancer. Clin. Cancer Res. 2003, 9, 1666–1672. [Google Scholar] [PubMed]
- Pass, H.I.; Brewer, G.J.; Dick, R.; Carbone, M.; Merajver, S. A phase II trial of tetrathiomolybdate after surgery for malignant mesothelioma: Final results. Ann. Thorac. Surg. 2008, 86, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Abelman, S.; Yano, N.; Ribeiro, J.R.; Singh, R.K.; Tipping, M.; Moore, R.G. Tetrathiomolybdate inhibits mitochondrial complex IV and mediates degradation of hypoxia-inducible factor-1α in cancer cells. Sci. Rep. 2015, 5, 14296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, M.; Nishiya, H.; Chiba, T.; Endoh, D.; Kon, Y.; Okui, T. Trientine, a copper-chelating agent, induced apoptosis in murine fibrosarcoma cells in vivo and in vitro. J. Vet. Med. Sci. 2007, 69, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, M.; Nakajima, T.; Kimura, H.; Watanabe, T.; Takashima, H.; Mitsumoto, Y.; Katagishi, T.; Okanoue, T.; Kagawa, K. The copper chelator trientine has an antiangiogenic effect against hepatocellular carcinoma, possibly through inhibition of interleukin-8 production. Int. J. Cancer 2002, 102, 445–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshii, J.; Yoshiji, H.; Kuriyama, S.; Ikenaka, Y.; Noguchi, R.; Okuda, H.; Tsujinoue, H.; Nakatani, T.; Kishida, H.; Nakae, D. The copper-chelating agent, trientine, suppresses tumor development and angiogenesis in the murine hepatocellular carcinoma cells. Int. J. Cancer 2001, 94, 768–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, J.-M.; Sun, L.-B.; Zheng, J.-S.; Wang, X.-X.; Chen, D.-X.; Li, N. Copper chelation by trientine dihydrochloride inhibits liver RFA-induced inflammatory responses in vivo. Inflamm. Res. 2016, 65, 1009–1020. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. EASL clinical practice guidelines: Wilson’s disease. J. Hepatol. 2012, 56, 671–685. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Codd, R. Copper (II)-based metal affinity chromatography for the isolation of the anticancer agent bleomycin from Streptomyces verticillus culture. J. Inorg. Biochem. 2012, 115, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Ehrenfeld, G.M.; Shipley, J.B.; Heimbrook, D.C.; Sugiyama, H.; Long, E.C.; Van Boom, J.H.; Van der Marel, G.A.; Oppenheimer, N.J.; Hecht, S.M. Copper-dependent cleavage of DNA by bleomycin. Biochemistry 1987, 26, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Solaiman, D.; Rao, E.A.; Antholine, W.; Petering, D.H. Properties of the binding of copper by bleomycin. J. Inorg. Biochem. 1980, 12, 201–220. [Google Scholar] [CrossRef]
- Matsui, H.; Kato, T.; Yamamoto, C.; Takita, T.; Takeuchi, T.; Umezawa, H.; Nagatsu, T. Inhibition of dopamine-β-hydroxylase, a copper enzyme, by bleomycin. J. Antibiot. 1980, 33, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, Y.; Ishizu, K.; Miyoshi, K. Studies of metallobleomycins by electronic spectroscopy, electron spin resonance spectroscopy, and potentiometric titration. J. Antibiot. 1979, 32, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, M.; Kumagai, T.; Hayashida, M.; Maruyama, M.; Matoba, Y. The 1.6-Å crystal structure of the copper (II)-bound bleomycin complexed with the bleomycin-binding protein from bleomycin-producing Streptomyces verticillus. J. Biol. Chem. 2002, 277, 2311–2320. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, H.; Hadi, S. Strand scission in DNA induced by curcumin in the presence of Cu(II). Cancer Lett. 1998, 124, 23–30. [Google Scholar] [CrossRef]
- Nair, J.; Strand, S.; Frank, N.; Knauft, J.; Wesch, H.; Galle, P.R.; Bartsch, H. Apoptosis and age-dependant induction of nuclear and mitochondrial etheno-DNA adducts in Long-Evans Cinnamon (LEC) rats: Enhanced DNA damage by dietary curcumin upon copper accumulation. Carcinogenesis 2005, 26, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, M.; Haneda, M.; Naruse, M.; Htay, H.H.; Tsubouchi, R.; Qiao, S.L.; Li, W.H.; Murakami, K.; Yokochi, T. Prooxidant activity of curcumin: Copper-dependent formation of 8-hydroxy-2′-deoxyguanosine in DNA and induction of apoptotic cell death. Toxicol. In Vitro 2004, 18, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Urbina-Cano, P.; Bobadilla-Morales, L.; Ramírez-Herrera, M.A.; Corona-Rivera, J.R.; Mendoza-Magaña, M.L.; Troyo-Sanromán, R.; Corona-Rivera, A. DNA damage in mouse lymphocytes exposed to curcumin and copper. J. Appl. Genet. 2006, 47, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Palanivelu, K. The effect of curcumin (turmeric) on Alzheimer’s disease: An overview. Ann. Indian Acad. Neurol. 2008, 11, 13. [Google Scholar] [CrossRef] [PubMed]
- Bharti, A.C.; Donato, N.; Singh, S.; Aggarwal, B.B. Curcumin (diferuloylmethane) down-regulates the constitutive activation of nuclear factor-κB and IκBα kinase in human multiple myeloma cells, leading to suppression of proliferation and induction of apoptosis. Blood 2003, 101, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Lou, J.R.; Zhang, X.-X.; Zheng, J.; Ding, W.-Q. Transient metals enhance cytotoxicity of curcumin: Potential involvement of the NF-κB and mTOR signaling pathways. Anticancer Res. 2010, 30, 3249–3255. [Google Scholar] [PubMed]
- Zhang, W.; Chen, C.; Shi, H.; Yang, M.; Liu, Y.; Ji, P.; Chen, H.; Tan, R.X.; Li, E. Curcumin is a biologically active copper chelator with antitumor activity. Phytomedicine 2016, 23, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zhou, Y.; Lind, S.E.; Ding, W.-Q. Clioquinol targets zinc to lysosomes in human cancer cells. Biochem. J. 2009, 417, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.-Q.; Liu, B.; Vaught, J.L.; Yamauchi, H.; Lind, S.E. Anticancer activity of the antibiotic clioquinol. Cancer Res. 2005, 65, 3389–3395. [Google Scholar] [CrossRef] [PubMed]
- Lind, S.E.; Park, J.S.; Drexler, J.W. Pyrithione and 8-hydroxyquinolines transport lead across erythrocyte membranes. Trans. Res. 2009, 154, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Prachayasittikul, V.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. 8-Hydroxyquinolines: A review of their metal chelating properties and medicinal applications. Drug Des. Dev. Ther. 2013, 7, 1157. [Google Scholar] [CrossRef] [PubMed]
- Palanimuthu, D.; Shinde, S.V.; Somasundaram, K.; Samuelson, A.G. In vitro and in vivo anticancer activity of copper bis(thiosemicarbazone) complexes. J. Med. Chem. 2013, 56, 722–734. [Google Scholar] [CrossRef] [PubMed]
- West, D.X.; Liberta, A.E.; Padhye, S.B.; Chikate, R.C.; Sonawane, P.B.; Kumbhar, A.S.; Yerande, R.G. Thiosemicarbazone complexes of copper(II): Structural and biological studies. Coord. Chem. Rev. 1993, 123, 49–71. [Google Scholar] [CrossRef]
- Paterson, B.M.; Donnelly, P.S. Copper complexes of bis(thiosemicarbazones): From chemotherapeutics to diagnostic and therapeutic radiopharmaceuticals. Chem. Soc. Rev. 2011, 40, 3005–3018. [Google Scholar] [CrossRef] [PubMed]
- Cater, M.A.; Pearson, H.B.; Wolyniec, K.; Klaver, P.; Bilandzic, M.; Paterson, B.M.; Bush, A.I.; Humbert, P.O.; La Fontaine, S.; Donnelly, P.S.; et al. Increasing intracellular bioavailable copper selectively targets prostate cancer cells. ACS Chem. Biol. 2013, 8, 1621–1631. [Google Scholar] [CrossRef] [PubMed]
- Park, K.C.; Fouani, L.; Jansson, P.J.; Wooi, D.; Sahni, S.; Lane, D.J.; Palanimuthu, D.; Lok, H.C.; Kovačević, Z.; Huang, M.L.; et al. Copper and conquer: Copper complexes of di-2-pyridylketone thiosemicarbazones as novel anti-cancer therapeutics. Metallomics 2016, 8, 874–886. [Google Scholar] [CrossRef] [PubMed]
- Ruschak, A.M.; Slassi, M.; Kay, L.E.; Schimmer, A.D. Novel proteasome inhibitors to overcome bortezomib resistance. J. Natl. Cancer Inst. 2011, 103, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhai, S.; Liu, X.; Li, L.; Wu, S.; Dou, Q.P.; Yan, B. A novel dithiocarbamate analogue with potentially decreased ALDH inhibition has copper-dependent proteasome-inhibitory and apoptosis-inducing activity in human breast cancer cells. Cancer Lett. 2011, 300, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helsel, M.E.; Franz, K.J. Pharmacological activity of metal binding agents that alter copper bioavailability. Dalton Trans. 2015, 44, 8760–8770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caragounis, A.; Du, T.; Filiz, G.; Laughton, K.M.; Volitakis, I.; Sharples, R.A.; Cherny, R.A.; Masters, C.L.; Drew, S.C.; Hill, A.F.; et al. Differential modulation of Alzheimer’s disease amyloid β-peptide accumulation by diverse classes of metal ligands. Biochem. J. 2007, 407, 435–450. [Google Scholar] [CrossRef] [PubMed]
- White, A.R.; Du, T.; Laughton, K.M.; Volitakis, I.; Sharples, R.A.; Xilinas, M.E.; Hoke, D.E.; Holsinger, R.D.; Evin, G.; Cherny, R.A.; et al. Degradation of the Alzheimer disease amyloid β-peptide by metal-dependent up-regulation of metalloprotease activity. J. Biol. Chem. 2006, 281, 17670–17680. [Google Scholar] [CrossRef] [PubMed]
- Allensworth, J.L.; Evans, M.K.; Bertucci, F.; Aldrich, A.J.; Festa, R.A.; Finetti, P.; Ueno, N.T.; Safi, R.; McDonnell, D.P.; Thiele, D.J. Disulfiram (DSF) acts as a copper ionophore to induce copper-dependent oxidative stress and mediate anti-tumor efficacy in inflammatory breast cancer. Mol. Oncol. 2015, 9, 1155–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safi, R.; Nelson, E.R.; Chitneni, S.K.; Franz, K.J.; George, D.J.; Zalutsky, M.R.; McDonnell, D.P. Copper signaling axis as a target for prostate cancer therapeutics. Cancer Res. 2014, 74, 5819–5831. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, R.; Madanmohan, S.; Kesavan, A.; Baskar, G.; Krishnamoorthy, Y.R.; Santosham, R.; Ponraju, D.; Rayala, S.K.; Venkatraman, G. Nanomedicine: Towards development of patient-friendly drug-delivery systems for oncological applications. Int. J. Nanomed. 2012, 7, 1043–1060. [Google Scholar]
- Dabrowiak, J.C. Metals in Medicine, 2nd ed.; Wiley: Hoboken, NJ, USA, 2017. [Google Scholar]
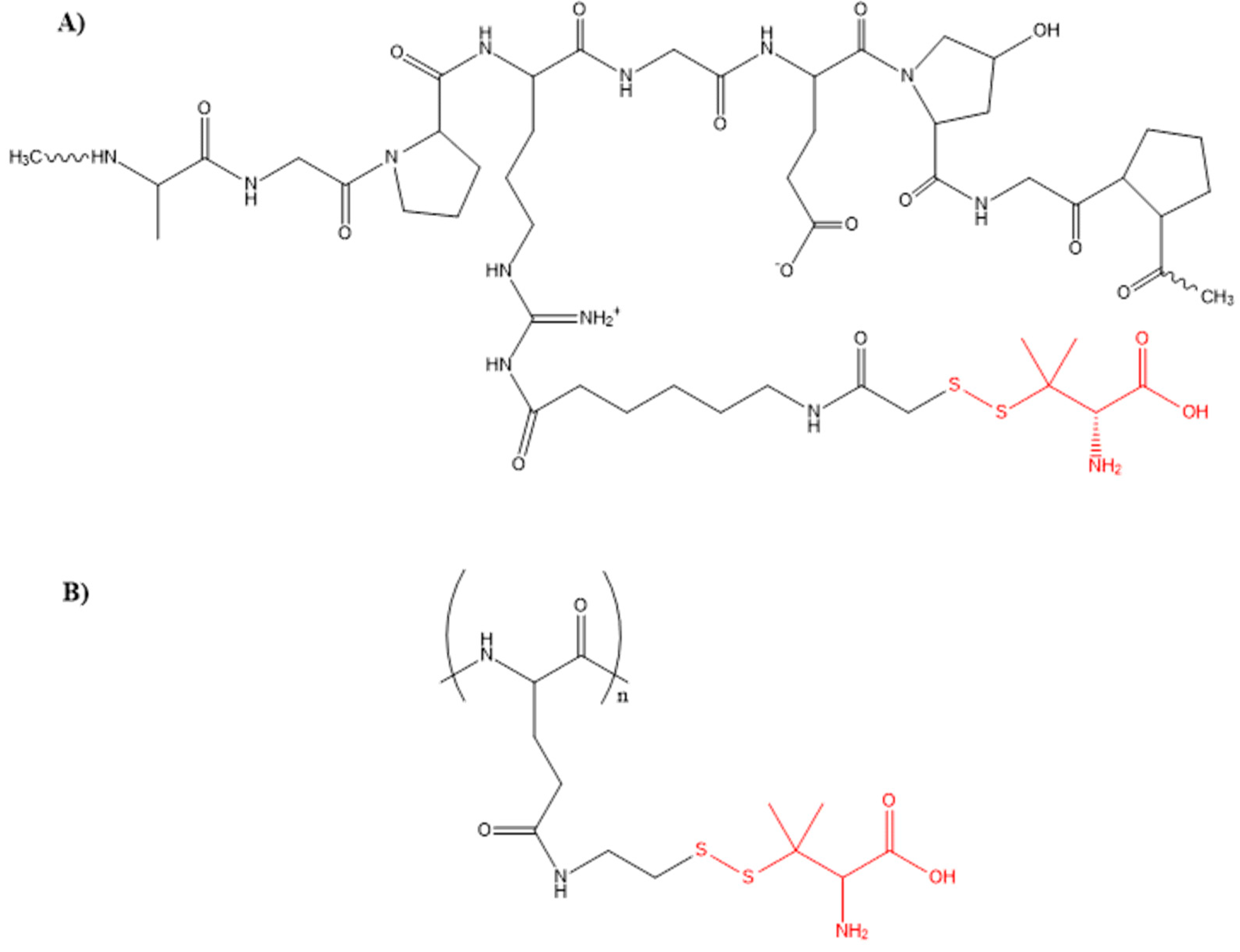
- Wadhwa, S.; Mumper, R.J. Intracellular delivery of the reactive oxygen species generating agent D-penicillamine upon conjugation to poly-l-glutamic acid. Mol. Pharm. 2010, 7, 854–862. [Google Scholar] [CrossRef] [PubMed]
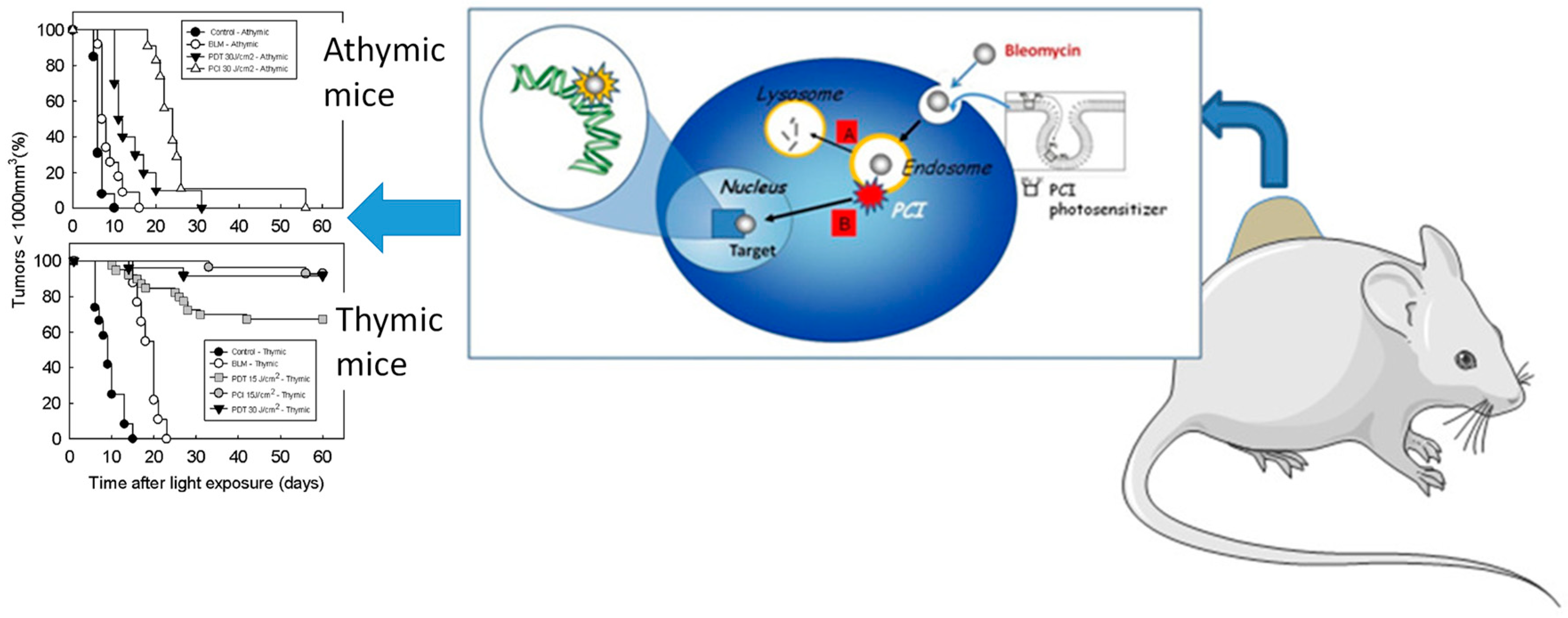
- Norum, O.-J.; Fremstedal, A.S.V.; Weyergang, A.; Golab, J.; Berg, K. Photochemical delivery of bleomycin induces T-cell activation of importance for curative effect and systemic anti-tumor immunity. J. Control. Release 2017, 268, 120–127. [Google Scholar] [CrossRef] [PubMed]
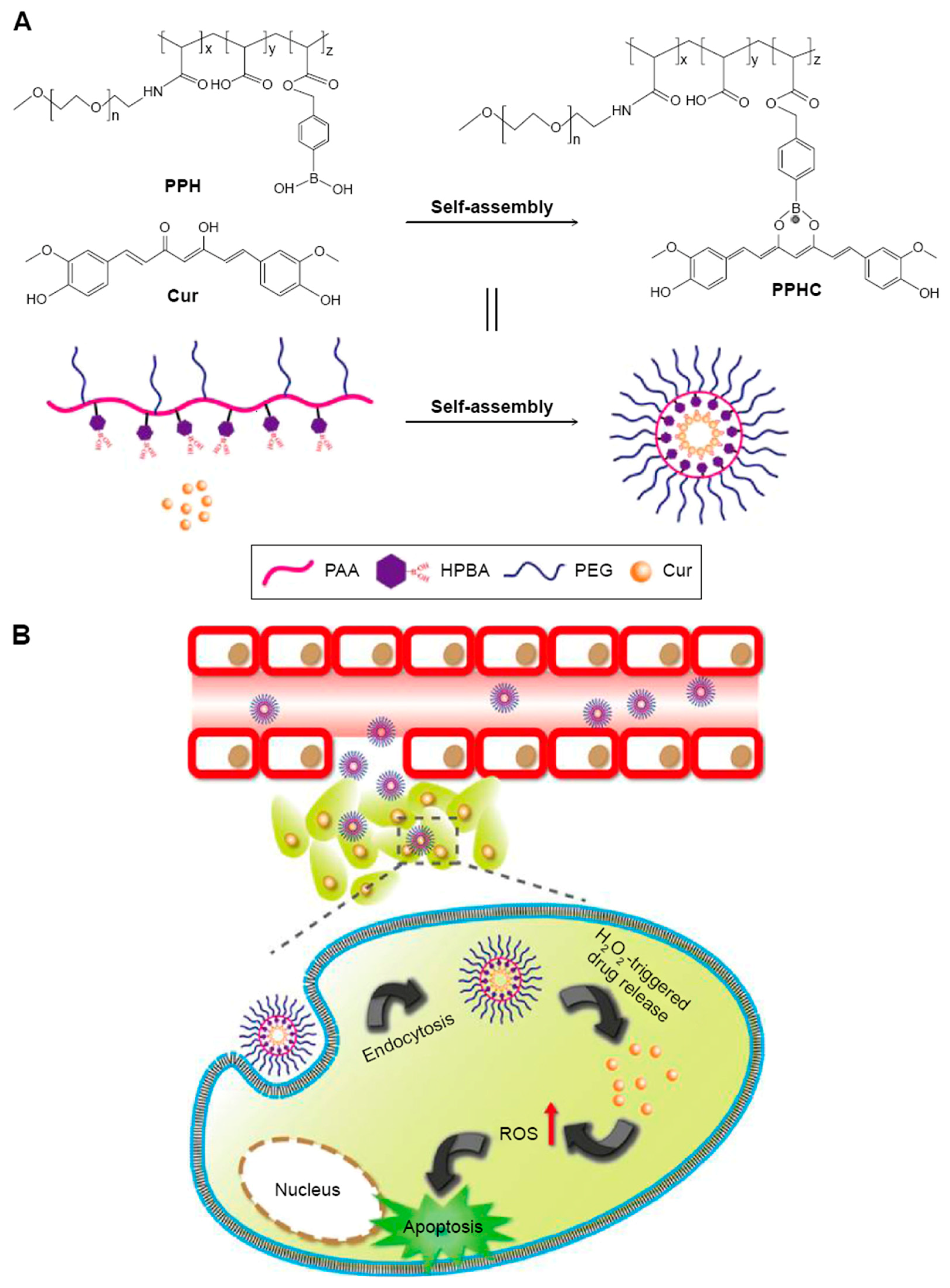
- Luo, C.-Q.; Xing, L.; Cui, P.-F.; Qiao, J.-B.; He, Y.-J.; Chen, B.-A.; Jin, L.; Jiang, H.-L. Curcumin-coordinated nanoparticles with improved stability for reactive oxygen species-responsive drug delivery in lung cancer therapy. Int. J. Nanomed. 2017, 12, 855. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, Y.; Zheng, S.; Weng, Z.; Ma, J.; Li, Y.; Xie, X.; Zheng, W. Detention of copper by sulfur nanoparticles inhibits the proliferation of A375 malignant melanoma and MCF-7 breast cancer cells. Biochem. Biophys. Res. Commun. 2016, 477, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Ishida, S.; McCormick, F.; Smith-McCune, K.; Hanahan, D. Enhancing tumor-specific uptake of the anticancer drug cisplatin with a copper chelator. Cancer Cell 2010, 17, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Lambert, I.; Sørensen, B. Facilitating the Cellular Accumulation of Pt-Based Chemotherapeutic Drugs. Int. J. Mol. Sci. 2018, 19, 2249. [Google Scholar] [CrossRef] [PubMed]
- Voss, F.K.; Ullrich, F.; Münch, J.; Lazarow, K.; Lutter, D.; Mah, N.; Andrade-Navarro, M.A.; von Kries, J.P.; Stauber, T.; Jentsch, T.J. Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science 2014, 344, 634–638. [Google Scholar] [CrossRef] [PubMed]
- Gradogna, A.; Gavazzo, P.; Boccaccio, A.; Pusch, M. Subunit-dependent oxidative stress sensitivity of LRRC8 volume-regulated anion channels. J. Physiol. 2017, 595, 6719–6733. [Google Scholar] [CrossRef] [PubMed]
- Jentsch, T.J.; Lutter, D.; Planells-Cases, R.; Ullrich, F.; Voss, F.K. VRAC: Molecular identification as LRRC8 heteromers with differential functions. Pflug. Arch. Eur. J. Phys. 2016, 468, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Cairo, G.; Bernuzzi, F.; Recalcati, S. A precious metal: Iron, an essential nutrient for all cells. Genes Nutr. 2006, 1, 25–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
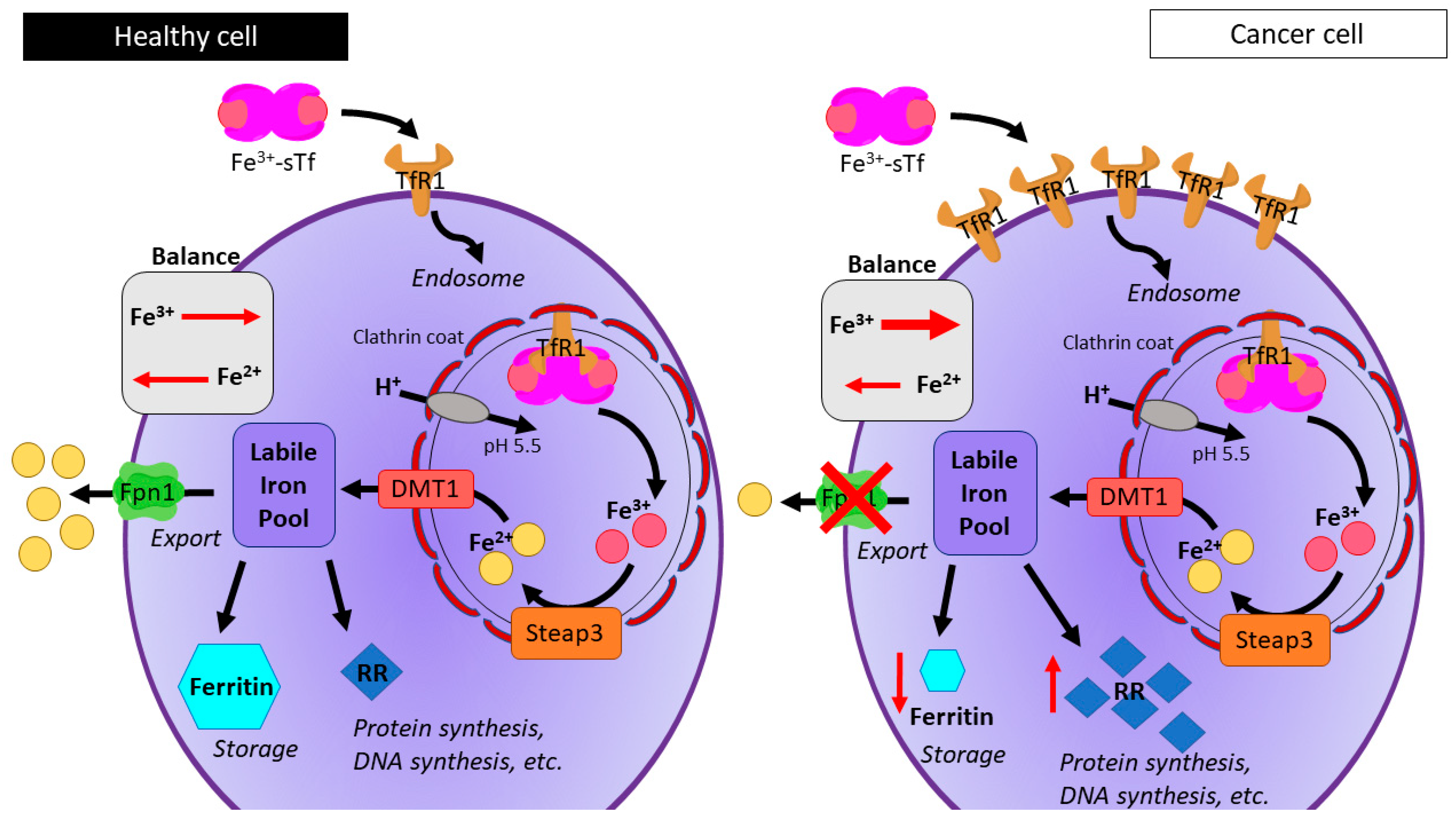
- Lane, D.J.R.; Merlot, A.M.; Huang, M.L.H.; Bae, D.H.; Jansson, P.J.; Sahni, S.; Kalinowski, D.S.; Richardson, D.R. Cellular iron uptake, trafficking and metabolism: Key molecules and mechanisms and their roles in disease. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 1130–1144. [Google Scholar] [CrossRef] [PubMed]
- Bertini, I.; Gray, H.B.; Stiefel, E.I.; Valentine, J.S. Biological Inorganic Chemistry: Structure and Reactivity; University Science Books: Mill Valley, CA, USA, 2007. [Google Scholar]
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Rev. Cancer 2013, 13, 342. [Google Scholar] [CrossRef] [PubMed]
- Huang, X. Iron overload and its association with cancer risk in humans: Evidence for iron as a carcinogenic metal. Mutat. Res. Fund. Mol. Mech. Mutagen. 2003, 533, 153–171. [Google Scholar] [CrossRef]
- Vashchenko, G.; MacGillivray, R. Multi-copper oxidases and human iron metabolism. Nutrients 2013, 5, 2289–2313. [Google Scholar] [CrossRef] [PubMed]
- Askwith, C.; Kaplan, J. Iron and copper transport in yeast and its relevance to human disease. Trends Biochem. Sci. 1998, 23, 135–138. [Google Scholar] [CrossRef]
- Brissot, P.; Ropert, M.; Le Lan, C.; Loréal, O. Non-transferrin bound iron: A key role in iron overload and iron toxicity. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Batey, R.; Fong, P.L.C.; Shamir, S.; Sherlock, S. A non-transferrin-bound serum iron in idiopathic hemochromatosis. Dig. Dis. Sci. 1980, 25, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.; Abeysinghe, R.; Marshall, L.; Hider, R.; Singh, S. Kinetics of removal and reappearance of non-transferrin-bound plasma iron with deferoxamine therapy. Blood 1996, 88, 705–713. [Google Scholar] [PubMed]
- Luria-Pérez, R.; Helguera, G.; Rodríguez, J.A. Antibody-mediated targeting of the transferrin receptor in cancer cells. Bol. Med. Hosp. Infant. Mex. 2016, 73, 372–379. [Google Scholar] [PubMed]
- Eckenroth, B.E.; Steere, A.N.; Chasteen, N.D.; Everse, S.J.; Mason, A.B. How the binding of human transferrin primes the transferrin receptor potentiating iron release at endosomal pH. Proc. Natl. Acad. Sci. USA 2011, 108, 13089–13094. [Google Scholar] [CrossRef] [PubMed]
- Steere, A.N.; Byrne, S.L.; Chasteen, N.D.; Mason, A.B. Kinetics of iron release from transferrin bound to the transferrin receptor at endosomal pH. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 326–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraiter, D.C.; Zak, O.; Aisen, P.; Crumbliss, A.L. A determination of the reduction potentials for diferric and C- and N-lobe monoferric transferrins at endosomal pH (5.8). Inorg. Chem. 1998, 37, 964–968. [Google Scholar] [CrossRef]
- Dhungana, S.; Taboy, C.H.; Zak, O.; Larvie, M.; Crumbliss, A.L.; Aisen, P. Redox properties of human transferrin bound to its receptor. Biochemistry 2004, 43, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Bou-Abdallah, F. Does iron release from transferrin involve a reductive process? Bioenergetics 2012, 1, e1111. [Google Scholar]
- Clardy, S.L.; Connor, J.R.; Beard, J. Restless Legs Syndrome; Elsevier Inc.: Amsterdam, The Netherlands, 2009; pp. 50–60. [Google Scholar]
- Wolff, N.A.; Garrick, M.D.; Zhao, L.; Garrick, L.M.; Ghio, A.J.; Thévenod, F. A role for divalent metal transporter (DMT1) in mitochondrial uptake of iron and manganese. Sci. Rep. 2018, 8, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiseman, H.; Halliwell, B. Damage to DNA by reactive oxygen and nitrogen species: Role in inflammatory disease and progression to cancer. Biochem. J. 1996, 313, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Hider, R.C.; Kong, X.L. Iron speciation in the cytosol: An overview. Dalton Trans. 2013, 42, 3220–3229. [Google Scholar] [CrossRef] [PubMed]
- Philpott, C.C.; Ryu, M.-S. Special delivery: Distributing iron in the cytosol of mammalian cells. Front. Pharmacol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Fenton, H.J.H. LXXIII.-Oxidation of tartaric acid in presence of iron. J. Chem. Soc. Trans. 1894, 65, 899–910. [Google Scholar] [CrossRef]
- Jang, S.; Imlay, J.A. Micromolar intracellular hydrogen peroxide disrupts metabolism by damaging iron-sulfur enzymes. J. Biol. Chem. 2007, 282, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Keyer, K.; Imlay, J.A. Superoxide accelerates DNA damage by elevating free-iron levels. Proc. Natl. Acad. Sci. USA 1996, 93, 13635–13640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, D.R.; Kalinowski, D.S.; Lau, S.; Jansson, P.J.; Lovejoy, D.B. Cancer cell iron metabolism and the development of potent iron chelators as anti-tumour agents. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 702–717. [Google Scholar] [CrossRef] [PubMed]
- Min Pang, B.S.; Connor, J.R. Role of Ferritin in Cancer Biology. J. Cancer Sci. Ther. 2015, 7, 155–160. [Google Scholar]
- Kabat, G.C.; Miller, A.B.; Jain, M.; Rohan, T.E. Dietary iron and haem iron intake and risk of endometrial cancer: A prospective cohort study. Br. J. Cancer. 2007, 98, 194. [Google Scholar] [CrossRef] [PubMed]
- Toyokuni, S. Iron-induced carcinogenesis: The role of redox regulation. Free Radic. Biol. Med. 1996, 20, 553–566. [Google Scholar] [CrossRef]
- Richmond, H.G. Induction of Sarcoma in the Rat by Iron—Dextran Complex. Br. Med. J. 1959, 1, 947. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corcé, V.; Gouin, S.G.; Renaud, S.; Gaboriau, F.; Deniaud, D. Recent advances in cancer treatment by iron chelators. Bioorg. Med. Chem. Lett. 2016, 26, 251–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prutki, M.; Poljak-Blazi, M.; Jakopovic, M.; Tomas, D.; Stipancic, I.; Zarkovic, N. Altered iron metabolism, transferrin receptor 1 and ferritin in patients with colon cancer. Cancer Lett. 2006, 238, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, H.; Fan, D.; Ozawa, S.; Yano, S.; Van Arsdell, M.; Viner, J.L.; Beers, R.; Pastan, I.; Fidler, I.J. Site-specific expression of transferrin receptor by human colon cancer cells directly correlates with eradication by antitransferrin recombinant immunotoxin. Int. J. Oncol. 2000, 17, 643–694. [Google Scholar] [CrossRef] [PubMed]
- Shindelman, J.E.; Ortmeyer, A.E.; Sussman, H.H. Demonstration of the transferrin receptor in human breast cancer tissue. Potential marker for identifying dividing cells. Int. J. Cancer 1981, 27, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Daniels, T.R.; Delgado, T.; Rodriguez, J.A.; Helguera, G.; Penichet, M.L. The transferrin receptor part I: Biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin. Immunol. 2006, 121, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, R.; Delia, D.; Schneider, C.; Newman, R.; Kemshead, J.; Greaves, M. Ubiquitous cell-surface glycoprotein on tumor cells is proliferation-associated receptor for transferrin. Proc. Natl. Acad. Sci. USA 1981, 78, 4515–4519. [Google Scholar] [CrossRef] [PubMed]
- Pinnix, Z.K.; Miller, L.D.; Wang, W.; D’Agostino, R.; Kute, T.; Willingham, M.C.; Hatcher, H.; Tesfay, L.; Sui, G.; Di, X.; et al. Ferroportin and Iron Regulation in Breast Cancer Progression and Prognosis. Sci. Transl. Med. 2010, 2, 43ra56. [Google Scholar] [CrossRef] [PubMed]
- Liotta, L.A.; Kohn, E.C. The microenvironment of the tumour–host interface. Nature 2001, 411, 375. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, D.; Goodison, S.; Nicholson, B.; Tarin, D.; Urquidi, V. Expression of matrix metalloproteinase 8 (MMP-8) and tyrosinase-related protein-1 (TYRP-1) correlates with the absence of metastasis in an isogenic human breast cancer model. Differentiation 2003, 71, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.R. Iron chelators as therapeutic agents for the treatment of cancer. Crit. Rev. Oncol. Hematol. 2002, 42, 267–281. [Google Scholar] [CrossRef]
- Buss, J.L.; Torti, F.M.; Torti, S.V. The role of iron chelation in cancer therapy. Curr. Med. Chem. 2003, 10, 1021–1034. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, D.S.; Richardson, D.R. The Evolution of Iron Chelators for the Treatment of Iron Overload Disease and Cancer. Pharmacol. Rev. 2005, 57, 547–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ninomiya, T.; Ohara, T.; Noma, K.; Katsura, Y.; Katsube, R.; Kashima, H.; Kato, T.; Tomono, Y.; Tazawa, H.; Kagawa, S.; et al. Iron depletion is a novel therapeutic strategy to target cancer stem cells. Oncotarget 2017, 8, 98405–98416. [Google Scholar] [CrossRef] [PubMed]
- Nurchi, V.M.; Crisponi, G.; Lachowicz, J.I.; Medici, S.; Peana, M.; Zoroddu, M.A. Chemical features of in use and in progress chelators for iron overload. J. Trace Elem. Med. Biol. 2016, 38, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Mobarra, N.; Shanaki, M.; Ehteram, H.; Nasiri, H.; Sahmani, M.; Saeidi, M.; Goudarzi, M.; Pourkarim, H.; Azad, M. A Review on Iron Chelators in Treatment of Iron Overload Syndromes. Int. J. Hematol. Oncol. Stem Cell Res. 2016, 10, 239–247. [Google Scholar] [PubMed]
- Neilands, J.B. Siderophores-Structure and function of microbial iron transport compounds. J. Biol. Chem. 1995, 270, 26723–26726. [Google Scholar] [CrossRef] [PubMed]
- Kontoghiorghes, G.J. New chelation therapies and emerging chelating drugs for the treatment of iron overload. Expert Opin. Emerg. Drugs 2006, 11, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Salis, O.; Bedir, A.; Kilinc, V.; Alacam, H.; Gulten, S.; Okuyucu, A. The anticancer effects of desferrioxamine on human breast adenocarcinoma and hepatocellular carcinoma cells. Cancer Biomark. 2014, 14, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Kicic, A.; Chua, A.C.; Baker, E. Desferrithiocin is a more potent antineoplastic agent than desferrioxamine. Br. J. Pharmacol. 2002, 135, 1393–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, T.; Ma, Y.; Kong, X.; Hider, R.C. Design of iron chelators with therapeutic application. Dalton Trans. 2012, 41, 6371–6389. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Yu, Y.; Ji, P.; He, H.; Qiao, C. Antitumor activity of endoperoxide-iron chelator conjugates—Design, synthesis and biological evaluation. Eur. J. Med. Chem. 2015, 102, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Steinhauser, S.; Heinz, U.; Bartholomä, M.; Weyhermüller, T.; Nick, H.; Hegetschweiler, K. Complex formation of ICL670 and related ligands with Fe(III) and Fe(II). Eur. J. Inorg. Chem. 2004, 2004, 4177–4192. [Google Scholar] [CrossRef]
- Ohyashiki, J.H.; Kobayashi, C.; Hamamura, R.; Okabe, S.; Tauchi, T.; Ohyashiki, K. The oral iron chelator deferasirox represses signaling through the mTOR in myeloid leukemia cells by enhancing expression of REDD1. Cancer Sci. 2009, 100, 970–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chantrel-Groussard, K.; Gaboriau, F.; Pasdeloup, N.; Havouis, R.; Nick, H.; Pierre, J.-L.; Brissot, P.; Lescoat, G. The new orally active iron chelator ICL670A exhibits a higher antiproliferative effect in human hepatocyte cultures than O-trensox. Eur. J. Pharmacol. 2006, 541, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.-R.; Lee, J.-W.; Jang, P.-S.; Chung, N.-G.; Cho, B.; Jeong, D.-C. Anti-leukemic properties of deferasirox via apoptosis in murine leukemia cell lines. Blood Res. 2015, 50, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Theerasilp, M.; Chalermpanapun, P.; Ponlamuangdee, K.; Sukvanitvichai, D.; Nasongkla, N. Imidazole-modified deferasirox encapsulated polymeric micelles as pH-responsive iron-chelating nanocarrier for cancer chemotherapy. RSC Adv. 2017, 7, 11158–11169. [Google Scholar] [CrossRef] [Green Version]
- Hrušková, K.; Potůčková, E.; Hergeselová, T.; Liptáková, L.; Hašková, P.; Mingas, P.; Kovaříková, P.; Šimůnek, T.; Vávrová, K. Aroylhydrazone iron chelators: Tuning antioxidant and antiproliferative properties by hydrazide modifications. Eur. J. Med. Chem. 2016, 120, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Abbate, V.; Hider, R.C. Iron-sensitive fluorescent probes: Monitoring intracellular iron pools. Metallomics 2015, 7, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Dean, K.M.; Qin, Y.; Palmer, A.E. Visualizing metal ions in cells: An overview of analytical techniques, approaches, and probes. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Melman, G.; LeTourneau, N.J.; Hays, A.M.; Melman, A. Synthesis and antiproliferating activity of iron chelators of hydroxyamino-1,3,5-triazine family. Bioorg. Med. Chem. Lett. 2010, 20, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Kalinowski, D.S.; Kovacevic, Z.; Siafakas, A.R.; Jansson, P.J.; Stefani, C.; Lovejoy, D.B.; Sharpe, P.C.; Bernhardt, P.V.; Richardson, D.R. Thiosemicarbazones from the old to new: Iron chelators that are more than just ribonucleotide reductase inhibitors. J. Med. Chem. 2009, 52, 5271–5294. [Google Scholar] [CrossRef] [PubMed]
- Mrozek-Wilczkiewicz, A.; Serda, M.; Musiol, R.; Malecki, G.; Szurko, A.; Muchowicz, A.; Golab, J.; Ratuszna, A.; Polanski, J. Iron chelators in photodynamic therapy revisited: Synergistic effect by novel highly active thiosemicarbazones. ACS Med. Chem. Lett. 2014, 5, 336–339. [Google Scholar] [CrossRef] [PubMed]
- Serda, M.; Kalinowski, D.S.; Mrozek-Wilczkiewicz, A.; Musiol, R.; Szurko, A.; Ratuszna, A.; Pantarat, N.; Kovacevic, Z.; Merlot, A.M.; Richardson, D.R.; et al. Synthesis and characterization of quinoline-based thiosemicarbazones and correlation of cellular iron-binding efficacy to anti-tumor efficacy. Bioorg. Med. Chem. Lett. 2012, 22, 5527–5531. [Google Scholar] [CrossRef] [PubMed]
- Finch, R.A.; Liu, M.-C.; Cory, A.H.; Cory, J.G.; Sartorelli, A.C. Triapine (3-aminopyridine-2-carboxaldehyde thiosemicarbazone; 3-AP): An inhibitor of ribonucleotide reductase with antineoplastic activity. Adv. Enzyme Regul. 1999, 39, 3–12. [Google Scholar] [CrossRef]
- Finch, R.A.; Liu, M.-C.; Grill, S.P.; Rose, W.C.; Loomis, R.; Vasquez, K.M.; Cheng, Y.-C.; Sartorelli, A.C. Triapine (3-aminopyridine-2-carboxaldehyde-thiosemicarbazone): A potent inhibitor of ribonucleotide reductase activity with broad spectrum antitumor activity. Biochem. Pharmacol. 2000, 59, 983–991. [Google Scholar] [CrossRef]
- Koppenol, W.; Hider, R. Iron and redox cycling. Do’s and don’ts. Free Radic. Biol. Med. 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Lovejoy, D.B.; Richardson, D.R. Novel di-2-pyridyl-derived iron chelators with marked and selective antitumor activity: In vitro and in vivo assessment. Blood. 2004, 104, 1450–1458. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; Kuo, C.F.; Majd, S. Nanoparticle-based delivery of an anti-proliferative metal chelator to tumor cells. In Proceedings of the 2017 39th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), Seogwipo, Korea, 11–15 July 2017; pp. 309–312. [Google Scholar]
- O’Neill, H.S.; Herron, C.C.; Hastings, C.L.; Deckers, R.; Lopez Noriega, A.; Kelly, H.M.; Hennink, W.E.; McDonnell, C.O.; O’Brien, F.J.; Ruiz-Hernández, E.; et al. A stimuli responsive liposome loaded hydrogel provides flexible on-demand release of therapeutic agents. Acta Biomater. 2017, 48, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Abayaweera, G.S.; Wang, H.; Shrestha, T.B.; Yu, J.; Angle, K.; Thapa, P.; Malalasekera, A.P.; Maurmann, L.; Troyer, D.L.; Bossmann, S.H. Synergy of Iron Chelators and Therapeutic Peptide Sequences Delivered via a Magnetic Nanocarrier. J. Funct. Biomater. 2017, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Agemy, L.; Friedmann-Morvinski, D.; Kotamraju, V.R.; Roth, L.; Sugahara, K.N.; Girard, O.M.; Mattrey, R.F.; Verma, I.M.; Ruoslahti, E. Targeted nanoparticle enhanced proapoptotic peptide as potential therapy for glioblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 17450–17455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croy, S.R.; Kwon, G.S. Polymeric Micelles for Drug Delivery. Curr. Pharm. Des. 2006, 12, 4669–4684. [Google Scholar] [CrossRef] [PubMed]
- Hume, D.A. The mononuclear phagocyte system. Curr. Opin. Immunol. 2006, 18, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Samad, A.; Sultana, Y.; Aqil, M. Liposomal drug delivery systems: An update review. Curr. Drug Deliv. 2007, 4, 297–305. [Google Scholar] [CrossRef] [PubMed]
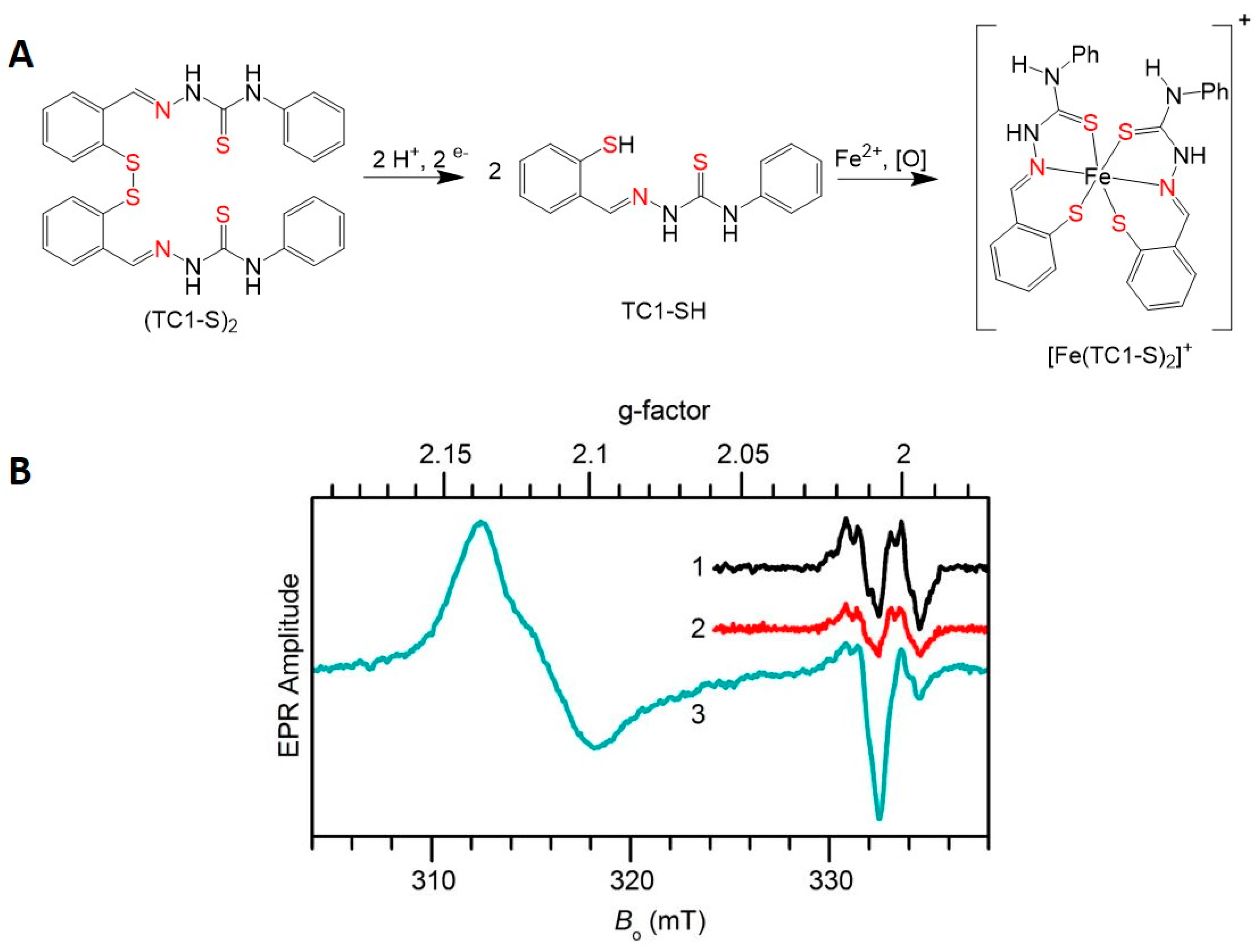
- Chang, T.M.; Tomat, E. Disulfide/thiol switches in thiosemicarbazone ligands for redox-directed iron chelation. Dalton Trans. 2013, 42, 7846–7849. [Google Scholar] [CrossRef] [PubMed]
- Akam, E.A.; Chang, T.M.; Astashkin, A.V.; Tomat, E. Intracellular reduction/activation of a disulfide switch in thiosemicarbazone iron chelators. Metallomics 2014, 6, 1905–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akam, E.A.; Tomat, E. Targeting Iron in Colon Cancer via Glycoconjugation of Thiosemicarbazone Prochelators. Bioconjug. Chem. 2016, 27, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Akam, E.A.; Utterback, R.D.; Marcero, J.R.; Dailey, H.A.; Tomat, E. Disulfide-masked iron prochelators: Effects on cell death, proliferation, and LTA hemoglobin production. J. Inorg. Biochem. 2018, 180, 186–193. [Google Scholar] [CrossRef] [PubMed]
- MacCormack, M.A. Photodynamic therapy. Adv. Dermatol. 2006, 22, 219–258. [Google Scholar] [CrossRef] [PubMed]
- Mrozek-Wilczkiewicz, A.; Malarz, K.; Rams-Baron, M.; Serda, M.; Bauer, D.; Montforts, F.-P.; Ratuszna, A.; Burley, T.; Polanski, J.; Musiol, R. Iron chelators and exogenic photosensitizers. Synergy through oxidative stress gene expression. J. Cancer. 2017, 8, 1979. [Google Scholar] [CrossRef] [PubMed]
- Kovacevic, Z.; Chikhani, S.; Lovejoy, D.B.; Richardson, D.R. Novel thiosemicarbazone iron chelators induce up-regulation and phosphorylation of the metastasis suppressor, NDRG1: A new strategy for the treatment of pancreatic cancer. Mol. Pharmacol. 2011, mol-111. [Google Scholar] [CrossRef]
- Nurtjahja-Tjendraputra, E.; Fu, D.; Phang, J.M.; Richardson, D.R. Iron chelation regulates cyclin D1 expression via the proteasome: A link to iron deficiency–mediated growth suppression. Blood 2007, 109, 4045–4054. [Google Scholar] [CrossRef] [PubMed]
- Kornmann, M.; Ishiwata, T.; Itakura, J.; Tangvoranuntakul, P.; Beger, H.G.; Korc, M. Increased cyclin D1 in human pancreatic cancer is associated with decreased postoperative survival. Oncology 1998, 55, 363–369. [Google Scholar] [CrossRef] [PubMed]
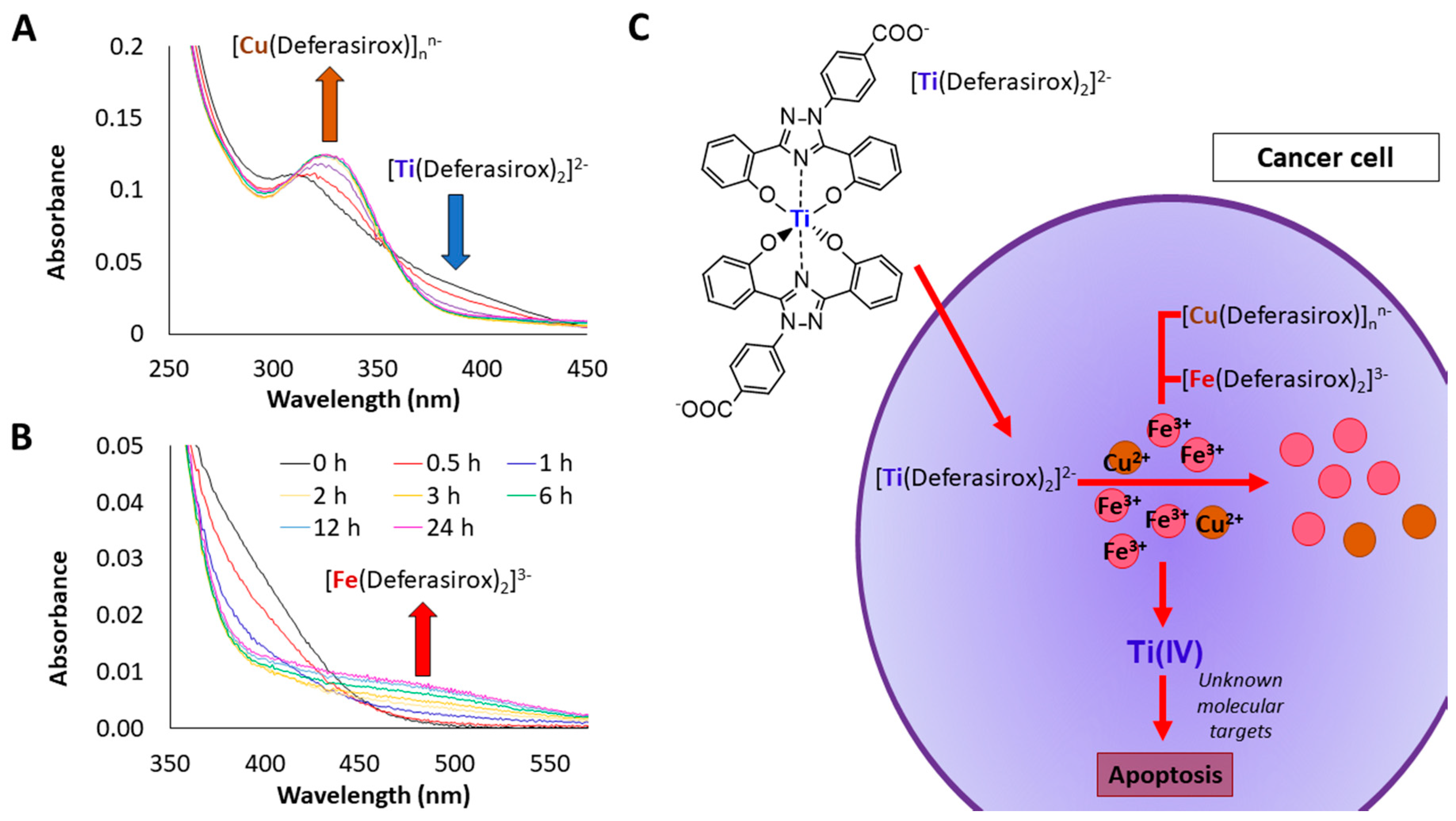
- Parks, T.B.; Cruz, Y.M.; Tinoco, A.D. Applying the Fe(III) Binding Property of a Chemical Transferrin Mimetic to Ti(IV) Anticancer Drug Design. Inorg. Chem. 2014, 53, 1743–1749. [Google Scholar] [CrossRef] [PubMed]
- Loza-Rosas, S.A.; Vázquez-Salgado, A.M.; Rivero, K.I.; Negrón, L.J.; Delgado, Y.; Benjamín-Rivera, J.A.; Vázquez-Maldonado, A.L.; Parks, T.B.; Munet-Colón, C.; Tinoco, A.D. Expanding the therapeutic potential of the iron chelator deferasirox in the development of aqueous stable Ti(IV) anticancer complexes. Inorg. Chem. 2017, 56, 7788–7802. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Garcia, J.D.; Gallegos-Villalobos, A.; Gonzalez-Espinoza, L.; Sanchez-Nino, M.D.; Villarrubia, J.; Ortiz, A. Deferasirox nephrotoxicity—The knowns and unknowns. Nat. Rev. Nephrol. 2014, 10, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Crisponi, G.; Nurchi, V.M.; Crespo-Alonso, M.; Sanna, G.; Zoroddu, M.A.; Alberti, G.; Biesuz, R. A Speciation Study on the Perturbing Effects of Iron Chelators on the Homeostasis of Essential Metal Ions. PLoS ONE 2015, 10, e0133050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.; Luo, Q.; Wu, K.; Li, X.; Wang, F.; Chen, Y.; Ma, X.; Wang, J.; Liu, J.; Xiong, S.; et al. The anticancer drug cisplatin can cross-link the interdomain zinc site on human albumin. Chem. Commun. 2011, 47, 6006–6008. [Google Scholar] [CrossRef] [PubMed]
- Larabee, J.L.; Hocker, J.R.; Hanas, J.S. Mechanisms of Aurothiomalate–Cys2His2 Zinc Finger Interactions. Chem. Res. Toxicol. 2005, 18, 1943–1954. [Google Scholar] [CrossRef] [PubMed]
- Loza-Rosas, S.A.; Saxena, M.; Delgado, Y.; Gaur, K.; Pandrala, M.; Tinoco, A.D. A ubiquitous metal, difficult to track: Towards an understanding of the regulation of titanium(IV) in humans. Metallomics 2017, 9, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; Loza Rosas, S.; Gaur, K.; Sharma, S.; Perez Otero, S.C.; Tinoco, A.D. Exploring titanium(IV) chemical proximity to iron(III) to elucidate a function for Ti(IV) in the human body. Coord. Chem. Rev. 2018, 363, 109–125. [Google Scholar] [CrossRef] [PubMed]
- Tinoco, A.D.; Saxena, M.; Sharma, S.; Noinaj, N.; Delgado, Y.; Gonzalez, E.P.Q.; Conklin, S.E.; Zambrana, N.; Loza-Rosas, S.A.; Parks, T.B. Unusual synergism of transferrin and citrate in the regulation of Ti(IV) speciation, transport, and toxicity. J. Am. Chem. Soc. 2016, 138, 5659–5665. [Google Scholar] [CrossRef] [PubMed]
- Loza Rosas, S.A.; Zayas-Ortiz, A.; Vazquez-Salgado, A.M.; Benjamin-Rivera, J.A.; Gaur, K.; Perez Otero, S.C.; Mendez-Fernandez, A.P.; Vazquez-Maldonado, A.L.; Alicea, N.; Tinoco, A.D. Transmetalation with a titanium(IV) compound transforms biofunctional copper into a cytotoxic agent. 2018; in preparation. [Google Scholar]
- Deda, D.K.; Cardoso, R.M.; Uchiyama, M.K.; Pavani, C.; Toma, S.H.; Baptista, M.S.; Araki, K. A reliable protocol for colorimetric determination of iron oxide nanoparticle uptake by cells. Anal. Bioanal. Chem. 2017, 409, 6663–6675. [Google Scholar] [CrossRef] [PubMed]
- Mounicou, S.; Szpunar, J.; Lobinski, R. Metallomics: The concept and methodology. Chem. Soc. Rev. 2009, 38, 1119–1138. [Google Scholar] [CrossRef] [PubMed]
- Patil, U.; Adireddy, S.; Jaiswal, A.; Mandava, S.; Lee, B.; Chrisey, D. In Vitro/In Vivo Toxicity Evaluation and Quantification of Iron Oxide Nanoparticles. Int. J. Mol. Sci. 2015, 16, 24417–24450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, G.; Carver, P.L. Role of divalent metals in infectious disease susceptibility and outcome. Clin. Microbiol. Infect. 2018, 24, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Nunes, J.A.; Batista, B.L.; Rodrigues, J.L.; Caldas, N.M.; Neto, J.A.G.; Barbosa, F. A Simple Method Based on ICP-MS for Estimation of Background Levels of Arsenic, Cadmium, Copper, Manganese, Nickel, Lead, and Selenium in Blood of the Brazilian Population. J. Toxicol. Environ. Health A 2010, 73, 878–887. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Chen, Z.; Xing, G.; Yuan, H.; Chen, C.; Zhao, F.; Zhang, C.; Zhao, Y. Ultrahigh reactivity provokes nanotoxicity: Explanation of oral toxicity of nano-copper particles. Toxicol. Lett. 2007, 175, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Medel, A.; Montes-Bayón, M.; Luisa Fernández Sánchez, M. Trace element speciation by ICP-MS in large biomolecules and its potential for proteomics. Anal. Bioanal. Chem. 2003, 377, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Herrera, C.; Pettiglio, M.A.; Bartnikas, T.B. Investigating the role of transferrin in the distribution of iron, manganese, copper, and zinc. J. Biol. Inorg. Chem. 2014, 19, 869–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, B.P.; Dwivedi, S.; Dhakad, U.; Murthy, R.C.; Choubey, V.K.; Goel, A.; Sankhwar, S.N. Status and Interrelationship of Zinc, Copper, Iron, Calcium and Selenium in Prostate Cancer. Indian J. Clin. Biochem. 2016, 31, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Matusiak, K.; Skoczen, A.; Setkowicz, Z.; Kubala-Kukus, A.; Stabrawa, I.; Ciarach, M.; Janeczko, K.; Jung, A.; Chwiej, J. The elemental changes occurring in the rat liver after exposure to PEG-coated iron oxide nanoparticles: Total reflection X-ray fluorescence (TXRF) spectroscopy study. Nanotoxicology 2017, 11, 1225–1236. [Google Scholar] [CrossRef] [PubMed]
- Kubala-Kukuś, A.; Banaś, D.; Braziewicz, J.; Majewska, U.; Pajek, M.; Wudarczyk-Moćko, J.; Antczak, G.; Borkowska, B.; Góźdź, S.; Smok-Kalwat, J. Analysis of Copper Concentration in Human Serum by Application of Total Reflection X-ray Fluorescence Method. Biol. Trace Elem. Res. 2014, 158, 22–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riemer, J.; Hoepken, H.H.; Czerwinska, H.; Robinson, S.R.; Dringen, R. Colorimetric ferrozine-based assay for the quantitation of iron in cultured cells. Anal. Biochem. 2004, 331, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Kundra, S.K.; Katyal, M.; Singh, R.P. Spectrophotometric determination of copper(I) and cobalt(II) with ferrozine. Anal. Chem. 1974, 46, 1605–1606. [Google Scholar] [CrossRef]
- Shimberg, G.D.; Pritts, J.D.; Michel, S.L.J. Methods Enzymology; David, S.S., Ed.; Academic Press: Cambridge, MA, USA, 2018; Volume 599, pp. 101–137. [Google Scholar]
- Abou-Shakra, F.R. Handbook of Analytical Separations; Wilson, I.D., Ed.; Elsevier Science B.V.: Amstedam, The Netherlands, 2003; Volume 4, pp. 351–371. [Google Scholar]
- Ammerman, J.; Huang, C.; Sailstad, J.; Wieling, J.; Whitmire, M.L.; Wright, D.; de Lisio, P.; Keenan, F.; McCurdy, E.; Woods, B.; et al. Technical aspects of inductively coupled plasma bioanalysis techniques. Bioanalysis 2013, 5, 1831–1841. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, L.V.; Glass, J.B. Methods Enzymology. Klotz, M.G., Ed.; Academic Press: Cambridge, MA, USA, 2011; Volume 486, pp. 483–506. [Google Scholar]
- Zhang, R.; Li, L.; Sultanbawa, Y.; Xu, Z.P. X-ray fluorescence imaging of metals and metalloids in biological systems. Am. J. Nucl. Med. Mol. Imaging 2018, 8, 169–188. [Google Scholar] [PubMed]
- Antosz, F.J.; Xiang, Y.; Diaz, A.R.; Jensen, A.J. The use of total reflectance X-ray fluorescence (TXRF) for the determination of metals in the pharmaceutical industry. J. Pharm. Biomed. Anal. 2012, 62, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Marcó, L.M.; Greaves, E.D.; Alvarado, J. Analysis of human blood serum and human brain samples by total reflection X-ray fluorescence spectrometry applying Compton peak standardization. Spectrochim. Acta Part B At. Spectrosc. 1999, 54, 1469–1480. [Google Scholar] [CrossRef]
- Wobrauschek, P. Total reflection X-ray fluorescence analysis—A review. X-Ray Spectrom. 2007, 36, 289–300. [Google Scholar] [CrossRef]
- Terzano, R.; Al Chami, Z.; Vekemans, B.; Janssens, K.; Miano, T.; Ruggiero, P. Zinc distribution and speciation within rocket plants (Eruca vesicaria L. Cavalieri) grown on a polluted soil amended with compost as determined by XRF microtomography and Micro-XANES. J. Agric. Food Chem. 2008, 56, 3222–3231. [Google Scholar] [CrossRef] [PubMed]
- Montarges-Pelletier, E.; Chardot, V.; Echevarria, G.; Michot, L.J.; Bauer, A.; Morel, J.L. Identification of nickel chelators in three hyperaccumulating plants: An X-ray spectroscopic study. Phytochemistry 2008, 69, 1695–1709. [Google Scholar] [CrossRef] [PubMed]
- Fahrni, C.J. Biological applications of X-ray fluorescence microscopy: Exploring the subcellular topography and speciation of transition metals. Curr. Opin. Chem. Biol. 2007, 11, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.Y.; Caruso, J.A.; Lai, B.; Matusch, A.; Becker, J.S. Trace metal imaging with high spatial resolution: Applications in biomedicine. Metallomics 2011, 3, 28–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, J.S.; Kumtabtim, U.; Wu, B.; Steinacker, P.; Otto, M.; Matusch, A. Mass spectrometry imaging (MSI) of metals in mouse spinal cord by laser ablation ICP-MS. Metallomics 2012, 4, 284–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köpf-Maier, P. Electron-spectroscopic imaging—A method for analysing the distribution of light elements in mammalian cells and tissues. Acta Histochem. 1991, 91, 25–37. [Google Scholar] [CrossRef]
- Kapp, N.; Studer, D.; Gehr, P.; Geiser, M. Electron Microscopy: Methods and Protocols; Kuo, J., Ed.; Humana Press: Totowa, NJ, USA, 2007; pp. 431–447. [Google Scholar]
- Morello, M.; Canini, A.; Mattioli, P.; Sorge, R.P.; Alimonti, A.; Bocca, B.; Forte, G.; Martorana, A.; Bemardi, G.; Sancesario, G. Sub-cellular localization of manganese in the basal ganglia of normal and manganese-treated rats—An electron spectroscopy imaging and electron energy-loss spectroscopy study. Neurotoxicology 2008, 29, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, N.; Podder, S.; Mitra, K.; Majumdar, S.; Nandi, D.; Chakravarty, A.R. Targeted photodynamic therapy in visible light using BODIPY-appended copper(II) complexes of a vitamin B6 Schiff base. Dalton Trans. 2018, 47, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Que, E.L.; Domaille, D.W.; Chang, C.J. Metals in neurobiology: Probing their chemistry and biology with molecular imaging. Chem. Rev. 2008, 108, 1517–1549. [Google Scholar] [CrossRef] [PubMed]
- Hernando, D.; Levin, Y.S.; Sirlin, C.B.; Reeder, S.B. Quantification of Liver Iron With MRI: State of the Art and Remaining Challenges. J. Magn. Reson. Imaging 2014, 40, 1003–1021. [Google Scholar] [CrossRef] [PubMed]
- Heffern, M.C.; Park, H.M.; Au-Yeung, H.Y.; Van de Bittner, G.C.; Ackerman, C.M.; Stahl, A.; Changa, C.J. In vivo bioluminescence imaging reveals copper deficiency in a murine model of nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2016, 113, 14219–14224. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, T.; Van de Bittner, G.C.; Gray, L.W.; Lutsenko, S.; Chang, C.J. Near-infrared fluorescent sensor for in vivo copper imaging in a murine Wilson disease model. Proc. Natl. Acad. Sci. USA 2012, 109, 2228–2233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aron, A.T.; Heffern, M.C.; Lonergan, Z.R.; Vander Wal, M.N.; Blank, B.R.; Spangler, B.; Zhang, Y.; Park, H.M.; Stahl, A.; Renslo, A.R.; et al. In vivo bioluminescence imaging of labile iron accumulation in a murine model of Acinetobacter baumannii infection. Proc. Natl. Acad. Sci. USA 2017, 114, 12669–12674. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaur, K.; Vázquez-Salgado, A.M.; Duran-Camacho, G.; Dominguez-Martinez, I.; Benjamín-Rivera, J.A.; Fernández-Vega, L.; Carmona Sarabia, L.; Cruz García, A.; Pérez-Deliz, F.; Méndez Román, J.A.; et al. Iron and Copper Intracellular Chelation as an Anticancer Drug Strategy. Inorganics 2018, 6, 126. https://doi.org/10.3390/inorganics6040126
Gaur K, Vázquez-Salgado AM, Duran-Camacho G, Dominguez-Martinez I, Benjamín-Rivera JA, Fernández-Vega L, Carmona Sarabia L, Cruz García A, Pérez-Deliz F, Méndez Román JA, et al. Iron and Copper Intracellular Chelation as an Anticancer Drug Strategy. Inorganics. 2018; 6(4):126. https://doi.org/10.3390/inorganics6040126
Chicago/Turabian StyleGaur, Kavita, Alexandra M. Vázquez-Salgado, Geraldo Duran-Camacho, Irivette Dominguez-Martinez, Josué A. Benjamín-Rivera, Lauren Fernández-Vega, Lesly Carmona Sarabia, Angelys Cruz García, Felipe Pérez-Deliz, José A. Méndez Román, and et al. 2018. "Iron and Copper Intracellular Chelation as an Anticancer Drug Strategy" Inorganics 6, no. 4: 126. https://doi.org/10.3390/inorganics6040126