Excited-State Relaxation in Luminescent Molybdenum(0) Complexes with Isocyanide Chelate Ligands


Department of Chemistry, University of Basel, St. Johanns-Ring 19, 4056 Basel, Switzerland
*
Author to whom correspondence should be addressed.
Inorganics 2020, 8(2), 14; https://doi.org/10.3390/inorganics8020014
Submission received: 20 January 2020
/
Revised: 13 February 2020
/
Accepted: 14 February 2020
/
Published: 17 February 2020
(This article belongs to the Special Issue Photochemistry & Photophysics of Transition Metal Complexes)
Abstract
:Diisocyanide ligands with a m-terphenyl backbone provide access to Mo0 complexes exhibiting the same type of metal-to-ligand charge transfer (MLCT) luminescence as the well-known class of isoelectronic RuII polypyridines. The luminescence quantum yields and lifetimes of the homoleptic tris(diisocyanide) Mo0 complexes depend strongly on whether methyl- or tert-butyl substituents are placed in α-position to the isocyanide groups. The bulkier tert-butyl substituents lead to a molecular structure in which the three individual diisocyanides ligated to one Mo0 center are interlocked more strongly into one another than the ligands with the sterically less demanding methyl substituents. This rigidification limits the distortion of the complex in the emissive excited-state, causing a decrease of the nonradiative relaxation rate by one order of magnitude. Compared to RuII polypyridines, the molecular distortions in the luminescent 3MLCT state relative to the electronic ground state seem to be smaller in the Mo0 complexes, presumably due to delocalization of the MLCT-excited electron over greater portions of the ligands. Temperature-dependent studies indicate that thermally activated nonradiative relaxation via metal-centered excited states is more significant in these homoleptic Mo0 tris(diisocyanide) complexes than in [Ru(2,2′-bipyridine)3]2+.
1. Introduction
Hexacarbonyl complexes of Cr0, Mo0, and W0 are prototypical coordination compounds obeying the 18-electron rule with a low-spin d6 valence electron configuration. Isocyanides (CNR) are formally isoelectronic with CO, and consequently it is unsurprising that hexakis(isocyanide) complexes of the abovementioned d6 metals as well as some heteroleptic complexes comprising both CO and CNR ligands have long been known [1,2,3,4,5,6,7]. The isocyanides are less π-accepting than CO, yet the ligand field remains very strong (even in the Cr0 complexes), and all 6 d-electrons are paired in the t2g-set of d-orbitals, which represent the HOMO in octahedral symmetry. In arylisocyanides, there is some π-conjugation between the C≡N group and the aryl π-system, and consequently antibonding ligand-based orbitals become the LUMO in hexakis(arylisocyanide) complexes of Cr0, Mo0, and W0 [1,2]. The resulting electronic structure with a metal-based HOMO and a ligand-centered LUMO is closely related to that encountered for isoelectronic RuII and OsII polypyridine complexes. Thus, in analogy to this well-known class of precious metal-based complexes, arylisocyanide complexes of Cr0, Mo0, and W0 have energetically low-lying metal-to-ligand charge transfer (MLCT) absorptions. An early investigation already reported luminescence from a 3MLCT state in W0 arylisocyanide complexes [2], and more recent work demonstrated that high luminescence quantum yields and long excited-state lifetimes are achievable by optimizing the ligand design [8,9,10]. Moreover, these W0 complexes with monodentate arylisocyanide ligands are very strong photoreductants, capable, for example, of reducing anthracene to its radical anion form. Many different kinds of metal complexes with isocyanide ligands have been explored over the past few decades [11,12,13,14,15], but metals with the d6 or d10 electron configurations are unique in their ability to show luminescence from a 3MLCT excited state.
Whilst structurally more flexible, multidentate isocyanide chelate ligands had been known for some time [16], we found that chelating diisocyanide ligands based on a m-terphenyl backbone permit the synthesis of homoleptic tris(diisocyanide) complexes of Cr0 and Mo0 that luminesce from 3MLCT excited states (Figure 1) [17]. The molecular and the electronic structures of these compounds are reminiscent of FeII and RuII polypyridine complexes, which have been investigated extensively in the past. Until now, no convincing case of steady-state MLCT luminescence from a FeII complex has been reported despite significant advances in extending their 3MLCT lifetimes in recent years [18,19,20,21,22,23]; hence, our Cr0 complex currently seems to be the only example of a first-row d6-metal complex showing MLCT luminescence in solution at room temperature under steady-state photo-irradiation [24]. The Mo0 diisocyanide complexes are not only emissive, but they can furthermore be employed in photoredox catalysis of thermodynamically challenging reductions, which cannot be performed with more widely known photoreductants such as fac-[Ir(ppy)3] (ppy = 2-phenylpyridine) [25]. Thus, the Mo0 diisocyanide complexes represent Earth-abundant alternatives to precious-metal based luminophores and photoredox catalysts, and in our view, there is interesting fundamental photophysics and photochemistry to be explored in this field [26].
Recently we reported that the [Mo(LtBu)3] complex exhibits much more favorable photophysical properties than the closely related [Mo(LMe)3] compound, and we demonstrated that [Mo(LtBu)3] is more widely applicable in photoredox catalysis due to greater photo-robustness [27]. The present article focuses on the origin of the photophysical differences between these two complexes and attempts to identify possible reasons for the very favorable luminescence behavior of [Mo(LtBu)3] in comparison to [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) and related RuII polypyridines. Herein, we provide the first analysis of relevant 3MLCT excited-state distortions in tris(diisocyanide) Mo0 complexes, and new temperature-dependent luminescence lifetime data give insight into thermally activated nonradiative relaxation via metal-centered excited states.
2. Results and Discussion
The [Mo(LMe)3] and [Mo(LtBu)3] complexes differ only by the substituents in ortho- and para-position to the isocyanide groups, yet their photoluminescence properties are very disparate (Table 1) [27]. Whilst the emissive excited state is of 3MLCT-type in both cases, the luminescence quantum yield (φem) for [Mo(LtBu)3] (in de-aerated solution at 20 °C) is an order of magnitude higher than for [Mo(LMe)3] under identical conditions. Similarly, the 3MLCT lifetime (τ) is roughly a factor of 10 longer for [Mo(LtBu)3] compared to [Mo(LMe)3]. (In a prior study we already noted that the tert-butyl decorated complex exhibits bi-exponential luminescence decays and transient absorption kinetics in all solvents investigated, and this is likely due to conformational equilibria in solution [27]).
The parallel combined trends in luminescence quantum yields and lifetimes indicate that the rate for nonradiative 3MLCT relaxation (knr) decreases by circa a factor of 10 between [Mo(LMe)3] and [Mo(LtBu)3], whereas the radiative relaxation rate (kr) remains similar. Using Equations (1) and (2), this effect can be quantified and the resulting rate constants can be compared to [Ru(dmb)3]2+ (dmb = 4,4′-dimethyl-2,2′-bipyridine) [28], which is an isoelectronic analogue of our molybdenum complexes (Table 2).
τ−1 = kr + knr
The radiative rate constants of the two Mo0 complexes are roughly a factor of 2 larger than for the RuII complex. This is in line with more strongly absorbing MLCT features for [Mo(LMe)3] and [Mo(LtBu)3] (with εmax up to 27,000 M−1 cm−1) compared to [Ru(dmb)3]2+ (εmax ≈ 16,500 M−1 cm−1, εmax is the extinction coefficient at the 1MLCT absorption band maximum) [27]. However, it should be kept in mind that these are 1MLCT absorption bands whereas the kr values are for 3MLCT relaxation. As anticipated above, knr is indeed 10 times smaller for [Mo(LtBu)3] than for [Mo(LMe)3] (last column in Table 2). In principle, a slower rate for nonradiative relaxation in the tert-butyl decorated complex is in line with the energy gap law [29], because this complex emits at somewhat shorter wavelengths (λmax = 585 nm, Table 1) than the methyl-substituted congener (λmax = 607 nm, Table 1). Yet, the factor of 10 difference in knr seems large in relation to the difference in 3MLCT excited-state energies (ca. 600 cm−1 when using the emission band maxima as a proxy).
Mere consideration of energy gaps is a very simplified view, and it is clear that the molecular distortions occurring in an excited state have a big influence on the rates for nonradiative relaxation [29]. The analysis of luminescence band shapes can provide deeper insight into excited-state distortions and nonradiative relaxation [30], and consequently it seemed meaningful to perform such analyses with the emission spectra of [Mo(LMe)3] and [Mo(LtBu)3] (Figure 2); such analyses had not been performed before on our Mo0 complexes. Ideally, vibrational fine structure would directly indicate the relevant distortion modes [31], but the lack thereof is common for MLCT luminescence and does not preclude emission band shape analysis [28].
Suitable models describe the emission spectrum as a superposition of vertical transitions between the electronically excited state and the ground state [32]. The intensity of each transition is determined by the Franck–Condon factor, which quantifies the overlap between the vibrational wave functions of the initial and the final state (Figure 3a). In this picture, the integral over all relevant vibronic transitions then makes up the experimentally observable emission band shape. Whilst several vibrations can in principle couple to an electronic transition [33], it is common to use single configurational coordinate models in which only one (weighted average) vibrational mode is considered, particularly when vibrationally unresolved (MLCT) luminescence spectra (at room temperature) are analyzed. In this limit, the emission intensity I(ν) at a given energy ν is described by Equation (3), where ħ·ωM is the average energy of the vibrational mode that couples to the luminescence transition [28,34].
E0 is the difference between the zero-point energies of the ground and the excited state, whereas SM is the Huang–Rhys parameter describing the extent of molecular distortion occurring between the two respective electronic states. The term Δν1/2 is the homogeneously broadened bandwidth associated with the vibronic transitions. For the simulation of the spectra, the quantum number νM runs over the number of relevant vibrational levels of ħ·ωM, which serve as final states in the electronic ground state.
For the simulations in Figure 2, we summed over νM values from 0 to 3 and adapted E0, SM and ħ·ωM to match the experimentally observed emission spectra as closely as possible. The outcomes are included in Figure 2 as dotted gray traces, and the fitting parameters are summarized in Table 3 along with those reported previously for the [Ru(dmb)3]2+ complex [28]. The experimental emission spectra are somewhat affected by an instrumental artefact at 15,400 cm−1 [24], which renders the simulations on the low energy side of the luminescence bands imperfect. Of key interest in this analysis are the Huang–Rhys parameter (SM) and the (average) vibrational frequency (ħ·ωM). The respective values for the [Ru(dmb)3]2+ complex are quite typical for RuII and OsII polypyridines [28,30,32,34,35]. The vibrational frequency ħ·ωM of ca. 1300 cm−1 for this class of compounds is usually interpreted as a dominance of polypyridine ring stretching modes in defining the relevant excited-state distortion of the emissive 3MLCT state relative to the electronic ground state. Huang–Rhys parameters in the range of 0.6 to 1.2 are very common for RuII and OsII polypyridines [28,30,32,34]. By contrast, our isoelectronic Mo0 complexes yield markedly lower Huang–Rhys parameters combined with a significantly higher average vibrational frequency (Table 3).
Specifically, our simulations provide ħ·ωM values near 1600 cm−1, suggesting that isocyanide C≡N stretch vibrations (ca. 1950 cm−1) [25,27] contribute substantially to the weighted average of all modes that are responsible for the excited-state distortion, presumably along with lower frequency aryl ring stretch modes. This interpretation is compatible with recent computational work on W0 complexes with monodentate arylisocyanide ligands, which demonstrated that distortion along a normal coordinate involving the C≡N stretch is important [10]. Furthermore, previous work discussed the coupling of the C≡N group to the aromatic π-system of arylisocyanides [1,2], and it seems plausible that any distortion along the C≡N coordinate will automatically also affect the aromatic π-system and associated ring stretching vibrations. On this basis, ħ·ωM values near 1600 cm−1 for the Mo0 complexes can be rationalized.
The Huang-Rhys parameters obtained for [Mo(LMe)3] and [Mo(LtBu)3] are considerably lower than that for [Ru(dmb)3]2+ (Table 3) and for many other RuII polypyridines [35]. This is a somewhat surprising finding, which presumably reflects the fundamentally dissimilar molecular structures of the diisocyanide and α-diimine ligands. Thus, in the Mo0 complexes, the 3MLCT excited-state distortion seems considerably weaker than in the RuII and OsII polypyridines, but this distortion occurs along vibrational modes with significantly higher average frequency. The combination of these two opposing effects results in rate constants for nonradiative relaxation (knr) that are within one order of magnitude the same for the Mo0 and [Ru(dmb)3]2+ complexes (last column of Table 2).
The energy gap E0 corresponds to the peak maximum of the first member of the vibrational progression in the ħ·ωM distortion mode [30], and as such does not strictly correspond to the 3MLCT energy (E00) used, for example, for the estimation of excited-state redox potentials from ground-state potentials [36]. However, the two quantities are related to one another by Equation (4), in which kB is Boltzmann’s constant and T is temperature [28].
This relationship yields E00 values (Table 3, third column) that are in line with those determined previously for [Mo(LMe)3] and [Mo(LtBu)3] using other methods (ca. 2.2 eV) [25,27]. Lastly, we note that the Δν1/2 values for our Mo0 diisocyanide complexes are similar to those obtained for the previously investigated RuII and OsII polypyridines [28,30,32,34,35].
Aside from the comparison between the spectral band fitting parameters obtained for the Mo0 and RuII complexes (Table 3), the comparison between the obtained parameter sets for [Mo(LMe)3] and [Mo(LtBu)3] is interesting. The emissive 3MLCT excited state is at slightly higher energy in the complex with tert-butyl-substituted ligands (ca. 300 cm−1) and its Huang–Rhys parameter is 35% lower than that of the complex with methyl-substituted ligands (Table 3). Both of these findings are in line with the higher luminescence quantum yield for [Mo(LtBu)3] compared to [Mo(LMe)3] (Table 1). The lower Huang–Rhys parameter for [Mo(LtBu)3] translates into a smaller distortion of the 3MLCT excited state relative to the 1A1g ground state along the nuclear coordinate Qe (Figure 3a), which in turn leads to weaker overlaps between vibrational functions of the 3MLCT and 1A1g states (grey shaded areas in Figure 3a). This makes nonradiative relaxation less efficient than in [Mo(LMe)3], where the excited-state distortion is stronger. The magnitude of the (equilibrium) distortion, ΔQe, is related to the Huang–Rhys parameter by Equation (5), where M is the reduced mass of the oscillator and ωM is the vibrational frequency.
Since multiple normal coordinates contribute to SM in our Mo0 complexes, and because we are lacking information regarding their relative importance, the ΔQe values for the relevant individual normal coordinates cannot be calculated here. If vibrational fine structure were observable in the emission spectra, this would be possible [33].
The above emission band shape analysis serves to rationalize nonradiative relaxation occurring directly from the emissive 3MLCT manifold to the electronic ground state. However, in RuII polypyridine complexes there is usually additional nonradiative relaxation from the 3MLCT via the 3T1g excited state (Figure 3b) [37,38]. Depending on ligand design [39], this metal-centered state can be energetically very close and nonradiative relaxation becomes very rapid, and this is the reason why [Ru(tpy)2]2+ (tpy = 2,2′:6′,2″-terpyridine) is essentially non-emissive in solution at room temperature [40]. Conversely, when the 3T1g state is located energetically sufficiently above the 3MLCT manifold, high luminescence quantum yields and long excited state lifetimes are achievable [41]. In [Ru(bpy)3]2+ that energy difference amounts to ca. 3600 cm−1 [42], but for emissive Mo0 isocyanide complexes this important aspect has not been explored before.
In order to gain insight into thermally activated 3MLCT relaxation via the 3T1g state, we therefore performed temperature-dependent luminescence lifetime studies (τ(T), open circles in Figure 4). The 3MLCT lifetimes of both [Mo(LMe)3] and [Mo(LtBu)3] decrease by roughly a factor of 4 between 283 and 338 K, in line with a thermally activated nonradiative decay process. The luminescence decays of [Mo(LtBu)3] remain bi-exponential at all temperatures measured (see above), and the τ(T)-values reported in Figure 4 are weighted averages from bi-exponential fits. Equation (6) and the simplified model illustrated by Figure 3b have been used previously to determine the 3MLCT-3T1g energy gap ΔE for [Ru(bpy)3]2+ [42], and the solid lines in Figure 4a,b is fits with the same model to the experimental data for our Mo0 complexes.
τ(T) = [kr + knr,1 + knr,2 · exp(−ΔE/k·T)]−1
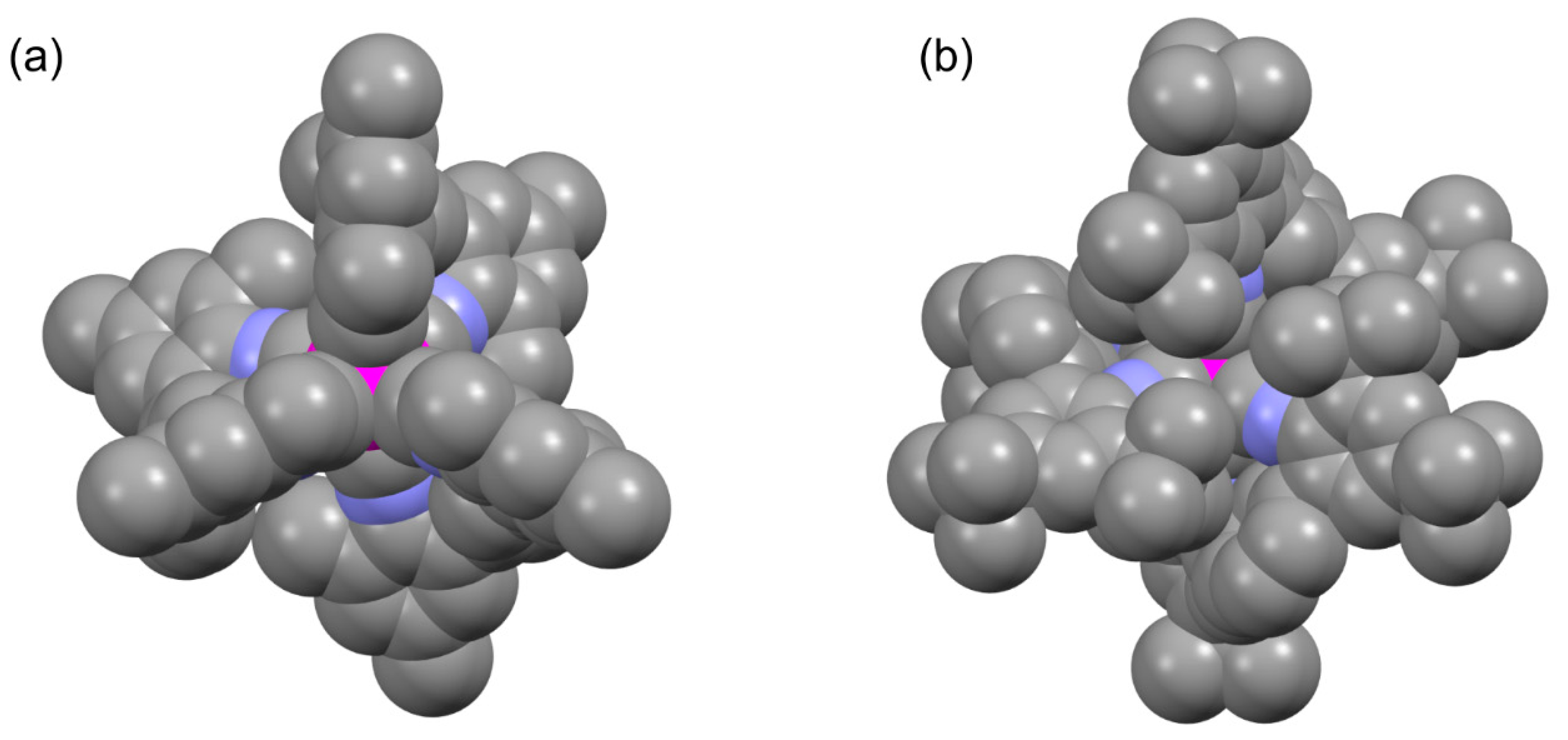
The sum kr + knr,1 (reflecting the total 3MLCT decay rate constant, Figure 3b) was fitted along with knr,2 and ΔE. The results from such 3-parameter fits are listed in Table 4 along with those reported previously for [Ru(bpy)3]2+ [42]. Of key interest is the comparison of ΔE-values. The 3MLCT-3T1g energy gap is largest in [Ru(bpy)3]2+ (3559 cm−1); hence, that compound features the highest barrier for thermally activated nonradiative relaxation via a metal-centered excited state. For the more strongly emissive [Mo(LtBu)3] complex ΔE is ca. 19% larger than for [Mo(LMe)3] (2934 vs. 2472 cm−1, last column in Table 4). Thus, the Mo0 complex with the tert-butylated ligand features less efficient nonradiative relaxation than the Mo0 complex with the methylated ligand, both via direct 3MLCT relaxation to the electronic ground state (Figure 3a) and via thermal activation of metal-centered excited states (Figure 3b). It seems plausible that the greater overall rigidity of the [Mo(LtBu)3] complex compared to [Mo(LMe)3] is responsible for that. An X-ray crystal structure of [Mo(LtBu)3] is not available, but when considering the X-ray structure of the analogous Cr0 compound in Figure 5b, it seems evident that the bulkier tert-butyl-substituents lead to a mutually more interlocked ligand framework than in the case of [Mo(LMe)3] (Figure 5a).
3. Materials and Methods
The [Mo(LMe)3] and [Mo(LtBu)3] complexes were available from two recent studies and were stored under an Argon atmosphere at 4 °C [25,27]. Samples of both complexes (25 µM in dry toluene) were degassed by three freeze-pump-thaw cycles prior to measurements. The new temperature-dependent luminescence lifetime data were obtained using an LP920-KS spectrometer from Edinburgh Instruments, employing a Nd:YAG laser (Quantel Brilliant b) with an OPO (Opotek) as excitation source. The excitation wavelength was 500 nm with a typical pulse energy of 7 mJ. Single-wavelength kinetics were recorded using a photomultiplier tube. Spectral band shape analysis occurred with the Igor Pro software (version 6.3.7.2). The method by Parker and Rees was applied when converting the emission spectra from wavelength to wavenumbers [43].
4. Conclusions
The new analyses and additional temperature-dependent lifetime data reported herein are useful to understand why [Mo(LtBu)3] exhibits much more favorable photophysical properties than [Mo(LMe)3]. Furthermore, the direct comparison between these tris(diisocyanide)molybdenum(0) complexes and the isoelectronic and structurally related tris(α-diimine)ruthenium(II) compounds made herein is insightful.
In both compound classes the 3MLCT relaxation is coupled to ring stretch vibrations (ca. 1300 cm−1) of the ligand backbone, but in the Mo0 diisocyanides there seems to be additional coupling to C≡N vibrations, manifesting in a higher average frequency (1600–1650 cm−1) of all modes responsible for excited-state distortion. The disadvantage of coupling to a higher frequency mode in the Mo0 complexes seems to be compensated by significantly smaller Huang–Rhys factors compared to RuII polypyridines. Thus, the combination of weaker distortion along higher frequency modes is likely responsible for the finding that the Mo0 diisocyanide complexes have similarly favorable luminescence properties as the [Ru(bpy)3]2+ parent compound. In future work, it will be desirable to complement these findings by time-dependent density functional theory calculations to get clearer insight into the relevant excited-state distortions.
The barrier for thermal 3MLCT deactivation via metal-centered excited states is 18–30% smaller in the two investigated Mo0 complexes than in [Ru(bpy)3]2+. For [Mo(LMe)3] that barrier is 19% lower than for [Mo(LtBu)3], and the spectral band shape analysis points to a significantly greater 3MLCT excited-state distortion in [Mo(LMe)3] relative to [Mo(LtBu)3]. These two new findings can explain why nonradiative relaxation is roughly 10 times slower in the tert-butylated than in the methylated complex. Differences in the rigidity of the molecular structures of these two complexes provide a plausible rationale for this behavior. The sterically more demanding tert-butyl substituents lead to a molecular structure of [Mo(LtBu)3], in which the three individual ligands are considerably more interlocked into one another than in [Mo(LMe)3] where only methyl-groups are present. The use of sterically demanding substituents at the ligand periphery that lead to a compact molecular structure could represent a more generally valid design principle for minimizing undesirable excited state distortions, particularly in photoactive complexes of Earth-abundant metals [44,45].
Author Contributions
P.H. performed research, analyzed data, and wrote the paper, O.S.W. conceived research, analyzed data, and wrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Swiss National Science Foundation through grant number 200021_178760.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Mann, K.R.; Cimolino, M.; Geoffroy, G.L.; Hammond, G.S.; Orio, A.A.; Albertin, G.; Gray, H.B. Electronic Structures and Spectra of Hexakisphenylisocyanide Complexes of Cr0, Mo0, W0, MnI, and MnII. Inorg. Chim. Acta 1976, 16, 97–101. [Google Scholar] [CrossRef]
- Mann, K.R.; Gray, H.B.; Hammond, G.S. Excited-State Reactivity Patterns of Hexakisarylisocyano Complexes of Chromium(0), Molybdenum(0), and Tungsten(0). J. Am. Chem. Soc. 1977, 99, 306–307. [Google Scholar] [CrossRef]
- King, R.B.; Saran, M.S. Isocyanide-Metal Complexes. II. CO and CN Stretching Modes in tert-Butyl Isocyanide Derivatives of Octahedral Metal-Carbonyls. Inorg. Chem. 1974, 13, 74–78. [Google Scholar] [CrossRef]
- Coville, N.J.; Albers, M.O. A Synthetic Route to M(CO)6−n(RNC)n (M = Cr, Mo, W; n = 1–6) from M(CO)6 and Isonitriles. Inorg. Chim. Acta 1982, 65, L7–L8. [Google Scholar] [CrossRef]
- Lyons, L.J.; Pitz, S.L.; Boyd, D.C. Electrochemical and IR Spectroelectrochemical Investigations of the Series Mo(CO)6−n(CNR)n (n = 1–6) (R = 2,8-Dimethylphenyl): In-situ Observation of fac-mer and cis-trans Isomerizations. Inorg. Chem. 1995, 34, 316–322. [Google Scholar] [CrossRef]
- Angelici, R.J.; Quick, M.H.; Kraus, G.A.; Plummer, D.T. Synthesis of Chelating Bidentate Isocyano and Cyano Ligands and Their Metal Complexes. Inorg. Chem. 1982, 21, 2178–2184. [Google Scholar] [CrossRef] [Green Version]
- Treichel, P.M.; Firsich, D.W.; Essenmacher, G.P. Manganese(I) and Chromium(0) Complexes of Phenyl Isocyanide. Inorg. Chem. 1979, 18, 2405–2409. [Google Scholar] [CrossRef]
- Sattler, W.; Henling, L.M.; Winkler, J.R.; Gray, H.B. Bespoke Photoreductants: Tungsten Arylisocyanides. J. Am. Chem. Soc. 2015, 137, 1198–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sattler, W.; Ener, M.E.; Blakemore, J.D.; Rachford, A.A.; LaBeaume, P.J.; Thackeray, J.W.; Cameron, J.F.; Winkler, J.R.; Gray, H.B. Generation of Powerful Tungsten Reductants by Visible Light Excitation. J. Am. Chem. Soc. 2013, 135, 10614–10617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvapilova, H.; Sattler, W.; Sattler, A.; Sazanovich, I.V.; Clark, I.P.; Towrie, M.; Gray, H.B.; Zalis, S.; Vlcek, A. Electronic Excited States of Tungsten(0) Arylisocyanides. Inorg. Chem. 2015, 54, 8518–8528. [Google Scholar] [CrossRef] [PubMed]
- Margulieux, G.W.; Weidemann, N.; Lacy, D.C.; Moore, C.E.; Rheingold, A.L.; Figueroa, J.S. Isocyano Analogues of [Co(CO)]4n: A Tetraisocyanide of Cobalt Isolated in Three States of Charge. J. Am. Chem. Soc. 2010, 132, 5033–5035. [Google Scholar] [CrossRef] [PubMed]
- Monti, F.; Baschieri, A.; Matteucci, E.; Mazzanti, A.; Sambri, L.; Barbieri, A.; Armaroli, N. A Chelating Diisocyanide Ligand for Cyclometalated Ir(III) Complexes with Strong and Tunable Luminescence. Faraday Discuss. 2015, 185, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Knorn, M.; Rawner, T.; Czerwieniec, R.; Reiser, O. Copper(phenanthroline)(bisisonitrile)+-Complexes for the Visible-Light-Mediated Atom Transfer Radical Addition and Allylation Reactions. ACS Catal. 2015, 5, 5186–5193. [Google Scholar] [CrossRef]
- Drance, M.J.; Sears, J.D.; Mrse, A.M.; Moore, C.E.; Rheingold, A.L.; Neidig, M.L.; Figueroa, J.S. Terminal Coordination of Diatomic Boron Monofluoride to Iron. Science 2019, 363, 1203–1205. [Google Scholar] [CrossRef] [PubMed]
- Cañada, L.M.; Kölling, J.; Teets, T.S. Blue-Phosphorescent Bis-Cyclometalated Iridium Complexes with Arylisocyanide Ancillary Ligands. Polyhedron 2020, 178, 114332. [Google Scholar] [CrossRef]
- Hahn, F.E. The Coordination Chemistry of Multidentate Isocyanide Ligands. Angew. Chem. Int. Ed. 1993, 32, 650–665. [Google Scholar] [CrossRef]
- Büldt, L.A.; Wenger, O.S. Chromium(0), Molybdenum(0), and Tungsten(0) Isocyanide Complexes as Luminophores and Photosensitizers with Long-Lived Excited States. Angew. Chem. Int. Ed. 2017, 56, 5676–5682. [Google Scholar] [CrossRef] [Green Version]
- Braun, J.D.; Lozada, I.B.; Kolodziej, C.; Burda, C.; Newman, K.M.E.; van Lierop, J.; Davis, R.L.; Herbert, D.E. Iron(II) Coordination Complexes with Panchromatic Absorption and Nanosecond Charge-Transfer Excited State Lifetimes. Nat. Chem. 2019, 11, 1144–1150. [Google Scholar] [CrossRef]
- Chábera, P.; Kjaer, K.S.; Prakash, O.; Honarfar, A.; Liu, Y.Z.; Fredin, L.A.; Harlang, T.C.B.; Lidin, S.; Uhlig, J.; Sundström, V.; et al. Fe-II Hexa N-Heterocyclic Carbene Complex with a 528 ps Metal-to-Ligand Charge-Transfer Excited-State Lifetime. J. Phys. Chem. Lett. 2018, 9, 459–463. [Google Scholar] [CrossRef]
- Steube, J.; Burkhardt, L.; Päpcke, A.; Moll, J.; Zimmer, P.; Schoch, R.; Wölper, C.; Heinze, K.; Lochbrunner, S.; Bauer, M. Excited-State Kinetics of an Air-Stable Cyclometalated Iron(II) Complex. Chem. Eur. J. 2019, 25, 11826–11830. [Google Scholar] [CrossRef]
- Duchanois, T.; Liu, L.; Pastore, M.; Monari, A.; Cebrian, C.; Trolez, Y.; Darari, M.; Magra, K.; Frances-Monerris, A.; Domenichini, E.; et al. NHC-Based Iron Sensitizers for DSSCs. Inorganics 2018, 6, 63. [Google Scholar] [CrossRef] [Green Version]
- Carey, M.C.; Adelman, S.L.; McCusker, J.K. Insights into the Excited State Dynamics of Fe(II) Polypyridyl Complexes from Variable-Temperature Ultrafast Spectroscopy. Chem. Sci. 2019, 10, 134–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenger, O.S. Is Iron the New Ruthenium? Chem. Eur. J. 2019, 25, 6043–6052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Büldt, L.A.; Guo, X.; Vogel, R.; Prescimone, A.; Wenger, O.S. A Tris(diisocyanide)chromium(0) Complex Is a Luminescent Analog of Fe(2,2’-Bipyridine)32+. J. Am. Chem. Soc. 2017, 139, 985–992. [Google Scholar] [CrossRef] [Green Version]
- Büldt, L.A.; Guo, X.; Prescimone, A.; Wenger, O.S. A Molybdenum(0) Isocyanide Analogue of Ru(2,2’-Bipyridine)32+: A Strong Reductant for Photoredox Catalysis. Angew. Chem. Int. Ed. 2016, 55, 11247–11250. [Google Scholar] [CrossRef]
- Büldt, L.A.; Wenger, O.S. Luminescent Complexes Made from Chelating Isocyanide Ligands and Earth-Abundant Metals. Dalton Trans. 2017, 46, 15175–15177. [Google Scholar] [CrossRef] [Green Version]
- Herr, P.; Glaser, F.; Büldt, L.A.; Larsen, C.B.; Wenger, O.S. Long-Lived, Strongly Emissive, and Highly Reducing Excited States in Mo(0) Complexes with Chelating Isocyanides. J. Am. Chem. Soc. 2019, 141, 14394–14402. [Google Scholar] [CrossRef]
- Damrauer, N.H.; Boussie, T.R.; Devenney, M.; McCusker, J.K. Effects of Intraligand Electron Delocalization, Steric Tuning, and Excited-State Vibronic Coupling on the Photophysics of Aryl-Substituted Bipyridyl Complexes of Ru(II). J. Am. Chem. Soc. 1997, 119, 8253–8268. [Google Scholar] [CrossRef]
- Caspar, J.V.; Meyer, T.J. Application of the Energy-Gap Law to Nonradiative Excited-State Decay. J. Phys. Chem. 1983, 87, 952–957. [Google Scholar] [CrossRef]
- Kober, E.M.; Caspar, J.V.; Lumpkin, R.S.; Meyer, T.J. Application of the Energy-Gap Law to Excited-State Decay of Osmium(II) Polypyridine Complexes: Calculation of Relative Nonradiative Decay-Rates from Emission Spectral Profiles. J. Phys. Chem. 1986, 90, 3722–3734. [Google Scholar] [CrossRef]
- Hauser, A.; Reber, C. Spectroscopy and Chemical Bonding in Transition Metal Complexes. Struct. Bond. 2016, 172, 291–312. [Google Scholar]
- Claude, J.P.; Meyer, T.J. Temperature-Dependence of Nonradiative Decay. J. Phys. Chem. 1995, 99, 51–54. [Google Scholar] [CrossRef]
- Wenger, O.S.; Güdel, H.U. Optical Spectroscopy of CrCl63− Doped Cs2NaScCl6: Broadband Near-Infrared Luminescence and Jahn-Teller Effect. J. Chem. Phys. 2001, 114, 5832–5841. [Google Scholar] [CrossRef]
- Treadway, J.A.; Strouse, G.F.; Ruminski, R.R.; Meyer, T.J. Long-Lived Near-Infrared MLCT Emitters. Inorg. Chem. 2001, 40, 4508–4509. [Google Scholar] [CrossRef]
- Ashford, D.L.; Glasson, C.R.K.; Norris, M.R.; Concepcion, J.J.; Keinan, S.; Brennaman, M.K.; Templeton, J.L.; Meyer, T.J. Controlling Ground and Excited State Properties through Ligand Changes in Ruthenium Polypyridyl Complexes. Inorg. Chem. 2014, 53, 5637–5646. [Google Scholar] [CrossRef]
- Roundhill, D.M. Photochemistry and Photophysics of Metal Complexes; Plenum Press: New York, NY, USA, 1994. [Google Scholar]
- Medlycott, E.A.; Hanan, G.S. Designing Tridentate Ligands for Ruthenium(II) Complexes with Prolonged Room Temperature Luminescence Lifetimes. Chem. Soc. Rev. 2005, 34, 133–142. [Google Scholar] [CrossRef]
- Dixon, I.M.; Heully, J.L.; Alary, F.; Elliott, P.I.P. Theoretical Illumination of Highly Original Photoreactive 3MC States and the Mechanism of the Photochemistry of Ru(II) Tris(Bidentate) Complexes. Phys. Chem. Chem. Phys. 2017, 19, 27765–27778. [Google Scholar] [CrossRef]
- Sun, Q.C.; Dereka, B.; Vauthey, E.; Daku, L.M.L.; Hauser, A. Ultrafast Transient IR Spectroscopy and DFT Calculations of Ruthenium(II) Polypyridyl Complexes. Chem. Sci. 2017, 8, 223–230. [Google Scholar] [CrossRef] [Green Version]
- Ponce, A.; Gray, H.B.; Winkler, J.R. Electron Tunneling Through Water: Oxidative Quenching of Electronically Excited Ru(tpy)22+ (tpy = 2,2′:6,2″-terpyridine) by Ferric Ions in Aqueous Glasses at 77 K. J. Am. Chem. Soc. 2000, 122, 8187–8191. [Google Scholar] [CrossRef] [Green Version]
- Abrahamsson, M.; Jäger, M.; Osterman, T.; Eriksson, L.; Persson, P.; Becker, H.C.; Johansson, O.; Hammarström, L. A 3.0 ms Room Temperature Excited State Lifetime of a Bistridentate RuII-polypyridine Complex for Rod-Like Molecular Arrays. J. Am. Chem. Soc. 2006, 128, 12616–12617. [Google Scholar] [CrossRef]
- Van Houten, J.; Watts, R.J. Temperature-Dependence of Photophysical and Photochemical Properties of Tris(2,2’-bipyridyl)ruthenium(II) Ion in Aqueous-Solution. J. Am. Chem. Soc. 1976, 98, 4853–4858. [Google Scholar] [CrossRef]
- Parker, C.A.; Rees, W.T. Correction of Fluorescence Spectra and Measurement of Fluorescence Quantum Efficiency. Analyst 1960, 85, 587–600. [Google Scholar] [CrossRef]
- Wenger, O.S. Photoactive Complexes with Earth-Abundant Metals. J. Am. Chem. Soc. 2018, 140, 13522–13533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Förster, C.; Heinze, K. Photophysics and Photochemistry with Earth-abundant Metals—Fundamentals and Concepts. Chem. Soc. Rev. 2020. [Google Scholar] [CrossRef]
Figure 1.
Molecular structures of Cr0 and Mo0 complexes with diisocyanide chelate ligands [17,24,25,27].

Figure 2.
Luminescence spectra of [Mo(LMe)3] and [Mo(LtBu)3] in n-hexane at 20 °C following excitation at 500 nm (solid black trace) [25,27], along with simulated emission spectra according to Equation (3) (dotted gray traces).

Figure 3.
(a) Vibrational overlap (gray shaded areas) leading to nonradiative excited-state decay. (b) Nonradiative deactivation of the 3MLCT excited state via thermal population of the 3T1g (metal-centered) excited state.
Figure 3.
(a) Vibrational overlap (gray shaded areas) leading to nonradiative excited-state decay. (b) Nonradiative deactivation of the 3MLCT excited state via thermal population of the 3T1g (metal-centered) excited state.

Figure 4.
Temperature-dependent luminescence lifetimes (circles) and fits with Equation (6) (solid lines) for (a) [Mo(LMe)3] and (b) [Mo(LtBu)3] in toluene. See text for further details.
Figure 4.
Temperature-dependent luminescence lifetimes (circles) and fits with Equation (6) (solid lines) for (a) [Mo(LMe)3] and (b) [Mo(LtBu)3] in toluene. See text for further details.
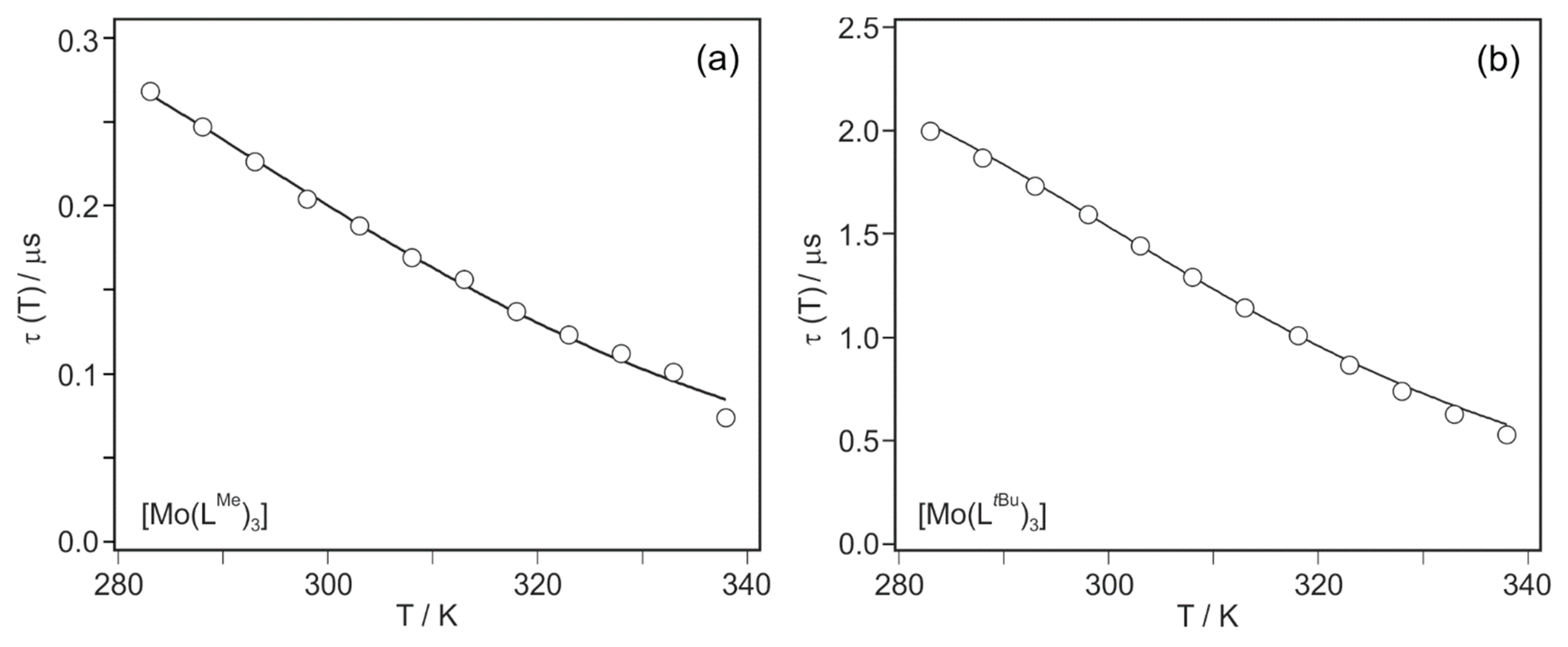
Figure 5.
Space-filling representations of X-ray crystal structures of (a) [Mo(LMe)3] and (b) [Cr(LtBu)3] [24,25]. An X-ray structure of [Mo(LtBu)3] is not available; hence, the Cr0 structure in (b) is used for comparison with [Mo(LMe)3].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Emission band maxima (λmax), luminescence quantum yields (φem), and 3MLCT lifetimes (τ) in de-aerated toluene at 20 °C [27].
Table 1.
Emission band maxima (λmax), luminescence quantum yields (φem), and 3MLCT lifetimes (τ) in de-aerated toluene at 20 °C [27].
| Compound | λmax/nm | φem | τ/ns |
|---|---|---|---|
| [Mo(LMe)3] | 607 | 0.023 | 166 |
| [Mo(LtBu)3] | 585 | 0.203 | 1110 (85%)/2330 (15%) 1 |
1 Bi-exponential decays are observed in all investigated solvents; see text for details.
Table 2.
Rate constants for radiative (kr) and nonradiative (knr) 3MLCT excited-state relaxation in solution at room temperature.
Table 2.
Rate constants for radiative (kr) and nonradiative (knr) 3MLCT excited-state relaxation in solution at room temperature.
| Compound | kr/105 s−1 | knr/105 s−1 |
|---|---|---|
| [Mo(LMe)3] 1 | 1.39 | 58.9 |
| [Mo(LtBu)3] 1,2 | 1.78 | 5.95 |
| [Ru(dmb)3]2+ 3,4 | 0.83 | 10.6 |
Table 3.
Emission spectral fitting parameters.
| Compound | E0/cm−1 | E00/cm−1 | ħ·ωM/cm−1 | SM | Δν1/2/cm−1 |
|---|---|---|---|---|---|
| [Mo(LMe)3] 1 | 16,700 | 18,100 | 1650 | 0.23 | 1800 |
| [Mo(LtBu)3] 1 | 17,150 | 18,400 | 1600 | 0.15 | 1700 |
| [Ru(dmb)3]2+ 2,3 | 15,980 | 17,310 | 1330 | 1.05 | 1750 |
1 In toluene. 2 In acetonitrile. 3 From [28].
Table 4.
Results from fits with Equation (6) to the temperature-dependent luminescence lifetime data in Figure 4.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Herr, P.; Wenger, O.S. Excited-State Relaxation in Luminescent Molybdenum(0) Complexes with Isocyanide Chelate Ligands. Inorganics 2020, 8, 14. https://doi.org/10.3390/inorganics8020014
AMA Style
Herr P, Wenger OS. Excited-State Relaxation in Luminescent Molybdenum(0) Complexes with Isocyanide Chelate Ligands. Inorganics. 2020; 8(2):14. https://doi.org/10.3390/inorganics8020014
Chicago/Turabian StyleHerr, Patrick, and Oliver S. Wenger. 2020. "Excited-State Relaxation in Luminescent Molybdenum(0) Complexes with Isocyanide Chelate Ligands" Inorganics 8, no. 2: 14. https://doi.org/10.3390/inorganics8020014
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.